

Abstract
Background
Th2 cytokines are not responsible for the on-going symptoms and pathology in children with severe therapy resistant asthma (STRA). Interleukin (IL)-33 induces airway hyperresponsiveness (AHR), but its role in airway remodelling and steroid resistance is unknown.
Objective
To investigate the relationship between IL-33 and airway remodelling in paediatric STRA.
Methods
IL-33 was quantified in neonatal mice given inhaled house dust mite (HDM), and the effect of blocking IL-13 on remodelling and IL-33 was assessed. HDM induced allergic airways disease (AAD) in neonatal ST2−/− mice lacking the IL-33 receptor was assessed, together with collagen production following IL-33 administration. Impact of steroid therapy on IL-33 levels in neonatal AAD was explored. IL-33 expression was quantified in endobronchial biopsies from children with STRA and related to remodelling, and collagen production by paediatric airway fibroblasts stimulated with IL-33 and budesonide quantified.
Results
Blocking IL-13 after AAD was established in neonatal mice did not reduce remodelling or IL-33 levels; AHR was only partially reduced. IL-33 promoted collagen synthesis both from paediatric asthmatic fibroblasts, and following intra-nasal administration in mice. Increased cellular expression of IL-33, but not IL-13, was associated with increased reticular basement membrane thickness in endobronchial biopsies from children with STRA, whilst remodelling was absent in HDM exposed ST2−/− mice. IL-33 was maintained whilst IL-13 was abrogated by steroid treatment in neonatal HDM exposed mice, and in endobronchial biopsies from children with STRA.
Conclusion
IL-33 is a relatively steroid resistant mediator that promotes airway remodelling in STRA, and is an important therapeutic target.
Keywords: Asthma, paediatric, airway remodelling, steroid resistance, IL-33, therapy
INTRODUCTION
Children with severe therapy resistant asthma (STRA) represent a small number of all asthmatics, yet they use a disproportionate amount of healthcare resources(1) and present a significant clinical challenge as they remain symptomatic despite maximal doses of conventional therapy.
The key pathophysiological abnormalities of asthma include airway hyperresponsiveness (AHR), inflammation and tissue remodelling, including increased thickness of the reticular basement membrane (RBM)(2), and increased smooth muscle mass(3). We have shown children with STRA have on-going symptoms, abnormal lung function and evidence of airway remodelling in the absence of the classical Th2 cytokines (interleukin (IL)-4, IL-5 and IL-13) that are thought to mediate allergic disease in adults(4). However, the high dose maintenance steroid therapy that is prescribed in severe disease may attenuate these relatively steroid sensitive cytokines(5-7). Critically, even in adults, severe asthma is recognised as being heterogeneous, with the Th2 pathway being active in only select subgroups, as demonstrated by the variable efficacy of anti-IL-13 antibody therapy(8). A significant limitation of current asthma therapies is their lack of effect on any features of airway remodelling, even though these structural changes contribute to disease chronicity. Specific steroid resistant mediators driving airway remodelling in STRA therefore need elucidation.
The importance of innate immune responses in asthma pathogenesis has become increasingly apparent(9;10), with particular emphasis on the innate cytokine IL-33(11). IL-33 is expressed in the epithelium and smooth muscle of adults with severe asthma(12;13). It is also sufficient to initiate allergic airway responses in adult mice lacking T and B cells via the induction of IL-13 producing innate helper cells(14;15). At present, little is known about the role of IL-33 in the pathophysiology of childhood asthma. Specifically, the contribution of IL-33 to airway remodelling and steroid resistance has not been investigated, although both are critical features of severe disease. Since the Th2 cytokines were not predominant in our patients with STRA(4), we hypothesised that IL-33 induces airway remodelling and is a steroid resistant mediator in paediatric STRA. We have investigated this using a neonatal mouse model of house dust mite (HDM) induced allergic airways disease (AAD), and endobronchial biopsies (EB) and cultured airway fibroblasts from children with STRA. We demonstrate for the first time the relationship between IL-33 and airway remodelling, and identify IL-33 as a therapeutic target for severe steroid resistant asthma.
METHODS
Animals and reagents
Female Balb/c mice and litters were maintained by in-house breeding in specific pathoge–free conditions and given food and water ad libitum (see online repository (OR)). Mice were matched for age and background strain. All procedures were conducted in accordance with the Animals (Scientific procedures) Act 1986. Recombinant mouse IL-33 (50mcg/kg/mouse) for intra-nasal administration was purchased from eBioscience (UK). Anti-IL-13 antibody was a gift from UCB® Celltech, UK, 10mg/kg was administered intra-peritoneally twice weekly. Relevant isotype-matched antibodies were used as controls. Budesonide Respules® (0.25mg/ml) were used for intra-nasal administration (0.6mg/kg).
Allergen challenge
From day 3 of life, pups received either intra-nasal house dust mite (HDM) extract (Greer, Lenior, USA) or PBS, three times a week, according to our published protocol(16) (see OR).
Measurement of airway hyperresponsiveness
Airways resistance was measured from 2 weeks of age using the flexivent small animal ventilator (Scireq) using our established protocols(16). See OR for details.
