

Abstract
Background
Caveolae are a nexus for protective signaling. Trafficking of caveolin to mitochondria is essential for adaptation to cellular stress though the trafficking mechanisms remain unknown. We hypothesized that G protein-coupled receptor/inhibitory G protein (Gi) activation leads to caveolin trafficking to mitochondria.
Methods
Mice were exposed to isoflurane or oxygen vehicle (30 min, ± 36 hr pertussis toxin pretreatment, an irreversible Gi inhibitor). Caveolin trafficking, cardioprotective ‘survival kinase’ signaling, mitochondrial function and ultrastructure were assessed.
Results
Isoflurane increased cardiac caveolae [n = 8/group; Data presented as Mean±SD for Ctrl versus Isoflurane; (caveolin- 1: 1.78 ± 0.12 versus 3.53 ± 0.77; p < 0.05); (caveolin-3: 1.68 ± 0.29 versus 2.67 ± 0.46; p < 0.05)] and mitochondrial caveolin levels [n = 16/group; (caveolin- 1: 0.87 ± 0.18 versus 1.89 ± .19; p < 0.05); (caveolin-3: 1.10 ± 0.29 versus 2.26 ± 0.28; p < 0.05)], and caveolin-enriched mitochondria exhibited improved respiratory function [n = 4/group; (State 3/Complex I: 10.67 ± 1.54 versus 37.6 ± 7.34; p < 0.05); (State 3/Complex II: 37.19 ± 4.61 versus 71.48 ± 15.28; p < 0.05)]. Isoflurane increased phosphorylation of survival kinases [n = 8/group; (protein kinase B: 0.63 ± 0.20 versus 1.47 ± 0.18; p < 0.05); (glycogen synthase kinase 3 beta: 1.23 ± 0.20 versus 2.35 ± 0.20; p < 0.05)]. The beneficial effects were blocked by pertussis toxin.
Conclusions
Gi proteins are involved in trafficking caveolin to mitochondria to enhance stress-resistance. Agents that target Gi activation and caveolin trafficking may be viable cardioprotective agents.
Introduction
Cardiac protective signaling involves transduction pathways involving membrane anchored receptors and effector molecules that ultimately impact mitochondrial function resulting in stress- resistance.1 Many elements of these pathways have been described; however, the crucial events linking the membrane to mitochondrial end-effects remain obscure.Studies reveal that the heart expresses a cadre of membrane receptors specifically activated by mediators released during stress/damage that trigger adaptive stress-resistance and cardiac protection.2
Caveolae are cholesterol and sphingolipid enriched invaginations of the plasma membrane3 and are considered a subset of lipid rafts.4 Caveolins, the structural proteins essential for caveolae formation, are present in three isoforms,5,6 and possess scaffolding domains that anchor and regulate a variety of proteins.7,8 Caveolin- 1 (Cav- 1) and -2 are expressed in multiple cell types, while caveolin- 3 (Cav-3) is found primarily in striated (skeletal and cardiac) muscle and certain smooth muscle cells.9 Caveolins are involved in multiple cellular processes including vesicular transport, cholesterol and calcium homeostasis,10-14 and signal transduction,15-18 and have been recently detected in mitochondria.19,20 Caveolins function as chaperones and scaffolds recruiting signaling molecules to caveolae to provide spatio-temporal regulation of signal transduction.16,21 G-protein coupled receptors (GPCRs) localize to caveolae, and caveolins regulate multiple GPCR-associated proteins (e.g., Gi, adenylyl cyclase, and effector kinases).22 We have shown that cardiac-specific Cav-3 expression not only mimics protective ischemic preconditioning via activation of GPCR/Gi linked signaling but also renders these mice resistant to pressure overload induced hypertrophy and heart failure.23,24
Recently, we have shown that a critical element in caveolin regulation of cardiac preconditioning is stress-dependent caveolin translocation from sarcolemma to mitochondria.20 Caveolae and mitochondria exist in close proximity, and preconditioning stimuli induce caveolar-mitochondrialnanocontacts and mitochondrial caveolin accumulation, stabilizing mitochondrial structure and function. The early events triggering caveolin-mitochondrial interaction remain unclear. Defining this mechanism may allow the generation of specific and targeted therapeutics to limit myocardial ischemic injury.
We here test the hypothesis that volatile anesthetic induced preconditioning involves Gi-dependent signaling and translocation of caveolin from sarcolemmal caveolae to mitochondria. We show that cardioprotective isoflurane (Iso) produces an accumulation of caveolin in mitochondria and improves mitochondrial function, similar to ischemic preconditioning. This effect was limited by pertussis toxin (PTX), an irreversible Gi inhibitor. Moreover, intrinsic tolerance to ischemia-reperfusion (IR) arising from cardiac-specific Cav-3 overexpression is also associated with mitochondrial caveolin enrichment, and these effects are attenuated by PTX treatment. A variety of experimental approaches were employed to define early molecular events regulating transfer of caveolin from membrane to mitochondria. Collectively, data identify Gi-coupled signaling as being essential to cardioprotective membrane-mitochondrial communication.
Materials and Methods
Animals
All animals were treated in compliance with the Guide for the Care and Use of Laboratory Animals, with animal use protocols approved by the VA San Diego Healthcare System Institutional Animal Care and Use Committee (San Diego, CA). Wild-type male C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). Transgenic mice with cardiac myocyte-specific overexpression of Cav-3 (Cav-3 OE, 12 week old, 20-25 grams body weight) were produced as previously reported.24 Animals were kept on a 12-hour light-dark cycle in a temperature-controlled room with ad libitum access to food and water. Animal assignment to specific experimental groups was blinded and randomized to treatment.
PTX treatment
All wild-type and Cav-3 OE mice randomly received intraperitoneal injection of either 100 µg/kg PTX (irreversible inhibitor of inhibitory G proteins)25 or saline 36 hrs before isoflurane/oxygen exposure and Langendorff perfusion experiments (Figure 1). To test that Gi activity was effectively blocked by PTX, we confirmed abolition of the bradycardia arising from 1.5 mg/kg acetylcholine (intraperitoneal) in the PTX treatment group.
Figure 1. Flow diagram of the experimental protocol.
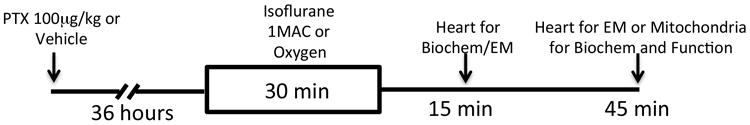
Mice were treated with pertussis toxin (PTX, 100 µg/kg intraperitoneal) or vehicle and allowed to recover for 36 hrs. After recovery, animals were exposed to isoflurane (1 MAC, minimum alveolar concentration) or oxygen for 30 min. Hearts were excised at 15 or 45 minutes following isoflurane for biochemical, mitochondrial or electron microscopy (EM) analysis.
In vivo isoflurane/oxygen exposure
Mice were anesthetized with sodium pentobarbital (80mg/kg intraperitoneal). A 20-gauge catheter was then inserted into the trachea and the lungs were mechanically ventilated (15 cm H2O peak inspiratory pressure, 100 breaths/min, 100% inspired oxygen) using a pressure-controlled TOPO ventilator (Kent Scientific Company, Torrington, CT). Core temperature was maintained at 36°C with a heating pad, and electro leads were placed to record heart rate. Mice received 100% oxygen in the control group or 1.4% isoflurane in oxygen. Following a 30 min isoflurane or oxygen exposure, hearts were excised after 15 or 45 min washout for electron microscopy (EM), biochemical or mitochondrial function analyses (Figure 1).
