

Abstract
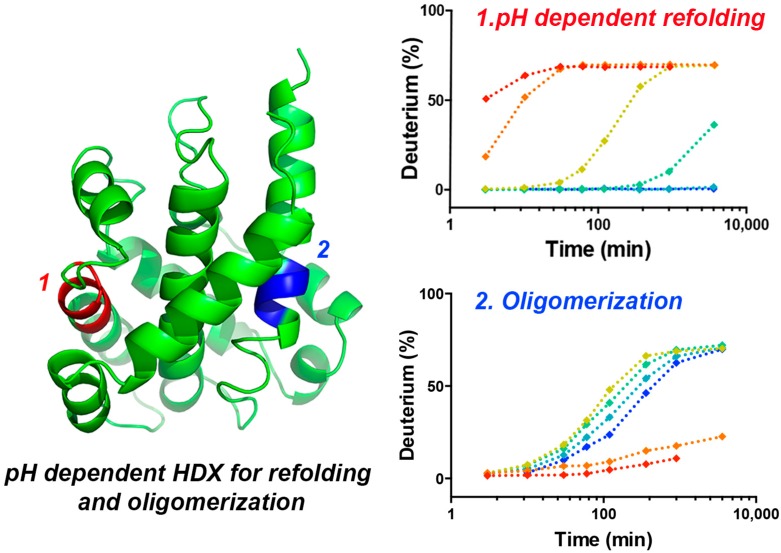
The translocation (T) domain of diphtheria toxin plays a critical role in moving the catalytic domain across the endosomal membrane. Translocation/insertion is triggered by a decrease in pH in the endosome where conformational changes of T domain occur through several kinetic intermediates to yield a final trans-membrane form. High-resolution structural studies are only applicable to the static T-domain structure at physiological pH, and studies of the T-domain translocation pathway are hindered by the simultaneous presence of multiple conformations. Here, we report the application of hydrogen–deuterium exchange mass spectrometry (HDX-MS) for the study of the pH-dependent conformational changes of the T domain in solution. Effects of pH on intrinsic HDX rates were deconvolved by converting the on-exchange times at low pH into times under our “standard condition” (pH 7.5). pH-Dependent HDX kinetic analysis of T domain clearly reveals the conformational transition from the native state (W-state) to a membrane-competent state (W+-state). The initial transition occurs at pH 6 and includes the destabilization of N-terminal helices accompanied by the separation between N- and C-terminal segments. The structural rearrangements accompanying the formation of the membrane-competent state expose a hydrophobic hairpin (TH8–9) to solvent, prepare it to insert into the membrane. At pH 5.5, the transition is complete, and the protein further unfolds, resulting in the exposure of its C-terminal hydrophobic TH8–9, leading to subsequent aggregation in the absence of membranes. This solution-based study complements high resolution crystal structures and provides a detailed understanding of the pH-dependent structural rearrangement and acid-induced oligomerization of T domain.
Diphtheria toxin enters the cell via the endosomal pathway.1 A key step in cell infection is the translocation of the catalytic domain from the endosome into the cytosol, accomplished by a conformational change of the translocation (T) domain.2 Many bacterial pore-forming proteins exist in both water-soluble and integral-membrane forms. Structural rearrangements from water-soluble to membrane proteins occur via multistep processes that include pH-dependent structural refolding; these steps enable the proteins to insert into lipid bilayers.3 Under the acidic conditions in the endosome, the T domain undergoes a series of conformational rearrangements that permit its insertion into the endosomal membrane to form an open-channel state that translocates the toxin’s catalytic (C) domain across the membrane into the cytosol. Once in the cytoplasm, the C domain catalyzes the transfer of the ADP-ribosyl group of NAD+ to elongation factor 2, blocking protein synthesis and killing the mammalian cell. The acid-induced membrane insertion of the T domain proceeds through two conformational transitions with staggered pH-dependencies:4 (1) formation of a membrane-competent state in bulk solution and (2) formation of an insertion-competent state at the membrane surface.
The crystallographic structure of water-soluble diphtheria toxin at neutral pH shows that the T domain consists of nine α-helices, with two of the most hydrophobic α-helical hairpins TH8 and TH9 forming a core that is covered by two layers of amphipathic helices to form a “sandwich-like” structure.5,6 Owing to the coexistence of multiple conformations along the translocation pathway,4 however, it is impossible to get a high resolution structure for T domain in the course of pH-induced refolding. Although numerous biophysical studies have been devoted to the structure and function of the T domain,4,7−16 a detailed characterization of conformational changes and dynamics at molecular level still has not been achieved.
Hydrogen−deuterium exchange coupled to mass spectrometry (HDX MS) has emerged as a powerful tool for probing protein dynamics.17−23 A fast amide hydrogen exchange rate indicates structural flexibility, whereas a slow exchange rate indicates that the amide is part of a relatively rigid, H-bonded structure. Changes in protein dynamics are often related to specific protein conformational changes induced by ligand binding,24 protein–protein interactions,25 changes in pH and salt concentrations,26 and formation of oligomers.27 Forest and co-workers28 demonstrated the use of differential HDX MS to study accessibility changes within the T domain by comparing the HDX results of the native state (pH 8) and a molten globule state (pH 4). However, the structural rearrangements during formation of the W+-state are likely to be subtle,15 and it is likely that the two pH points selected in this early study (pH 8 and 4) are insufficient for characterizing a detailed picture of low pH-induced structural change of the T domain. In addition, the earlier study probably misidentified a molten globule state, formed at pH < 5, as a main membrane-binding species.