Inflammation and cell recovery
Bronchoalveolar lavage (BAL) was performed with PBS via a tracheal cannula as previously described(16). Lavage fluid was centrifuged and cell pellet re-suspended in 0.5ml complete media. After lavage, 2 lobes from the right lung were dissected and processed as previously published to obtain lung cells(16). The total cell yield of BAL and lung cells was quantified by hemocytometer (Immune Systems) (see OR).
Quantification of cytokines
Lung tissue was homogenized and the supernatant was collected (see OR). Cytokines were analyzed in lung homogenate supernatants. Paired antibodies for mouse interleukin (IL)-4 and IL-5 (PharMingen) and IL-13 and IL-25 (eBioscience) and IL-33 (R&D Systems) were used in standardized sandwich ELISAs according to the manufacturer’s protocol.
Airway remodelling
Lung sections stained with sirius red and Gordon and Sweet’s silver stain as previously described(16) were used to quantify peri-bronchiolar collagen and reticulin respectively using computer aided image analysis (Leica QWin v3). (See OR)
RNA extraction and real-time PCR
Total RNA was extracted from 50–100 mg lung tissue using the Qiagen RNeasy Mini Kit. cDNA was synthesized from 1 μg of total RNA and analyzed by PCR on a CFX96 Real-Time PCR detection system (Biorad). Reactions were run using Taqman probes (Applied Biosystems) and SsoFast Probe Supermix (Biorad) (see OR).
Human data
Subjects
School-aged children (6 to 16 years) with STRA(4) that were undergoing a clinically indicated bronchoscopy, were recruited. Their clinical details have been published(4). They were prescribed ≥ 800mcg/day budesonide (or equivalent). Children with mild/moderate asthma (inhaled corticosteroids <800mcg/day) and age matched, non-asthmatic control subjects having a bronchoscopy for another indication were also recruited. The study was approved by the institutional ethics committee, and written, informed parental consent and child assent was obtained. (See OR for clinical details of all subjects).
Histopathology
Endobronchial biopsies (EB) were processed to paraffin. 5μm sections were stained with haematoxylin and eosin and used to assess morphology and quantify RBM thickness(4) and smooth muscle mass(3). Sections were stained by immunohistochemistry for IL-13 and IL-33. Human polyclonal rabbit anti-IL-13 antibody (Peprotech), dilution 1:50, and Human Clone IL3305B mouse monoclonal antibody (Alexis Biochemicals) used at a dilution of 1:1000 in PBS and 1% BSA using an avidin/biotin staining. Subepithelial cells were quantified and expressed per mm2 of tissue.
Fibroblast culture and stimulation
Primary paediatric bronchial fibroblasts were isolated by outgrowth culture, and used between passages 3-6. Fibroblasts were grown to 80% confluence. Recombinant human IL-33 was used between 10 and 100ng/ml, and budesonide at a final concentration of 10−8M. In co-stimulations, budesonide was added to cultures for 2 hours prior to the addition of rIL-33. Conditioned media were taken and cell lysates prepared in RIPA buffer (see OR).
Western blotting and densitometry
Cell lysates were cleared by centrifugation at 10000g, 10min. 5μg protein/well in LDS sample buffer (Invitrogen) was loaded onto 4-12% Bis-Tris gels (Bio-Rad), and run at 150V in a Criterion Cell (Bio-Rad). α-SMA primary antibody (ab5694, Abcam) was added at 1/5000 dilution in blocking buffer, incubated overnight at 4°C. HRP-conjugated anti-rabbit antibody (CST) was used at 1/2000 dilution, and membranes developed with ECL substrate (Pierce) for film exposure (Amersham). (See OR)
Soluble Collagen measurement
Secreted collagen in conditioned media was assayed using the Sircol kit (Biocolor), according to the manufacturer’s instructions.
Statistical analysis
Non-parametric tests (Mann Whitney U) were used to detect differences between groups, using GraphPad Prism 4 software, and statistical significance accepted when p<0.05. Correlations were assessed using the Spearman correlation coefficient (see OR).
RESULTS
IL-13 and IL-33 show divergent responses with increasing duration of intra-nasal HDM exposure in neonatal mice
Since IL-13 has been shown to be necessary and sufficient to induce all features of AAD in adult mice(17), we investigated the relationship between IL-13 and IL-33 during the development of neonatal AAD. The longitudinal production of IL-13 and IL-33 in response to allergen in neonatal mice of increasing age (figure 1A) was strikingly different. IL-13 production started at week 1, peaked during week 2 then declined with increasing age (figure 1B). IL-33 production was similar in allergen exposed and control neonatal mice at week 1, but increased with age in response to HDM exposure (figure 1C). Levels of IL-33 remained elevated in HDM exposed mice after 4 weeks without allergen challenge (figure E1 OLR). In contrast, the Th2 cytokines IL-4, IL-5 and IL-13 reduced to baseline levels after 4 weeks without allergen challenge(16). A single re-challenge with HDM after 4 weeks without allergen exposure in neonatal mice (figure 1D) re-established AHR (figure 1E), a significant further increase in IL-33 (figure 1F), and elevated Th2 cytokines IL-13 (figure 1G), IL-4 and IL-5 (figure E1 OLR). These data suggest IL-33 remains elevated and may mediate the persistent airway remodelling observed in the absence of continued allergen exposure(16).