Ischemia-reperfusion in Langendorff perfused hearts
Mice (n= 11-13/group) were anesthetized with sodium pentobarbital (60 mg/kg, intraperitoneal) before heart excision, aortic cannulation, and Langendorff perfusion of the coronary circulation.26,27 All hearts were perfused at a pressure of 80 mmHg with modified Krebs-Henseleit buffer bubbled with 95% oxygen/5% carbon dioxide at 37°C (giving a pH of 7.4), and containing (in mM): sodium chloride, 120; sodium bicarbonate, 25; potassium chloride, 4.7; calcium chloride, 2.5; magnesium chloride, 1.2; potassium phosphate monobasic, 1.2; D-glucose, 15; and EDTA, 0.5. After 20 min normoxic stabilization at intrinsic heart rates, ventricular pacing at 7 hertz was initiated. After a further 15 minutes, baseline measures were made and hearts subjected to 25 minutes of global normothermic ischemia followed by 45 minutes aerobic reperfusion.
Sucrose-density membrane fractionation
Whole hearts (n=8/group) were fractionated using sucrose density gradients as previously reported.28 Fractions 4 through 6 were buoyant membrane fractions (BF) enriched in caveolae, caveolins, and proteins associated with caveolins. Fractions 9 through 12 were defined as non-buoyant heavy membrane fractions (HF).
Immunoblot Analysis
Whole cardiac lysates or cellular fractions were separated by SDS-PAGE with 10% polyacrylamide precast gels (Invitrogen, Carlsbad, CA) and transferred to polyvinylidene difluoride membranes by electroelution. Membranes were blocked in 20 mmol/L tris bluffered saline Tween (1%) containing 3% bovine serum albumin and incubated with primary antibody overnight at 4°C. Primary antibodies employed included: Cav- 1, GM130, calreticulin, lamin A/C, histone, protein kinase B (Akt), phosphorylated Akt (Ser 473), glycogen synthase kinase-3β (GSK3β), and phosphorylated GSK3β Ser 9)(Cell Signaling Technology Incorporated, Danvers, MA); Cav-3 (BD Bioscience, San Jose, CA and Santa Cruz Biotechnology Incorporated, Dallas, TX); glyceraldehyde 3-phosphate dehydrogenase (GeneTex Incorporated, Irvine, CA); cytochrome C (Imgenex Company, San Diego, CA); transferrin and prohibitin (Abcam, Cambridge, MA). Blots were visualized using secondary antibodies from Santa Cruz Biotechnology and enhanced chemiluminescence reagent from Lumigen (Southfield, MI). All displayed bands migrated to molecular mass standards.
Isolation of cardiac mitochondria
Mice (n=8/group) were sacrificed as above and hearts removed. Ventricles were placed in ice-cold mitochondrial isolation medium (MIM: 0.3 M sucrose, 10 mM HEPES, 250 µM EDTA), minced and homogenized with a Tissuemiser (Fisher Scientific, Waltham, MA). Homogenates were rinsed in MIM and samples centrifuged at 600 g to clear nuclear/membrane debris. The resulting supernatant was spun at 8000 g for 15 minutes, and the pellet re-suspended in MIM with 1 mM bovine serum albumin and re-spun at 8000 g for 15 minutes (with this latter step repeated). To isolate pure mitochondria the washing steps were repeated with MIM in a final 2-ml re-suspension of the pellet in mitochondrial re-suspension buffer (MRB; 500 mM EDTA, 250 mM mannitol, and 5 mM HEPES). The mitochondria were layered on top of a 30% Percoll/70% MRB solution. The Percoll gradient was spun at 95,000 g for 30 min. The mitochondrial band was removed from the gradient, and volume was increased 10-fold with MRB and the Percoll removed by centrifugation at 8000 g for 15 min. The final mitochondrial pellet was resuspended in MRB prior to analysis.
Mitochondrial respiration
Mitochondrial oxygen consumption was measured using a Clark-type O2 electrode (Oxygraph; Hansatech, Norfolk, United Kingdom) during sequential additions of substrates and inhibitors to crude mitochondria. Mitochondria (100–200 μg protein) were added to the oxymetry chamber in a 300 μl solution containing 100 mM potassium chloride, 75 mM mannitol, 25 mM sucrose, 5 mM phosphoric acid, 0.05 mM EDTA, and 10 mM tris-hydrochloride, pH 7.2 at 37°C. After 2 min equilibration, 5 mM pyruvate and 5 mM malate were added and oxygen consumption followed for 1–2 min (state 4). Adenosine diphosphate (250 μM) was added to measure state 3 (phosphorylating) respiration. To switch from nicotinamide adenine dinucleotide- to flavin adenine dinucleotide-linked respiration, we first eliminated complex I through inhibition of back electron transfer using 0.5 mM rotenone and triggered complex II activity by addition of 10 mM succinate. Oxygen utilization traces and rate determinations were obtained using Oxygraph software and normalized to protein.
Electron microscopy
Whole hearts were perfused with standard Karnovsky's fix of 4% paraformaldehyde and 1.5% glutaraldehyde in 0.1M cacodylate buffer. The samples were further post-fixed in 1% osmium tetroxide and en bloc stained with uranyl acetate. After dehydration, hearts were embedded in a longitudinal orientation in LX-112 (Ladd Research, Williston, VT) and polymerized at 60°C for 48 hours. Blocks were trimmed to regions of matching longitudinal orientation and thin sectioned. Sections were stained in uranyl acetate and lead citrate and were observed with an electron microscope (JOEL 1200 EX-II, JEOL USA, Peabody, MA; or Philips CM-10, Philips Electronic Instruments, Mahwah, NY).
Statistical analysis
All data were analyzed using GraphPad Prism 6 software (GraphPad Software, Inc., San Diego, CA). The data are depicted as mean ±SD and in all cases p<0.05 was considered statistically significant. Data were tested for normal distribution and analyzed with an ordinary two-way analysis of variance followed by Tukey's post-hoc (two-tailed) comparison or by repeated measure two-way analysis of variance followed by post-hoc Bonferroni (two-tailed) comparison. N values were determined based on prior experience with biochemical analyses and perfused heart studies.