Our study, which builds on Forest’s work, addresses the conformational dynamics of water-soluble diphtheria toxin T domain as a function of pH (7.5–5.0) in the absence of a membrane by hydrogen–deuterium exchange mass spectrometry (HDX MS). The changes in HDX dynamics from a standard state (7.5) to five lower pH states (7.0, 6.5, 6.0, 5.5, and 5.0) provide detailed information on the kinetics of conformational transition of T domain as well as the structural rearrangements accompanying each transition. In fact, we identified the pH range of structural transition and mapped the regions involved in the pH-induced conformational transitions onto the crystal structure of soluble T domain. In addition, we also identified the regions involved in oligomerization at low pH in the absence of the membrane. Knowledge of how the water-soluble protein refolds and crosses lipid bilayers should not only provide insight into the T-domain transitions, but also uncover general principles of protein transportation across biological membranes and demonstrate new approaches to study the transport.
Materials and Methods
Materials
All chemicals, proteases, and solvents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Deuterium oxide was purchased from Cambridge Isotope Laboratories Inc. (Andover, MA). The POROS 20 AL beads used to pack immobilized porcine pepsin columns were purchased from Applied Biosystems (Grand Island, NY).
Purification of Diphtheria Toxin T Domain
Diphteria toxin T domain was prepared as described previously.29 Briefly, BL23DELysS cells transformed using a pET15b plasmid-containing T domain gene were grown to OD600 = 0.6, induced with 0.8 mM IPTG, and grown overnight at 24 °C. After harvesting, the cells were lysed by sonification, cell debris was spun down, and soluble DTT was bound to Ni-NTA (Qiagen, MA). After several washes, the protein was eluted from the resin with 0.5 M imidazole in the binding buffer and additionally purified by size-exclusion FPLC on a Superose 12 column in 50 mM sodium-phosphate buffer, pH 8. T domain-containing fractions were stored at −80 °C until further use.
pH-Dependent Hydrogen/Deuterium Exchange Mass Spectrometry
To perform pH-dependent HDX, the pHs of various DTT solutions were adjusted to 7.5, 7.0, 6.5, 6.0, 5.5, and 5.0 by mixing appropriately 50 mM sodium phosphate buffer (pH 7.5) with 50 mM sodium acetate buffer (pH 4). All pH values were corrected (pD = pHread + 0.4)30 from the actual pH readings of a SevenEasy pH meter (Mettler Toledo, Columbus, OH). The HDX was initiated by mixing 1 μL of DTT protein solution (75 uM) with 19 μL of D2O exchange buffer (final D2O content was 95%, final protein concentration was 3.75 μM in labeling solution), or H2O buffer for t0 samples, and incubated for a predetermined set of times under each pH condition (Table 1) at 4 °C. Following a specific incubation time, 30 μL of ice-cold 3 M urea/1% (v/v) trifluoroacetic acid (TFA) was rapidly added to 20 μL of the incubated protein solution to quench the HDX reaction and denature the protein structure. The quenched protein mixture was immediately passed through a custom-built, immobilized pepsin column (2 mm × 2 cm) at a flow rate of 200 μL/min, and the resulting peptic peptides were captured on a C8 cartridge (2.1 mm × 1.5 cm, Agilent, Santa Clara, CA) for desalting; the total time for digestion and desalting was 3 min. Peptides were than separated by using a C18 HPLC column (1.9 μm Hypersil Gold, Thermo Fisher, Waltham, MA) over a 5 min linear gradient (4–40% CH3CN, 0.1% (v/v) formic acid). Valves, columns, and tubing for protein digestion and peptide separation were submerged in an ice–water bath to minimize back-exchange. Mass analysis was performed with an Orbitrap mass spectrometer (LTQ Orbitrap XL, Thermo Fisher, Waltham, MA) equipped with an electrospray ionization source operated at a capillary temperature of 225 °C and a spray voltage of 3.5 kV. Each measurement was made in triplicate.
Table 1. Hydrogen Deuterium Exchange Reaction Time Tablea.
