

Abstract
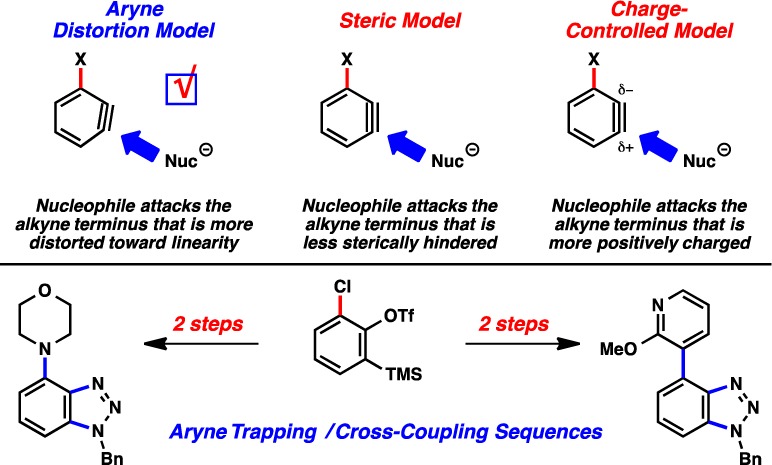
The distortion/interaction model has been used to explain and predict reactivity in a variety of reactions where more common explanations, such as steric and electronic factors, do not suffice. This model has also provided new fundamental insight into regioselectivity trends in reactions of unsymmetrical arynes, which in turn has fueled advances in aryne methodology and natural product synthesis. This article describes a systematic experimental and computational study of one particularly important class of arynes, 3-halobenzynes. 3-Halobenzynes are useful synthetic building blocks whose regioselectivities have been explained by several different models over the past few decades. Our efforts show that aryne distortion, rather than steric factors or charge distribution, are responsible for the regioselectivities observed in 3-haloaryne trapping experiments. We also demonstrate the synthetic utility of 3-halobenzynes for the efficient synthesis of functionalized heterocycles, using a tandem aryne-trapping/cross-coupling sequence involving 3-chlorobenzyne.
Introduction
The fundamental understanding of molecular reactivity continues to fuel countless aspects of scientific discovery. One model for understanding reactivity that has recently received great attention is the distortion/interaction model.1−6 The premise of this model, which is also known as the activation–strain model according to Bickelhaupt,7 divides the activation energy of a bimolecular process into two components: the energy needed to distort reactants to the transition state geometry, and the energy of interaction between the distorted fragments. The distortion/interaction model has provided fundamental new insight into chemical reactivity and has been used to understand and predict reactivities and selectivities in an array of chemical processes, including Diels–Alder, 1,3-dipolar and bioorthogonal cycloadditions,1 palladium-catalyzed cross-couplings,2 C–H functionalizations,3 and epoxidation reactions.4
We have recently explored the application of the distortion/interaction model to explain regioselectivity patterns observed in the reactions of certain arynes, especially heterocyclic arynes such as indolynes.5,6 Although benzynes have been historically avoided because of their high reactivities, a revival of interest in their chemistry has occurred in recent decades, and benzyne itself may now be exploited in a variety of efficient transformations.8 Garnering an improved understanding of the reactivity of substituted benzynes not only should facilitate their use in complexity-generating reactions, but also may explain reactivity trends observed over several decades of prior study.
One particular class of substituted benzynes known to react with significant regioselectivities are 3-substituted benzynes 1 (Figure 1).8,9 More specifically, when X is an inductively electron-withdrawing group (e.g., methoxy or halide), nucleophilic attack at C1 is preferred.10 This leads to the formation of meta-substituted products 2 rather than ortho-substituted adducts 3. This has been explained by several models. In the Charge-Controlled Model,11 the X group polarizes the triple bond, and nucleophilic addition occurs at the site of greatest positive charge.12 A related model, based on Natural Bond Orbital (NBO) electron densities of in-plane π-orbitals, has been advocated by Ikawa, Akai, and co-workers.9c,9d Alternatively, nucleophilic attack at C1 might be dictated by steric effects, i.e., the Steric Model.3 Finally, our groups have shown that the regioselectivities of reactions of hetarynes and other arynes are affected when a substituent causes a geometrical distortion such that the geometry of the aryne resembles the transition state for nucleophilic attack on one of the carbons, referred to as the Aryne Distortion Model.5 Each of these models provides a useful mnemonic to predict aryne regioselectivities, but the importance of the different factors emphasized by each of these models has not been unambiguously determined. We report a systematic experimental and theoretical study of 3-substituted arynes 1, where X = halide or methoxy, which demonstrates that regioselectivities for these arynes are predominantly controlled by aryne distortions. Moreover, we showcase the synthetic utility of 3-haloarynes for the efficient synthesis of heterocyclic compounds using a tandem aryne trapping/cross-coupling sequence.
Figure 1.
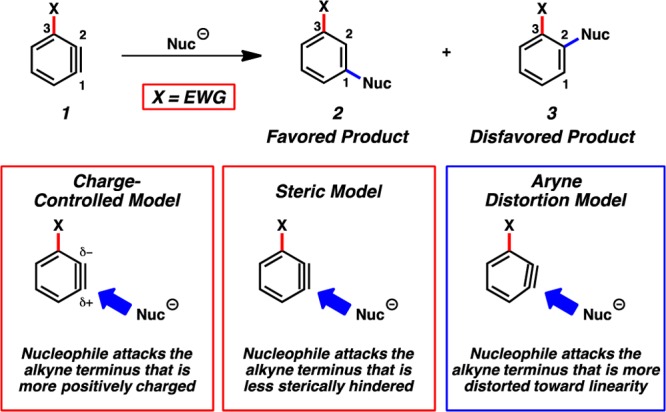
Charge-controlled, steric, and aryne distortion models.
Results and Discussion
Aryne Distortion Versus Steric Factors
We first performed computational geometry optimizations of the 3-substituted benzynes 1a–1e shown in Figure 2. Calculations were carried out using DFT methods (B3LYP/6-311+G(d,p) and LANL2DZ for Br and I atoms). We also studied these arynes and reactants with M06-2X and MP2, and these results are reported and discussed in the Supporting Information (SI).13−16 The SI also provides structural and charge information for each substituted benzyne. Methoxybenzyne (1a) is well known to react with a high degree of regioselectivity for attack at C18 and serves as a useful point of comparison to the corresponding haloarynes 1b–1e. Regarding the Aryne Distortion Model, a simplified view of this model allows one to make predictions based on an analysis of an optimized geometry of the aryne.5b,17 The 3-methoxybenzyne (1a) shows significant distortion; there is a 15° difference in internal angles between C1 and C2. Nucleophilic addition is favored at the more linear terminus, C1, as the transition state distortion energy for attack at this site is lowest.4 Halobenzynes 1b–1e are all distorted in a similar manner and are all predicted to undergo preferential attack at C1. The degree of regioselectivity is expected to decrease as a function of the electronegativity of the halide going from 3-fluorobenzyne (1b) to 3-iodobenzyne (1e), as a consequence of decreased distortion. Although reactions of 3-haloarynes are well known in the literature,1,18 a systematic study of reactions involving 1b–1e has not been performed previously.