Figure 1.

(A) Experimental plan of intermittent HDM or saline exposure. (B-C) IL-13 and IL-33 in lung homogenates (LH) from HDM and control mice. (D) Experimental plan to assess effects of allergen exposure for 3 weeks followed by absence of allergen challenge for 28 days in neonatal mice. (E) Airway resistance at 30mg/ml methacholine in neonatal mice following 3 weeks of allergen exposure (4 hours after last challenge) and 28 days without allergen challenge. (Lung IL-33 and IL-13 after 3 weeks of allergen exposure (4 hours after last dose) and 28 days without allergen (F&G). *p<0.05, **p<0.01, ***p<0.001. Data representative of 2 experiments, n=4-6 for controls, n=6-8 for HDM.
Therapeutic blocking of IL-13 in neonatal HDM induced AAD does not reduce airway remodelling or IL-33 levels and only partly abrogates AHR
We have previously shown persistent airway pathology in paediatric STRA despite the absence of IL-13(4). Blocking the action of IL-13 in our neonatal mouse model of HDM induced AAD after disease was established (figure 2A) only partly reduced AHR (figure 2B&C), and had no impact on airway inflammation (figure E2 OLR) or airway remodelling, as demonstrated by peri-bronchiolar reticulin or collagen deposition (figure 2D&E). We next found that lung IL-33 levels remained elevated despite blocking IL-13 therapeutically (figure 2G). In contrast, blocking IL-13 using a prevention regimen, before AAD was established, completely abrogated AHR to control levels and significantly reduced airway remodelling (figure E2 OLR). This suggests IL-33 may contribute towards maintaining the critical features of asthma including AHR, inflammation and airway remodelling in the absence of IL-13.
Figure 2.

(A) IL-13 was blocked in HDM exposed neonatal mice from week 3 to 5. (B-C) Airway resistance in HDM exposed mice after blocking with anti-IL-13 antibody. (D-E) peri-bronchiolar reticulin and collagen deposition, (F-G) IL-5 and IL-33 levels after blocking IL-13. Data representative of 2 experiments, n=4-6 for controls, n=6-8 for HDM, horizontal bars represent median. NS = not significant.
Allergen challenge in neonatal ST2−/− mice does not generate AHR or airway remodelling, whilst intra-nasal IL-33 induces AHR and promotes pulmonary collagen secretion
The relationship between IL-33, AHR and airway remodelling was investigated by administration of high dose allergen to neonatal mice lacking a functional receptor for IL-33 (ST2−/− mice) for 5 weeks (figure 3A). HDM exposure from day 3 of life failed to cause AHR (figure 3B), increased goblet cell hyperplasia (data not shown) or increased peri-bronchiolar collagen deposition in ST2−/− mice (figure 3C). However, there was increased total BAL inflammation (figure E3 OLR) suggesting this is not IL-33 dependent.
Figure 3.

(A) High dose intra-nasal HDM or saline exposure from day 3 of life for 5 weeks in ST2−/− or WT Balb/c mice. (B) AHR in WT and ST2−/− mice exposed to HDM. (C) Peri-bronchiolar collagen deposition in WT and ST2−/− mice exposed to HDM. (Data representative of 2 experiments, n=4-8 per group). (D) Intra-nasal administration of IL-33 to 6 week old mice for 2 weeks (n=4-6 per group). (E) Airway resistance at week 1 and 2 weeks after IL-33 administration. (F) Collagen levels in lung homogenates. (G) Fibronectin mRNA after 2 weeks of rIL-33. NS: not significant, **p<0.01.
In order to assess whether IL-33 may contribute to AHR and airway remodelling, intra-nasal IL-33 was administered to 6 week old Balb/c mice for 2 weeks (figure 3D). AHR was significantly increased compared to control, and was significantly higher at week 2 than week 1 (figure 3E). Lung collagen levels were significantly increased after 2 weeks of IL-33 administration (figure 3F), and at the same time-point fibronectin mRNA was significantly up-regulated (figure 3G). Eosinophilic inflammation (figure E3 OLR) and levels of the Th2 cytokines IL-5, 4 and 13 also increased after rIL-33 (figure E3 OLR). These data suggest that IL-33 promotes AHR, and that it contributes to collagen production (which is the main component of the thickened subepithelial reticular basement membrane in asthmatics(18)) either directly or via the Th2 cytokines and eosinophilic inflammation.
IL-33 expression is significantly increased in submucosal inflammatory cells in paediatric STRA
We determined IL-33 and IL-13 expression in EB from severe asthmatic children. Whilst IL-13+ cells were similar in the submucosa of asthmatics and controls (figure 4A, C & E), IL-33+ cells were significantly increased in the submucosa of asthmatics compared to controls (figure 4B, D & F). In contrast, IL-33 expression was similar in the epithelium and smooth muscle (figure E4 OLR) of asthmatics and controls. These data suggest children with STRA who are on high dose steroid therapy have persistent symptoms and poor control despite an absence of tissue IL-13, but they have increased IL-33 expression which may be a more important mediator of disease pathophysiology.
Figure 4.