| HDX
reaction time under various pH conditions |
||||||
|---|---|---|---|---|---|---|
| exchange time corrected to standard condition (pH 7.5, 4 °C) | pH 7.5 (Standard) | pH 7.0 (× 100.5) | pH 6.5 (× 101) | pH 6.0 (× 101.5) | pH 5.5 (× 102) | pH 5.0 (× 102.5) |
| 3 s | -- | 10 s | 32 s | 100 s | 5 min 16 s | 16 min 40 s |
| 10 s | 10 s | 32 s | 100 s | 5 min 16 s | 16 min 40 s | 52 min 42 s |
| 30 s | 30 s | 1 min 35 s | 5 min | 15 min 49 s | 50 min | 2 h 38 min |
| 1 min | 1 min | 3 min 10 s | 10 min | 31 min 37 s | 1 h 40 min | 5 h 16 min |
| 2 min | 2 min | 6 min 19 s | 20 min | 1 h 3 min | 3 h 20 min | 10 h 32 min |
| 6 min | 6 min | 18 min 58 s | 1 h | 3 h 9 min | 10 h | 1 d 7 h |
| 15 min | 15 min | 47 min 26 s | 2 h 30 min | 7 h 54 min | 1 d 1 h | 3 d 7 h |
| 1 h | 1 h | 3 h 9 min | 10 h | 31 h 37 min | 4 d 4 h | -- |
The exchange times are corrected to standard condition at pH 7.5 and 4 °C shown in the first column of the table. The numbers in the brackets in columns 2 to 7 are the time conversion factors for corresponding pH conditions.
Fungal XIII Digestion
In addition to digesting the protein with pepsin, digestion with Fungal XIII was performed in solution to increase sequence coverage. Briefly, 20 μL of protein solution after HDX was mixed with 30 μL of Fungal XIII solution (1 mg/mL, in 0.1% formic acid, 0 °C) and held at 4 °C for 2 min before conducting the peptide separation and MS analysis described above.
Peptide Identification and HDX Data Analysis
LC-MS/MS analysis to identify the peptides from digestion was performed in a data-dependent mode of an Orbitrap mass spectrometer, and the top six most abundant ions were selected for CID fragmentation. MS/MS product-ion spectra were submitted to Mascot (Matrix Science, London, UK) for peptide identification. Peptide spectra with a Mascot score of 20 or greater and with no ambiguous hits in a decoy (reversed sequence) database were manually inspected, and the verified peptides were selected as the HDX peptide set. HDX spectra were processed with HDX Workbench software.31 All HDX data were normalized to 100% deuterium content, and no correction was made for back exchange because relative trends provided the needed information. To compare the difference in HDX at various pH conditions, the averaged deuterium uptake percentage at standard buffer condition (pH 7.5) was calculated following 10 s, 30 s, and 1, 2, 6, and 15 min of HDX incubation, and the deuterium uptake percentages at other pHs following adjusted incubation times were calculated and subtracted from those at the standard condition.
pH Dependence of Amide Hydrogen–Deuterium Exchange Rate
Determining the pH dependence of the amide hydrogen exchange rate was previously described by a time-window expansion method to deconvolve the pH effect from protein conformational changes according to eq 1(32)
| 1 |
where A is the frequency factor, and Ea is the activation energy of the dominate base-catalyzed amide hydrogen exchange reaction in the range of pH 5–10. When the temperature was constant, a simple equation (eq 2) can be derived from eq 1 to calculate the ratio of amide hydrogen exchange rate constants that are applicable to two pH conditions:
| 2 |
According to eq 2, the intrinsic HDX rate decreases 107.5–x-fold when decreasing the pH from pH 7.5 to pH x (x = 7.0, 6.5, 6.0, 5.5, 5.0). Thus, to have the same amount of amide hydrogen exchanged at all pH conditions, one needs to deconvolve this effect by converting the exchange times referenced to a standard condition (pH 7.5, 4 °C) as shown in Table 1. After time conversion, the HDX time points at all conditions were converted to 0 s, 3 s, 10 s, 30 s, 1 min, 2 min, 6 min, 15 min, and 1 h.
Results and Discussion
HDX Dynamics of Diphtheria Toxin T Domain at Neutral pH
We first investigated the dynamics of native diphtheria toxin by performing HDX experiments at neutral pH (pH 7.5). Under these conditions, the protein is in membrane-incompetent conformation, which corresponds to the one existing prior to endosomal acidification. HDX provides useful insight into protein structure because the rate of HDX is affected by hydrogen bonding and solvent accessibility of the backbone amides.17,23 In this study, HDX and peptic digestion yielded 123 peptides covering 100% of the T domain sequence across all pH conditions and all on-exchange times. The large number of peptides generated from online pepsin digestion results in multiple overlapping peptides covering most T-domain regions, providing additional information for HDX over small sequence intervals. The only exception is found in region TH2 and the loop connecting TH2 and TH3 (residue 221–241), where only one long peptide spanning the entire region was generated by pepsin digestion. The resistance to pepsin digestion may be due to the locally low hydrophobicity and the tendency of pepsin to cleave after bulky hydrophobic amino acid residues. Interestingly, a previous circular dichroism (CD) and molecular dynamics (MD) study suggested that formation of a water-competent state at low pH is accompanied by partial loss of secondary structure owing to unfolding of helices TH1 and TH2.15 Thus, it is necessary to obtain localized HDX information for TH2. To this end, we carried out a complementary set of HDX experiments using Fungal protease type XIII, an acidic protease that was previously demonstrated to improve digestion efficiency.33 Fortunately, protease type XIII digestion successfully generated 11 overlapping peptides covering region 221–241.