Figure 2.
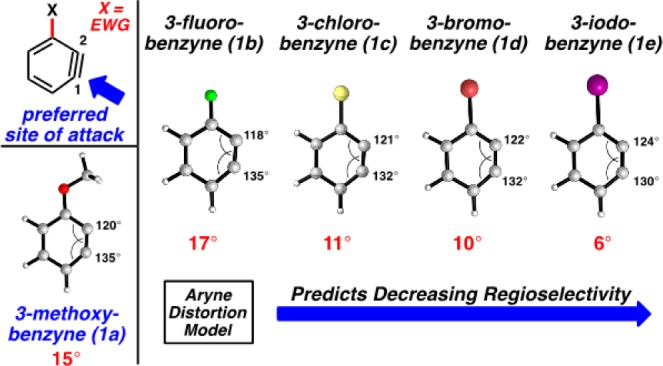
Geometry-optimized structures of 1a–1e (B3LYP) and regioselectivity predictions for nucleophilic attack based on the aryne distortion model.
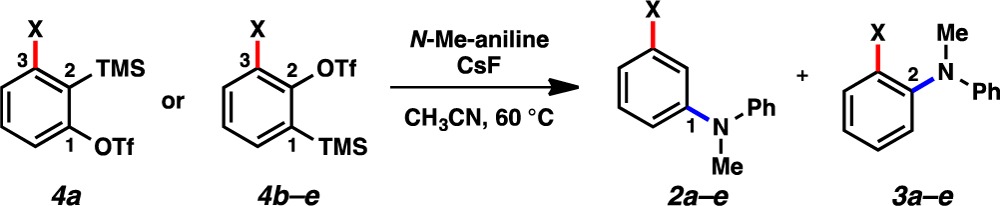
The regioselectivities of reactions of 1a–1e using N-methylaniline as the trapping agent were determined using both computations and experiments (Table 1). The results vary from exclusive attack at C1 for OMe and F, high selectivity with Cl, and less pronounced selectivity with Br and I. Transition state modeling was performed using DFT calculations (B3LYP) for the addition of N-methylaniline to C1 or C2 for each aryne. The ΔΔG⧧ values predict that nucleophilic addition to 3-methoxybenzyne (1a) and 3-fluorobenzyne (1b) should be highly regioselective (entries 1 and 2). Decreased regioselectivity was predicted for 1c–1e (entries 3–5), consistent with the Aryne Distortion Model (see Figure 2). After accessing suitable silyl triflate precursors 4a–4e,19,20 we verified the computational predictions experimentally.21 The Steric Model was deemed inconsequential based on our results and a comparison to A-values, which are 0.15 (fluoride), 0.43 (chloride), 0.38 (bromide), and 0.43 (iodide).22 The highest selectivity for attack at C1 is observed for the smallest substituent, fluoro 4b. Consequently steric effects are not dictating regioselectivities in reactions of 3-halobenzynes. Previous studies of 3-silylarynes have also shown that steric effects can be outweighed by other factors (i.e., distortion),6,9a,9b despite the fact that trialkylsilyl groups have A-values greater than 2.
Table 1. Addition of N-Methylaniline to Various Benzynesa.
| entry | 4a–e | computed ΔΔG⧧ (ratio 2:3) | experimental yield (ratio 2:3)b |
|---|---|---|---|
| 1a | 4a, X = OMe | 5.2 kcal/mol (>500:1) | 94% (2a formed exclusively) |
| 2 | 4b, X = F | 4.1 kcal/mol (>500:1) | 80% (2b formed exclusively) |
| 3 | 4c, X = Cl | 2.4 kcal/mol (>57:1) | 66% (>20:1) |
| 4 | 4d, X = Br | 1.4 kcal/mol (>11:1) | 67% (13:1) |
| 5 | 4e, X = I | 1.7 kcal/mol (>19:1) | 57% (9:1) |
Conditions: see Supporting Information. Computed ratios obtained from Boltzmann factors using B3LYP/6-31G(d) free energies including conductor-like polarizable continuum model (CPCM) solvation by MeCN.
Experimental yields and ratios are the average of three experiments and were determined by 1H NMR analysis using hexamethylbenzene as an external standard.
The same conclusion was drawn for the trapping of arynes 1a–1e in azide cycloaddition reactions (Table 2).23 Consistent with computational predictions, 3-methoxybenzyne (1a) and 3-fluorobenzyne (1b) react with high regioselectivity (entries 1 and 2, respectively). A sequential decrease in regioselectivity was observed for reactions of arynes 1c–1e, as the electron-withdrawing effects of the halide substituents decrease from F to Cl to Br to I (entries 3–5, respectively).24
Table 2. Cycloaddition of Benzylazide with Various Benzynesa.
| entry | 4a–e | computed ΔΔG⧧ (ratio 5:6) | experimental yield (ratio 5:6)b |
|---|---|---|---|
| 1a | 4a, X = OMe | 3.4 kcal/mol (>292:1) | 94% (5a formed exclusively) |
| 2 | 4b, X = F | 2.5 kcal/mol (>71:1) | 68% (5b formed exclusively) |
| 3 | 4c, X = Cl | 1.4 kcal/mol (>10:1) | 53% (>16:1) |
| 4 | 4d, X = Br | 1.2 kcal/mol (>8:1) | 45% (12:1) |
| 5 | 4e, X = I | 0.9 kcal/mol (>5:1) | 43% (6:1) |
Conditions: see Supporting Information. Computed ratios obtained from Boltzmann factors using B3LYP/6-31G(d) free energies including CPCM solvation by MeCN; methylazide was used as a model for benzylazide to simplify computational studies.
Experimental yields and ratios are the average of three experiments and were determined by 1H NMR analysis using hexamethylbenzene as an external standard.
The Role of Charges
Having ruled out the importance of the Steric Model, we next assessed the role of charges, which have often been used to explain aryne regioselectivities.3 Indeed it is natural to think of the greater negative charge at the carbon with the smallest angle because that carbon will have more s character in the orbital involved in the in-plane π bond. However, we will argue here that this charge polarization is insufficient to account for observed regioselectivities.