(A) Subepithelial IL-13+ cells in endobronchial biopsies (EB) from children with severe asthma, compared to mild-moderate asthma and age matched controls (n=13 severe asthma, n=6 mild asthma, n=5 control). (B) Subepithelial IL-33+ cells in EB from school-aged children with severe asthma compared to children with mild asthma and non-asthmatic controls (n=50 severe asthma, n=7 mild asthma, n=11 control). (C&E) EB from a school-aged asthmatic stained for IL-13 (magnification: ×200 and ×400). (D&F) EB from school-aged asthmatic stained for IL-33, including smooth muscle (black arrows) (magnification: ×200 and ×400).
IL-33 promotes collagen secretion from paediatric asthmatic airway fibroblasts
Since children with STRA had increased IL-33, and airway remodelling is a key feature of the disease(4), we explored the relationship between IL-33 expression and airway remodelling in children with STRA. There was a positive correlation between mucosal IL-33 expression and RBM thickness in children with STRA (figure 5A); there were too few non-asthmatic controls to explore any relationship in this group. In order to investigate the functional relationship between IL-33 and remodelling, we assessed the response of airway fibroblasts from children with STRA to IL-33 in vitro. Cultured fibroblasts produced increased collagen following stimulation with IL-33. Although the addition of budesonide significantly decreased collagen secretion, increased secretion was maintained with both budesonide and IL-33 (figure 5B). The time at which maximal effects of IL-33 were seen in asthmatic children varied from 24 up to 72 hours after stimulation (figure E5 OLR). Collagen secretion in response to IL-33 was only apparent in fibroblasts cultured from asthmatics, whilst fibroblasts from healthy adults failed to show any response to IL-33 (figure 5C). The relationship between IL-33 and remodelling only applied to a specific feature of airway remodelling, increased RBM thickness, and not to smooth muscle mass (figure E5 OLR). Paediatric asthmatic fibroblasts stimulated with IL-33 did not express alpha smooth muscle actin, showing they had not developed a myofibroblast phenotype (figure E5 OLR). In addition, cultured asthmatic fibroblasts expressed the receptor for IL-33 (figure 5D). These data show IL-33 promotes increased RBM thickness in paediatric STRA by causing collagen secretion from paediatric asthmatic fibroblasts, and that IL-33 is relatively steroid resistant since induction of collagen secretion is maintained despite the addition of budesonide.
Figure 5.

(A) Correlation between IL-33+ cells per area of submucosa and RBM thickness in endobronchial biopsies from children with severe therapy resistant asthma (STRA), (n=50, Spearman correlation coefficient, r=0.38). (B) In vitro collagen secretion from paediatric airway fibroblasts at baseline, and after stimulation with rIL-33 and budesonide alone and together. Each dot represents one child (circle and triangle = severe asthma, square = mild asthma). (C) Collagen secretion from adult normal human lung fibroblasts (NHLF) 24 hours after stimulation (each dot represents average data from n=2 subjects). (D) ST2 staining of paediatric and (E) normal adult lung fibroblasts.