Results
Inhibition of Gi proteins via PTX reduces isoflurane-induced caveolar formation
Previous data indicate that isoflurane exposure of myocytes and myocardium results in a rapid and significant increase in caveolae formation.28 The triggering event for this shift in membrane dynamics is unknown. We confirmed that isoflurane increases caveolae formation within 45 min of exposure, demonstrating Cav-3 and -1 (caveolar marker proteins) enrichment in buoyant fractions localized to caveolae (Figure 2, A and B quantified pooled buoyant and heavy membrane fractions). Treatment with PTX attenuated the isoflurane-induced increase in Cav-3 and -1 together with localization to buoyant fractions. A two-way analysis of variance was conducted that examined the effect of isoflurane and PTX treatment on caveolin-1 & -3 expression in heavy and buoyant fractions (all groups n = 8). There was a statistically significant interaction between the effects of isoflurane and PTX treatment on Cav-1 and -3 expression [Cav-1 BF F (1,28) = 24.73, p <0.0001; Cav- 1 HF F (1,28) = 188.1, p <0.0001; Cav-3 BF F (1,28) = 7.16, p = 0.012; Cav-3 HF F (1,28) = 26.94, p <0.0001]. Post hoc analysis using the Tukey's post hoc criterion for significance indicated that the Cav-1 and -3 to GAPDH ratios (Arbitrary Units) were significantly different in the comparisons Ctrl:Ctrl vs Ctrl:Iso [Cav-1 BF (M = 1.79 vs 3.53, SD = 0.11 vs 0.77, p<0.0001); Cav- 1 HF (M = 1.82 vs 3.72, SD = 0.19 vs 0.12, p<0.0001); Cav-3 BF (M = 1.68 vs 2.67, SD = 0.29 vs 0.46, p<0.0001); Cav-3 HF (M = 1.28 vs 3.03, SD = 0.45 vs 0.32, p<0.0001)], and Ctrl:Iso vs PTX:Iso [Cav- 1 BF (M = 3.52 vs 1.83, SD = 0.77 vs 0.32, p<0.0001); Cav- 1 HF (M = 3.72 vs 2.19, SD = 0.12 vs 0.19, p<0.0001); Cav-3 BF (M = 2.67 vs 1.78, SD = 0.46 vs 0.33, p = 0.0001); Cav-3 HF (M = 3.03 vs 1.83, SD = 0.32 vs 0.41, p<0.0001)], with our Ctrl:Iso group having the highest expression.
Figure 2. Pertussis toxin (PTX) inhibits isoflurane-dependent upregulation of caveolin/caveolae.
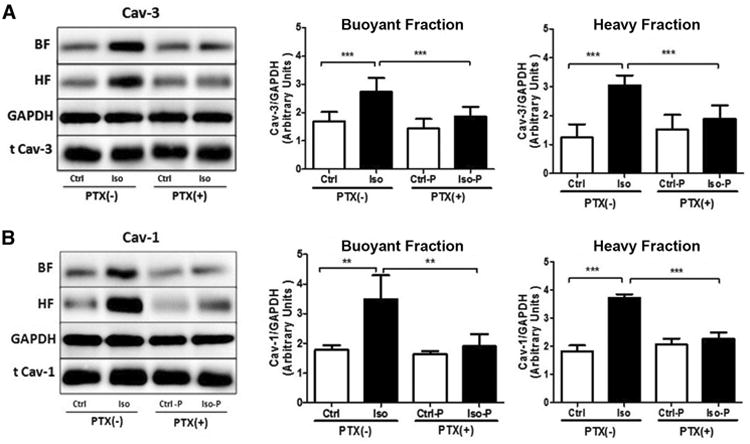
Mice were exposed to isoflurane or oxygen for 30 min ± PTX treatment. After 15 min washout, excised hearts underwent sucrose density fractionation and buoyant (BF) and heavy (HF) membrane fractions were pooled and analyzed for caveolin-3 (Cav-3) (A) and caveolin-1 (Cav- 1) (B) expression. Cav-3 and Cav- 1 was increased in BFs and HFs after Iso exposure, while this effect was blocked by PTX. Data are from 8 mice per group. **P<0.01, ***P<0.001 relative to respective control. Ctrl = control; Iso = isoflurane; Ctrl-P = control with PTX treatment; Iso-P = isoflurane with PTX treatment; GAPDH=glyceraldehyde 3-phosphate dehydrogenase.
Isoflurane modifies key phospho-kinases via Gi signaling and cardiac myocyte ultrastructure
Increased phosphorylation of Akt and GSK3β is reported with a variety of protective interventions, and these kinases may be key downstream mediators of GPCR/Gi signaling.29 Cardioprotectiveisoflurane substantially up-regulated Akt and GSK3β phosphorylation, with these effects negated by pretreatment with PTX (Figure 3A-C). A two-way ANOVA was conducted that examined the effect of isoflurane and PTX treatment on phosphorylation of Akt and GSK3β (all groups n = 8). There was a statistically significant interaction between the effects of isoflurane and PTX treatment on pAkt/tAkt and pGSK3β/tGSK3β expression [pAkt/tAktF (1,28) = 78.21, p< 0.0001; pGSK3β/tGSK3β F (1,28) = 113.0, p< 0.0001]. Post hoc analysis using the Tukey's post hoc criterion for significance indicated that the pAkt/Akt and pGSK3β/GSK3β ratios (Arbitrary Units) were significantly different in the comparisons Ctrl:Ctrl vs Ctrl:Iso [pAkt/tAkt (M = 0.63 vs 1.47, SD = 0.20 vs 0.18, p< 0.0001); pGSK3β/tGSK3β (M = 1.23 vs 2.34, SD = 0.20 vs 0.20, p< 0.0001)], and Ctrl:Iso vs PTX:Iso [pAkt/tAkt (M = 1.47 vs 0.48, SD = 0.18 vs 0.12, p< 0.0001); pGSK3β/tGSK3β (M = 2.34 vs 1.01, SD = 0.20 vs 0.18, p< 0.0001)], with our Ctrl:Iso group having the highest expression. This is consistent with PTX-sensitivity of protective ischemic and pharmacologic preconditioning responses.30,31
Figure 3. Pertussis toxin (PTX) inhibits isoflurane-dependent kinase signaling.
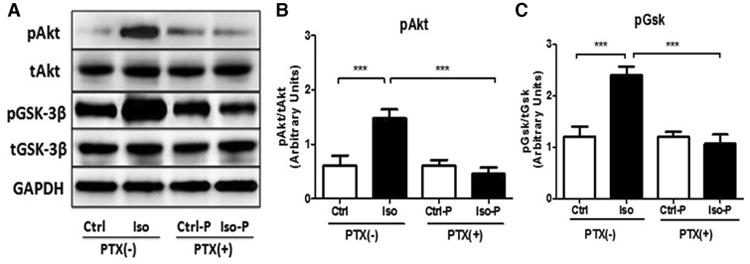
A. Whole-heart homogenates showed elevated phosphorylation of Akt and GSK3β after 30 min isoflurane exposure and 15 min washout. Phosphorylation of Akt (B, Ser 473) and GSK3β (C, Ser 9) was attenuated by PTX (n=8 each group). ***P<0.001 relative to respective control. Ctrl = control; Iso = isoflurane; Ctrl-P = control with PTX treatment; Iso-P = isoflurane with PTX treatment; pAKT = phosphorylated protein kinase B; tAKT = total protein kinase B; pGSK-3β = phosphorylated glycongen synthase kinase 3 beta; tGSK-3β = total glycongen synthase kinase 3 beta; GAPDH=glyceraldehyde 3-phosphate dehydrogenase.
These biochemical data were confirmed by EM (in perfusion fixed hearts at the same exposure and washout times as biochemical studies), which showed isoflurane-induced increases in caveolaeformation (Figure 4A and 4B). Pretreatment with PTX in control and isoflurane exposed animals dramatically altered mitochondrial andsarcomeric structure in both groups, with limited changes in caveolar/membrane morphology after isoflurane exposure (Figure 4C and 4D).
Figure 4. Pertussis toxin (PTX) attenuates isoflurane-dependent caveolae formation and impacts mitochondrial morphology.