The charge on an atom is not an observable parameter, and there have been different definitions made of charges on atoms. Each of these depends on the definition of the boundaries separating atoms in molecules. Charges derived from a natural bond orbital analysis have been found to be very useful,25 and we use NBO charges here. Figure 3 shows computed NBO charges of o-benzyne (7), which were obtained computationally using B3LYP/6-311+G(d,p). The charges found on the triple bond carbons of 7 are +0.02. This charge is negligible, and the high electrophilic reactivity of benzyne is not a result of charge effects. For comparison, NBO charges were computed at the same level of theory for acetone. A charge of 0.57 was found for the electrophilic carbon, in agreement with its polarized double bond. Acetone is, however, much less reactive than the nonpolar benzyne, so the magnitude of charge is not an index of reactivity. We next studied the charges of substituted arynes that might have polarized triple bonds to determine if the charges are related to regioselectivity.
Figure 3.
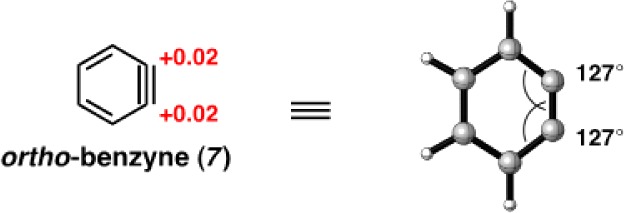
Geometry-optimized structures and NBO charges for o-benzyne (7) (B3LYP).
Since many authors use charges for qualitative interpretations, it behooves us to provide a quantitative assessment of such a model and not just invoke the view that theoreticians deny the validity of atomic charges. The charges of 3-fluorobenzyne (1b) are shown in Figure 4. The geometry-optimized structure reveals NBO charges of +0.14 and −0.11 for C1 and C2, respectively. To determine if this charge polarization could be responsible for the observed regioselectivities, a simple Coulombic interaction model was devised. A point charge of −1 was placed in the benzyne plane at a distance of 2.4 Å from C1. This model is an exaggeration in the localization of charge, but it is the extreme case of an anionic nucleophile. The position of the nucleophile charge bisects the C6–C1–C2 angle at C1. The distance between the point charge and C2 is 3.1 Å in this model. Coulomb’s law was used to compute the net interaction energy between the point negative charge and benzyne with these charges.26 This gives an attractive energy of 0.2 kcal/mol at C1 using a dielectric constant of 36, which is appropriate for acetonitrile. The corresponding analysis with a point negative charge 2.4 Å from C2 and a dielectric constant of 36 gives an interaction energy of 0.0 kcal/mol. The electrostatic energy difference for the two modes of attack differ by 0.2 kcal/mol, whereas moderate to high regioselectivities are observed in reactions of all 3-halobenzynes, in addition to computed energy differences that are typically several kcal/mol. We conclude that electrostatic effects are nearly negligible, and in any case too small to explain the regioselectivities. Furthermore, the explanation of regioselectivities of reactions such as cycloadditions by electrostatic effects have long been discredited.27
Figure 4.
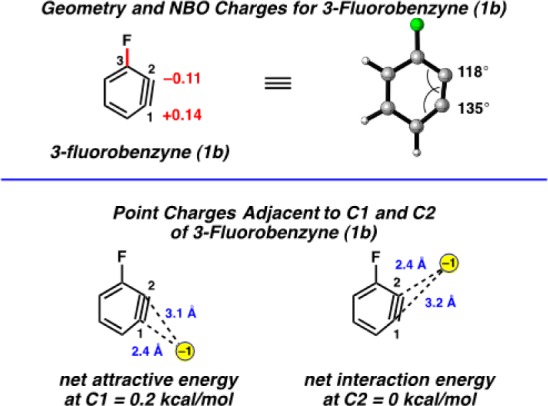
Geometry-optimized structure and NBO charges for 3-fluorobenzyne (1b) (B3LYP) and point charge analysis.
We also calculated the electrostatic potentials for interaction of a charge with the full 3-fluorobenzyne (1b) to compare to our simple Coulombic model, again with a dielectric constant of 36, for acetonitrile. These values are 0.0 kcal/mol at C1 and repulsive by 0.2 kcal/mol at C2. The 0.2 kcal/mol preference for attack at C1 is favored in both models and is not enough to explain the observed regioselectivities. We also performed calculations of this type for 3-chlorobenzyne (1c). While our simple Coulombic model predicts a 0.1–0.2 kcal/mol preference for attack at C1 (with the charge placed anywhere from 2.0 to 2.4 Å from the carbon being attacked), the full electrostatic potential calculation predicts a modest 0.2–0.3 kcal/mol preference for attack at C1.
Two additional calculations involving 1b were performed to probe the origin of the small charge polarization shown from NBO charges or electrostatic potentials (Figure 5). First, we replaced the F substituent with H, but maintained the geometry of 1b. Despite not having the electron-withdrawing substituent, significant charge polarization was observed (+0.10 for C1 and −0.05 for C2) in the distorted molecule. The negative charge is preferred on C2, the carbon with the smaller angle and greater s character in the in-plane π bond. Additionally, calculations were performed on 1b, but with the geometry restricted to that of benzyne (i.e., 127° internal angle at C1 and C2). Charges of +0.07 and −0.05 for C1 and C2, respectively, were observed. Here the charge polarization due to pure induction without rehybridization is one-half that observed when there is geometrical relaxation. Figure 5 also shows color-coded electrostatic potentials on the isodensity surfaces for benzyne (7) and 3-fluorobenzyne (1b), in addition to those for benzyne constrained to the 3-fluorobenzyne geometry and for 3-fluorobenzyne constrained to the benzyne geometry. These geometrical constraints have a meaningful influence on the relative electrostatic potential at C1 and C2.
Figure 5.
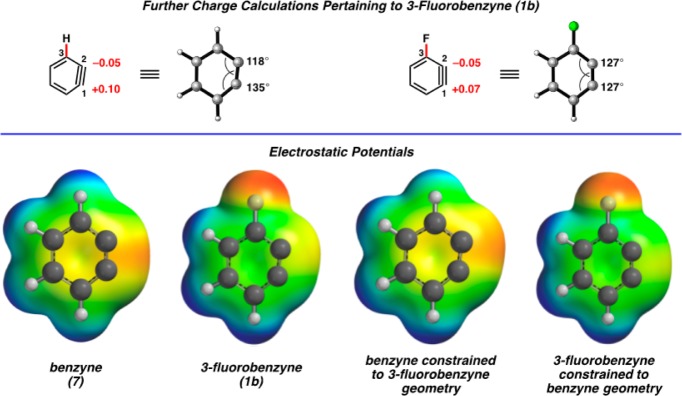
NBO charges for 1b separated based on distortion or inductive effects. Electrostatic potentials of benzyne (7) and 3-fluorobenzyne (1b). Also shown are electrostatic potentials for benzyne with 3-fluorobenzyne geometry and 3-fluorobenzyne with benzyne geometry (red indicates the lowest electrostatic potential energy, whereas blue indicates the highest).
