

Abstract
We analyzed the effects of 14 different missense mutations in the RING domain of BRCA1 on the function of the protein in the control of centrosome number in tissue culture cells. Whereas 2 of the 14 BRCA1 variant proteins were neutral in the centrosome duplication assay, missense mutations of zinc-coordinating residues (C24R, C27A, C39Y, H41F, C44F and C47G) and mutations encoding BRCA1 variants M18T and I42V resulted in BRCA1 proteins that caused centrosome amplification. BRCA1 variant proteins I21V, I31M, L52F and D67Y had an intermediate effect on centrosome duplication. In addition, one of the variants, L52F, caused a peculiar phenotype with amplified centrosomes but the centrioles remained paired. By comparison, other BRCA1 variants that caused centrosome amplification had clustering of supernumerary centrosomes with unpaired centrioles. This surprising phenotype suggests that the BRCA1 protein regulates two functions in the control of centrosome duplication: regulation of centrosome number and regulation of centriole pairing. The L52F is unusual as it is defective in only one of these processes. This study analyzes the function of BRCA1 missense mutations in the control of centrosome duplication, a critical step in the maintenance of genetic stability of mammary epithelial cells, and indicates a new function of BRCA1 in the control of centriole pairing.
Keywords: BRCA1, breast cancer, centrosomes, RING domain
Introduction
BRCA1 is a tumor suppressor gene associated with familial breast and ovarian cancer. Among familial cases of breast cancer, approximately 23% of the patients are born with a mutant BRCA1 allele (Sinilnikova et al., 2006), and the tumors are associated with a loss of the second allele (Miki et al., 1994). The most common deleterious mutations of BRCA1 are nonsense and frame-shift mutations that result in a truncated protein. However, not all BRCA1 variants have clear consequences. 36% of all BRCA1 mutations are missense mutations (Ruffner et al., 2001). In the BIC database (http://research.nhgri.nih.gov/projects/bic/), 567 of the 1638 total mutations are missense variants of BRCA1. Individuals who have a BRCA1 missense mutation present a diagnostic dilemma, as the resultant BRCA1 protein may or may not be functional. Among the 567 BRCA1 missense mutations in the BIC database, 14 are deleterious and 27 are recorded as neutral. Though the BIC database may not have the latest results, only 7% of the BRCA1 missense mutations have a determined clinical phenotype. Apart from these several missense mutations of BRCA1 with defined clinical phenotype, the numbers of individuals with a specific variant are too few, and genetic segregation analysis fails to determine the consequences.
BRCA1 is known to be required in several cellular processes including: transcription, cell-cycle check point control, DNA damage repair and control of centrosome number (Chapman and Verma, 1996; Monteiro et al., 1996; Haile and Parvin, 1999; Moynahan et al., 1999; Xu et al., 1999; MacLachlan et al., 2000). None of these processes, however, explain why BRCA1 mutations are specific for breast and ovarian cancers. BRCA1 depletion in mammary-derived cells leads to the formation of supernumerary centrosomes (Xu et al., 1999; Starita et al., 2004). Centrosomes are non-membranous organelles that are important for the formation of the bipolar mitotic spindle that insures the proper segregation of chromosomes into two daughter cells (Nigg, 2002; Sluder and Nordberg, 2004). Having exactly two centrosomes in dividing cells is essential for the formation of bipolar spindles, and thus for the appropriate segregation of chromosomes into progeny cells. Centrosome amplification will lead to improper chromosome segregation, aneuploidy, genomic instability and thus ultimately may lead to cancer. BRCA1 regulates centrosome duplication through its E3 ubiquitin ligase activity where it ubiquitinates gamma tubulin (Starita et al., 2004) and a gamma tubulin adapter protein (Sankaran et al., 2007), and thereby prevents centrosome reduplication within the same cell cycle (Kais and Parvin, 2008).