We selected the relative extents of HDX for a combination of 41 peptic peptides and 11 protease XIII digested peptides as representative peptides; the basis for selection is to obtain sufficient short and overlapping peptides to cover most of the protein sequence. HDX at specific incubation times are then mapped onto the crystal structure of T domain,5 as shown in Figure 1. The X-ray crystal structure shows that the T domain is composed of three layers of α-helices and flexible surface loops connecting the helices.5 As expected, we found rapid HDX for the regions that are dynamic and lack structure (loops), whereas regions with highly defined secondary structures (helices) are less solvent-exposed and more H-bonded, generally exchanging slowly (<50% after 1 h of incubation) and showing a higher degree of protection. Regions that are most protected from HDX are the C-terminal TH8 (residues 328–335) and TH9 (residues 360−367) located in the center core of T domain, where less than 5% of exchange occurs during the entire time course of the experiment. Moreover, helices in close contact with the hydrophobic core of the T domain (e.g., TH3 (residues 241–249, 250–255) and TH5 (residues 281−284)) are also highly protected from exchange, and the maximal exchange is less than 5% after 1 h of incubation. In addition, peptides derived from the helices remote to the center core of T domain (TH4, residues 261–273; TH6, residues 298−304; TH7, residues 305−315), despite having secondary structure, are very dynamic and must frequently undergo local unfolding. As a result, TH4 and TH6 exchange much more rapidly than TH3, 5, and TH8–9, with over 50% of amide hydrogens exchanged after 1 h of incubation.
Figure 1.
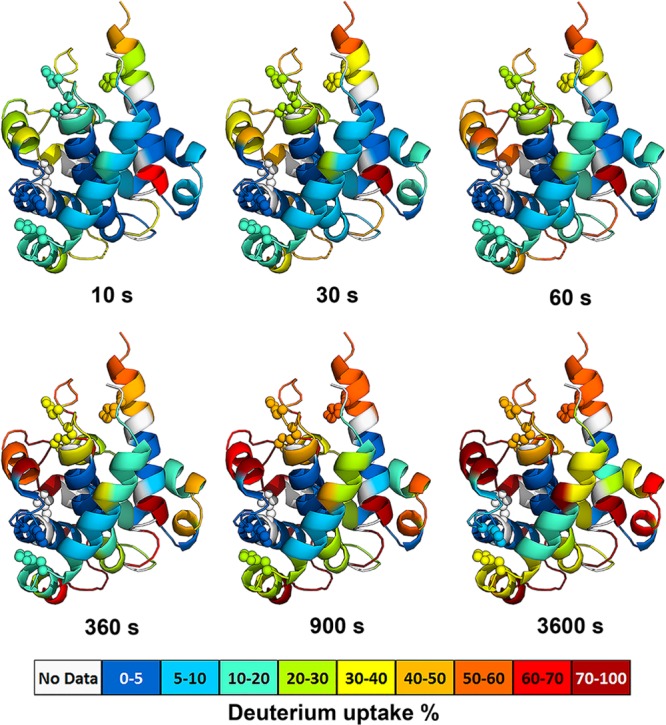
HDX dynamics of monomeric DTT at physiological pH condition (pH 7.5). The ribbon diagrams are colored according to the relative percentage of deuterium uptake of peptides mapped onto the structure of T domain5 (PDB: 1MDT). Histidine residues are shown as spheres. Each color-coded structure corresponds to a selected on-exchange time at pH 7.5. The color code is shown at the bottom of the figure. Sequences that could not be detected by HDX are colored in gray. All time points were corrected to standard condition at pH 7.5 and 4 °C.
pH-Dependent HDX Analysis of T Domain Reveals Two Conformational Transitions
The X-ray crystal structure of the water-soluble T domain at physiological pH provides a starting point for studying its low pH-induced conformational change. To get a detailed picture of how pH mediates the conformational switching of the T domain, we performed a comprehensive pH-dependent HDX analysis of the T domain at six pHs ranging from pH 7.5 to 5.0 with an interval of 0.5. These pH values were selected to be associated with the pH range in the endosome. Because the intrinsic HDX rate also changes with pH, we need to deconvolve the pH effect by converting the on-exchange times at low pH conditions into those at a standard condition, as first demonstrated by Humaro,32 and elaborated later by Griffin.26 Owing to the wide range of pHs and, thus, the large time–conversion factor used in this study, the longest incubation time at pH 7.5 was restricted to 1 h, and the corresponding incubation time (13 d, 4 h) at pH 5.0 was eliminated because the T domain monomer is vulnerable to long-term storage in solution and is prone to aggregate, as shown in Table 1.
The shifts in the centroid mass of isotopic patterns of peptic peptides at all on-exchange time points under six pH conditions were measured, and the deuterium incorporation percentage was plotted as a function of incubation time (after applying the pH-dependent time correction). A complete set of deuterium incorporation plots are shown in Supporting Information Figure S1, and selected deuterium incorporation plots are shown in Figure 2. Upon close inspection of the selected pH-dependent deuterium incorporation plots, we found three distinct HDX patterns: (1) N-terminal and middle region helices TH1–8 (Figure 2a–h,j–k), (2) C-terminal interhelical loop TL8–9 and hydrophobic TH9 (Figure 2l–n), and (3) unstructured loops TL7–8 and TL9 (Figure 2i and o). For the first pattern, all helices remain folded in their native state (W-state) between pH 7.5 and pH 6.5 (blue, cyan, and green curves), but become flexible and undergo two conformational transitions at pH 6.0 (yellow curve) and pH 5.5 (orange curves), characterized by two major increases in HDX. Other studies reported that, between pH 7.5 and pH 6.0, the T domain undergoes conformational rearrangement into a more solvent-exposed membrane competent (W+) state,4,15,34 and our results are consistent with those outcomes. Interestingly, the transition midpoint between the native and the membrane-competent state was calculated to be pH 6.2,2,4,15 a pH condition close to one used in this study (pH 6.0). Thus, one may infer that the extent of HDX represents the combined structural dynamics of both native conformation (W) and the membrane-competent (W+) conformation.
