

Abstract
Enhanced fucosylation has been suggested as a marker for serologic monitoring of liver disease and hepatocellular carcinoma (HCC). We present a workflow for quantitative site-specific analysis of fucosylation and apply it to a comparison of hemopexin (HPX) and complement factor H (CFH), two liver-secreted glycoproteins, in healthy individuals and patients with liver cirrhosis and HCC. Label-free LC-MS quantification of glycopeptides derived from these purified glycoproteins was performed on pooled samples (2 pools/group, 5 samples/pool) and complemented by glycosidase assisted analysis using sialidase and endoglycosidase F2/F3, respectively, to improve resolution of glycoforms. Our analysis, presented as relative abundance of individual fucosylated glycoforms normalized to the level of their nonfucosylated counterparts, revealed a consistent increase in fucosylation in liver disease with significant site- and protein-specific differences. We have observed the highest microheterogeneity of glycoforms at the N187 site of HPX, absence of core fucosylation at N882 and N911 sites of CFH, or a higher degree of core fucosylation in CFH compared to HPX, but we did not identify changes differentiating HCC from matched cirrhosis samples. Glycosidase assisted LC-MS-MRM analysis of individual patient samples prepared by a simplified protocol confirmed the quantitative differences. Transitions specific to outer arm fucose document a disease-associated increase in outer arm fucose on both bi- and triantennary glycans at the N187 site of HPX. Further verification is needed to confirm that enhanced fucosylation of HPX and CFH may serve as an indicator of premalignant liver disease. The analytical strategy can be readily adapted to analysis of other proteins in the appropriate disease context.
Glycosylation is a common and highly diverse modification of proteins.1,2 N-Glycans, the focus of our discussion, are added to proteins through an amide linkage to the Asn (N) side chain in the sequence Asn-X-Ser/Thr, where X is any amino acid except Pro, by a series of reactions catalyzed by a complex enzymatic machinery localized in the endoplasmic reticulum (ER) and Golgi compartments.3 According to UniProtKB/Swiss-Prot, the majority of liver secreted proteins is N-glycosylated at one or more sequons. All the N-linked glycans have a common core structure, but the extension of the core by specific glycosyltransferases leads to substantial diversity of monosaccharides and their linkages in the mature glycans. N-Glycans associated with proteins are therefore heterogeneous, and their composition changes in disease context.4,5 However, the details of quantitative changes in sequon occupancy and glycan microheterogeneity in disease context are known only for a very limited set of proteins. It is therefore of considerable interest to characterize and quantify the glycoforms of liver secreted glycoproteins in the context of liver disease.6
Aberrant glycosylation, in general, and increased fucosylation, in particular, are increasingly recognized as an indicator of liver disease progression to hepatocellular carcinoma (HCC), the most common type of liver cancer.7−19 More than 80% of HCC cases have underlying liver cirrhosis which masks the initial symptoms of HCC development, substantially remodels composition of liver secreted proteins, and thus represents a major challenge for early detection of HCC.20 Despite an extensive search for a reliable HCC biomarker, only alpha-fetoprotein (AFP) is currently used in some countries for serologic monitoring of HCC,20 but only two out of four HCC subtypes are positive for AFP21 which leads to relatively high false negative rates of HCC detection and limits the usefulness of AFP as a diagnostic marker. Recent studies therefore attempt to identify additional proteins and their disease specific glycoforms, accompanying liver disease progression.11,18
A large portion of the reported serologic studies of liver secreted N-linked glycoproteins has been done on detached glycans.11,13,19,22−25 These informative analyses show that changes in specific glycans, primarily fucosylated N-glycans, accompany development of liver disease.7,13,17 However, the studies of detached glycans in complex samples have a common limitation in the undefined changes of composition of the carrier proteins; the changes in protein concentration can contribute significantly to the observed changes in composition of detached N-glycans because glycosylation is protein-specific.24,26 Some reports focus on characterization of isolated proteins,10,11,19 but even these analyses average in most cases across multiple N-glycosylation sites which limits specificity of the observed changes.19,27 This is the major reason why we focus on quantification of site specific protein glycoforms.