These findings show that the aryne distortion and inductive effects are synergistic factors contributing to the overall charge polarization. Of course, there is no geometrical distortion until a substituent is added. This geometrical distortion and the small charge polarization are caused by the electronegativity of the substituent. According to Bent’s rule,28 the bond from C2 to C3 of the aryne will involve a hybrid orbital on C2 with more p character than that on C3. This releases electron density to C3 and its attached electronegative atom. This decreases the C2 bond angle from its natural 127° (see Figure 3) toward 90°. The in-plane π orbital between C1 and C2 is polarized toward C2 since C2 will have more s character.
A similar charge analysis was performed for 3-trimethylsilylbenzyne (8), as shown in Figure 6.6,29 Notably, the distortion and partial charges for 8 are reversed in comparison to 3-haloarynes. The charges at C1 and C2 are −0.04 and +0.09, respectively. By replacing the trimethylsilyl group with H, but maintaining the geometric constraints found in 8, the charge distribution was found to be similar (−0.03 and +0.07 at C1 and C2, respectively). Most of the polarization is the result of distortion and rehybridization codified in Bent’s rule. Finally, we gauged the electronic influence of the trimethylsilyl group on charge by performing calculations on 8 but with the undistorted geometry of benzyne. Only small charges of −0.01 and +0.04 at C1 and C2, respectively, were observed.
Figure 6.
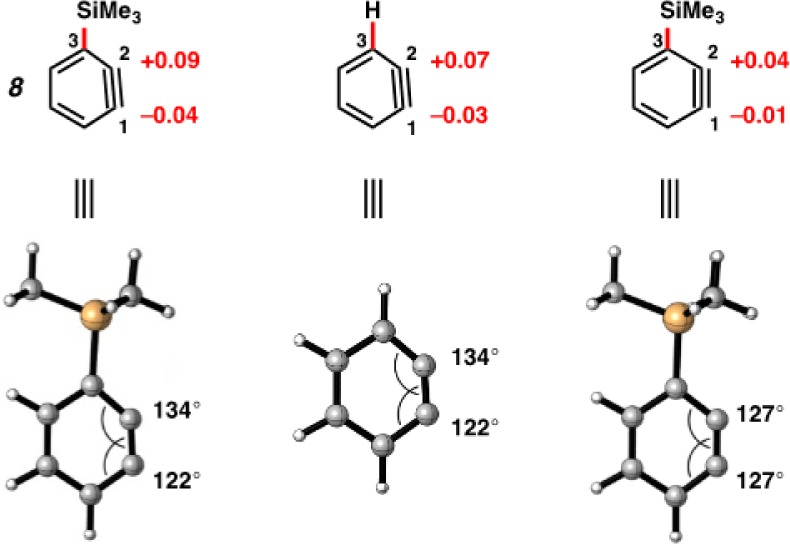
Geometry-optimized structure and NBO charges for 3-trimethylsilylbenzyne (8), in addition to charge distribution due to distortion or inductive effects.
These results underscore that the geometrical distortion present in unsymmetrical arynes, and rehybridization that accompanies this distortion, largely contributes to observed charge polarization. The small degree of charge polarization is not the sole cause of the observed regioselectivites, and we conclude that the Charge-Controlled Model is not sufficient to explain the regioselectivities observed in these unsymmetrical aryne reactions, particularly in the case of 3-halobenzynes. It does, of course, give a qualitatively correct prediction about selectivity, and might be considered a useful mnemonic for this reason. However, it is an example of “the right answer for the wrong reason”.
Ikawa, Akai, and co-workers have shown that there is a qualitative correspondence between the NBO electron density of the in-plane aryne π-orbital and the regioselectitivity of nucleophilic attack.9c,9d Attack occurs at the site of lower NBO electron density. This is presumably related to the lesser closed-shell repulsion that occurs upon overlap of the occupied orbitals of the nucleophile and aryne.
Transition State Analysis and Aryne Distortion
In previous articles,5 we have shown that the reactant distortion controls regioselectivites by influencing the distortion energies for attack at C1 vs C2. Figure 7 shows the geometry of the transition state for methyl azide attack on benzyne (7). As described earlier, the nucleophilic attack of N1 of the azide occurs at the relatively more linear angle on the benzyne where the π orbital has more p character. The 131° angle is similar to that in benzyne itself (i.e., 127°). The weaker interaction is at the carbon with the angle of 121°.
Figure 7.
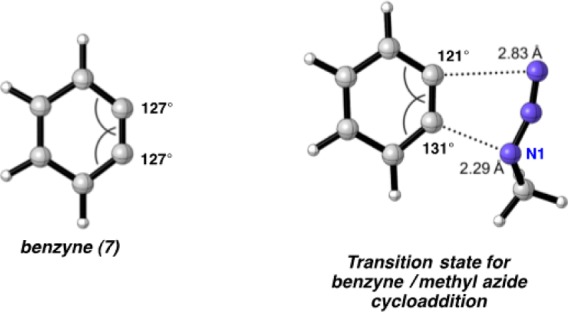
Benzyne internal angles and transition state for methyl azide/benzyne cycloaddition.
The regioselectivity trends for the reactions of halobenzynes are explained by analysis of the competing transitions states, as shown in Figure 8 for 3-fluorobenzyne (1b) and 3-chlorobenzyne (1c). In the case of 3-fluorobenzyne (1b), TS1 and TS3 are favored over TS2 and TS4, respectively. The aryne distortion5 in each of the favored transition states closely resembles the distortion already present in the ground state of aryne 1b. Initial bond formation occurs at C1; the π orbital at this site possesses greater p character due to the aryne distortion. In the preferred transition states TS1 and TS3, the distortion caused by fluorine is slightly increased by the attacking azide, but fluorine and the azide are distorting in conflicting manners for the disfavored transition state, TS4.
Figure 8.

Competing transition states for the addition of N-methylaniline and methyl azide to 3-fluorobenzyne (1b) and 3-chlorobenzyne (1c). Transition states were located using B3LYP/6-311+G(d).