Control (A), isoflurane (B), PTX+control (C), and PTX+isoflurane (D) treated mice were allowed 15 min washout following isoflurane/oxygen exposure. Animals were perfusion fixed and hearts processed for electron microscopy (EM) analysis. Isoflurane increased membrane invaginations consistent with caveolae (white arrowheads, B), and this was blocked with PTX. PTX treatment resulted in loss of mitochondrial structure in control and isoflurane groups (* and arrow represent altered mitochondrial morphology and potential mitophagy, respectively).
Isoflurane exposure induces Cav-3 and Cav-1 localization to mitochondria
We have shown that mitochondrial localization of caveolin is crucial to cellular stress adaptation and thatischemic preconditioning increases mitochondria-localized caveolin.20 The early signaling events inducing thiscaveolin trafficking are unknown. After exposing animals to isoflurane or oxygenin the presence or absence of PTX, we assessed caveolin enrichment in purified mitochondrial fractions. To assess purity of the isolates we probed formembrane (Na+/K+-ATPase), golgi (GM130), clathrin-dependent endocytosis (transferrin), endoplasmic reticulum (calreticulin), nuclear (lamin A/C, histone), and mitochondrial (prohibitin, cytochrome C) protein markers. All markers were detected in whole heart lysates (H); however mitochondrial fractions (M) were highly enriched in mitochondrial markers with only trace contamination of clathrin and nuclei (Figure 5A). Isoflurane exposure increased localization of Cav-3 and -1 to the mitochondrial fractionin a PTX-sensitive manner (Figure 5B-D). A two-way ANOVA was conducted that examined the effect of isoflurane and PTX treatment on Cav- 1/prohibitin and Cav-3/prohibitin (all groups n =16). There was a statistically significant interaction between the effects of isoflurane and PTX treatment on Cav- 1/prohibitin and Cav-3/prohibitinexpression [Cav- 1/prohibitin F (1,60) = 114.0, p<0.0001; Cav-3/prohibitin F (1,60) = 84.1, p<0.0001]. Post hoc analysis using the Tukey's post hoc criterion for significance indicated that the Cav- 1/prohibitin and Cav-3/prohibitin ratios (Arbitrary Units) were significantly different in the comparisons Ctrl:Ctrl vs Ctrl:Iso [Cav- 1/prohibitin (M = 0.87 vs 1.89, SD = 0.18 vs 0.19, p <0.0001); Cav-3/prohibitin (M = 1.10 vs 2.26, SD = 0.29 vs 0.28, p <0.0001)], and Ctrl:Iso vs PTX:Iso [Cav- 1/prohibitin (M = 1.89 vs 0.94, SD = 0.19 vs 0.20, p <0.0001); Cav-3/prohibitin (M = 2.26 vs 0.90, SD = 0.28 vs 0.27, p <0.0001)], with our Ctrl:Iso group having the highest expression.
Figure 5. Pertussis toxin (PTX) inhibits isoflurane-dependent caveolin trafficking to mitochondria.
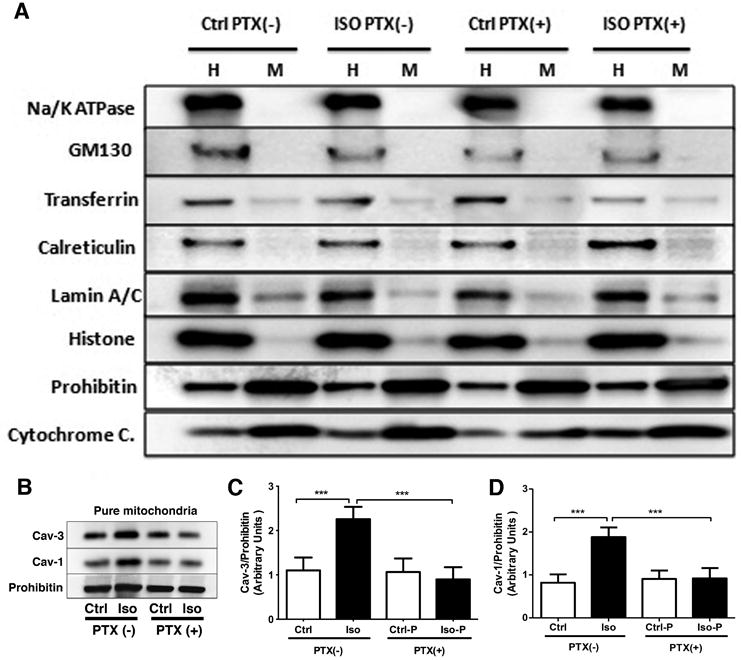
Mitochondria were isolated after 30 min isoflurane and 45 min washout (controls were exposed to oxygen). Whole heart homogenate (H) and pure mitochondria (M) were probed for cell compartment markers. Purified mitochondria (A, high enrichment of mitochondrial markers with only trace contamination of nuclei and clathrin) were enriched in Cav- 1 and -3 following isoflurane, and trafficking was inhibited by PTX (B-D). Data are from 16 mice per group. ***P<0.001 relative to respective control. Ctrl = control; Iso = isoflurane; Ctrl-P or Ctrl-PTX = control with PTX treatment; Iso-P or Iso-PTX = isoflurane with PTX treatment.
Isoflurane-dependent changes in mitochondrial structure and function are PTX-sensitive
We performed EM at the 45 min washout time-point correlating to increased mitochondrial localization of caveolin after isoflurane exposure. Control hearts (Figure 6A) showed normal sub-sarcolemmal and interfibrillar mitochondria. Isoflurane treatment did not substantially modify these populations of mitochondria (Figure 6B), with the figure inset showing internalized caveolae structures in close proximity to mitochondria, indicative of trafficking. PTX treatment in control and isoflurane exposed hearts dramatically modified mitochondrial structure (i.e., elongation and large fused mitochondria that appear to hinder caveolar traffic; Figure 6C-E). These images also reveal disruption of the cyto- architecture of the cardiac myocyte. To determine if PTX-induced changes in mitochondrial structure impact mitochondrial function, respiratory function was analyzed in purified mitochondria (all groups n = 4). No significant interaction was observed in resting or state 4 respiration with complex I (malate/pyruvate, mal/pyr) substrates [F (1,12) = 0.47, p = 0.50].Isoflurane significantly enhanced state 3 respiration with complex 1 and complex II (succinate, succ) substrates, and this effect was blocked by PTX pretreatment (Figure 6F). There was a statistically significant interaction between the effects of isoflurane and PTX treatment on State 3 (mal/pyr) and State 3 (succ) respiration [State 3 (mal/pyr) F (1,12) = 48.89, p <0.0001; State 3 (succ) F (1,12) = 8.37, p = 0.0135]. Post hoc analysis using the Tukey's post hoc criterion for significance indicated that the State 3 (mal/pyr) and State 3 (succ) ratios (Arbitrary Units) were significantly different in the comparisons Ctrl:Ctrl vs Ctrl:Iso [State 3 (mal/pyr) (M = 10.67 vs 37.60, SD = 1.52 vs 7.34, p 4.61 vs 15.28, p = 0.0006)], Ctrl:Iso vs PTX:Iso [State 3 (mal/pyr) (M = 37.6 vs 3.67, SD = 7.34 vs 0.90, p <0.0001); State 3 (succ) (M = 71.48 vs 27.11, SD = 15.28 vs 1.90, p <0.0001)], and Ctrl:Ctrl vs PTX:Ctrl [State 3 (succ) only (M = 37.19 vs 18.10, SD = 4.61 vs 6.85, p = 0.0404)] with our Ctrl:Iso group having the highest respiration.