Glycoproteins exist as multiple glycoforms due to variability in glycosylation site occupancy (percentage of a site occupied by N-glycan) and microheterogeneity of glycan structures at each glycosylation site. Increasing evidence documents that intramolecular glycosylation is not uniform; glycoproteins carry different glycoforms at different sites of N-glycan attachment.28,29 Reported site-specific changes in protein glycosylation in cancer diseases substantiate the need for their quantitative analysis in terms of disease detection and classification.16,28,30 In the case of liver disease, changes in both outer arm and core fucosylation were reported9−13,16−19 and AFP-L3, the core fucosylated form of AFP, was introduced as an improved diagnostic test of HCC.31 Here, we examine site specific glycoforms of two heme-binding liver secreted glycoproteins by glycosidase-assisted liquid chromatography tandem mass spectrometry (LC-MS). We document quantitative changes of the resolved site-specific linkage isoforms of fucosylated hemopexin (HPX) and complement factor H (CFH) which provides new insights into liver disease processes and may ultimately improve noninvasive disease monitoring.
Methods
Study Population
HCC patients (n = 10), cirrhotic patients (n = 10), and healthy individuals (n = 10) were enrolled into the study in collaboration with the Department of Hepatology and Liver Transplantation, Georgetown University Hospital, Washington, D.C, under protocols approved by the Institutional Review Board. The diagnosis of HCC was made by the attending physician based on liver imaging and/or biopsy. All the HCC patients had early stage disease (stage 1 and 2) according to the seventh edition of the American Joint Committee on Cancer Staging manual. All the patients (HCC and cirrhosis) had chronic hepatitis C virus infection as the primary diagnosis. All participants were matched on age, race (60% Caucasian, 40% African American), and gender (80% males); HCC and cirrhosis groups were further matched on MELD score and prothrombin time represented as International Normalized Ratio (INR) (Table 1).
Table 1. Basic Characteristics of Patient Groupsa.
| healthy | healthy | cirrhosis | cirrhosis | HCC | HCC | |
|---|---|---|---|---|---|---|
| 1 | 2 | 1 | 2 | 1 | 2 | |
| male [%] | 80 | 80 | 80 | 80 | 80 | 80 |
| race (CA/AA) | 3/2 | 3/2 | 3/2 | 3/2 | 3/2 | 3/2 |
| age | 56 ± 1 | 57 ± 4 | 55 ± 2 | 56 ± 4 | 57 ± 2 | 55 ± 4 |
| MELD | n/a | n/a | 10 ± 6 | 11 ± 5 | 10 ± 5 | 10 ± 4 |
| INR | n/a | n/a | 1.2 ± 0.3 | 1.2 ± 0.2 | 1.2 ± 0.3 | 1.2 ± 0.2 |
| albumin [g/dL] | n/a | n/a | 3.4 ± 0.7 | 3.3 ± 0.4 | 3.2 ± 0.6 | 3.5 ± 0.8 |
| AFP [ng/mL] | n/a | n/a | 15.5 ± 64.8 | 8.4 ± 15.3 | 22.3 ± 11.0 | 16.2 ± 9.2 |
| bilirubin [mg/dL] | n/a | n/a | 1.5 ± 1.1 | 2.5 ± 1.7 | 2.1 ± 1.0 | 1.5 ± 0.5 |
| AST/ALT | n/a | n/a | 1.3 ± 0.7 | 1.9 ± 0.3 | 1.3 ± 0.5 | 1.4 ± 1.1 |
| alkaline phosphatase [IU/L] | n/a | n/a | 143 ± 30 | 114 ± 40 | 132 ± 75 | 94 ± 119 |
| creatinine [mg/dL] | n/a | n/a | 0.9 ± 0.1 | 0.8 ± 0.1 | 0.7 ± 0.0 | 0.8 ± 0.3 |
| BUN [mg/dL] | n/a | n/a | 11 ± 1 | 9 ± 3 | 10 ± 3 | 13 ± 2 |
| WBC count [103/mm3] | n/a | n/a | 8.0 ± 2.1 | 5.7 ± 1.7 | 5.0 ± 0.9 | 5.9 ± 1.4 |
| lymphocytes [%] | n/a | n/a | 30.2 ± 9.5 | 37.3 ± 5.0 | 25.9 ± 16.1 | 32.0 ± 23.2 |
| neutrophils [%] | n/a | n/a | 57.5 ± 12.6 | 47.0 ± 4.7 | 60.3 ± 13.9 | 52.3 ± 28.8 |
| platelet count [103/mm3] | n/a | n/a | 70 ± 17 | 143 ± 52 | 86 ± 73 | 78 ± 24 |
| ascites (yes/no) | n/a | n/a | 2/3 | 2/3 | 2/3 | 1/4 |
Values are expressed as median ± interquartile range. There are no significant differences among groups in the listed parameters (P > 0.05). Abbreviations: HCC, hepatocellular carcinoma; CA, Caucasian; AA, African American; MELD, Model for End-Stage Liver Disease; INR, International Normalized Ratio for prothrombin time; AFP, Alpha-fetoprotein; AST/ALT, aspartate/alanine transaminase ratio; BUN, blood urea nitrogen; WBC, white blood cell.