The reactions involving 3-chlorobenzyne (1c) are analogous. TS5 and TS7 are favored over TS6 and TS8, respectively due to the distortion present in 1c. As the atomic radius and A-values for Cl (79 pm and 0.43, respectively) are significantly larger compared to those of F (42 pm and 0.15, respectively), steric effects should be considered as well in the disfavored transition states, TS6 and TS8. Comparisons of TS2 and TS6 show that the trajectories for approach of the N-Me-aniline nucleophile is nearly identical; additionally, the forming C–N bond distances are nearly the same in both cases (2.12 and 2.16 Å, respectively). As such, there is no evidence for steric repulsion by chlorine in TS2 or TS6. The comparison of TS4 and TS8 reveals slightly different transition states, but the shorter distance of the forming C–N bond at C2 in TS8 (2.52 Å in TS4 vs and 2.35 Å in TS8) suggests that steric effects are not a major controlling factor in the reaction of 3-chlorobenzyne. Moreover, as mentioned earlier, if steric factors were the guiding factor in reactions of 3-halobenzynes, a higher preference for reaction at C1 would be expected in trapping experiments of 3-chlorobenzyne (1c) than with 3-fluorobenzyne (1b). Experimentally and computationally the opposite trend is observed. We can conclude that, although steric factors and charge distribution can make small contributions to the observed regioselectivities, the aryne distortion and the associated transition state distortion play key roles in determining regioselectivity in these trapping experiments.
Efficient Synthesis of Heterocyclic Scaffolds
Although trapping experiments of 3-haloarynes have been reported,8,18 the general synthetic utility of these species has remained underexplored. We hypothesized that 3-halosilyl triflates (and, in turn, the corresponding arynes) could serve as valuable building blocks for the synthesis of functionalized heterocycles. Specifically, it was envisioned that a sequence involving aryne cycloaddition30 and subsequent metal-catalyzed cross-coupling31 could allow for the conversion of aryne precursors 4 to decorated heterocycles 10 in just two steps. In this scenario, the halide would first be used to govern aryne distortion; cycloaddition of the aryne would then give rise to a heterocyclic product 9 with regiocontrol. Finally, the halide would be used as a cross-coupling partner to construct a new C–C or C–N bond and deliver products 10.
We elected to synthesize C4-substituted benzotriazoles as a means to validate the sequence suggested in Figure 9. C4-substituted benzotriazoles have been studied as drug candidates, for example in the search for JNK1 inhibitors.32 As described earlier, the 3-haloarynes readily undergo cycloaddition with benzylazide to give benzotriazole products bearing halide substituents with significant regioselectivities (see Table 2). As a challenging test for the cross-coupling part of the sequence, we chose chlorobenzotriazole 5c as the test substrate. Although cross-couplings of aryl chlorides are generally less common compared to couplings of aryl bromides and iodides, conditions for aryl chloride couplings are available. In fact, nickel catalysis can be used for aryl chloride couplings using conventional ligands, including readily available phosphines.33 As shown in Figure 10, 5c could be employed in the Ni-catalyzed Suzuki–Miyaura coupling with heteroaryl boronic acids.34 The transformation proceeds in the green solvent 2-Me-THF, and gives products 11a and 11b in good yields. Additionally, the Ni-catalyzed amination35 of 5c proceeded smoothly to produce aminobenzotriazoles 12a and 12b, also in synthetically useful yields. It should be emphasized that the Ni-catalyzed C–C and C–N bond formations (a) utilize air-stable precatalysts, and thus are carried out on the benchtop, (b) are tolerant of the benzotriazole motif, and (c) are tolerant of a variety of other heterocycles, as demonstrated by the formation of products 11a, 11b, 12a, and 12b. Therefore, our results not only validate the utility of 3-haloarynes for the construction of functionalized heterocycles, but also showcase the growing value of nickel catalysis in modern cross-coupling reactions.33
Figure 9.
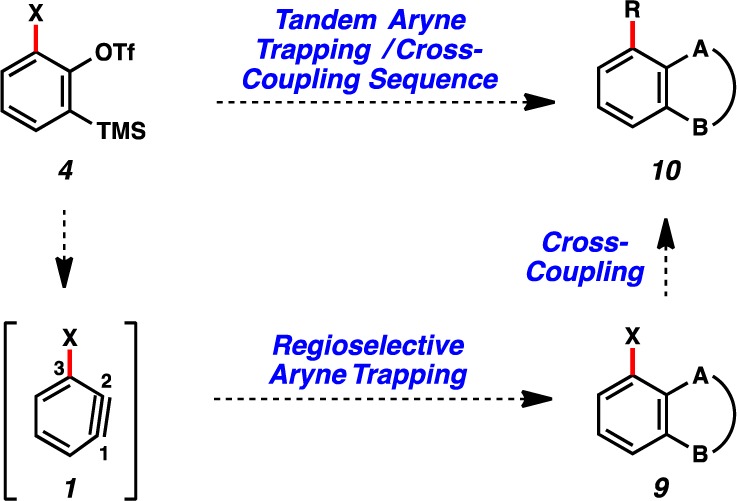
Tandem aryne trapping/cross-coupling sequence.
Figure 10.
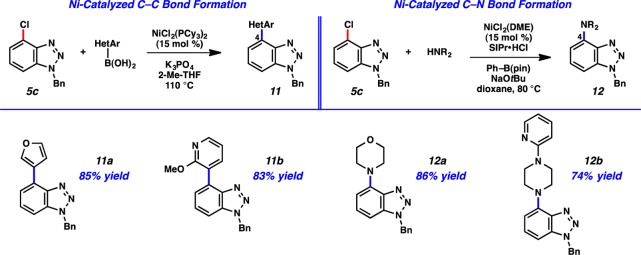
Nickel-catalyzed C–C and C–N bond-forming reactions for the synthesis of functionalized benzotriazoles 11 and 12.
Conclusion
We have compared three commonly used models for rationalizing regioselectivity in reactions of 3-haloarynes. Our experimental and computational study shows that regioselectivity in these systems is explained by the Aryne Distortion Model. Moreover, by virtue of the tandem aryne trapping/cross-coupling sequence developed, we have demonstrated the synthetic utility of 3-haloarynes for the assembly of functionalized heterocyclic compounds. We expect that these studies of reactivity, regioselectivity, and synthetic applications will help propel the use of unsymmetrical arynes in complexity-generating transformations.
Acknowledgments
The authors are grateful to the NIH-NIGMS (R01 GM090007 to N.K.G.), the National Science Foundation (CHE-1361104 to K.N.H.), Boehringer Ingelheim, Bristol–Myers Squibb, DuPont, Eli Lilly, Amgen, AstraZeneca, Roche, the A. P. Sloan Foundation, the S. T. Li Foundation, the Dreyfus Foundation, the University of California, Los Angeles, and the UCLA Cota Robles Fellowship Program (J.M.M.) for financial support. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (OCI-1053575) along with the UCLA Institute of Digital Research and Education (IDRE). We thank the Garcia-Garibay laboratory (UCLA) for access to instrumentation. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the National Center for Research Resources (S10RR025631).