IL-33 is less steroid sensitive than IL-13 in paediatric severe therapy resistant asthma
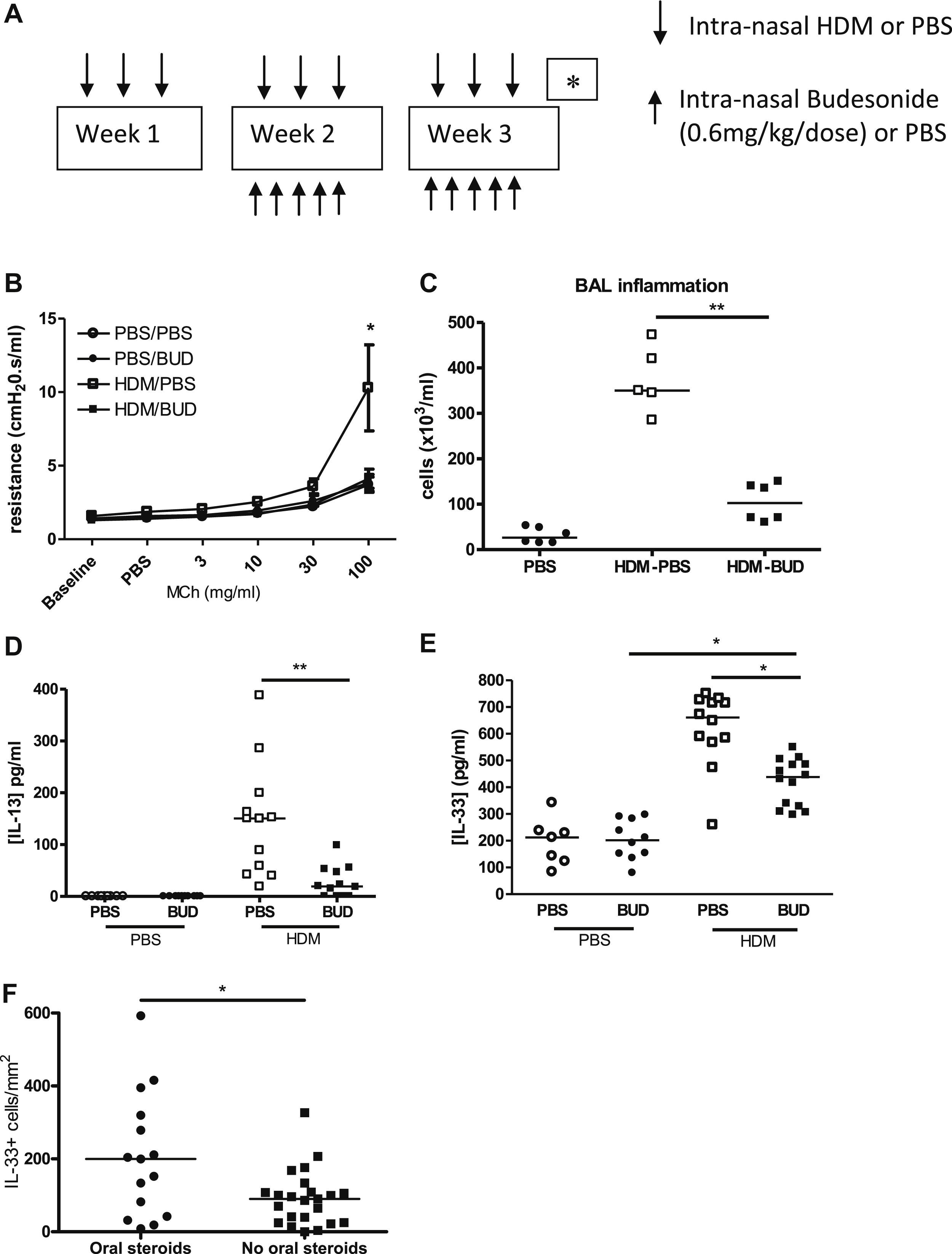
IL-13 expression was not increased in biopsies from children with STRA (figure 4A), possibly because of high dose steroid therapy. In order to determine how IL-13 and IL-33 were affected by steroids, budesonide was administered intra-nasally to HDM exposed neonatal mice before AAD was established (figure E6 OLR). This resulted in a complete reduction in AHR and inflammation (figure E6). Levels of IL-13 were significantly reduced whilst IL-33 remained elevated in HDM exposed mice treated with budesonide compared to controls (figure E6). In contrast, when budesonide was administered therapeutically, after AAD was established (figure 6A), AHR was only partially reduced (figure 6B), although inflammation was still attenuated (figure 6C). Critically, levels of IL-13 were significantly reduced in budesonide treated mice (figure 6D) whilst levels of IL-33 remained elevated (figure 6E). In addition, peri-bronchiolar collagen deposition was unchanged in HDM exposed mice that were treated with budesonide (figure 6F). These data show for the first time that IL-33 is less steroid sensitive than IL-13. There was tissue IL-33 expression in STRA children despite high dose inhaled steroid therapy, and expression was even higher in those additionally prescribed oral corticosteroids (figure E6).
Figure 6.

(A) Administration of HDM or saline from day 3 of life (↓), and intra-nasal budesonide (↑) (0.6mg/kg/dose) between week 3 and 6. (B) AHR, (C) inflammation in BAL fluid, (D) levels of IL-13 and (E) IL-33 and (F) peri-bronchiolar collagen deposition in budesonide treated and HDM exposed mice. Data representative of 2 experiments, n=4-6 PBS, n=6-8 HDM, horizontal bars represent median, vertical bars represent IQR. ns; not significant, *p<0.05, **p<0.01).
DISCUSSION
We have shown for the first time that IL-33 is increased in a neonatal mouse model of HDM induced AAD, and in biopsies from children with STRA. Moreover, IL-33 promotes a specific feature of airway remodelling, increased RBM thickness, via collagen secretion from asthmatic airway fibroblasts in vitro, and following direct in vivo administration of IL-33 to mice. Of note, we have shown that AHR and airway remodelling are unaffected when IL-13 is blocked therapeutically in the neonatal mouse model, while levels of IL-33 remain elevated. Critically, the role of IL-33 is of particular importance in severe asthma, since we have shown for the first time that its expression in paediatric asthmatic biopsies, and function in the neonatal mouse model, is less sensitive to steroid therapy than IL-13.
