

Abstract
Injury to the blood-brain barrier (BBB) is a key feature of intracerebral hemorrhage (ICH) and may contribute to perihematomal cell injury. Pretreatment with the heme oxygenase (HO)-1 inducer hemin improves barrier function and neurological outcome in experimental models of traumatic and ischemic CNS injury. Since hemin is already in clinical use to treat acute porphyrias, this translational study was designed to test its effect on BBB function when initiated after ICH in two mouse models. At a dose similar to those used in most preconditioning studies (26 mg/kg i.p.), post-hemorrhage treatment with hemin reduced parenchymal extravasation of Evans blue by about three-quarters in both the blood injection and collagenase ICH models. Similar efficacy was observed when treatment was begun at one or three hours. At the lower dose that is currently in clinical use (4 mg/kg beginning at 3 hours), hemin also improved barrier function in both models, as assessed by both Evans blue and FITC-dextran leakage; however, it was somewhat less potent, reducing Evans blue leakage by about half. This dose was nevertheless sufficient to attenuate striatal cell loss and accelerate neurological recovery. Consistent with prior observations, striatal HO-1 expression was increased by hemin, and was localized to perivascular cells. These results suggest that hemin may be an effective therapy for ICH with a clinically relevant time window. Further study of the repurposing of this old drug seems warranted.
Keywords: Heme, Heme oxygenase, Preconditioning, Stroke
Introduction
Hemin, the ferric form of heme with a chloride ligand, accumulates in intracranial hematomas (Letarte et al., 1993). It may contribute to cell injury in adjacent tissue by a direct cytotoxic effect and also by release of redox-active iron with its breakdown (Robinson et al., 2009). However, at nontoxic concentrations, hemin delivers a potent preconditioning stimulus to cultured cells that protects them from subsequent exposure to toxic concentrations of hemin and other oxidants (Balla et al., 1992; Li et al., 2009; Regan et al., 2000). This effect, which requires a preconditioning interval of several hours (Balla et al., 1993), is mediated at least in part by induction of heme oxygenase (HO)-1 (Belcher et al., 2006; Li et al., 2009). In vivo, systemic pretreatment with hemin was protective in multiple acute injury models, including brain (Takizawa et al., 1998; Zhang et al., 2008), heart (Clark et al., 2000; Hangaishi et al., 2000), kidney (Demirogullari et al., 2006), liver (Xue et al., 2007), and gut (Attuwaybi et al., 2004) ischemia/reperfusion, spinal cord trauma (Yamauchi et al., 2004), colitis (Zhong et al., 2010), and pancreatitis (Habtezion et al., 2011). Hemin is excluded from most organs after parenteral injection due to rapid binding to hemopexin or albumin (Linden et al., 1987), and its primary site of action appears to be the microvasculature (Belcher et al., 2006; Holzen et al., 2008; Lindenblatt et al., 2004). In the CNS, this resulted in a significant attenuation of blood-spinal cord barrier permeability in a mouse weight-drop contusion model (Yamauchi et al., 2004).
Hematin, which is ferric heme with a hydroxide ligand rather than chloride, is currently FDA-approved to treat neurovisceral symptoms associated with the acute porphyrias. A related compound, hemin arginate, is marketed for the same indication in Europe. Although the putative mechanism of action in these diseases is feedback inhibition of δ-aminolevulinic acid synthase rather than HO-1 induction (Anderson et al., 2005), the safety of ferric heme has already been established by decades of clinical use. It is somewhat surprising, then, that the therapeutic potential of hemin or hematin postconditioning has not been addressed in translational studies to date. One possible explanation is an inadequate time window. Since hemin is not directly protective, but rather upregulates an endogenous defense against oxidative stress and inflammation, its onset of action is likely to be delayed for several hours. It therefore may not be an ideal agent for treating rapidly-progressing insults such as CNS ischemia or trauma.
Two factors suggest that parenteral hemin may be a particularly attractive drug therapy for intracerebral hemorrhage (ICH). First, the heme-mediated component of injury after ICH is delayed for at least 1–2 days in experimental models (Xi et al., 1998), providing an adequate time window to manifest the protective effects of hemin conditioning. This delay represents the interval required for both erythrocyte lysis and hemoglobin oxidation to methemoglobin, which has a lower affinity for its heme moieties and releases them to lipid and protein binding sites (Bunn and Jandl, 1968). Second, blood-brain barrier breakdown is a prominent feature of ICH, is hypothesized to contribute to its poor neurological outcome, and is a likely target of systemic hemin therapy (Keep et al., 2008; Yamauchi et al., 2004). However, no publication to date has tested hemin in any model that is relevant to spontaneous ICH. The goal of this translational project was to assess the effect of hemin therapy when treatment was begun after ICH, specifically testing the hypotheses that it would improve blood-brain barrier function and neurological outcome.
Materials and Methods
Experimental Animals
All experiments were conducted using protocols that were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee. Swiss-Webster mice (138 male and 98 female) were purchased from Taconic. They were housed in a facility that is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, and were used for experiments when 11–13 weeks old. Animals were provided with food and water ad libitum throughout the course of all experiments.