Figure 6. Pertussis toxin (PTX) impacts mitochondrial structure and function.
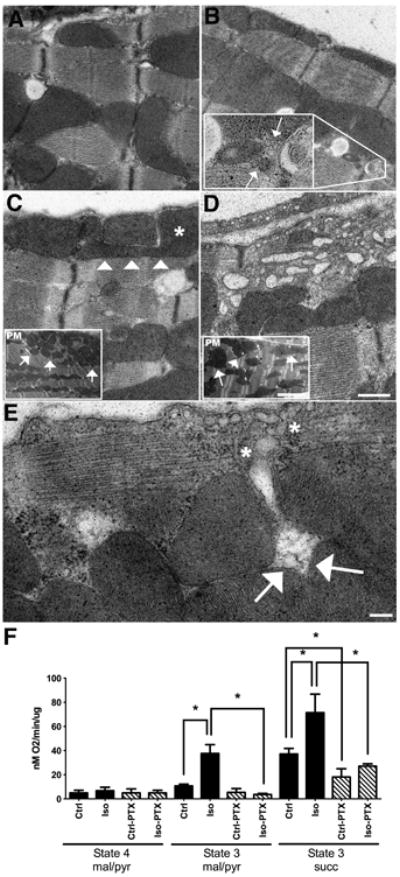
A. Control cardiac tissue showing both subsarcolemmal (SSM) and interfibrillary (IFM) populations and normal ultrastructural morphology. B. Isoflurane treated hearts showing the two distinct healthy mitochondrial populations. Inset localizes increased internalized caveolae (arrows) near IFM mitochondria, indicating trafficking from the PM to internal compartments. C-D. PTX treated control and isoflurane heart ultrastructure, respectively. Arrowheads and * mark an atypical shaped SSM, which appears to “block” a Z-line transport zone. Limited internalized caveolae are seen in either C or D. The blockade effect of PTX treatment is seen dramatically in D where vesicles appear trapped between the PM and SSM. Insets demonstrate the clumping of SSM and irregular shape and distribution of IFM. E. Higher magnification of the PM and SSM following PTX treatment. Internalized putative caveolae vesicles (*) are trapped by the smaller mitochondria (arrows). Mag bars A-D = 2 μm. Insets = 500nM E = 200nM. F. Mice were exposed to O2 or isoflurane (30 min) ± PTX treatment, and hearts were excised after 45 min washout for preparation of crude mitochondria for respiratory function studies. State 4 respiration was assessed with complex 1 substrates. State 3 was assessed with complex I (malate/pyruvate, mal/pyr) and II (succinate, succ) substrates. Isoflurane enhanced state 3 respiration was attenuated with PTX treatment. Data are from 4 mice per group. *P<0.05, relative to respective control. Ctrl = control; Iso = isoflurane; Ctrl-PTX = control with PTX treatment; Iso-PTX = isoflurane with PTX treatment.
Cardiac function in Cav-3 OE, which are endogenously adapted to ischemia-reperfusion injury, is sensitive to PTX treatment
We have previously shown that Cav-3 OE protects myocardium from ischemia-reperfusion injury and mimics a preconditioning like phenotype with increased survival kinase activation linked to Gi signaling.24 These hearts also exhibit increased localization of Cav-3 to mitochondria.20 Here we show that the functional protection from ischemia in Cav-3 OE hearts is sensitive to PTX treatment, with IR tolerance returned to levels observed in wild-type hearts. Post-ischemic left ventricular end diastolic pressure was increased whereas developed pressure declined to levels observed in wild-type C57Bl/6 mice following 45 minutes of reperfusion (Figure 7A-B). There was a statistically significant interaction in the repeated measure 2-way analysis of variance between the groups (Cav-3 OE, n = 13; Cav-3 OE + PTX, n = 12; C57Bl/6J, n = 11) for the left ventricular end diastolic pressure and left ventricular developed pressure [left ventricular end diastolic pressure F (20,330) = 5.144, p <0.0001; ventricular developed pressure F (20,330) = 1.648, p = 0.0405]. Post hoc analysis using the Bonferroni post hoc criterion for significance are depicted in Figure 7.
Figure 7. Pertussis toxin (PTX) blunts intrinsic IR tolerance with cardiac specific Cav-3 overexpression.
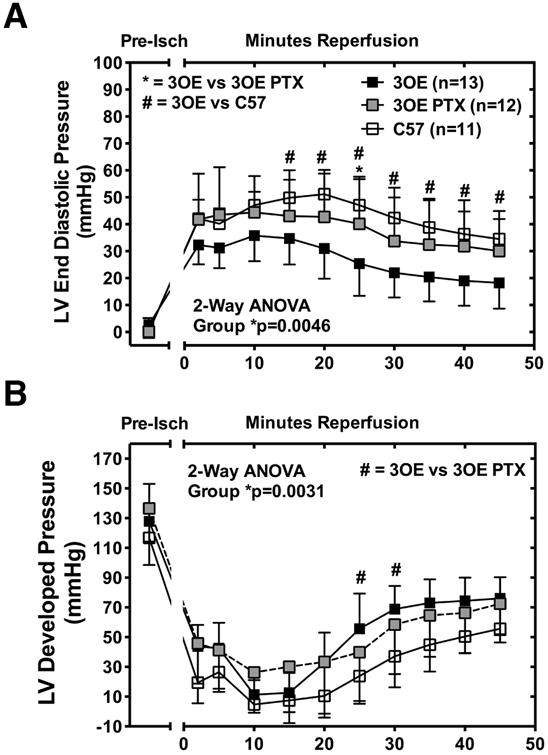
Hearts from Cav-3 OE mice treated with PTX (3OE PTX) show higher LVEDP (A, left ventricular end diastolic pressure) over 45 min reperfusion and lower LVDP (B, left ventricular developed pressure) compared to hearts from untreated Cav-3 OE (3OE) mice. Data were analyzed with repeated measure 2-Way ANOVA and Bonferroni post-hoc comparison. Significance was accepted when *p<0.05; #p<0.05. Data presented as mean±SD, n = 11-13/group, C57 = C57Bl/6 mice; Pre-Isch = pre-ischemia.
Discussion
In this study, we observed that isoflurane exposure rapidly alters plasma membrane ultrastructure, enhancing caveolae formation through increased trafficking of caveolin proteins to the membrane. Subsequently, isoflurane modified mitochondrial structure and function in a Gi-dependent manner. Thus, under baseline conditions there is a homeostatic interaction of membrane and mitochondrial compartments that can be dramatically enhanced by protective stimuli inducing acute and adaptive stress-resistance. This effect of isoflurane appears dependent on Gi signaling and is involved in regulating membrane morphology, mitochondrial structure, and myocyte cyto-architecture (Figure 8). Our findings provide novel mechanistic insight into phenomenology described in the literature over the last 20 years showing the importance of Gi signaling to cardiac protection induced by volatile anesthetics as well as other pharmacologic agents, and suggest that this initial trigger event is necessary and sufficient to initiate changes in membrane morphology that ultimately impact mitochondrial function.