Figure 2.

HDX kinetic curves of peptides HDX under all pH conditions. HDX kinetic curves show three characteristic patterns: (1) Low D% at pH ≥ 6.5, high D% at pH ≤ 5.5, and intermediate D% at pH 6, (a–h, j–k); (2) no differences for all pH conditions, (i,o); (3) increase in D% when increasing pH to 6.5, protection in HDX at pH ≤ 5.5, (l–n). All time points were corrected to standard condition at pH 7.5 and 4 °C. Secondary features were added to all kinetic curves: for example, a) TH1 represents helix1 in T domain; (i) solid line represents loop between helix 7 and 8.
To investigate this possibility, we carefully inspected the mass spectra of peptides across the entire T domain sequences at pH 6. No HDX pattern shows a bimodal distribution, suggesting that the majority of the T domain has already transitioned into the membrane-competent state at pH 6.0. At pH 5.5, the transition into the membrane-competent state is complete, and the N-terminal helices unfold more (show less protection to HDX), further exposing the hydrophobic core and allowing formation of oligomer. The direct evidence for the oligomer is found in the second pattern of HDX kinetics observed in the C-terminal interhelical loop TL8–9 and TH9, which undergoes a similar conformational transition at pH 6.0, but then shows HDX protection at pH 5.5 and pH 5.0, presumably owing to subsequent formation of oligomers. Unlike the structured helices, the third type of HDX was found for most interhelical loops. Owing to the fast HDX for flexible loops and the lack of structural protection from adjacent helices, the HDX kinetics curves in most interhelical loops reach plateaus at early time points at pH 7.5 and do not change at low pH when the structure unfolds.
To elucidate the structural transition at each lower pH, we compared the time-averaged deuterium uptake percentage in the same peptide between the standard pH and the low pH of seven exchange time points. The differences in averaged deuterium uptake percentage of selected peptides were mapped onto the crystal structure of the T domain, as shown in Figure 3. In general, a positive value represents an increase in deuterium exchange (less protected) in that region of the T domain at low pH, whereas a negative value represents a decrease in deuterium exchange (increase in protection). Previous studies demonstrated that the acid-induced conformational rearrangement and the insertion of T domain occur in two steps of protonation.4,34 The initial protonation occurs in bulk solution,4 which results in the structural conversion from a native state (W) to a membrane-competent state (W+).
Figure 3.

Averaged deuterium uptake differences between a low pH state and standard state (pH 7.5)mapped onto crystal structure of DTT (PDB: 1MDT). The color code (see legend) represents the differential HDX between a low pH condition and the standard pH condition; the regions in the crystal structure colored in white are not detected by HDX. Protonation of histidine residues (shown as spheres) is implicated in modulating pH-dependent refolding of the T-domain.
There have been many structural and biological studies addressing the important role of histidine protonation in the formation of a membrane-competent state (W+),8,15,29,34,35 a critical step in inducing membrane binding at pH 6. As expected, after decreasing the pH to 6.0, a modest increase in HDX occurs mainly in the interface between N-terminal TH1–4 and C-terminal hydrophobic TH8–9, corresponding to the partial unfolding of N-terminal layer triggered by the repulsive electrostatic forces between protonated histidine residues (shown as spheres in Figure 1 and Figure 3), and the initial separation between the N- and C- segments. In addition, the decrease in HDX protection may also be due to the loss of secondary structure, measured by CD and, based upon MD simulations, a loss that may be due to unfolding of TH1 and TH2.15 In the presence of the membrane, the second protonation step would promote the insertion of the TH8–9 helical hairpin into lipid bilayers. In the absence of the membrane, however, the T domain aggregates easily and forms high oligomers at pH ≤ 5.5, as shown by size-exclusion chromatography and UV-absorbance measurements (Supporting Information Figure S2). Note that the chromatography experiments were carried out at a higher protein concentration (∼20–30 μM), which would explain the formation of the dimer already at pH 6, while the first signs of oligomerization by HDX were not observed until pH 5.5, as described in the next section. Indeed, our HDX results reveal a significant decrease in HDX for the C-terminal TH9 after decreasing the pH to 5.5, indicating the formation of an oligomer. The strong increase in deuterium uptake in TH 1–4 suggests the N-terminal region further unfolds and releases the initially buried hydrophobic C-terminal TH8–9. The increase in deuterium exchange for TH 8 is possibly due to the loss of contact from nearby N-terminal helices.