Although there has been evidence from experimental models that allergic asthma is mediated by Th2 cytokines, asthma is now recognised as being a heterogeneous disease(22). This is particularly important for paediatric asthma, since we have previously found little evidence of Th2 mediators in children with STRA, after careful exclusion of modifiable factors such as poor adherence and persistent allergen exposure that may contribute to severe disease(19). Therefore, we explored the role of the innate cytokine IL-33 in mediating severe disease. IL-33 is a member of the IL-1 family, and is the ligand of ST2(20), and has been shown to induce the production of innate helper cells which in turn secrete IL-13 and IL-5 in adult mouse models of AAD(14). We have previously shown early release of IL-13 in a neonatal model of AAD(16) before development of an adaptive immune response as demonstrated by the presence of either increased serum total or HDM-specific IgE(16). This reinforces the idea that innate immune mechanisms are important in the initiation of early onset allergic disease. We measured both IL-33 and IL-25 since both have been shown to induce innate helper cells and AHR in adult mice(14). IL-25 was unchanged following neonatal HDM exposure (supplementary figure E7), however IL-33 levels rapidly increased. This finding is supported by reports of the association between IL-33 and childhood asthma in genome wide association studies(21). However, unlike IL-13, which peaked at two to three weeks after allergen exposure in neonatal mice, IL-33 production increased with age and allergen exposure. Although the role of IL-13 in initiating AHR in AAD has been widely investigated, its role in disease maintenance has been questioned(22;23). We confirmed that disease maintenance was not IL-13 dependent since AHR was only partially abrogated when IL-13 was neutralised during established disease. This has also been shown following exposure of adult mice to inhaled HDM(23). In addition, expression of IL-13 in airway biopsies from children with STRA was not increased compared to age-matched non-asthmatic controls. In contrast, levels of IL-33 were maintained during therapeutic blockade of IL-13 in our neonatal model, and tissue expression of IL-33 in paediatric STRA was increased compared to non-asthmatic controls. Persistence of IL-33 during established disease was further supported in our neonatal mouse model because elevated levels of IL-33 were maintained in HDM exposed neonatal mice up to 4 weeks without allergen challenge, and this coincided with a persistence of peri-bronchiolar collagen deposition at 4 weeks without allergen challenge(16). We have also shown that mucosal infiltrating inflammatory cells are the likely source of IL-33 in severe paediatric asthma, rather than epithelial cells or smooth muscle as has been reported in adult severe asthmatics(12;13).
Although IL-33 has been shown to induce AHR in models of acute allergen exposure(14;15) and infection(24), and the IL-33/ST2 signalling pathway has been shown to activate airway eosinophils in an autocrine and paracrine manner(25), this is the first report of a relationship between IL-33 and airway remodelling in asthma. Increased thickness of the RBM is a specific feature of airway remodelling in asthma that is independent of disease severity(2) and is present in childhood (median age 10 years)(2;26) and preschool wheezers (median age 3 years)(27-29). RBM thickening results from increased deposition of collagen fibrils in the subepithelial basement membrane which ultra-structurally resemble collagen III(18). Critically, there are currently no available therapies that target airway remodelling in asthma(30). This is a significant limitation specifically in STRA, where persistent lung function abnormalities and symptoms may be due to structural airway changes. We have shown that IL-33 induces collagen release from airway fibroblasts cultured from paediatric asthmatics, and this likely explains the association between increased IL-33 expression in submucosal cells and increased RBM thickness in biopsies from children with STRA. Importantly, IL-33 also resulted in increased pulmonary collagen production following intra-nasal administration in mice. Interestingly, the distribution of collagen deposition in the lung following intra-nasal rIL-33 was in between the broncho-vascular bundle, whereas collagen deposition following HDM exposure is peri-bronchiolar. This suggests the distribution of the cells promoting fibrosis dependent on IL-33 may be predominantly in that region. It has been argued that increased RBM thickness may be protective in asthma, since it may cause increased airways stiffness which in turn prevents excessive AHR(31). However, in our paediatric cohort RBM thickness was not associated with bronchodilator responsiveness(4) and in adults increased RBM thickness has been reported as a predictor of poor steroid response(32), suggesting RBM thickness may be adverse.
An important finding from our study was the lack of a response to IL-33 from healthy adult lung fibroblasts. Although an inherent functional abnormality of the asthmatic airway epithelium has been reported,(33) these novel data suggest other structural airway cells, including fibroblasts, may also have different functional characteristics that are specific to asthma. An acknowledged limitation is the absence of fibroblast cultures from healthy children. However performance of bronchoscopy and EB for research in healthy children is not ethically acceptable. IL-33 has been associated with fibrosis in other lung diseases such as pulmonary fibrosis in systemic sclerosis(34), and with fibrosis in other organs, such as cardiac fibrosis(35). IL-33 has also been shown as a critical regulator/enhancer of TNF-α-induced functions in rheumatoid arthritis synovial fibroblasts(36).
We have shown that IL-33 promotes increased collagen production, but in contrast to adult studies(12) we have not shown an association between IL-33 expression and smooth muscle in airway biopsies. The absence of alpha smooth muscle actin release after IL-33 stimulation of asthmatic fibroblast cultures suggests they were unlikely to have a myofibroblast phenotype.
Increased IL-33 expression was present in our patients with STRA despite high dose steroid therapy. In contrast, IL-13 was not detectable, suggesting IL-13 is steroid sensitive whilst IL-33 may be a relatively more steroid resistant mediator. Of note, IL-33 is not known to have a classical steroid response element. Hence, we administered steroids to neonatal mice exposed to HDM. We used inhaled budesonide in high dose to optimally reflect treatment in severe asthma. IL-13 was completely abrogated following budesonide administration when administered both prophylactically (before AAD was established) and therapeutically (after fully established AAD). In contrast, IL-33 remained higher in HDM exposed, budesonide treated mice than controls in both regimens, and in the therapeutic regimen AHR was only partially reduced, while peri-bronchiolar collagen deposition was unaffected by budesonide administration. Since only partial effects of budesonide were seen in the mice when used therapeutically, this suggests the neonatal model of HDM exposure is closely aligned with human severe asthma. In further support of IL-33 being less steroid sensitive than IL-13, we found children that were prescribed regular oral steroids and high dose inhaled steroids had significantly higher mucosal IL-33 expression than those just prescribed high dose inhaled steroids. We therefore provide compelling data in vitro and in vivo in patients and mouse models that IL-33 is a relatively steroid resistant cytokine involved in airway remodelling.