In an attempt to determine the significance of BRCA1 missense variants, some studies examined the effect of these mutations on the ubiquitin ligase activity of the protein. The missense mutation of zinc-coordinating residues, that affect the proper folding of the RING domain, inhibited the BRCA1–BARD1 interaction as well as the BRCA1-dependent ubiquitin ligase activity of the protein (Ruffner et al., 2001; Morris et al., 2006). The inhibition of the ubiquitin ligase activity of BRCA1 was not only restricted to mutations of the zinc-coordinating residues, but also M18T, T37R and L52F were inactive for the ubiquitin ligase (Morris et al., 2006). It is known that the N-terminal region of BRCA1 interacts with several proteins (Wu et al., 1996; Jensen et al., 1998; Houvras et al., 2000; Pao et al., 2000) suggesting that this domain of BRCA1 participates in several other functions in addition to its known ubiquitin ligase activity. In a previous study, we investigated the effect of missense mutations in the N-terminal region of BRCA1 on its function in homologous recombination. In addition to the zinc-coordinating residues, the variants M18T and T37R were inactive in the homologous recombination process (Ransburgh et al., 2010).
In this study, we assess the effects of different BRCA1 missense mutations on centrosome duplication, a cellular process that is regulated by BRCA1 in mammary epithelial cells, and we discover variants that are deleterious in this biological process. Comparing the results of this study with prior analysis of homologous recombination, it is clear that different amino acid residues of BRCA1 differentially contribute to regulation of homologous recombination and to centrosome control.
Results and discussion
In this study, we tested the effects of 14 BRCA1 point mutations (M18T, I21V, C24R, C27A, I31M, T37R, C39Y, H41R, I42V, C44F, C47G, L52F, D67Y and R71G) on centrosome number in cultured cells. These mutations except one, C27A, have been identified in individuals with a family history of breast cancer and are listed in the BIC database. All of these variants are located in BRCA1 RING domain, and most have been assayed for enzymatic ubiquitin ligase activity of the protein (Morris et al., 2006). In a previous study, we tested BRCA1 variants for their effect on homologous recombination (Ransburgh et al., 2010). In this paper, we study the effects of these same missense mutations of BRCA1 in a tissue culture-based functional assay for centrosome control (Figure 1a). Consistent with our previous results, depletion of BRCA1 using a small-interfering RNA specific for the 30-UTR of the endogenous BRCA1 mRNA resulted in centrosome amplification. When BRCA1 was depleted by RNA interference, consistently 410% of cells had aberrant centrosome numbers (Figure 1a). Thus, we use 10% as the value for an abnormal phenotype. By comparison, transfection of a control resulted in a range of 2.0–5.5% (mean ¼ 3.6%) of cells with aberrant centrosome number. We thus set the high end of normal at 5.5%. Upon addition of the plasmid for expression of wild-type BRCA1 protein, the centrosome defect was corrected. Depleting BRCA1 and expressing BRCA1 variants by transfecting the different plasmids that express full-length mutant BRCA1 protein affected centrosome number differently. Some missense mutants (M18T, C24R, C27A, C39Y, H41R, I42V, C44F and C47G) caused centrosome amplification, while others had an intermediate phenotype (D67Y, I31M, I21V or L52F) or no effect on centrosome control (T37R and R71G) (Figure 1a).
Figure 1.