Supporting Information Available
Detailed experimental procedures, characterization data for all new compounds, computational data, and Cartesian coordinates of all structures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† J.M.M. and J.L.M. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For the application of distortion energies to regioselectivity of cycloaddition reactions, see:; a Ess D. H.; Houk K. N. J. Am. Chem. Soc. 2007, 129, 10646–10647. [DOI] [PubMed] [Google Scholar]; b Ess D. H.; Houk K. N. J. Am. Chem. Soc. 2008, 130, 10187–10198. [DOI] [PubMed] [Google Scholar]; c Lam Y.-h.; Cheong P. H.-Y.; Blasco Mata J. M.; Stanway S. J.; Gouverneur V.; Houk K. N. J. Am. Chem. Soc. 2009, 131, 1947–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hayden A. E.; Houk K. N. J. Am. Chem. Soc. 2009, 131, 4084–4089. [DOI] [PubMed] [Google Scholar]; e Schoenebeck F.; Ess D. H.; Jones G. O.; Houk K. N. J. Am. Chem. Soc. 2009, 131, 8121–8133. [DOI] [PubMed] [Google Scholar]; f Osuna S.; Houk K. N. Chem.—Eur. J. 2009, 15, 13219–13231. [DOI] [PubMed] [Google Scholar]; g Paton R. S.; Kim S.; Ross A. G.; Danishefsky S. J.; Houk K. N. Angew. Chem., Int. Ed. 2011, 50, 10366–10368. [DOI] [PubMed] [Google Scholar]; h Lan Y.; Wheeler S. E.; Houk K. N. J. Chem. Theory Comput. 2011, 7, 2104–2111. [DOI] [PubMed] [Google Scholar]; i Liang Y.; Mackey J. L.; Lopez S. A.; Liu F.; Houk K. N. J. Am. Chem. Soc. 2012, 134, 17904–17907. [DOI] [PubMed] [Google Scholar]; j Gordon C. G.; Mackey J. L.; Jewett J. C.; Sletten E. M.; Houk K. N.; Bertozzi C. R. J. Am. Chem. Soc. 2012, 134, 9199–9208. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Lopez S. A.; Munk M. E.; Houk K. N. J. Org. Chem. 2013, 78, 1576–1582. [DOI] [PubMed] [Google Scholar]; l Lopez S. A.; Houk K. N. J. Org. Chem. 2013, 78, 1778–1783. [DOI] [PubMed] [Google Scholar]; m Kamber D. N.; Nazarova L. A.; Liang Y.; Lopez S. A.; Patterson D. M.; Shih H.-W.; Houk K. N.; Prescher J. A. J. Am. Chem. Soc. 2013, 135, 13680–13683. [DOI] [PubMed] [Google Scholar]; n Liu F.; Paton R. S.; Kim S.; Liang Y.; Houk K. N. J. Am. Chem. Soc. 2013, 135, 15642–15649. [DOI] [PubMed] [Google Scholar]; o Yang J.; Liang Y.; Šečkutė J.; Houk K. N.; Devaraj N. K. Chem.—Eur. J. 2014, 20, 3365–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]; p Hong X.; Liang Y.; Griffith A. K.; Lambert T. H.; Houk K. N. Chem. Sci. 2014, 5, 471–475. [Google Scholar]; q Liu F.; Liang Y.; Houk K. N. J. Am. Chem. Soc. 2014, 136, 11483–11493. [DOI] [PubMed] [Google Scholar]; r Cao Y.; Liang Y.; Zhang L.; Osuna S.; Hoyt A.-L. M.; Briseno A. L.; Houk K. N. J. Am. Chem. Soc. 2014, 136, 10743–10751. [DOI] [PubMed] [Google Scholar]; s Hong X.; Liang Y.; Brewer M.; Houk K. N. Org. Lett. 2014, 16, 4260–4263. [DOI] [PubMed] [Google Scholar]
- For the application of distortion energies to regioselectivity of palladium-catalyzed cross coupling reactions, see:; a Legault C. Y.; Garcia Y.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2007, 129, 12664–12665. [DOI] [PubMed] [Google Scholar]; b Garcia Y.; Schoenebeck F.; Legault C. Y.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2009, 131, 6632–6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the application of distortion energies to understanding selectivities in C–H functionalization reactions, see:; a Zou L.; Paton R. S.; Eschenmoser A.; Newhouse T. R.; Baran P. S.; Houk K. N. J. Org. Chem. 2013, 78, 4037–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Green A. G.; Liu P.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2014, 136, 4575–4583. [DOI] [PubMed] [Google Scholar]
- For the application of the distortion/interaction model to explain stereoselectivity in epoxidation reactions, see:Kolakowski R. V.; Williams L. J. Nat. Chem. 2010, 2, 303–307. [DOI] [PubMed] [Google Scholar]
- For early studies and reviews regarding the aryne distortion model, see:; a Cheong P. H.-Y.; Paton R. S.; Bronner S. M.; Im G.-Y. J.; Garg N. K.; Houk K. N. J. Am. Chem. Soc. 2010, 132, 1267–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Im G.-Y. J.; Bronner S. M.; Goetz A. E.; Paton R. S.; Cheong P. H.-Y.; Houk K. N.; Garg N. K. J. Am. Chem. Soc. 2010, 132, 17933–17944. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bronner S. M.; Goetz A. E.; Garg N. K. Synlett 2011, 18, 2599–2604. [Google Scholar]; d Goetz A. E.; Garg N. K. J. Org. Chem. 2014, 79, 846–851. [DOI] [PubMed] [Google Scholar]