ICH Models
Mice were anesthetized with 2% isoflurane in oxygen and were placed into a stereotactic frame (David Kopf Instruments). Collagenase (Type VIIS, Sigma-Aldrich, St. Louis, MO) and blood injections followed previously described procedures (Chen et al., 2011). A 28–33 g needle attached to a glass Hamilton syringe was inserted into the right striatum at the following stereotaxic coordinates relative to bregma: 0.5 mm rostral, 2.5 mm lateral, 3.5 mm below the skull surface. After 10 min, collagenase was injected in 1 μl artificial CSF (NaCl 148 mmol/L, KCl 3 mmol/L, CaCL2 1 mmol/L, MgCL2 0.8 mmol/L, Na2HPO4 0.8 mmol/L, and NaH2PO4 0.2 mmol/L) over 1 minute. Due to variability in the potency of different collagenase lots, the dose of each lot was adjusted to that which produced ~50% reduction in striatal cell viability via MTT assay (Chen-Roetling et al., 2013). Dose range was 0.014–0.074 units, and all direct comparisons were made between groups receiving the same collagenase dose. For striatal autologous blood injection, blood was collected from a tail vein and injected at the above coordinates at a rate of 1 μl/minute. Ten minutes after completion of collagenase or blood injection, the needle was slowly removed and the wound was closed with sutures. Surgical control mice were subjected to needle trauma only. Mice were recovered in a warm environment.
Hemin injection
Hemin (Frontier Scientific, Logan, UT) was prepared under reduced light immediately prior to use as a 2 mM solution in sodium phosphate buffer, pH 7.4. It was further diluted in sterile saline and was injected i.p. at one or three hours after striatal collagenase or blood injection, at the following doses: 1) 26 mg/kg, repeated 24 hours later; 2) 4 mg/kg, repeated 24 and 48 hours later. Pilot studies demonstrated that a single repeat dose of 4 mg/kg at 24 hours was sufficient to increase perihematomal cell viability in the blood injection model as quantified by MTT assay, so the third dose was omitted in those experiments to minimize animal discomfort. Control mice were injected with an equal volume of vehicle. In order to control for reduced edema and behavioral deficits in female mice as previously reported by Nakamura et al. (Nakamura et al., 2004), gender ratios were balanced in hemin and vehicle groups.
Assessment of Blood-Brain Barrier Permeability
Protein permeability was quantified by Evans blue, which binds to albumin. Mice (5–8/condition) were injected with 2% Evans blue in sterile saline, 4 ml/kg i.p. Three hours later, under isoflurane anesthesia, they were perfused with 50 ml PBS through the left ventricle to remove intravascular dye, and were euthanized by cervical dislocation. Striata were dissected free and weighed; Evans blue extraction and assay followed the method of Uyama et al. (Uyama et al., 1988). Tissue was homogenized in 200 μl 50% trichloracetic acid. After debris removal by centrifugation, the supernatant was collected and diluted 1:3 in 100% ethanol. Evans blue fluorescence (ex: 620 nm, em: 680 nm) was then measured using a Perkin Elmer fluorescence spectrometer. In order to quantify leakage of lower molecular weight molecules, additional mice (4–7/condition) were injected with 2% fluorescein isothiocyanate (FITC)-dextran (MW 3–5 kDa, 4 mg/kg, Sigma-Aldrich) rather than Evans blue. Five minutes later, mice were euthanized and striata were removed, homogenized in PBS, and centrifuged. The supernatant fluorescence (ex: 490 nm, em: 520 nm) was then measured. Mean fluorescence intensity (MFI) was normalized to that in vehicle-treated controls injected with collagenase (= 100).
Striatal Water Content
At three days after ICH, injected and contralateral striata (n = 10) were excised and weighed. After drying in an oven at 95°C for 24 hours, tissue samples were weighed again. Water content was calculated by subtracting dry weight from wet weight. The water content of the contralateral striatum was subtracted from that of the corresponding injected striatum to yield the value produced by ICH-mediated injury.
MTT Striatal Viability Assay
The close correlation of this method with stereology-based cell counts of tissue sections has recently been described (Chen-Roetling et al., 2013). At 5 days after striatal blood or collagenase injection, mice (6–8/condition) were deeply anesthetized with isoflurane and euthanized by cervical dislocation. Injected and contralateral striata were rapidly removed, minced with forceps, and dissociated by trituration in Hanks Balanced Salt Solution supplemented with 27.8 mM glucose, 20.5 mM sucrose, and 4.2 mM sodium bicarbonate. One ml of 0.25 mg/ml MTT was added, and the cell suspension was incubated at 37°C for four minutes. After centrifugation (1380 x g, 2 minute), the supernatant was discarded, and formazan was immediately extracted in 2 ml isopropanol. The absorbance of this solution was then quantified at 562 nm, and was normalized to the absorbance of the solution obtained from the contralateral striatum.
Behavioral Testing
Neurological outcome was assessed in mice (11–12/condition) receiving striatal collagenase injections followed by treatment with vehicle or hemin (4 mg/kg i.p.) at 3 hours, 27 hours, and 51 hours. Testing was conducted on the same mice at 3, 5, 10, 14, 21 and 28 days after ICH, using a previously described protocol (Chen-Roetling et al., 2013; Chen et al., 2011).