Identification of BRCA1 missense mutations that affect regulation of centrosome number. (a) Results of the centrosome amplification assay with BRCA1 substitution variants. Control (lane 1) or BRCA1 30-UTR small-interfering RNA (lanes 2–17) were transfected into Hs578T cells along with GFP-centrin 2 and the indicated BRCA1 mutant expressing plasmid or empty vector. The histogram shows the percentage of Hs578T cells with centrosome amplifications after the substitution of BRCA1 with the indicated BRCA1 variant. Less than 5.5% of the cells with amplified centrosomes was considered normal in this assay, and greater than 10% with amplified centrosomes was considered defective for centrosome regulation. The percentage given for each variant represents the average of three independent repeats of the assay with more than 100 cells counted for each mutant/experiment. The control small-interfering RNA was tested a total of 11 times and the BRCA1si with co-transfected vector plasmid was tested eight times. (b) A histogram showing the number of cells with centrosome amplification (left, same value as in panel a) that also had paired centrioles upon the introduction of the L52F variant as compared with the M18T variant. (c) Immunoblots showing the expression of BRCA1 substitution variants in Hs578T cells. Cells were processed according to the schedule of the centrosome assay and the lysates were analyzed for the expression of BRCA1 (top) or the loading control alpha-tubulin (bottom). In lanes 1, 10 and 17 cells were transfected with the control small-interfering RNA and with empty plasmid vector. In lanes 2, 11 and 18 cells were transfected with BRCA1 30-UTR-specific small-interfering RNA and with empty plasmid vector. In lanes 3–9, 12–16 and 18–21 cells were transfected with BRCA1 30-UTR small-interfering RNA small-interfering RNA along with the indicated substituting BRCA1 variant. Densitometric analysis is included in supplementary Figure S2.
In most cases, when there was centrosome amplification the centrioles detected in the assay were all clustered. Strikingly, expression of the L52F mutant resulted in a phenotype that differed from the other mutants (Figure 2). In 56% of the cells with centrosome amplification, the expression of this mutant resulted in paired centrioles instead of the commonly seen centriole clustering phenotype (Figure 1b). This result indicated that BRCA1 might have a second function at the centrosome controlling centriole separation. The BRCA1(L52F) variant was functional for controlling centriole pairing, but was defective in controlling centrosome duplication.
Figure 2.
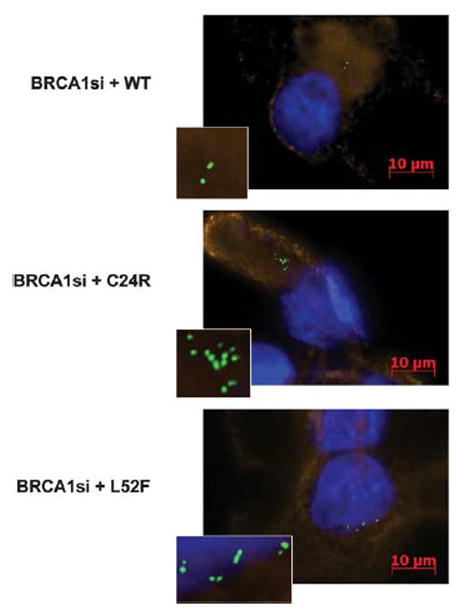
Centriole amplification and control of pairing upon the expression of BRCA1 variants. BRCA1 depletion caused centrosome amplification. Introduction of wild-type BRCA1 back to the breast cells restored the normal numbers of centrosomes per cell (top). Depleting BRCA1 and introducing a BRCA1 variant that is deleterious to the centrosome function results in centrosome amplification that is mostly seen as clustering of 44 centrioles per cell (middle). Depleting BRCA1 and introducing the L52F mutant resulted in centrosome amplification with a distinct pairing of the centrioles that was distinct from the other variants tested (bottom). Small-interfering RNA, GFP-centrin plasmid and BRCA1 point mutant plasmid were transfected into Hs578T breast cancer cells in the presence of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Cells were fixed 48 h post-transfection with methanol, stained in DAPI and then incubated with wheat germ agglutinin conjugated with Texas red (Invitrogen) to stain the cell membranes. Cells were washed and mounted in ProLong Gold antifade (ProLong, South Plainfield, NJ, USA). Centrioles were then counted by fluorescence microscopy using a Zeiss Axiovert 200M microscope. BRCA1 expression plasmid, pcDNA-5HA-BRCA1, as previously described (Scully et al., 1997; Chiba and Parvin, 2002) and site-specific point mutations identified on the Breast Cancer Information Core (BIC) were generated using the QuikChange kit from Stratagene (Santa Clara, CA, USA). GFP-Centrin 2 plasmid was used to mark the centrioles (D’Assoro et al., 2001). The 30-untranslated region (UTR) of BRCA1 was targeted with small-interfering RNA with the sequence 50-GCUCCUCUCACUCUUCAGU-30 specific for nucleotides 80 780–80 798 of the BRCA1 gene (accession number AY273801). The control oligonucleotide targets luciferase mRNA (50–30, UCGAAGUAUUCCGCGUACGTT)
Some of the residues that we tested are important in coordinating the zinc atoms that control the structure of the RING domain of BRCA1 (C24R, C27A, C39Y, H41R, C44F and C47G) (Brzovic et al., 2003). Substitution of any of these residues would likely have a major effect on the structure of the protein that would affect the formation of the BRCA1/BARD1 heterodimer, a complex with an E3 ubiquitin ligase activity. Consistent with this expectation, replacing the endogenous BRCA1 with any of these variant BRCA1 proteins resulted in an increase in the number of cells with supernumerary centrosomes. In addition to the six variants tested at zinc-coordinating residues, two other variants were clearly deleterious in the centrosome duplication assay where more than 10% of the cells had abnormal centrosome numbers: M18T and I42V. Replacement of the endogenous BRCA1 with BRCA1 (M18T) resulted in 13.6% of the cells with abnormal centrosome amplification, and the BRCA1(I42V) resulted in 10.8% of the cells with supernumerary centrosomes.