Isolation of Glycoproteins from Plasma
Blood samples were collected using EDTA Vacutainer tubes (BD Diagnostics, Franklin Lakes, NJ); plasma was collected according to the manufacturer’s protocol within 6 h of blood draw and was stored at −80 °C until use. The samples of each study group were divided into two subsets (n = 5 each) and, when needed, samples were pooled by equal volume. HPX and CFH were isolated from plasma by hemin affinity chromatography as described previously32 with slight modifications. Briefly, 200 μL of plasma was diluted 1:2 with PBS, loaded to 200 μL of hemin–agarose suspension (Sigma-Aldrich, St. Louis, MO), and incubated overnight at 4 °C. Bound glycoproteins were eluted with 0.2 M citric acid, pH 2.0, neutralized with 1 M Tris-HCl, pH 9.5, precipitated with methanol/chloroform as described,33 solubilized in solvent A (2% acetonitrile (ACN), 0.1% TFA), and separated on an mRP Hi-Recovery Protein 4.6 × 50 mm C18 column (Agilent Technologies, Santa Clara, CA) heated to 40 °C at a flow rate of 0.75 mL/min as follows: 0–5 min 1% B, 10 min 35% B, 25 min 45% B, 30 min 100% B, 31 min 100% B, 33 min 1% B, 45 min 1% B (B: 98% ACN, 0.08% TFA). The chromatogram was monitored at 214 and 280 nm, and HPX and CFH were collected manually (Figure S-1, Supporting Information). Purified proteins were dried in a CentriVap vacuum concentrator (Labconco, Kansas City, MO), reconstituted in 50 μL of 50 mM NH4HCO3, pH 8.0, with 0.05% RapiGest (Waters, Milford, MA), and stored at −20 °C until use.
Proteolytic and Glycosidase Digests
Reconstituted proteins were reduced with 5 mM DTT for 60 min at 60 °C and alkylated with 15 mM iodoacetamide for 30 min in the dark. Trypsin (Promega, Madison, WI) digestion (2.5 ng/μL) was carried out at 37 °C in Barocycler NEP2320 (Pressure BioSciences, South Easton, MA) for 1 h. Tryptic peptides derived from 2 μg of purified glycoprotein were desialylated with 100 U of α(2-3,6,8)-neuraminidase (New England BioLabs, Ipswich, MA) in 50 mM sodium acetate, 5 mM CaCl2, pH 5.5, at 37 °C for 20 h. For the analysis of core fucosylation, tryptic peptides corresponding to 2 μg of purified glycoprotein were vacuum evaporated, reconstituted in 50 mM sodium acetate, pH 4.5, and digested with 1 μL of each endoglycosidase F2 and F3 from Elizabethkingia miricola (Sigma-Aldrich) at 37 °C for 12 h.