Adhesive removal test
Adhesive dots (3 mm diameter) were cut from electrical tape and attached to the left or right forepaw. The interval until the mouse noticed the dot and the subsequent time needed for removal were recorded by a blinded observer. Mean times from the right (ipsilateral to hemorrhage) forepaw was subtracted from that of the left to calculate an asymmetry score (MacLellan et al., 2006). Four training sessions were conducted prior to data collection.
Elevated body swing test
After habituation in a Plexiglass box for 10 minutes, the mouse was held about 3 cm from the base of its tail, and lifted so that its head was about 3 cm above the box floor. Left and right swings were recorded by a blinded observer over 45 seconds and the percentage of left swings was calculated. A right striatal lesion produces a leftward swinging bias (Liu et al., 2008).
Spontaneous locomotor activity
A video camera (Sony DCR-HC62) was positioned perpendicular to the long axis of a standard cage housing one mouse. Activity was recorded for three hours (12–3PM). Videos were then analyzed with HomeCageScan (Version 3.0, Clever Systems Inc., Reston, VA USA), quantifying the following behaviors: walking, feeding, vertical hanging, rearing, and jumping. The time spent in each behavior was summed to yield an index of total activity.
Immunoblotting and immunostaining
Anti-HO-1 (Enzo Life Sciences, Inc, Farmingdale, NY, USA, Catalog # ADI-SPA-895) was used at a dilution of 1:4000 for immunoblotting (n = 4/condition) and 1:4000 or 1:8000 for immunostaining (n = 3/condition). Both methods have been described in detail in prior publications (Chen-Roetling et al., 2013; Chen-Roetling et al., 2012).
Statistical Analysis
Differences between three or more groups were analyzed with one way analysis of variance (ANOVA) and the Bonferroni multiple comparisons test. Differences between two groups were analyzed with an unpaired t test. Behavioral testing data collected at multiple time points over a 4 week interval was analyzed by two-way ANOVA. GraphPad Prism software was used for all analyses.
Results
Mortality
Striatal injections were generally well-tolerated. All mice injected with autologous blood (n = 69) survived until completion of the experiment. Eight of 143 mice injected with collagenase died within 48 hours. Two had been treated with hemin alone, one with tin protoporphyrin IX alone, and five with vehicle.
Systemic hemin therapy reduces blood-brain barrier permeability after ICH
In initial experiments, mice were treated with 40 μmoles/kg (26 mg/kg) hemin i.p. after striatal blood or collagenase injection (Figs. 1, 2). This dose replicated that used by Belcher et al. (Belcher et al., 2006) to increase vascular HO-1 expression and reduce inflammation in mice. Consistent with prior observations (MacLellan et al., 2008), striatal autologous blood injection produced less Evans blue leakage than collagenase. Hemin therapy attenuated BBB disruption at 3 days when administered 1 hour and 25 hours after blood injection; a single dose did not have a significant effect. A similar degree of barrier protection was observed when ICH was produced by striatal collagenase injection rather than blood. Delaying the first hemin injection until 3 hours after ICH induction did not significantly diminish this effect.
Fig. 1.

Systemic hemin therapy decreases blood-brain barrier disruption after intracerebral hemorrhage. A) ICH was induced by striatal autologous blood injection. Mice were treated with 26 mg/kg hemin i.p. 1 hour later, as a one-time dose (n = 6), or repeated in 24 hours (n = 6); controls received vehicle i.p. only. Bars represent mean fluorescence intensity (MFI, ± S.E.M.) of Evans blue at 3 days after hemorrhage induction. B) MFI at 3 days after striatal collagenase injection in mice receiving 26 mg/kg hemin at 1 h (n = 7) or 3 h (n = 5), and again 24 h later, or an equal volume of vehicle. *P < 0.05, **P < 0.01 v. mean value in vehicle-treated controls.
Fig. 2.
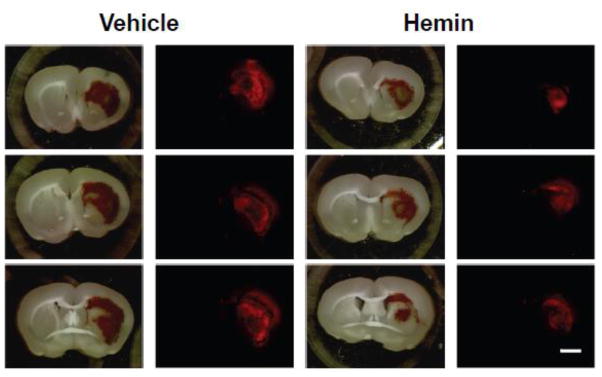
Representative brain slices 3 days after striatal collagenase injection and 3 hours after Evans blue injection, from mice treated with vehicle or hemin 26 mg/kg i.p. at 3 and 27 hours after collagenase. Red Evans blue fluorescence (Em:680nm) is reduced in hemin-treated slices.
Efficacy of a dosing schedule already in clinical use
Although a hemin dose approximating 26 mg/kg hemin has frequently been employed in animal studies (Attuwaybi et al., 2004; Belcher et al., 2006; Hangaishi et al., 2000; Takizawa et al., 1998), the dose of ferric heme that is currently approved for human use is 1–4 mg/kg administered daily for 3–14 days, with a recommended daily dose of 3–4 mg/kg (Anderson et al., 2005) and a maximum of 6 mg/kg/24h. To further test the translational relevance of hemin therapy, additional experiments were conducted using a similar dose (4 mg/kg). Evans blue leakage into the striatal parenchyma in mice treated with this lower hemin dose was somewhat greater than at the higher dose but was about half of that observed in vehicle-treated mice (Fig. 3). In order to determine if this regimen also reduced extravasation of lower molecular weight compounds, additional mice were injected with FITC-dextran (MW 3–5 kDa) prior to perfusion rather than Evans blue. Decreased striatal FITC-dextran fluorescence was also observed in hemin-treated mice in both models.