- For the application of the aryne distortion model to 3-silylbenzynes, see:Bronner S. M.; Mackey J. L.; Houk K. N.; Garg N. K. J. Am. Chem. Soc. 2012, 134, 13966–13969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a van Zeist W.-J.; Bickelhaupt F. M. Org. Biomol. Chem. 2010, 8, 3118–3127. [DOI] [PubMed] [Google Scholar]; b Fernández I.; Cossío F. P.; Bickelhaupt F. M. J. Org. Chem. 2011, 76, 2310–2314. [DOI] [PubMed] [Google Scholar]; c Fernández I.; Bickelhaupt F. M. J. Comput. Chem. 2012, 33, 509–516. [DOI] [PubMed] [Google Scholar]; d Fernández I.; Sola M.; Bickelhaupt F. M. Chem.—Eur. J. 2013, 19, 7416–7422. [DOI] [PubMed] [Google Scholar]; e Fernández I.; Bickelhaupt F. M. Chem. Soc. Rev. 2014, 43, 4953–4967. [DOI] [PubMed] [Google Scholar]
- For pertinent reviews, see refs (5c,5d) and the following:; a Pellissier H.; Santelli M. Tetrahedron 2003, 59, 701–730. [Google Scholar]; b Wenk H. H.; Winkler M.; Sander W. Angew. Chem., Int. Ed. 2003, 42, 502–528. [DOI] [PubMed] [Google Scholar]; c Sanz R. Org. Prep. Proced. Int. 2008, 40, 215–291. [Google Scholar]; d Chen Y.; Larock R. C. In Modern Arylation Methods; Ackermann L., Ed.; Wiley-VCH: Weinheim, 2009; pp 401–473. [Google Scholar]; e Tadross P. M.; Stoltz B. M. Chem. Rev. 2012, 112, 3550–3577. [DOI] [PubMed] [Google Scholar]; f Gampe C. M.; Carreira E. M. Angew. Chem., Int. Ed. 2012, 51, 3766–3778. [DOI] [PubMed] [Google Scholar]; g Yoshida H.; Takaki K. Synlett 2012, 23, 1725–1732. [Google Scholar]; h Dubrovskiy A. V.; Markina N. A.; Larock R. C. Org. Biomol. Chem. 2013, 11, 191–218. [DOI] [PubMed] [Google Scholar]; i Wu C.; Shi F. Asian J. Org. Chem. 2013, 2, 116–125. [Google Scholar]; j Hoffmann R. W.; Suzuki K. Angew. Chem., Int. Ed. 2013, 52, 2655–2656. [DOI] [PubMed] [Google Scholar]; k Bhunia A.; Biju A. T. Synlett 2014, 25, 608–614. [Google Scholar]; l Bhunia A.; Yetra S. R.; Biju A. T. Chem. Soc. Rev. 2012, 41, 3140–3152. [DOI] [PubMed] [Google Scholar]
- For studies of 3-silylarynes or 3-borylbenzynes, which are not the major focus of the current study, see ref (6) and the following:; a Ikawa T.; Nishiyama T.; Shigeta T.; Mohri S.; Morita S.; Takayanagi S.-i.; Terauchi Y.; Morikawa Y.; Takagi A.; Ishikawa Y.; Fujii S.; Kita Y.; Akai S. Angew. Chem., Int. Ed. 2011, 50, 5674–5677. [DOI] [PubMed] [Google Scholar]; b Ikawa T.; Takagi A.; Goto M.; Aoyama Y.; Ishikawa Y.; Itoh Y.; Fujii S.; Tokiwa H.; Akai S. J. Org. Chem. 2013, 78, 2965–2983. [DOI] [PubMed] [Google Scholar]; c Takagi A.; Ikawa T.; Kurita Y.; Saito K.; Azechi K.; Egi M.; Itoh Y.; Tokiwa H.; Kita Y.; Akai S. Tetrahedron 2013, 69, 4338–4352. [Google Scholar]; d Takagi A.; Ikawa T.; Saito K.; Masuda S.; Ito T.; Akai S. Org. Biomol. Chem. 2013, 11, 8145–8150. [DOI] [PubMed] [Google Scholar]
- For a recent study of a 3-alkoxycyclohexyne, see:Medina J. M.; McMahon T. C.; Jiménez-Osés G.; Houk K. N.; Garg N. K. J. Am. Chem. Soc. 2014, 136, 14706–14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples involving the Charge Distribution and Steric Models to rationalize regioselectivities in trapping experiments of 3-substituted arynes, see:; a Kessar S. V. In Comprehensive Organic Synthesis; Trost B. M., Fleming I., Eds.; Pergamon Press: Oxford, England, 1991; Vol. 4, pp 483–515. [Google Scholar]; b Liu Z.; Larock R. C. J. Org. Chem. 2006, 71, 3198–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Liu Z.; Larock R. C. Org. Lett. 2003, 5, 4673–4675. [DOI] [PubMed] [Google Scholar]; d Tadross P. M.; Gilmore C. D.; Bugga P.; Virgil S. C.; Stoltz B. M. Org. Lett. 2010, 12, 1224–1227. [DOI] [PubMed] [Google Scholar]; e Yoshida H.; Sugiura S.; Kunai A. Org. Lett. 2002, 4, 2767–2769. [DOI] [PubMed] [Google Scholar]; f Hamura T.; Ibusuki Y.; Sato K.; Matsumoto T.; Osamura Y.; Suzuki K. Org. Lett. 2003, 5, 3551–3554. [DOI] [PubMed] [Google Scholar]
- In a somewhat related sense, it has also been argued that the presence of an adjacent inductively withdrawing group causes regioselectivity in aryne reactions due to stabilization of the developing carbanion, which would be formed upon nucleophilic addition to the substituted benzyne. However, arynes undoubtedly react through very early transition states with low enthalpic barriers, so this explanation is inadequate.
- Calculations shown are at the B3LYP/6-311+G(d,p) level of theory and LANL2DZ for the Br and I atoms with tight convergence criteria and an ultrafine integration grid. Angles were also calculated at this level of theory. See the SI for calculations of angles of reactants and TSs with M06-2X and MP2. Because the potential energy surface of the reaction of 1b has no maximum, a variational transition state search was performed to locate a saddle point. Although B3LYP overestimates the regioselectivities, it predicts the correct trend in reactivity.