In summary, we are able to demonstrate that the structural destabilization in the N-terminal segment of the T domain likely unpacks the overall folded structure, leading to separation between the N-terminal and C-terminal segments at pH 6.0, as previously suggested on the basis of fluorescence experiments and MD simulations.15 At pH 5.5, the T domain further unfolds, resulting in the exposure of its initially buried C-terminal hydrophobic helices (TH8–9) and subsequent oligomerization.
Formation of the Oligomerization Interface T Domain at pH 5.5
Careful analysis of the data presented in Figure 2i–n indicates that the T domain forms an oligomer at pH 5.5, and suggests the likely interface that leads to this process. Specifically, the exchange of the outside helix TH9 appears to be lower than that of the internal helix TH8. This apparent contradiction can be rationalized by assuming that the additional protection for TH9 is coming from the interaction with another TH9 helix, belonging to another T domain molecule in the oligomer. Many previous studies have suggested that the most hydrophobic helices TH8 and TH9 form a transmembrane structure upon insertion into the membrane bilayer;4,9,13 thus, it is not surprising that, in the absence of the membranes, these helixes will nucleate oligomerization via a hydrophobic effect. To examine the dimerization interface of the T domain dimer formed at pH 5.5, we compared HDX of the T domain between pH 7.5 and pH 5.5. As shown in Figure 2, TH9 (residues 360–367) and the interhelical loop (residues 351–355 and 356–360) undergo slower HDX at pH 5.5 and pH 5.0, indicating formation of a new structural feature involving these regions. That feature is likely oligomer formation.
Unexpectedly, TH8, despite its hydrophobic features and involvement in membrane insertion, does not show any protection in HDX kinetics at pH 5.5. A careful inspection of the mass spectra, however, reveals a distinct HDX pattern for TH 8, as is shown in Figure 4. Unlike peptides 351–355 (loop), 356–360 (loop), and 360–367 (TH9), TH8 (residues 333–338, and residues 338–350) after 2 min (adjust time) of exposure to the deuterated buffer showed bimodal distributions in their mass spectra at pH 5.5 and pH 5.0. The major higher mass distribution in TH 8 (residues 333–338 and 338–350) suggests that the majority of T domain further unfolds at pH 5.5, whereas the minor lower mass distribution corresponds to a small population of the T domain that shows HDX protection at pH 5.5, indicating the involvement of oligomerization. In addition, the relative population of the lower mass distribution increases when the pH was dropped to 5.0, where the acidic pH further induces the formation of the oligomer.
Figure 4.

Selected mass spectra of peptides in the aggregation interface after 2 min (adjusted time) exposure to deuterated buffer at pH 7.5 (blue), pH 6.0 (yellow), pH 5.5 (orange), and pH 5.0 (red).
In this way, we are able to demonstrate that, at pH 5.5, the T domain further unfolds, allowing the C-terminal hydrophobic hairpin (TH 8, residues 333–350; TH 9, residues 360–367) and the interhelical loop TL8–9 (residues 351–360) to be exposed to aqueous environment, creating a hydrophobic interface for T domain aggregation in the absence of membranes. Our conclusion is consistent with previous reports that showed acid-induced aggregation at or below pH 5.14,16,28
Although the usual explanation for bimodal behavior in HDX is an EX1 regime for exchange, we do not consider the bimodal distribution seen for peptides covering residues 333 to 350 to be due to EX1 behavior. If EX1 exchange were occurring, we would expect the abundance of the low mass component of the distribution to decrease as pH decreases. Instead, the low mass component of the bimodal distribution increases, consistent with an alternate explanation that the increasing protection is due to oligomerization.
Conclusions
In this study, we demonstrated the use of HDX MS to gain insight into the pH-dependent conformational changes of diphtheria toxin T domain. Effects of pH on intrinsic HDX rates were deconvolved by converting the on-exchange times at low pH conditions into times under our “standard condition” (pH 7.5). The results from this solution-based study extend our understanding from the high-resolution static crystal structures by providing a more detailed picture of the pH-dependent structural rearrangement that precedes membrane insertion. As illustrated in Figure 5, when the solution pH reaches 6, the T domain undergoes a conformational refolding into a membrane-competent state (W+), triggered by the protonation of histidine residues. The repulsive electrostatic static interaction between these protonated histidine resides results in the loss of contact between N- and C-terminal segments, and the exposure of the hydrophobic hairpin (TH8–9) to the aqueous environment. At pH 5.5, the T domain further unfolds to expose the C-terminal hydrophobic hairpin (TH 8, residues 333–350; TH 9, residues 360–367) and the interhelical loop (TL 8–9), providing a hydrophobic surface for subsequent oligomerization.
Figure 5.
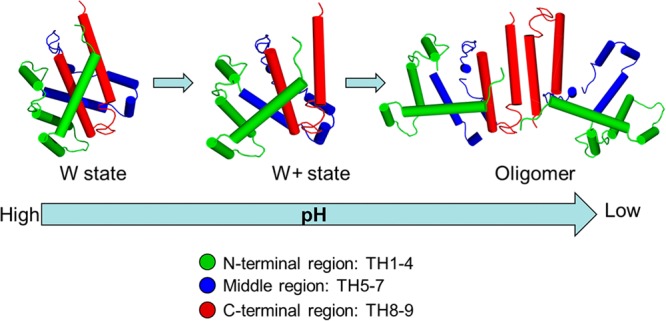
Cartoon illustration of pH-dependent conformational change of DTT in aqueous solution. N-terminal region, middle region, and C-terminal region are shown in green, blue, and red, respectively.