Fig. 3.

Low-dose hemin therapy protects the blood-brain barrier after ICH. Mice received 4 mg/kg hemin or vehicle control at 3, 27, and 51 hours after striatal blood or collagenase injection. Barrier function was assessed at 3 days after blood or collagenase injection by Evans blue assay (A, n = 6 for collagenase groups and n = 7 for blood injection groups) or FITC-dextran assay (MW 3–5 kDa, n = 6 for collagenase hemin group, n = 4 for collagenase vehicle group, n = 7 for blood injection groups). Sham mice (n = 5) received anesthesia and needle trauma only. *P < 0.05, **P < 0.01, ***P < 0.001 v. mean value in vehicle-treated controls.
Hemin reduces perihematomal cell injury
Striatal cell viability at 5 days after collagenase injection followed by vehicle treatment was 38.0±3.4% of contralateral (Fig. 4), as measured by MTT assay, which correlates well with histological and fluorescent methods (Chen-Roetling et al., 2013). It was increased to 52.6±3.9% by low-dose hemin treatment. Striatal blood injection followed by vehicle reduced viability to 57.1±3.3% of contralateral, and was increased to 80.2±4.3% by hemin treatment.
Fig 4.

Hemin increases perihematomal cell viability. A) ICH was modeled in 2–3 month old mice by striatal injection of autologous blood (n = 6) or collagenase (n=8 for hemin group and 7 for vehicle group), followed 3 hours later by treatment with 4 mg/kg hemin i.p. or vehicle. Hemin or vehicle was repeated 24 hours later for blood injection groups and 24 and 48 hours later for collagenase groups. Five days after ICH, striatal cell viability was quantified by MTT assay. B) Representative brain slices were stained with MTT 5 days after striatal injection of autologous blood, followed by hemin or vehicle treatment as above. Purple staining indicates cell viability.
Systemic hemin therapy improves neurological outcome after striatal hemorrhage
Since hemin had a similar effect on barrier function and cell viability in the collagenase and blood injection models, neurological outcome over four weeks was assessed using only the former, which produces a more sustained deficit than blood injection (MacLellan et al., 2008). Mice treated with hemin (4 mg/kg) recovered more rapidly, as determined by elevated body swing test and adhesive contact time (Fig. 5). A trend toward more rapid adhesive removal was also observed that did not reach statistical significance. Activity level as detected by video analysis was also greater over the 28 day testing interval after ICH in hemin-treated mice, compared with vehicle-treated controls.
Fig. 5.

Systemic hemin therapy improves neurological function after striatal hemorrhage. Mice received 4 mg/kg hemin (n = 11) or vehicle (n = 12) i.p at 3, 27, and 51 hours after collagenase injection. Bars represent mean values (±S.E.M.) at indicated days after ICH for: A) percentage left swings in elevated body swing test; B, C) adhesive tape contact and removal times (seconds); D) mean activity time (sum of seconds spent walking, feeding, vertical hanging, rearing and jumping during 3 hour video recording). Hemin vs. vehicle 2-way ANOVA P values; A, 0.0011; B, 0.0001; C, 0.2628; D, 0.001.
Hemin decreases striatal edema
Collagenase injection resulted in a significant increase in striatal water content 3 days later in vehicle-treated mice. This was reduced by treatment with 4 mg/kg hemin beginning at 3 hours after collagenase and repeated at 27 and 51 hours (Fig. 6).
Fig. 6.
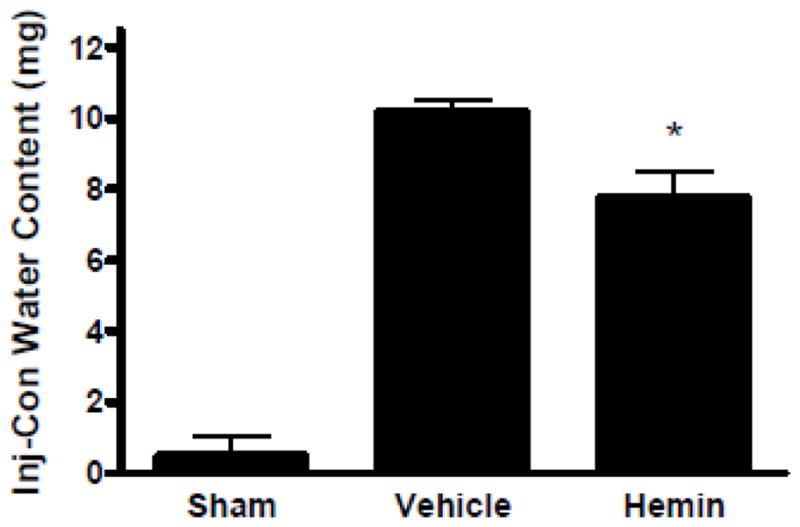
Low-dose hemin therapy decreases perihematomal edema after ICH. Mice received 4 mg/kg hemin or vehicle control at 3, 27, and 51 hours after collagenase injection. Striatal water content was quantified three days after ICH. *P < 0.05 v. mean value in vehicle-treated controls. n=10 for hemin and vehicle group, n = 5 for sham group.