Figure 8. Summary diagram.
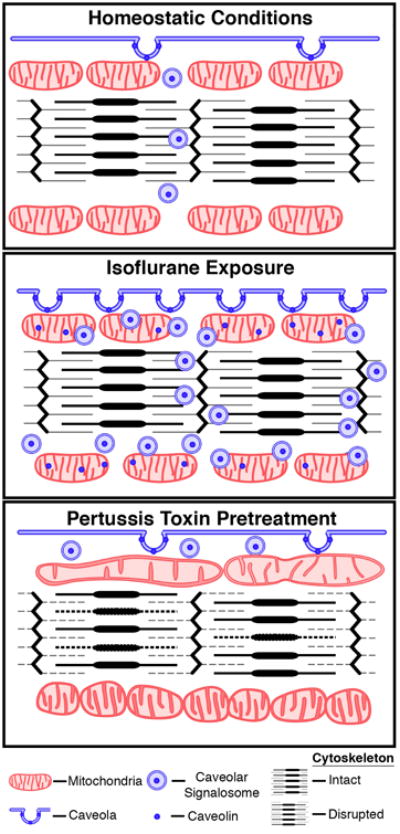
Cartoon depicting homeostatic, isoflurane-induced, and pertussis toxin (PTX)-sensitive trafficking of caveolin/caveolae and alterations in membrane/mitochondrial dynamics.
We recently demonstrated the importance of caveolae-mitochondria interactions to cellular stress resistance, with protective stimuli triggering dynamic physical association of caveolae and sub-sarcolemmal mitochondria and translocation of caveolins to mitochondria.20 Increased mitochondrial localization of caveolin results in mitochondrial structural and functional stability that ultimately promotes adaptation/resistance to stress. This phenomenon was observed not only in heart but also in cancer cells and C. elegans, suggesting an important generalized mechanism for stress adaptation. However, what is unknown is the mechanism by which these events are initially triggered.
Caveolae play an important role in physiologic functions and previous studies32 have shown that: 1) caveolin is both necessary and sufficient to protect heart from IR injury; 2) preconditioning via transient ischemia and exposure to volatile anesthetics enhances the number of membrane caveolae; and 3) cardiac myocyte-specific overexpression of Cav-3 enhances intrinsic IR tolerance. Anesthetic preconditioning is an effective means of protecting the myocardium against prolonged ischemia.33,34 We have shown that caveolae and Cav-3 are essential to isoflurane-induced cardioprotection in adult cardiac myocytes from rats and in situ hearts of Cav-3 knockout mice.35 Isoflurane produces this effect by enhancing myocyte caveolin and caveolae expression. However, it is unknown how isoflurane increases caveolin/caveolae and thus induces cardiac protection.
General anesthetics have been reported to alter the functionality of GPCR signaling systems.36,37 We show here that anesthetic-dependent responses are sensitive to PTX, supporting involvement of Gi signaling in isoflurane induced membrane and mitochondrial control. Inhibition of Gi signaling with a targeted peptide inhibitor results in increased apoptosis in response to ischemic stress, suggesting loss of Gi signaling leads to mitochondrial specific cardiac injury.38 Evidence also suggests that activation of vesicle formation and trafficking is sensitive to Gi antagonism in end othelial cells, and this is caveolin-dependent and linked to distal survival kinase signaling.39 Such findings suggest that alterations in plasma membrane dynamics with volatile anesthetic may be the result of receptor-dependent Gi activation. Our EM analysis with PTX treatment shows effects on structure of mitochondria in the control setting suggesting that basal Gi tone in the heart may have a major impact on structure and function, an idea advanced in 1990 when ischemic preconditioning, which activates Gi signaling, was shown to preserve myocyte ultrastructure whereas lethal injury resulted in ultrastructural damage.40 It is possible that Gi signaling works constitutively through caveolins/caveolae and cytoskeletal regulation to maintain basal structure that is perturbed when Gi signaling is inhibited. The consequences of structural changes on function are complicated and therefore must be evaluated by multiple techniques. We do observe basal localization of caveolin to mitochondria (this localization is enhanced by isoflurane) and PTX causes a slight but not significant reduction in mitochondrial caveolin in controls. It is possible that a small change in caveolin protein may have a large impact on mitochondrial structure. Isoflurane dramatically elevates mitochondrial respiration potentially through modulation of caveolin enrichment in mitochondria; this effect is completely blocked by PTX. The effect of PTX on control mitochondria is not quite as dramatic though some effect is present suggesting that PTX may have direct impact on mitochondrial structure/function possibly through a slight basal decrease in caveolin.
Caveolins can modulate signal transduction by interacting with multiple GPCRs, Gα subunits of heterotrimeric G-proteins, Src kinases, PI3K, endothelial nitric oxide synthase, protein kinase C isoforms, extracellular signal-regulated kinase 1 and 2, and superoxide dismutase (among other regulatory proteins). Many of these proteins may be regulated via binding to scaffolding domains of caveolin41. Recent data shows spatial organization of signaling molecules within caveolar microdomains, and the interaction of signaling molecules with caveolins helps determine the resistance of the heart to IR. A large number of GPCRs have been localized to membrane caveolae. As implied in the caveolin signaling hypothesis, caveolae bring downstream effectors in proximity to GPCRs to promote receptor-, tissue-, and cell-specific signal transduction.18,42,43 These effectors are thought to reside in caveolae due to direct interaction with caveolins (via the caveolin scaffolding domain) or by other caveolae-associated proteins. In addition, volatile anesthetics modulate adenosine triphosphate-sensitive K+channel activity,44,45 the generation of reactive oxygen species, and mitochondrial permeability transition pore opening.46 In the current study, we found that isoflurane cardioprotection is accompanied by caveolin translocation to mitochondria after 30 min isoflurane exposure and 45 min washout, a process that may be critical to effective stress adaptation. We have reported that Cav-3 OE mice are resistant to IR injury, manifest enhanced Gi signaling, and have increased localization of caveolin in their cardiac mitochondria.20,24
Though we establish a role for Gi signaling as an early control point for anesthetic-induced alterations in membrane caveolae, a number of important questions remain unresolved, warranting further investigation. While we observe trafficking of caveolin to mitochondria, it is unclear what mitochondrial proteins caveolin interacts with or influences to alter structure and function. Defining the complement of mitochondrial caveolin binding partners may help to define mechanisms and novel targets for stress adaptation. Our EM evidence suggests PTX treatment leads to altered myocyte cyto- architecture and mitochondrial morphology. Studies to define the link between G-protein signaling and the cytoskeleton have been suggested though not in the context of myocardial stress resistance.47 Additionally, in isolated lung it has been shown that the phosphorylation of Cav- 1 by Src plays a critical role in trafficking of caveolae48 and is consistent with our previous observations regarding the interplay between isoflurane induced protection, Src, and Cav- 1.28 We do not delineate between Cav- 1 versus Cav-3 specific effects on mitochondrial function but focus on early upstream events that regulate trafficking of caveolins to mitochondria. It is possible that the two caveolin isoforms may have differential effects on mitochondrial structure and function; such characterization is the focus of our future work.
In summary, the current results show that Gi signaling plays an essential role in isoflurane- dependent changes in IR tolerance and caveolar- mitochondrial communication. Modulation of caveolae-mitochondrial interactions, and their regulation by upstream Gi-related signaling, may represent novel targets for promoting myocardial resistance to ischemia-reperfusion injury.