In summary, we have shown for the first time that IL-33 promotes increased RBM thickness, and that IL-33 alone, in the absence of IL-13 seems sufficient to maintain remodelling despite steroid treatment. IL-33 may be the first therapeutic target identified that specifically attenuates airway remodelling in the severe, steroid resistant asthma phenotype, and it seems directly applicable as a target for children.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Key messages.
IL-33 promotes a specific feature of airway remodelling, increased reticular basement membrane thickness, in paediatric STRA
IL-33 is a less steroid sensitive cytokine than IL-13 and persists in paediatric STRA
Acknowledgements
We are grateful for Lorraine Lawrence for histology support. We are grateful to UCB® Celltech for the gift of the anti-IL-13 antibody. We are grateful to Dr Andrew McKenzie for the ST2 knock-out mice.
SS was supported by an Intermediate Clinical Fellowship from the Wellcome Trust, UK. CML is a Wellcome Senior Fellow in Basic Biomedical Sciences, AB is an NIHR Senior Clinical Investigator. The project was supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
Abbreviations
- STRA
severe therapy resistant asthma
- RBM
reticular basement membrane
- IL
interleukin
- HDM
house dust mite
- AAD
allergic airways disease
- OR
online repository
- AHR
airway hyperresponsiveness
- EB
endobronchial biopsies
References
- (1).Szefler SJ, Zeiger RS, Haselkorn T, Mink DR, Kamath TV, Fish JE, et al. Economic burden of impairment in children with severe or difficult-to-treat asthma. Ann Allergy Asthma Immunol. 2011;107(2):110–9. doi: 10.1016/j.anai.2011.04.008. [DOI] [PubMed] [Google Scholar]
- (2).Payne DN, Rogers AV, Adelroth E, Bandi V, Guntupalli KK, Bush A, et al. Early thickening of the reticular basement membrane in children with difficult asthma. Am J Respir Crit Care Med. 2003;167(1):78–82. doi: 10.1164/rccm.200205-414OC. [DOI] [PubMed] [Google Scholar]
- (3).Regamey N, Ochs M, Hilliard TN, Muhlfeld C, Cornish N, Fleming L, et al. Increased airway smooth muscle mass in children with asthma, cystic fibrosis, and non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2008;177(8):837–43. doi: 10.1164/rccm.200707-977OC. [DOI] [PubMed] [Google Scholar]
- (4).Bossley CJ, Fleming L, Gupta A, Regamey N, Frith J, Oates T, et al. Pediatric severe asthma is characterized by eosinophilia and remodeling without T(H)2 cytokines. J Allergy Clin Immunol. 2012;129(4):974–82. doi: 10.1016/j.jaci.2012.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Liou CJ, Huang WC. Dehydroepiandrosterone suppresses eosinophil infiltration and airway hyperresponsiveness via modulation of chemokines and Th2 cytokines in ovalbumin-sensitized mice. J Clin Immunol. 2011;31(4):656–65. doi: 10.1007/s10875-011-9529-3. [DOI] [PubMed] [Google Scholar]
- (6).Heijink IH, Kauffman HF, Vellenga E, Veltman-Starkenburg CA, Postma DS, de Monchy JG. Effect of ciclesonide treatment on allergen-induced changes in T cell regulation in asthma. Int Arch Allergy Immunol. 2008;145(2):111–21. doi: 10.1159/000108136. [DOI] [PubMed] [Google Scholar]
- (7).Choi IS, Cui Y, Koh YA, Lee HC, Cho YB, Won YH. Effects of dehydroepiandrosterone on Th2 cytokine production in peripheral blood mononuclear cells from asthmatics. Korean J Intern Med. 2008;23(4):176–81. doi: 10.3904/kjim.2008.23.4.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365(12):1088–98. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- (9).Barlow JL, McKenzie AN. Nuocytes: expanding the innate cell repertoire in type-2 immunity. J Leukoc Biol. 2011;90(5):867–74. doi: 10.1189/jlb.0311160. [DOI] [PubMed] [Google Scholar]
- (10).Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18(5):673–83. doi: 10.1038/nm.2731. [DOI] [PubMed] [Google Scholar]
- (11).Lloyd CM. IL-33 family members and asthma - bridging innate and adaptive immune responses. Curr Opin Immunol. 2010;22(6):800–6. doi: 10.1016/j.coi.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. 2009;183(8):5094–103. doi: 10.4049/jimmunol.0802387. [DOI] [PubMed] [Google Scholar]
- (13).Prefontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125(3):752–4. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- (14).Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. 2012;129(1):191–8. doi: 10.1016/j.jaci.2011.09.041. [DOI] [PubMed] [Google Scholar]