Hemin increases striatal HO-1 expression
Consistent with observations by Yamauchi et al. in the murine spinal cord (Yamauchi et al., 2004), hemin therapy produced a modest but significant increase in total striatal HO-1 expression, as detected by Western blot density (Fig. 7A). Immunostaining of tissue sections indicated that this increase was localized to vascular cells (Fig. 7B), as was also noted by Yamauchi et al.
Fig. 7.

Hemin treatment increases HO-1 expression in the mouse striatum. A) Mice (4/condition) received 26 mg/kg hemin i.p for 1 or 2 days or an equal volume of vehicle. Striatal HO-1 expression was assessed 24 hours after last hemin dose. Actin is gel-loading control. Bar order correlates with lane order. B) Representative striatal sections from mice treated with hemin or vehicle as above for 2 days, stained with antibody to HO-1, showing cross sectional (B1–B4) and longitudinal (B5,B6) views of blood vessels. Immunoreactivity is indicated by brown staining; counterstain is cresyl violet. Scale bar = 10 μm, B1-4 and B5-6 share scale bars. *P < 0.05 vs. expression in vehicle-treated controls.
Effect of nonferric hemin analog on BBB function
Tin protoporphyrin IX (SnPPIX) is structurally similar to hemin, but with tin replacing ferric iron. It is not a HO substrate, but rather acts as a competitive inhibitor. Treatment with SnPPIX alone (4.6 mg/kg, equimolar to 4 mg/kg hemin, three daily doses) had no effect on Evans blue extravasation after ICH (Fig. 8). In mice treated with hemin plus SnPPIX, a weaker and more variable reduction in Evans blue leakage was observed compared with that in mice treated with 4 mg/kg hemin alone. However, a significant effect was still observed when compared with the mean signal in vehicle-treated mice.
Fig. 8.
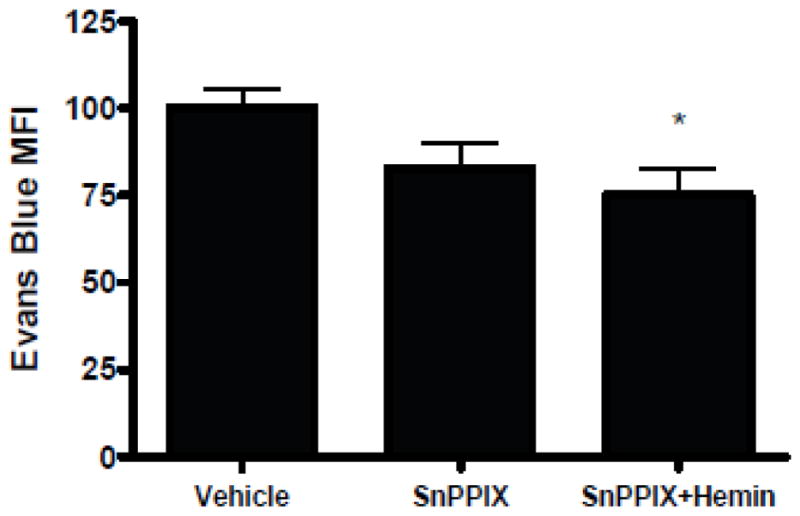
Effect of HO-1 inhibitor on barrier function after ICH. Bars represent mean striatal Evans blue fluorescence intensity three days after collagenase-induced ICH in mice treated at 3, 27, and 51 hours with hemin 4 mg/kg or equimolar tin protoporphyrin IX (SnPPIX, 4.6 mg/kg). *P < 0.05 vs. value in vehicle-treated controls, n = 7 for SnPPIX group, n = 8 for vehicle and hemin + SnPPIX groups).
Discussion
The results of this translational project identify hemin as a promising therapeutic candidate for ICH. Several prior studies had demonstrated the beneficial effects of hemin preconditioning in vitro (Balla et al., 1992; Li et al., 2009; Regan et al., 2000) and also in various acute injury models in vivo, including stroke (Takizawa et al., 1998), global CNS ischemia (Zhang et al., 2008), and spinal cord trauma (Yamauchi et al., 2004). The present study is the first to indicate that hemin is also protective when therapy is initiated hours after an acute CNS injury. At the higher dose that is similar to that used in most preconditioning experiments, a robust decrease in Evans blue leakage into the striatal parenchyma was observed at 3 days in both the collagenase and blood injection ICH models. At the lower dose that is currently approved for clinical use to treat the acute porphyrias, significant improvement in barrier function persisted, and was associated with cytoprotection and attenuation of neurological deficits over the four week interval after hemorrhage.