- a Becke A. D. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]; c Vosko S. H.; Wilk L.; Nusair M. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar]; d Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar]
- a Hay P. J.; Wadt W. R. J. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar]; b Wadt W. R. J.; Hay P. J. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar]; c Hay P. J.; Wadt W. R. J. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam M. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
- a Bronner S. M.; Goetz A. E.; Garg N. K. J. Am. Chem. Soc. 2011, 133, 3832–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Goetz A. E.; Bronner S. M.; Cisneros J. D.; Melamed J. M.; Paton R. S.; Houk K. N.; Garg N. K. Angew. Chem., Int. Ed. 2012, 51, 2758–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For select examples of studies pertaining to 3-haloarynes, see ref (17a) and the following:; a Biehl E. R.; Nieh E.; Hsu K. C. J. Org. Chem. 1969, 34, 3595–3599. [Google Scholar]; b Moreau-Hochu M. F.; Caubere P. Tetrahedron 1977, 33, 955–959. [Google Scholar]; c Ghosh T.; Hart H. J. Org. Chem. 1988, 53, 3555–3558. [Google Scholar]; d Hart H.; Ghosh T. Tetrahedron Lett. 1988, 29, 881–884. [Google Scholar]; e Wickham P. P.; Reuter K. H.; Senanayake D.; Guo H.; Zalesky M.; Scott W. J. Tetrahedron Lett. 1993, 34, 7521–7524. [Google Scholar]; f Gokhale A.; Scheiss P. Helv. Chem. Acta 1998, 81, 251–267. [Google Scholar]; g Goetz A. E.; Garg N. K. Nat. Chem. 2013, 5, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Hendrick C. E.; McDonald S. L.; Wang Q. Org. Lett. 2013, 15, 3444–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Kirkham J. D.; Delaney P. M.; Ellames G. J.; Row E. C.; Harrity J. P. A. Chem. Commun. 2010, 46, 5154–5156. [DOI] [PubMed] [Google Scholar]
- Using Kobayashi’s strategy, silyl triflates readily undergo conversion to the corresponding arynes upon treatment with fluoride sources:Himeshima Y.; Sonoda T.; Kobayashi H. Chem. Lett. 1983, 12, 1211–1214. [Google Scholar]
- Silyl triflate 4a was obtained from commercial sources. The syntheses of silyl triflates 4b–4e are described in the SI. For an alternative synthesis of 4c, see:; a Hall C.; Henderson J. L.; Ernouf G.; Greaney M. F. Chem. Commun. 2013, 49, 7602–7604. [DOI] [PubMed] [Google Scholar]; For the prior synthesis of 4d, see:; b Dai M.; Wang Z.; Danishefsky S. J. Tetrahedron Lett. 2008, 49, 6613–6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the addition of heteroatom nucleophiles to benzynes generated from silyl triflate precursors, see ref (11d).
- Although the atomic radii increase steadily from F to I, the corresponding A-values only increase from F to Cl and then remain nearly constant for Cl, Br, and I. This is due to the increased C–X bond length as atom size increases.
- a Shi F.; Waldo J. P.; Chen Y.; Larock R. C. Org. Lett. 2008, 10, 2409–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Campbell-Verduyn L.; Elsinga P. H.; Mirfeizi L.; Dierckx R. A.; Feringa B. L. Org. Biomol. Chem. 2008, 6, 3461–3463. [DOI] [PubMed] [Google Scholar]
- Given that reaction yields are not quantitative, and that computational free energies have some standard error, the predicted and experimental ratios should be taken with a grain of salt. Nonetheless, the general trends seen in the experimental and computational data of Tables 1 and 2 entirely correlate to predictions made by the aryne distortion model (i.e., highest selectivity is observed and calculated for the most distorted 3-haloaryne, 1b, whereas the lowest selectivity is observed and calculated with the least distorted 3-haloaryne, 1e).
- Weinhold F.; Landis C. R. Discovering Chemistry with Natural Bond Orbitals; John Wiley & Sons, Inc.: Hoboken, NJ, 2012. [Google Scholar]
- The net attractive energy was calculated by separately determining the Coulombic interactions between the point charge and C1 and C2, using the aforementioned calculated NBO charges for 1b, and then adding these values.
- Houk K. N. Acc. Chem. Res. 1975, 8, 361–369. [Google Scholar]
- Bent H. Chem. Rev. 1961, 61, 275–311. [Google Scholar]
- Although not the main focus of this study, it should be noted that steric effects are important in reactions of other unsymmetrical arynes, such as 3-silylbenzynes; for further discussion, see refs (6) and (9).
- An array of aryne cycloaddition reactions are available in the literature using silyl triflate precursors; see ref (8).
- a Hassan J.; Sevignon M.; Gozzi C.; Schulz E.; Lemaire M. Chem. Rev. 2002, 102, 1359–1469. [DOI] [PubMed] [Google Scholar]; b Topics in Current Chemistry, Vol. 219; Miyaura N., Ed.; Springer Verlag: New York, 2002. [Google Scholar]; c Metal-Catalyzed Cross-Coupling Reactions; Diederich F., Meijere A., Eds.; Wiley-VCH: Weinheim, 2004. [Google Scholar]; d Corbet J.; Mignani G. Chem. Rev. 2006, 106, 2651–2710. [DOI] [PubMed] [Google Scholar]; e Negishi E. Bull. Chem. Soc. Jpn. 2007, 80, 233–257. [Google Scholar]; f Application of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective; Shen H. C., Crawley M. L., Trost B. M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2012. [Google Scholar]
- Gong L.; Jahangir A.; Reuter D. C. U.S. Patent US2010160360A1, 2010.
- For recent reviews regarding Ni-catalyzed cross-couplings, see:; a Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.-M.; Garg N. K.; Percec V. Chem. Rev. 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li B.-J.; Yu D.-G.; Sun C.-L.; Shi Z.-J. Chem.—Eur. J. 2011, 17, 1728–1759. [DOI] [PubMed] [Google Scholar]; c Mesganaw T.; Garg N. K. Org. Process Res. Dev. 2013, 17, 29–39. [Google Scholar]; d Tasker S. Z.; Standley E. A.; Jamison T. F. Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For our previous studies of Ni-catalyzed Suzuki–Miyaura couplings, see:; a Quasdorf K. W.; Tian X.; Garg N. K. J. Am. Chem. Soc. 2008, 130, 14422–14423. [DOI] [PubMed] [Google Scholar]; b Quasdorf K. W.; Riener M.; Petrova K. V.; Garg N. K. J. Am. Chem. Soc. 2009, 131, 17748–17749. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Quasdorf K. W.; Antoft-Finch A.; Liu P.; Silberstein A. L.; Komaromi A.; Blackburn T.; Ramgren S. D.; Houk K. N.; Snieckus V.; Garg N. K. J. Am. Chem. Soc. 2011, 133, 6352–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ramgren S. D.; Hie L.; Ye Y.; Garg N. K. Org. Lett. 2013, 15, 3950–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hie L.; Chang J. J.; Garg N. K. J. Chem. Ed. 2014, 10.1021/ed500158p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For our previous studies of Ni-catalyzed amination reactions, see:; a Mesganaw T.; Silberstein A. L.; Ramgren S. D.; Fine Nathel N. F.; Hong X.; Liu P.; Garg N. K. Chem. Sci. 2011, 2, 1766–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ramgren S. D.; Silberstein A. L.; Yang Y.; Garg N. K. Angew. Chem., Int. Ed. 2011, 50, 2171–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hie L.; Ramgren S. D.; Mesganaw T.; Garg N. K. Org. Lett. 2012, 14, 4182–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Fine Nathel N. F.; Kim J.; Hie L.; Jiang X.; Garg N. K. ACS Catal. 2014, 4, 3289–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.