- (15).Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. 2012;129(1):216–27. doi: 10.1016/j.jaci.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Saglani S, Mathie SA, Gregory LG, Bell MJ, Bush A, Lloyd CM. Pathophysiological Features of Asthma Develop in Parallel in House Dust Mite Exposed Neonatal Mice. Am J Respir Cell Mol Biol. 2009;41(3):281–9. doi: 10.1165/rcmb.2008-0396OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev. 2004;202:175–90. doi: 10.1111/j.0105-2896.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- (18).Saglani S, Molyneux C, Gong H, Rogers A, Malmstrom K, Pelkonen A, et al. Ultrastructure of the reticular basement membrane in asthmatic adults, children and infants. Eur Respir J. 2006;28(3):505–12. doi: 10.1183/09031936.06.00056405. [DOI] [PubMed] [Google Scholar]
- (19).Sharples J, Gupta A, Fleming L, Bossley CJ, Bracken-King M, Hall P, et al. Long-term effectiveness of a staged assessment for paediatric problematic severe asthma. Eur Respir J. 2012;40(1):264–7. doi: 10.1183/09031936.00209511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liew FY. IL-33: a Janus cytokine. Ann Rheum Dis. 2012;71(Suppl 2):i101–i104. doi: 10.1136/annrheumdis-2011-200589. [DOI] [PubMed] [Google Scholar]
- (21).Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211–21. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Leigh R, Ellis R, Wattie J, Donaldson DD, Inman MD. Is interleukin-13 critical in maintaining airway hyperresponsiveness in allergen-challenged mice? Am J Respir Crit Care Med. 2004;170(8):851–6. doi: 10.1164/rccm.200311-1488OC. [DOI] [PubMed] [Google Scholar]
- (23).Tomlinson KL, Davies GC, Sutton DJ, Palframan RT. Neutralisation of interleukin-13 in mice prevents airway pathology caused by chronic exposure to house dust mite. PLoS One. 2010;5(10) doi: 10.1371/journal.pone.0013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–8. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol. 2010;185(6):3472–80. doi: 10.4049/jimmunol.1000730. [DOI] [PubMed] [Google Scholar]
- (26).Barbato A, Turato G, Baraldo S, Bazzan E, Calabrese F, Tura M, et al. Airway inflammation in childhood asthma. Am J Respir Crit Care Med. 2003;168(7):798–803. doi: 10.1164/rccm.200305-650OC. [DOI] [PubMed] [Google Scholar]
- (27).Baraldo S, Turato G, Bazzan E, Ballarin A, Damin M, Balestro E, et al. Noneosinophilic asthma in children: relation with airway remodelling. Eur Respir J. 2011;38(3):575–83. doi: 10.1183/09031936.00168210. [DOI] [PubMed] [Google Scholar]
- (28).Saglani S, Payne DN, Zhu J, Wang Z, Nicholson AG, Bush A, et al. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am J Respir Crit Care Med. 2007;176(9):858–64. doi: 10.1164/rccm.200702-212OC. [DOI] [PubMed] [Google Scholar]
- (29).Halwani R, Al-Muhsen S, Hamid Q. Airway remodeling in asthma. Curr Opin Pharmacol. 2010;10(3):236–45. doi: 10.1016/j.coph.2010.06.004. [DOI] [PubMed] [Google Scholar]
- (30).Durrani SR, Viswanathan RK, Busse WW. What effect does asthma treatment have on airway remodeling? Current perspectives. J Allergy Clin Immunol. 2011;128(3):439–48. doi: 10.1016/j.jaci.2011.06.002. [DOI] [PubMed] [Google Scholar]
- (31).Milanese M, Crimi E, Scordamaglia A, Riccio A, Pellegrino R, Canonica GW, et al. On the functional consequences of bronchial basement membrane thickening. J Appl Physiol. 2001;91(3):1035–40. doi: 10.1152/jappl.2001.91.3.1035. [DOI] [PubMed] [Google Scholar]
- (32).Bourdin A, Kleis S, Chakra M, Vachier I, Paganin F, Godard P, et al. Limited Short-term Steroid Responsiveness Is Associated With Thickening of Bronchial Basement Membrane in Severe Asthma. Chest. 2012;141(6):1504–11. doi: 10.1378/chest.11-0232. [DOI] [PubMed] [Google Scholar]
- (33).Kicic A, Hallstrand TS, Sutanto EN, Stevens PT, Kobor MS, Taplin C, et al. Decreased fibronectin production significantly contributes to dysregulated repair of asthmatic epithelium. Am J Respir Crit Care Med. 2010;181(9):889–98. doi: 10.1164/rccm.200907-1071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yanaba K, Yoshizaki A, Asano Y, Kadono T, Sato S. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol. 2011;30(6):825–30. doi: 10.1007/s10067-011-1686-5. [DOI] [PubMed] [Google Scholar]
- (35).Zhu J, Carver W. Effects of interleukin-33 on cardiac fibroblast gene expression and activity. Cytokine. 2012;58(3):368–79. doi: 10.1016/j.cyto.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kunisch E, Chakilam S, Gandesiri M, Kinne RW. IL-33 regulates TNF-alpha dependent effects in synovial fibroblasts. Int J Mol Med. 2012;29(4):530–40. doi: 10.3892/ijmm.2012.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.