Acknowledgments
Grant Support: This work was supported by grants from the National Institutes of Health (Bethesda, Maryland) from the National Heart, Lung, Blood Institute to Dr. Hemal H. Patel (HL091071, HL107200) and Dr. David M. Roth (HL066941, HL115933) and the National Institute for Neurological Disorders and Stroke to Dr. Brian P. Head (NS073653); the United States Department of Veterans Affairs (Washington District of Columbia), to Dr. Hemal H. Patel (BX001963), Dr. David M. Roth (BX000783), and Dr. Brian P. Head (BX001225).
Footnotes
Presentation: The data in this manuscript were presented at the American Society of Anesthesiologists annual meeting in San Francisco, CA in October 15, 2013.
Disclosures: The authors declare no competing interests.
References
- 1.Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–53. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 2.Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011;301:H1723–41. doi: 10.1152/ajpheart.00553.2011. [DOI] [PubMed] [Google Scholar]
- 3.Palade G. Fine structure of blood capillaries. J Appl Physiol. 1953;24:1424. [Google Scholar]
- 4.Pike LJ. Lipid rafts: Bringing order to chaos. J Lipid Res. 2003;44:655–67. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Parton RG, Way M, Zorzi N, Stang E. Caveolin-3 associates with developing T-tubules during muscle differentiation. J Cell Biol. 1997;136:137–54. doi: 10.1083/jcb.136.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chun M, Liyanage UK, Lisanti MP, Lodish HF. Signal transduction of a G protein-coupled receptor in caveolae: Colocalization of endothelin and its receptor with caveolin. Proc Nat Acad Sci USA. 1994;91:11728–32. doi: 10.1073/pnas.91.24.11728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feron O, Balligand JL. Caveolins and the regulation of endothelial nitric oxide synthase in the heart. Cardiovasc Res. 2006;69:788–97. doi: 10.1016/j.cardiores.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 8.Sargiacomo M, Scherer PE, Tang Z, Kübler E, Song KS, Sanders MC, Lisanti MP. Oligomeric structure of caveolin: Implications for caveolae membrane organization. Proc Nat Acad Sci USA. 1995;92:9407–11. doi: 10.1073/pnas.92.20.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, Chu C, Kohtz DS, Lisanti MP. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem. 1996;271:15160–5. doi: 10.1074/jbc.271.25.15160. [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto T. Calcium pump of the plasma membrane is localized in caveolae. J Cell Biol. 1993;120:1147–57. doi: 10.1083/jcb.120.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujimoto T, Nakade S, Miyawaki A, Mikoshiba K, Ogawa K. Localization of inositol 1,4,5-trisphosphate receptor-like protein in plasmalemmal caveolae. J Cell Biol. 1992;119:1507–13. doi: 10.1083/jcb.119.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scriven DR, Klimek A, Lee KL, Moore ED. The molecular architecture of calcium microdomains in rat cardiomyocytes. Ann N Y Acad Sci. 2002;976:488–99. doi: 10.1111/j.1749-6632.2002.tb04783.x. [DOI] [PubMed] [Google Scholar]
- 13.Jones KA, Jiang X, Yamamoto Y, Yeung RS. Tuberin is a component of lipid rafts and mediates caveolin- 1 localization: Role of TSC2 in post-Golgi transport. Exp Cell Res. 2004;295:512–24. doi: 10.1016/j.yexcr.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 14.Peng Y, Akmentin W, Connelly MA, Lund-Katz S, Phillips MC, Williams DL. Scavenger receptor BI (SR-BI) clustered on microvillar extensions suggests that this plasma membrane domain is a way station for cholesterol trafficking between cells and high-density lipoprotein. Mol Biol Cell. 2004;15:384–96. doi: 10.1091/mbc.E03-06-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lisanti MP, Scherer PE, Tang Z, Sargiacomo M. Caveolae, caveolin and caveolin-rich membrane domains: A signalling hypothesis. Trends Cell Biol. 1994;4:231–5. doi: 10.1016/0962-8924(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 16.Williams TM, Lisanti MP. The caveolin proteins. Genome Biol. 2004;5:214. doi: 10.1186/gb-2004-5-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinberg SF, Brunton LL. Compartmentation of g protein-coupled signaling pathways in cardiac myocytes. Annu Rev Pharmacol Toxicol. 2001;41:751–73. doi: 10.1146/annurev.pharmtox.41.1.751. [DOI] [PubMed] [Google Scholar]
- 18.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–79. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 19.Li WP, Liu P, Pilcher BK, Anderson RG. Cell-specific targeting of caveolin- 1 to caveolae, secretory vesicles, cytoplasm or mitochondria. J Cell Sci. 2001;114:1397–408. doi: 10.1242/jcs.114.7.1397. [DOI] [PubMed] [Google Scholar]
- 20.Fridolfsson HN, Kawaraguchi Y, Ali SS, Panneerselvam M, Niesman IR, Finley JC, Kellerhals SE, Migita MY, Okada H, Moreno AL, Jennings M, Kidd MW, Bonds JA, Balijepalli RC, Ross RS, Patel PM, Miyanohara A, Chen Q, Lesnefsky EJ, Head BP, Roth DM, Insel PA, Patel HH. Mitochondria-localized caveolin in adaptation to cellular stress and injury. FASEB J. 2012;26:4637–49. doi: 10.1096/fj.12-215798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaul PW, Anderson RG. Role of plasmalemmal caveolae in signal transduction. Am J Physiol. 1998;275:L843–51. doi: 10.1152/ajplung.1998.275.5.L843. [DOI] [PubMed] [Google Scholar]
- 22.Patel HH, Murray F, Insel PA. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol. 2008;48:359–91. doi: 10.1146/annurev.pharmtox.48.121506.124841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horikawa YT, Panneerselvam M, Kawaraguchi Y, Tsutsumi YM, Ali SS, Balijepalli RC, Murray F, Head BP, Niesman IR, Rieg T, Vallon V, Insel PA, Patel HH, Roth DM. Cardiac-specific overexpression of caveolin- 3 attenuates cardiac hypertrophy and increases natriuretic Peptide expression and signaling. J Am Coll Cardiol. 2011;57:2273–83. doi: 10.1016/j.jacc.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsutsumi YM, Horikawa YT, Jennings MM, Kidd MW, Niesman IR, Yokoyama U, Head BP, Hagiwara Y, Ishikawa Y, Miyanohara A, Patel PM, Insel PA, Patel HH, Roth DM. Cardiac-specific overexpression of caveolin- 3 induces endogenous cardiac protection by mimicking ischemic preconditioning. Circulation. 2008;118:1979–88. doi: 10.1161/CIRCULATIONAHA.108.788331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tong H, Rockman HA, Koch WJ, Steenbergen C, Murphy E. G protein-coupled receptor internalization signaling is required for cardioprotection in ischemic preconditioning. Circ Res. 2004;94:1133–41. doi: 10.1161/01.RES.0000126048.32383.6B. [DOI] [PubMed] [Google Scholar]