Further investigation is required to define the optimal hemin dose and therapeutic window after ICH. It is noteworthy that the tissue Evans blue signal was stronger in mice receiving the low dose schedule (4 mg/kg x 3) than the higher dose (26 mg/kg x 2). The maximum dose of hematin that currently can be administered to porphyria patients over a 24 hour period is 6 mg/kg. This limit is apparently based on a case report (Dhar et al., 1978) of reversible acute renal failure in a patient with mild baseline renal insufficiency who rapidly received an intravenous bolus of 1000 mg (12.2 mg/kg). It is possible that this or a higher dose would be tolerated if administered slowly or by an alternate route, or if accompanied by hydration and other supportive care to minimize the likelihood of acute kidney injury. In rodent studies, doses exceeding 10 mg/kg i.p. are routinely employed, without any reports of nephrotoxicity. Conversely, such doses have actually protected rodent kidneys from a variety of acute insults (Chok et al., 2009; Kang et al., 2013; Kim et al., 2006), while slowing or preventing loss of function in chronic renal failure models (Desbuards et al., 2009; Jadhav et al., 2009).
Hemin is a very potent HO-1 inducer and is also the preferred substrate of this enzyme. Its benefit is usually attributed to HO-1 catalytic activity and the antioxidant and anti-inflammatory properties of two breakdown products, bilirubin and carbon monoxide. Hemin catabolism also releases ferrous iron and so has the potential for toxicity, particularly to cell populations with weak antioxidant defenses such as neurons. The conflicting biological activity of these breakdown products may account for the variable effect of HO-1 in hemorrhagic CNS injury models. Wang and Doré reported that HO-1 knockout decreased perihematomal injury and inflammation after collagenase-induced ICH, (Wang and Doré, 2007). Conversely, vasospasm after subarachnoid hemorrhage was reduced by HO-1 gene transfer or treatment with the HO-1 protein fused to a polyarginine transduction domain (Ogawa et al., 2011; Ono et al., 2002), and was increased by antisense targeting of HO-1 (Suzuki et al., 1999). The latter observations and the present results are consistent with the hypothesis that HO-1 improves the function of vascular cells after a hemorrhagic CNS injury. However, parenchymal overexpression of HO-1 after ICH may be deleterious, perhaps by subjecting neurons or other vulnerable cell populations to elevated levels of redox active iron (Huang et al., 2002; Koeppen et al., 2004).
Concomitant administration of the HO-1 inhibitor tin protoporphyrin IX diminished the barrier protection provided by hemin, but did not completely reverse it. While this may reflect incomplete HO-1 inhibition, participation of other mechanisms in the protection provided by hemin cannot be excluded. Hemin is a highly reactive compound and has pleiotropic effects in vivo. In a microarray study, Correa-Costas and colleagues reported that i.p. injection of 25 mg/kg hemin increased expression of 346 genes in the mouse kidney within 24 hours (Correa-Costa et al., 2012). These included genes that regulate transcription, development, signal transduction, DNA repair, MAPKKK cascade, cell differentiation, coagulation cascade and amino acid metabolism. When followed by renal ischemia-reperfusion, hemin pretreatment specifically increased expression of genes relevant to vascular smooth muscle contraction, arachidonic acid metabolism, and metal ion response, while decreasing expression of genes linked to apoptosis, toll-like receptor signaling, hypoxia response, sodium ion transport, cell proliferation and cytoskeletal organization. These changes in the transcriptome may be mediated in part by the signaling function of catalytically active or inactive HO-1 protein, as reported in cell culture after HO-1 gene transfer or hemin treatment (Abraham et al., 2003; Lin et al., 2007). Alternatively, they may be due to direct regulation by hemin of transcription factors and signal transducers, including HRI kinase, Bach1, and the Ras-MAPK pathway (Mense and Zhang, 2006).
A compound with such a broad spectrum of biological activity would usually not be considered a serious candidate for clinical development, due to the high likelihood of multiple adverse effects. However, a favorable toxicity profile has already been established for hemin and hematin, despite relatively little effort to optimize their delivery and stability. Most adverse effects have been attributed to in vitro reaction products rather than ferric heme per se, and include local thrombophlebitis and a mild coagulopathy; both can be reduced or eliminated by preparing hemin liganded to arginine (Tenhunen et al., 1987). It is quite possible that the pleiotropic effects of hemin, whether downstream to or independent of HO-1 catalytic activity, may actually be advantageous if several injury cascades are mitigated in parallel. It is noteworthy that the focus on mechanism-specific therapies for stroke over the past two decades has not yet yielded an effective therapy, suggesting that targeting a single pathway may not be sufficient. While hemin is a very poor mechanistic probe, the present results demonstrate efficacy after experimental ICH with a clinically relevant time window. Further studies on the repurposing of this old drug to treat hemorrhagic stroke therefore seem warranted.
Highlights.
Effect of hemin therapy after intracerebral hemorrhage (ICH) was evaluated.
Hemin reduced blood-brain barrier disruption in two mouse models.
Increased perihematomal cell viability and neurologic recovery were also observed.
Further study of the repurposing of this old drug to treat ICH is warranted.