Expression of four variants revealed intermediate phenotypes where more than 5.5% of the cells had abnormal centrosome numbers. Expression of D67Y, I31M, L52F and I21V resulted in a moderate effect on centrosome amplification with 6.4, 6.6, 9.0 and 8.8% of the cells having extra centrioles, respectively. By comparison, expression of BRCA1 variants T37R and R71G resulted in no effect on centrosome numbers (Figure 1a).
To rule out the possibility that the centrosome phenotype was cell-line specific, we tested selected mutants (M18T, T37R, I42V and L52F) for effects on centrosome number in the MCF7 cell line (Supplementary Figure S1A). Expression of BRCA1 (M18T) and BRCA1(I42V), similar to the results in the HS578T cell line, resulted in centrosome amplification in 11.2 and 12.9% of the cells, respectively. L52F resulted in the same intermediate phenotype seen before with 7.5% of the cells showing amplification, and 45% of the cells with centrosome amplifications displayed the paired centriole phenotype (Supplementary Figure S1B). Expression of the T37R mutant in MCF7 cells resulted in the same neutral phenotype seen in Hs578T cells. These results suggested that the effect of the expression of the BRCA1 mutants on centrosome number was not cell-line specific, and these mutants result in the same phenotype in a comparable number of cells among different mammary epithelial cell lines.
It is unlikely that the quantitative level of control of centrosome duplication due to expression of a BRCA1 variant protein was due to the expression level of the mutant protein. In the immunoblots from the transfected cells, the BRCA1 protein was modestly depleted (Figure 1c, lanes 2, 11, 18), and the expression level of each mutant was comparable (Figure 1c, lanes 4–9, 12–16,19–21, Supplementary Figure S2). The level of depletion of BRCA1 by transfection of the small-interfering RNA was modest, probably due to poor transfection rates with the Hs578T cell lines used in this assay. As the detection of the centrioles using green fluorescent protein (GFP)-centrin was also dependent on transfection, only the transfected cells were counted, and thus the resulting centrosome number was robust.
We compared the results of centrosome control variants with the results of the homologous recombination assay (Ransburgh et al., 2010). Even though all of the tested variants were clustered in the BRCA1 RING domain, some variants differentially affected the two assays (Table 1). M18T mutant was deleterious in both assays. By comparison, BRCA1(I42V) functioned normally in the homologous recombination assay but was defective in the centrosome assay. I42V has been shown to have a normal ubiquitin ligase activity (Morris et al., 2006), leading to the inference that mutation of I42V blocks the protein–protein interaction of BRCA1 with another protein that is required for the BRCA1 function in the control of centrosome number. Remarkably, the T37R variant, which is just five amino acid residues from isoleucine-42 and which reportedly has no ubiquitin ligase activity (Morris et al., 2006), was deleterious in homologous recombination but functioned normally in the control of centrosome number. The amino acid-specific resolution between the homologous recombination assay and the centrosome regulation assay suggest an intricate network of protein–protein interactions that differ between the two BRCA1-dependent processes.