Although pH-dependent studies of HDX at varying pHs are not common, this approach can be readily applied to the analysis of other proteins that change conformation with pH. In the future, we plan to test the applicability of fast photochemical oxidation of proteins (FPOP)36,37 with and without lipids to provide even more detailed insights on this important phenomenon. We expect that FPOP will be less sensitive to pH than HDX. We also plan to study mutants to uncover the role of His protonation on the pH-induced structural changes of T domain in diphtheria toxin T domain.
Supporting Information Available
HDX kinetic curves of all peptic peptides and size-exclusion chromatograph. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding was provided by the National Institutes of Health, NIGMS 8 P41 GM103422–35 to M.L.G. and GM069783 to A.S.L.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hoch D. H.; Romero-Mira M.; Ehrlich B. E.; Finkelstein A.; DasGupta B. R.; Simpson L. L. (1985) Channels formed by botulinum, tetanus, and diphtheria toxins in planar lipid bilayers: relevance to translocation of proteins across membranes. Proc. Natl. Acad. Sci. U.S.A. 82, 1692–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin A. S. (2013) pH-triggered conformational switching along the membrane insertion pathway of the diphtheria toxin T-domain. Toxins 5, 1362–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M. W.; Feil S. C. (2005) Pore-forming protein toxins: from structure to function. Prog. Biophys. Mol. Biol. 88, 91–142. [DOI] [PubMed] [Google Scholar]
- Kyrychenko A.; Posokhov Y. O.; Rodnin M. V.; Ladokhin A. S. (2009) Kinetic intermediate reveals staggered pH-dependent transitions along the membrane insertion pathway of the diphtheria toxin T-domain. Biochemistry 48, 7584–7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett M. J.; Eisenberg D. (1994) Refined structure of monomeric diphtheria toxin at 2.3 A resolution. Protein Sci. 3, 1464–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M. S.; Blanke S. R.; Collier R. J.; Eisenberg D. (1995) Structure of the isolated catalytic domain of diphtheria toxin. Biochemistry 34, 773–781. [DOI] [PubMed] [Google Scholar]
- Chenal A.; Prongidi-Fix L.; Perier A.; Aisenbrey C.; Vernier G.; Lambotte S.; Haertlein M.; Dauvergne M. T.; Fragneto G.; Bechinger B.; Gillet D.; Forge V.; Ferrand M. (2009) Deciphering membrane insertion of the diphtheria toxin T domain by specular neutron reflectometry and solid-state NMR spectroscopy. J. Mol. Biol. 391, 872–883. [DOI] [PubMed] [Google Scholar]
- Rodnin M. V.; Kyrychenko A.; Kienker P.; Sharma O.; Posokhov Y. O.; Collier R. J.; Finkelstein A.; Ladokhin A. S. (2010) Conformational switching of the diphtheria toxin T domain. J. Mol. Biol. 402, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh K. J.; Zhan H.; Cui C.; Hideg K.; Collier R. J.; Hubbell W. L. (1996) Organization of diphtheria toxin T domain in bilayers: a site-directed spin labeling study. Science (New York, N.Y.) 273, 810–812. [DOI] [PubMed] [Google Scholar]
- Oh K. J.; Senzel L.; Collier R. J.; Finkelstein A. (1999) Translocation of the catalytic domain of diphtheria toxin across planar phospholipid bilayers by its own T domain. Proc. Natl. Acad. Sci. U.S.A. 96, 8467–8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G.; London E. (2005) Behavior of diphtheria toxin T domain containing substitutions that block normal membrane insertion at Pro345 and Leu307: control of deep membrane insertion and coupling between deep insertion of hydrophobic subdomains. Biochemistry 44, 4488–4498. [DOI] [PubMed] [Google Scholar]
- Ren J.; Kachel K.; Kim H.; Malenbaum S. E.; Collier R. J.; London E. (1999) Interaction of diphtheria toxin T domain with molten globule-like proteins and its implications for translocation. Science (New York, N.Y.) 284, 955–957. [DOI] [PubMed] [Google Scholar]
- Kachel K.; Ren J.; Collier R. J.; London E. (1998) Identifying transmembrane states and defining the membrane insertion boundaries of hydrophobic helices in membrane-inserted diphtheria toxin T domain. J. Biol. Chem. 273, 22950–22956. [DOI] [PubMed] [Google Scholar]
- Zhan H.; Oh K. J.; Shin Y. K.; Hubbell W. L.; Collier R. J. (1995) Interaction of the isolated transmembrane domain of diphtheria toxin with membranes. Biochemistry 34, 4856–4863. [DOI] [PubMed] [Google Scholar]