Acknowledgments
This study was supported by a grant from the National Institutes of Health (NS079500) to R.F.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham NG, et al. Human heme oxygenase: cell cycle-dependent expression and DNA microarray identification of multiple gene responses after transduction of endothelial cells. J Cell Biochem. 2003;90:1098–1111. doi: 10.1002/jcb.10736. [DOI] [PubMed] [Google Scholar]
- Anderson KE, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439–50. doi: 10.7326/0003-4819-142-6-200503150-00010. [DOI] [PubMed] [Google Scholar]
- Attuwaybi BO, et al. Heme oxygenase-1 induction by hemin protects against gut ischemia/reperfusion injury. J Surg Res. 2004;118:53–7. doi: 10.1016/j.jss.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Balla G, et al. Ferritin: A cytoprotective strategem of endothelium. J Biol Chem. 1992;267:18148–18153. [PubMed] [Google Scholar]
- Balla J, et al. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci U S A. 1993;90:9285–9. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcher JD, et al. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Invest. 2006;116:808–16. doi: 10.1172/JCI26857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunn HF, Jandl JH. Exchange of heme along hemoglobins and between hemoglobin and albumin. J Biol Chem. 1968;243:465–475. [PubMed] [Google Scholar]
- Chen-Roetling J, et al. A rapid fluorescent method to quantify neuronal loss after experimental intracerebral hemorrhage. J Neurosci Methods. 2013;216:128–36. doi: 10.1016/j.jneumeth.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Roetling J, et al. Effect of iron chelators on methemoglobin and thrombin preconditioning. Transl Stroke Res. 2012;3:452–9. doi: 10.1007/s12975-012-0195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, et al. Increased striatal injury and behavioral deficits after intracerebral hemorrhage in hemopexin knockout mice. J Neurosurg. 2011;114:1159–67. doi: 10.3171/2010.10.JNS10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chok MK, et al. Renoprotective potency of heme oxygenase-1 induction in rat renal ischemia-reperfusion. Inflamm Allergy Drug Targets. 2009;8:252–9. doi: 10.2174/187152809789352186. [DOI] [PubMed] [Google Scholar]
- Clark JE, et al. Heme oxygenase-1-derived bilirubin ameliorates postischemic myocardial dysfunction. Am J Physiol Heart Circ Physiol. 2000;278:H643–51. doi: 10.1152/ajpheart.2000.278.2.H643. [DOI] [PubMed] [Google Scholar]
- Correa-Costa M, et al. Transcriptome analysis of renal ischemia/reperfusion injury and its modulation by ischemic pre-conditioning or hemin treatment. PLoS One. 2012;7:e49569. doi: 10.1371/journal.pone.0049569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirogullari B, et al. A comparative study of the effects of hemin and bilirubin on bilateral renal ischemia reperfusion injury. Nephron Exp Nephrol. 2006;103:e1–5. doi: 10.1159/000090113. [DOI] [PubMed] [Google Scholar]
- Desbuards N, et al. Heme oxygenase-1 inducer hemin attenuates the progression of remnant kidney model. Nephron Exp Nephrol. 2009;113:e35–44. doi: 10.1159/000228081. [DOI] [PubMed] [Google Scholar]
- Dhar GJ, et al. Transitory renal failure following rapid administration of a relatively large amount of hematin in a patient with acute intermittent porphyria in clinical remission. Acta Med Scand. 1978;203:437–43. doi: 10.1111/j.0954-6820.1978.tb14903.x. [DOI] [PubMed] [Google Scholar]
- Habtezion A, et al. Panhematin provides a therapeutic benefit in experimental pancreatitis. Gut. 2011;60:671–9. doi: 10.1136/gut.2010.217208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangaishi M, et al. Induction of heme oxygenase-1 can act protectively against cardiac ischemia/reperfusion in vivo. Biochem Biophys Res Commun. 2000;279:582–8. doi: 10.1006/bbrc.2000.3973. [DOI] [PubMed] [Google Scholar]
- Holzen JP, et al. Influence of heme oxygenase-1 on microcirculation after kidney transplantation. J Surg Res. 2008;148:126–35. doi: 10.1016/j.jss.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Huang FP, et al. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- Jadhav A, et al. Hemin therapy attenuates kidney injury in deoxycorticosterone acetate-salt hypertensive rats. Am J Physiol Renal Physiol. 2009;296:F521–34. doi: 10.1152/ajprenal.00510.2007. [DOI] [PubMed] [Google Scholar]
- Kang K, et al. Heme oxygenase 1 modulates thrombomodulin and endothelial protein C receptor levels to attenuate septic kidney injury. Shock. 2013;40:136–43. doi: 10.1097/SHK.0b013e31829d23f5. [DOI] [PubMed] [Google Scholar]
- Keep RF, et al. Blood-brain barrier function in intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:73–7. doi: 10.1007/978-3-211-09469-3_15. [DOI] [PubMed] [Google Scholar]
- Kim JH, et al. Heme oxygenase-1 protects rat kidney from ureteral obstruction via an antiapoptotic pathway. J Am Soc Nephrol. 2006;17:1373–81. doi: 10.1681/ASN.2005091001. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, et al. Heme oxygenase in experimental intracerebral hemorrhage: the benefit of tin-mesoporphyrin. J Neuropathol Exp Neurol. 2004;63:587–597. doi: 10.1093/jnen/63.6.587. [DOI] [PubMed] [Google Scholar]