Glycopeptide Analysis by Nano LC-MS/MS
Glycopeptide separation (without glycosidase treatment) was achieved on a Tempo Capillary LC equipped with HiPLC-nanoflex (Eksigent, Framingham, MA) using a nano cHiPLC trap, 200 μm × 0.5 mm, and analytical ChromXP C18-CL, 3 μm, 300 Å columns (Eksigent, Framingham, MA) interfaced with 5600 TripleTOF (AB Sciex, Framingham, MA). A 10 min trapping step using 2% ACN, 0.1% formic acid at 3 μL/min was followed by chromatographic separation at 0.3 μL/min as follows: starting conditions 5% ACN, 0.1% formic acid; 1–35 min, 5–50% ACN, 0.1% formic acid; 35–37 min, 50–95% ACN, 0.1% formic acid; 37–40 min 95% ACN, 0.1% formic acid followed by equilibration to starting conditions for an additional 20 min. For all runs, we have injected 1 μL (2 pmol) of sample directly after enzymatic digestion. Analysis used an Information Dependent Acquisition (IDA) workflow with one full scan (400–1600 m/z) and 50 MS/MS fragmentations of major multiply charged precursor ions with rolling collision energy. Mass spectra were recorded in the MS range of 400–1600 m/z and MS/MS spectra in the range of 100–1800 m/z with resolution of 30 000 and mass accuracy up to 2 ppm using the following experimental parameters: declustering potential, 80 V; curtain gas, 15; ion spray voltage, 2300 V; ion source gas 1, 20; interface heater, 180 °C; entrance potential, 10 V; collision exit potential, 11 V; exclusion time, 5 s; collision energy was set automatically according to m/z of the precursor. Data were processed using ProteinPilot 4.0 software (AB Sciex, Framingham, MA); glycopeptides were screened by GlycoPeptideSearch,34,35 and all assignments were manually verified. Identified glycopeptides were quantified using peak area from the extracted ion chromatogram (XIC) of the precursor ion. Peak integration was performed manually using MultiQuant 2.0 software (AB Sciex) using a 50 mDa window around the theoretical monoisotopic precursor m/z. Internal peptides derived from HPX (GGYTLVSGYPK) and CFH (SSNLIILEEHLK) were used for normalization.
Determination of Glycosylation Site Occupancy
Occupancy of glycosylation sites was quantified by comparison of XIC precursor ion intensities of deglycosylated [18O]-labeled and nonglycosylated peptides acquired on the TripleTOF 5600 mass analyzer using an IDA workflow following PNGaseF deglycosylation under H2[18O] as described.16
MRM Quantification of Glycopeptides
HPX and CFH were enriched from individual patient samples (50 μL of plasma) on hemin–agarose as described above, followed by desalting (100 μg of protein in 1 mL of 0.1% TFA) on SPE cartridge Empore C18-SD 44 mm/1 mL (3M, Saint Paul, MN) activated with 1 mL of 50% ACN and equilibrated with 1 mL of 0.1% TFA. The SPE column was washed with 3 mL of 0.1% TFA, eluted with 1 mL of 40% ACN, evaporated using a vacuum concentrator, and dissolved for digestion in 25 mM NH4HCO3 to a final concentration of 1 μg/μL. MRM quantification of desialylated samples (see above) was performed as described previously36 with the following modifications: RP nanoLC chromatography was interfaced with a 6500 Q-TRAP mass analyzer (AB Sciex, Framingham, MA) with conditions set to curtain gas, 10; ion spray voltage, 2300 V; ion source gas, 20; interface heater temperature, 180 °C; entrance potential, 10 V; and collision exit potential, 13. Chromatographic conditions were as follows: starting conditions 2% ACN, 0.1% formic acid; 0–1 min, 2–16.7% ACN, 0.1% formic acid; 1–10 min, 16.7–26.5% ACN, 0.1% formic acid; 10–13 min 26.5–98% ACN, 0.1% formic acid; 13–17 min, 98% ACN, 0.1% formic acid followed by equilibration to starting conditions for additional 12 min.