- Kurnikov I. V.; Kyrychenko A.; Flores-Canales J. C.; Rodnin M. V.; Simakov N.; Vargas-Uribe M.; Posokhov Y. O.; Kurnikova M.; Ladokhin A. S. (2013) pH-triggered conformational switching of the diphtheria toxin T-domain: the roles of N-terminal histidines. J. Mol. Biol. 425, 2752–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palchevskyy S. S.; Posokhov Y. O.; Olivier B.; Popot J. L.; Pucci B.; Ladokhin A. S. (2006) Chaperoning of insertion of membrane proteins into lipid bilayers by hemifluorinated surfactants: application to diphtheria toxin. Biochemistry 45, 2629–2635. [DOI] [PubMed] [Google Scholar]
- Englander S. W.; Kallenbach N. R. (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 16, 521–655. [DOI] [PubMed] [Google Scholar]
- Bai Y.; Milne J. S.; Mayne L.; Englander S. W. (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L.; Pan J.; Liu Y. H. (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 40, 1224–1234. [DOI] [PubMed] [Google Scholar]
- Chalmers M. J.; Busby S. A.; Pascal B. D.; He Y.; Hendrickson C. L.; Marshall A. G.; Griffin P. R. (2006) Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 78, 1005–1014. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan C. R., and Engen J. R. (2009) Investigating solution-phase protein structure and dynamics by hydrogen exchange mass spectrometry, Current protocols in protein science (Coligan J. E.,. et al. , Eds.) pp 11–17, Chapter 17, Unit 17.16, John Wiley & Sons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales T. E.; Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chalmers M. J.; Stayrook K. R.; Burris L. L.; Garcia-Ordonez R. D.; Pascal B. D.; Burris T. P.; Dodge J. A.; Griffin P. R. (2010) Hydrogen/deuterium exchange reveals distinct agonist/partial agonist receptor dynamics within vitamin D receptor/retinoid X receptor heterodimer. Structure (London, England: 1993) 18, 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chik J. K.; Schriemer D. C. (2003) Hydrogen/deuterium exchange mass spectrometry of actin in various biochemical contexts. J. Mol. Biol. 334, 373–385. [DOI] [PubMed] [Google Scholar]
- Goswami D.; Devarakonda S.; Chalmers M. J.; Pascal B. D.; Spiegelman B. M.; Griffin P. R. (2013) Time window expansion for HDX analysis of an intrinsically disordered protein. J. Am. Soc. Mass Spectrom. 24, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Rempel D. L.; Zhang J.; Sharma A. K.; Mirica L. M.; Gross M. L. (2013) Pulsed hydrogen-deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Abeta) peptide aggregation. Proc. Natl. Acad. Sci. U.S.A. 110, 14604–14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man P.; Montagner C.; Vitrac H.; Kavan D.; Pichard S.; Gillet D.; Forest E.; Forge V. (2010) Accessibility changes within diphtheria toxin T domain when in the functional molten globule state, as determined using hydrogen/deuterium exchange measurements. FEBS J. 277, 653–662. [DOI] [PubMed] [Google Scholar]
- Rodnin M. V.; Kyrychenko A.; Kienker P.; Sharma O.; Vargas-Uribe M.; Collier R. J.; Finkelstein A.; Ladokhin A. S. (2011) Replacement of C-terminal histidines uncouples membrane insertion and translocation in diphtheria toxin T-domain. Biophys. J. 101, L41–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasoe P. K.; Long F. A. (1960) Use of glass electrodes to measure acidities in deuterium oxide1,2. J. Phys. Chem. 64, 188–190. [Google Scholar]
- Pascal B. D.; Willis S.; Lauer J. L.; Landgraf R. R.; West G. M.; Marciano D.; Novick S.; Goswami D.; Chalmers M. J.; Griffin P. R. (2012) HDX workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 23, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coales S. J.; E S. Y.; Lee J. E.; Ma A.; Morrow J. A.; Hamuro Y. (2010) Expansion of time window for mass spectrometric measurement of amide hydrogen/deuterium exchange reactions. Rapid Commun. Mass Spectrom, 24, 3585–3592. [DOI] [PubMed] [Google Scholar]
- Hamuro Y.; Coales S. J.; Molnar K. S.; Tuske S. J.; Morrow J. A. (2008) Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun. Mass Spectrom. 22, 1041–1046. [DOI] [PubMed] [Google Scholar]
- Perier A.; Chassaing A.; Raffestin S.; Pichard S.; Masella M.; Menez A.; Forge V.; Chenal A.; Gillet D. (2007) Concerted protonation of key histidines triggers membrane interaction of the diphtheria toxin T domain. J. Biol. Chem. 282, 24239–24245. [DOI] [PubMed] [Google Scholar]
- Vargas-Uribe M.; Rodnin M. V.; Kienker P.; Finkelstein A.; Ladokhin A. S. (2013) Crucial role of H322 in folding of the diphtheria toxin T-domain into the open-channel state. Biochemistry 52, 3457–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambly D. M.; Gross M. L. (2005) Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 16, 2057–2063. [DOI] [PubMed] [Google Scholar]
- Gau B. C.; Sharp J. S.; Rempel D. L.; Gross M. L. (2009) Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem. 81, 6563–6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.