- Letarte PB, et al. Hemin: levels in experimental subarachnoid hematoma and effects on dissociated vascular smooth muscle cells. J Neurosurg. 1993;79:252–255. doi: 10.3171/jns.1993.79.2.0252. [DOI] [PubMed] [Google Scholar]
- Li RC, et al. Heme-hemopexin complex attenuates neuronal cell death and stroke damage. J Cereb Blood Flow Metab. 2009;29:953–64. doi: 10.1038/jcbfm.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem. 2007;282:20621–33. doi: 10.1074/jbc.M607954200. [DOI] [PubMed] [Google Scholar]
- Linden IB, et al. Fate of haem after parenteral administration of haem arginate to rabbits. J Pharm Pharmacol. 1987;39:96–102. doi: 10.1111/j.2042-7158.1987.tb06952.x. [DOI] [PubMed] [Google Scholar]
- Lindenblatt N, et al. Vascular heme oxygenase-1 induction suppresses microvascular thrombus formation in vivo. Arterioscler Thromb Vasc Biol. 2004;24:601–6. doi: 10.1161/01.ATV.0000118279.74056.8a. [DOI] [PubMed] [Google Scholar]
- Liu DZ, et al. Src kinase inhibition decreases thrombin-induced injury and cell cycle re-entry in striatal neurons. Neurobiol Dis. 2008;30:201–11. doi: 10.1016/j.nbd.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLellan CL, et al. Gauging recovery after hemorrhagic stroke in rats: implications for cytoprotection studies. J Cereb Blood Flow Metab. 2006;26:1031–42. doi: 10.1038/sj.jcbfm.9600255. [DOI] [PubMed] [Google Scholar]
- MacLellan CL, et al. Intracerebral hemorrhage models in rat: comparing collagenase to blood infusion. J Cereb Blood Flow Metab. 2008;28:516–25. doi: 10.1038/sj.jcbfm.9600548. [DOI] [PubMed] [Google Scholar]
- Mense SM, Zhang L. Heme: a versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006;16:681–92. doi: 10.1038/sj.cr.7310086. [DOI] [PubMed] [Google Scholar]
- Nakamura T, et al. Intracerebral hemorrhage in mice: model characterization and application for genetically modified mice. J Cereb Blood Flow Metab. 2004;24:487–94. doi: 10.1097/00004647-200405000-00002. [DOI] [PubMed] [Google Scholar]
- Ogawa T, et al. Protein therapy using heme-oxygenase-1 fused to a polyarginine transduction domain attenuates cerebral vasospasm after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2011;31:2231–42. doi: 10.1038/jcbfm.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S, et al. Heme oxygenase-1 gene therapy for prevention of vasospasm in rats. J Neurosurg. 2002;96:1094–102. doi: 10.3171/jns.2002.96.6.1094. [DOI] [PubMed] [Google Scholar]
- Regan RF, et al. Heme oxygenase-1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci Lett. 2000;282:1–4. doi: 10.1016/s0304-3940(00)00817-x. [DOI] [PubMed] [Google Scholar]
- Robinson SR, et al. Hemin toxicity: a preventable source of brain damage following hemorrhagic stroke. Redox Rep. 2009;14:228–35. doi: 10.1179/135100009X12525712409931. [DOI] [PubMed] [Google Scholar]
- Suzuki S, et al. Heme oxygenase-1 gene induction as an intrinsic regulation against delayed cerebral vasospasm in rats. J Clin Invest. 1999;104:59–66. doi: 10.1172/JCI5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa S, et al. Induction of heme oxygenase protein protects neurons in cortex and striatum, but not hippocampus, against transient forebrain ischemia. J Cereb Blood Flow Metab. 1998;18:559–569. doi: 10.1097/00004647-199805000-00011. [DOI] [PubMed] [Google Scholar]
- Tenhunen R, et al. Haem arginate: a new stable haem compound. J Pharm Pharmacol. 1987;39:780–6. doi: 10.1111/j.2042-7158.1987.tb05119.x. [DOI] [PubMed] [Google Scholar]
- Uyama O, et al. Quantitative evaluation of vascular permeability in the gerbil brain after transient ischemia using Evans blue fluorescence. J Cereb Blood Flow Metab. 1988;8:282–4. doi: 10.1038/jcbfm.1988.59. [DOI] [PubMed] [Google Scholar]
- Wang J, Doré S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007;130:1643–52. doi: 10.1093/brain/awm095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi GH, et al. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg. 1998;89:991–996. doi: 10.3171/jns.1998.89.6.0991. [DOI] [PubMed] [Google Scholar]
- Xue H, et al. Heme oxygenase-1 induction by hemin protects liver cells from ischemia/reperfusion injury in cirrhotic rats. World J Gastroenterol. 2007;13:5384–90. doi: 10.3748/wjg.v13.i40.5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, et al. Hemin induces heme oxygenase-1 in spinal cord vasculature and attenuates barrier disruption and neutrophil infiltration in the injured murine spinal cord. J Neurotrauma. 2004;21:1017–30. doi: 10.1089/0897715041651042. [DOI] [PubMed] [Google Scholar]
- Zhang B, et al. Effects of heme oxygenase 1 on brain edema and neurologic outcome after cardiopulmonary resuscitation in rats. Anesthesiology. 2008;109:260–8. doi: 10.1097/ALN.0b013e31817f5c2e. [DOI] [PubMed] [Google Scholar]
- Zhong W, et al. Hemin exerts multiple protective mechanisms and attenuates dextran sulfate sodium-induced colitis. J Pediatr Gastroenterol Nutr. 2010;50:132–9. doi: 10.1097/MPG.0b013e3181c61591. [DOI] [PubMed] [Google Scholar]