Table 1.
Summary comparing the effects of the different variants on homologous recombination and centrosome duplication
| BRCA1 missense change | Functional analysis of BRCA1 variant protein
|
|
|---|---|---|
| Homologous recombination | Centrosome number | |
| M18T | Deleterious | Deleterious |
| I21V | As wild-type | Intermediate |
| C24R | Deleterious | Deleterious |
| C27A | Deleterious | Deleterious |
| I31M | As wild-type | Intermediate |
| T37R | Deleterious | As wild-type |
| C39Y | Deleterious | Deleterious |
| H41R | Deleterious | Deleterious |
| I42V | As wild-type | Deleterious |
| C44F | Deleterious | Deleterious |
| C47G | Deleterious | Deleterious |
| L52F | As wild-type | Intermediate |
| D67Y | As wild-type | Intermediate |
| R71G | As wild-type | As wild-type |
In this study we (1) developed an assay for determining the effect of specific single amino acid variants of BRCA1 on centrosome regulation; (2) identified a second function of BRCA1 in controlling the pairing of centrioles; and (3) identified specific residues required for BRCA1 control of homologous recombination versus centrosome regulation. Of the 14 mutants that have been analyzed in this study C44F, a zinc-coordinating residue, and R71G have been classified as deleterious clinically. D67Y on the other hand has been classified as neutral (Easton et al., 2007), and the rest fall into the category of variants of unknown clinical consequence. Our assay showed C44F to be deleterious to the control of centrosome duplication and D67Y to be of intermediate phenotype. With regard to the D67Y, it might be judged to be at the high end of normal, as the centrosome amplification due to its expression was modest. These results with these two variants of known cancer predisposition indicate a correlation between this centrosome duplication assay and cancer predisposition. R71G, however, had full activity in the centrosome assay. This mutant is cancer associated due to a defect in splicing that cannot be identified by our assay that utilizes already spliced complementary DNA (Vega et al., 2001). The BRCA1(R71G) protein, once made, apparently has normal function with regard to centrosome duplication (this study) and homologous recombination (Ransburgh et al., 2010). Thus, combining the functional analysis revealed by the two cellular assays, the centrosome duplication assay and the homologous recombination assay, with the genetic and the clinical analysis of the mutants could potentially be important in the counseling process of women holding such missense mutations. As an example, the M18T variant trends to deleterious based on genetic segregation analysis, but the measure is not sufficiently significant to make a clinical prediction (Easton et al., 2007). When taking into account the defect of BRCA1(M18T) in controlling centrosome duplication and in regulating homologous recombination, it is likely that this variant is in fact a cancer predisposing mutant. Functional assays that are capable of measuring the effect of variants on the protein activity can help define the pathogenicity or neutrality of a specific variant, and thus are important initial steps in helping physicians reach a clear understanding of the risk level of carrying a certain mutation in the BRCA1 gene. Clearly, more work is required to determine whether the results from BRCA1 biological functional assays translate into clinical predictors of tumor likelihood, but an important first step is cataloging many of these variants in functional assays, such as centrosome duplication control.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant CA111480 (JDP).
Footnotes
Conflict of interest
The authors report no conflict of interest.