- 26.Headrick JP, Willems L, Ashton KJ, Holmgren K, Peart J, Matherne GP. Ischaemic tolerance in aged mouse myocardium: The role of adenosine and effects of A1 adenosine receptor overexpression. J Physiol. 2003;549:823–33. doi: 10.1113/jphysiol.2003.041541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reichelt ME, Willems L, Hack BA, Peart JN, Headrick JP. Cardiac and coronary function in the Langendorff-perfused mouse heart model. Exp Physiol. 2009;94:54–70. doi: 10.1113/expphysiol.2008.043554. [DOI] [PubMed] [Google Scholar]
- 28.Patel HH, Tsutsumi YM, Head BP, Niesman IR, Jennings M, Horikawa Y, Huang D, Moreno AL, Patel PM, Insel PA, Roth DM. Mechanisms of cardiac protection from ischemia/reperfusion injury: A role for caveolae and caveolin- 1. FASEB J. 2007;21:1565–74. doi: 10.1096/fj.06-7719com. [DOI] [PubMed] [Google Scholar]
- 29.Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: A common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15:69–75. doi: 10.1016/j.tcm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Schultz JeJ, Hsu AK, Nagase H, Gross GJ. TAN-67, a delta 1-opioid receptor agonist, reduces infarct size via activation of Gi/o proteins and KATP channels. Am J Physiol Heart Circ Physiol. 1998;274:H909–14. doi: 10.1152/ajpheart.1998.274.3.H909. [DOI] [PubMed] [Google Scholar]
- 31.Schultz JEJ, Hsu AK, Barbieri JT, Li PL, Gross GJ. Pertussis toxin abolishes the cardioprotective effect of ischemic preconditioning in intact rat heart. Am J Physiol Heart Circ Physiol. 1998;275:H495–H500. doi: 10.1152/ajpheart.1998.275.2.H495. [DOI] [PubMed] [Google Scholar]
- 32.Roth DM, Patel HH. Role of caveolae in cardiac protection. Pediatr Cardiol. 2011;32:329–33. doi: 10.1007/s00246-010-9881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaugg M, Lucchinetti E, Uecker M, Pasch T, Schaub MC. Anaesthetics and cardiac preconditioning. Part I Signalling and cytoprotective mechanisms. Br J Anaesth. 2003;91:551–65. doi: 10.1093/bja/aeg205. [DOI] [PubMed] [Google Scholar]
- 34.Kersten JR, Schmeling TJ, Pagel PS, Gross GJ, Warltier DC. Isoflurane mimics ischemic preconditioning via activation of KATP channels: Reduction of myocardial infarct size with an acute memory phase. Anesthesiology. 1997;87:361–70. doi: 10.1097/00000542-199708000-00024. [DOI] [PubMed] [Google Scholar]
- 35.Horikawa YT, Patel HH, Tsutsumi YM, Jennings MM, Kidd MW, Hagiwara Y, Ishikawa Y, Insel PA, Roth DM. Caveolin- 3 expression and caveolae are required for isoflurane-induced cardiac protection from hypoxia and ischemia/reperfusion injury. J Mol Cell Cardiol. 2008;44:123–30. doi: 10.1016/j.yjmcc.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minami K, Uezono Y. The effects of anesthetics on G-protein-coupled receptors. Masui. 2005;54:118–25. [PubMed] [Google Scholar]
- 37.Ludwig LM, Patel HH, Gross GJ, Kersten JR, Pagel PS, Warltier DC. Morphine enhances pharmacological preconditioning by isoflurane: Role of mitochondrial K(ATP) channels and opioid receptors. Anesthesiology. 2003;98:705–11. doi: 10.1097/00000542-200303000-00019. [DOI] [PubMed] [Google Scholar]
- 38.DeGeorge BR, Jr, Gao E, Boucher M, Vinge LE, Martini JS, Raake PW, Chuprun JK, Harris DM, Kim GW, Soltys S, Eckhart AD, Koch WJ. Targeted inhibition of cardiomyocyte Gi signaling enhances susceptibility to apoptotic cell death in response to ischemic stress. Circulation. 2008;117:1378–87. doi: 10.1161/CIRCULATIONAHA.107.752618. [DOI] [PubMed] [Google Scholar]
- 39.Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE, Malik AB. Endothelial cell-surface gp60 activates vesicle formation and trafficking via G(i)-coupled Src kinase signaling pathway. J Cell Biol. 2000;150:1057–70. doi: 10.1083/jcb.150.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murry CE, Richard VJ, Reimer KA, Jennings RB. Ischemic preconditioning slows energy metabolism and delays ultrastructural damage during a sustained ischemic episode. Circ Res. 1990;66:913–31. doi: 10.1161/01.res.66.4.913. [DOI] [PubMed] [Google Scholar]
- 41.Krajewska WM, Maslowska I. Caveolins: Structure and function in signal transduction. Cell Mol Biol Lett. 2004;9:195–220. [PubMed] [Google Scholar]
- 42.Insel PA, Patel HH. Do studies in caveolin-knockouts teach us about physiology and pharmacology or instead, the ways mice compensate for ‘lost proteins’? Br J Pharmacol. 2007;150:251–4. doi: 10.1038/sj.bjp.0706981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ostrom RS, Bundey RA, Insel PA. Nitric oxide inhibition of adenylyl cyclase type 6 activity is dependent upon lipid rafts and caveolin signaling complexes. J Biol Chem. 2004;279:19846–53. doi: 10.1074/jbc.M313440200. [DOI] [PubMed] [Google Scholar]
- 44.Marinovic J, Bosnjak ZJ, Stasnicka A. Preconditioning by isoflurane induces lasting sensitization of the cardiac sarcolemmal adenosine triphosphate-sensitive potassium channel by a protein kinase C-delta-mediated mechanism. Anesthesiology. 2005;103:540–7. doi: 10.1097/00000542-200509000-00017. [DOI] [PubMed] [Google Scholar]
- 45.Ludwig LM, Weihrauch D, Kersten JR, Pagel PS, Warltier DC. Protein kinase C translocation and Src protein tyrosine kinase activation mediate isoflurane-induced preconditioning in vivo: Potential downstream targets of mitochondrial adenosine triphosphate-sensitive potassium channels and reactive oxygen species. Anesthesiology. 2004;100:532–9. doi: 10.1097/00000542-200403000-00011. [DOI] [PubMed] [Google Scholar]
- 46.Krolikowski JG, Bienengraeber M, Weihrauch D, Warltier DC, Kersten JR, Pagel PS. Inhibition of mitochondrial permeability transition enhances isoflurane-induced cardioprotection during early reperfusion: The role of mitochondrial KATP channels. Anesth Analg. 2005;101:1590–6. doi: 10.1213/01.ANE.0000181288.13549.28. [DOI] [PubMed] [Google Scholar]
- 47.Schappi JM, Krbanjevic A, Rasenick MM. Tubulin, actin and heterotrimeric G proteins: Coordination of signaling and structure. Biochim Biophys Acta. 2013;1838:674–81. doi: 10.1016/j.bbamem.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu G, Schwartz DE, Shajahan AN, Visintine DJ, Salem MR, Crystal GJ, Albrecht RF, Vogel SM, Minshall RD. Isoflurane, but not sevoflurane, increases transendothelial albumin permeability in the isolated rat lung: Role for enhanced phosphorylation of caveolin- 1. Anesthesiology. 2006;104:777–85. doi: 10.1097/00000542-200604000-00023. [DOI] [PubMed] [Google Scholar]