References
- Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D, III, Fukuda M, et al. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitinligase complex. Proc Natl Acad Sci USA. 2003;100:5646–5651. doi: 10.1073/pnas.0836054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MS, Verma IM. Transcriptional activation by BRCA1. Nature. 1996;382:678–679. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]
- Chiba N, Parvin JD. The BRCA1 and BARD1 association with the RNA polymerase II holoenzyme. Cancer Res. 2002;62:4222–4228. [PubMed] [Google Scholar]
- D’Assoro AB, Stivala F, Barrett S, Ferrigno G, Salisbury JL. GFP-centrin as a marker for centriole dynamics in the human breast cancer cell line MCF-7. Ital J Anat Embryol. 2001;106:103–110. [PubMed] [Google Scholar]
- Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, et al. A systematic genetic assessment of 1433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–883. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haile DT, Parvin JD. Activation of transcription in vitro by the BRCA1 carboxyl-terminal domain. J Biol Chem. 1999;274:2113–2117. doi: 10.1074/jbc.274.4.2113. [DOI] [PubMed] [Google Scholar]
- Houvras Y, Benezra M, Zhang H, Manfredi JJ, Weber BL, Licht JD. BRCA1 physically and functionally interacts with ATF1. J Biol Chem. 2000;275:36230–36237. doi: 10.1074/jbc.M002539200. [DOI] [PubMed] [Google Scholar]
- Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16:1097–1112. doi: 10.1038/sj.onc.1201861. [DOI] [PubMed] [Google Scholar]
- Kais Z, Parvin JD. Regulation of centrosomes by the BRCA1-dependent ubiquitin ligase. Cancer Biol Ther. 2008;7:1540–1543. doi: 10.4161/cbt.7.10.7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLachlan TK, Somasundaram K, Sgagias M, Shifman Y, Muschel RJ, Cowan KH, et al. BRCA1 effects on the cell cycle and the DNA damage response are linked to altered gene expression. J Biol Chem. 2000;275:2777–2785. doi: 10.1074/jbc.275.4.2777. [DOI] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- Monteiro AN, August A, Hanafusa H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc Natl Acad Sci USA. 1996;93:13595–13599. doi: 10.1073/pnas.93.24.13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JR, Pangon L, Boutell C, Katagiri T, Keep NH, Solomon E. Genetic analysis of BRCA1 ubiquitin ligase activity and its relationship to breast cancer susceptibility. Hum Mol Genet. 2006;15:599–606. doi: 10.1093/hmg/ddi476. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer. 2002;2:815–825. doi: 10.1038/nrc924. [DOI] [PubMed] [Google Scholar]
- Pao GM, Janknecht R, Ruffner H, Hunter T, Verma IM. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc Natl Acad Sci USA. 2000;97:1020–1025. doi: 10.1073/pnas.97.3.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransburgh DJ, Chiba N, Ishioka C, Toland AE, Parvin JD. Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res. 2010;70:988–995. doi: 10.1158/0008-5472.CAN-09-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffner H, Joazeiro CA, Hemmati D, Hunter T, Verma IM. Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc Natl Acad Sci USA. 2001;98:5134–5139. doi: 10.1073/pnas.081068398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran S, Crone DE, Palazzo RE, Parvin JD. BRCA1 regulates gamma-tubulin binding to centrosomes. Cancer Biol Ther. 2007;6:1853–1857. doi: 10.4161/cbt.6.12.5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Anderson SF, Chao DM, Wei W, Ye L, Young RA, et al. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc Natl Acad Sci USA. 1997;94:5605–5610. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinilnikova OM, Mazoyer S, Bonnardel C, Lynch HT, Narod SA, Lenoir GM. BRCA1 and BRCA2 mutations in breast and ovarian cancer syndrome: reflection on the Creighton University historical series of high risk families. Fam Cancer. 2006;5:15–20. doi: 10.1007/s10689-005-2571-7. [DOI] [PubMed] [Google Scholar]
- Sluder G, Nordberg JJ. The good, the bad and the ugly: the practical consequences of centrosome amplification. Curr Opin Cell Biol. 2004;16:49–54. doi: 10.1016/j.ceb.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, Schlegel BP, et al. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol. 2004;24:8457–8466. doi: 10.1128/MCB.24.19.8457-8466.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega A, Campos B, Bressac-De-Paillerets B, Bond PM, Janin N, Douglas FS, et al. The R71G BRCA1 is a founder Spanish mutation and leads to aberrant splicing of the transcript. Hum Mutat. 2001;17:520–521. doi: 10.1002/humu.1136. [DOI] [PubMed] [Google Scholar]
- Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, et al. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999;3:389–395. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.