Statistical Analysis
Our study is a three-armed case-control study among healthy controls, HCV-related cirrhosis without HCC, and HCV-related cirrhosis with HCC groups. We have matched the three groups on age, gender, and race. The two liver disease groups were additionally matched on INR and MELD score (index of liver function damage). Quantitative analysis of fucosylation of HPX and CFH in pooled samples was done by a one-way ANOVA and t test for pairwise comparisons.37 MRM analysis used individual samples from the pooled analysis (2 × 5 samples per group), and all groups were matched as described above (Table 1). Normality of distribution of the MRM data sets was confirmed for 7 of 11 glycopeptides which were analyzed further by a t test and one-way ANOVA adjusted by Bonferroni methods. Data that did not show a normal distribution were analyzed by nonparametric tests (Kruskal–Wallis test, Mann–Whitney U test) to confirm validity of the one-way ANOVA findings. All reported p values are two sided. Statistical analyses were performed with SAS releases 9.3 (SAS Institute, Cary, NC).
Results and Discussion
Enhanced fucosylation has been proposed as a marker of liver disease progression to HCC, but a large portion of the studies has been done on detached glycans isolated from crude protein mixtures (like serum) or partially purified secreted glycoproteins.9−13,19,22−25 These studies provide valuable information about the disease-related changes in glycan distribution, but quantitative information about protein- and site-specific changes is rarely reported. In the present study, we document a workflow allowing quantitative site- and glycan-specific analysis of fucosylation in the context of liver disease. Our aim is to provide further insight into changes in site-specific protein glycoforms by improved quantitative analysis based on glycosidase assisted LC-MS/MS and LC-MS-MRM.
Fucosylation changes with the progression of liver disease and specific changes are expected at the stage of cirrhosis and HCC.17,19 We performed our analysis of fucosylation on plasma samples of patients with liver cirrhosis and HCC, matched on the extent of liver damage (Table 1), and we compared them to samples obtained from healthy volunteers. This is essential for unbiased quantification of HCC-related glycoforms. Two abundant liver-secreted glycoproteins, HPX and CFH, were selected for analysis. Fucosylated HPX, a 60 kDa heme-binding glycoprotein with five N-glycosylation sites, has been suggested as a candidate HCC marker by previous studies,9,11,38 but site-specific analysis of its glycoforms has not been reported. CFH, a 140 kDa plasma glycoprotein with nine N-glycosylation sites, is a major regulator of the alternative complement pathway39 that mediates the escape of malignant cells from complement-cytotoxicity.40−43 No information in this regard is available for HCC, and we are not aware of any study reporting glycosylation changes in CFH in the context of liver disease.
Both glycoproteins were isolated from human plasma by heme affinity chromatography followed by protein RP-HPLC as described.32 We do this because HPX has the highest reported affinity toward heme (Kd < 1 pmol/L)44,45 and direct binding of CFH to heme has also been reported.46 A typical RP-HPLC chromatogram shows that HPX and CFH were major components of the heme bound fraction and were free of major contaminants after the RP isolation (Figure S-1, Supporting Information). Tryptic glycopeptides derived from HPX and CFH are listed in Table S-1 (Supporting Information). All NXS/T sequons in both proteins are reported to be glycosylated.45,47,48 We analyzed three HPX glycopeptides, corresponding to glycosites N64, N187, and N453, and four CFH glycopeptides, corresponding to sites N217, N882, N911, and N1029. We did not attempt to analyze doubly glycosylated tryptic peptides and CFH peptides corresponding to glycosites N529, N718, and N1095 which were out of fragmentation range of our LC-MS/MS instrumentation.
Site occupancy is an important quantitative parameter which can change in the context of disease.49 We have determined occupancy in pooled samples of the three patient groups. Our analysis shows that all analyzed sites were highly occupied without significant differences among patient groups (Table S-2, Supporting Information). This is important to know because a change in occupancy would affect the quantitative comparison of glycoforms.
Analysis of Glycopeptides in Pooled Samples before Glycosidase Digestion
We have first analyzed tryptic peptides of HPX and CFH isolated from pooled samples (5 samples per pool, 2 pools per group) prior to glycosidase digest (Table S-3, Supporting Information). All glycosites on both proteins were dominated by fully sialylated biantennary glycans with minor contribution of undersialylated biantennary forms (nomenclature is in agreement with the NIBRT GlycoBase).50 Triantennary complex glycans were detected at a subset of the sites (N187 of HPX and N882, 911, and 1029 of CFH) while tetra-antennary sialylated glycoforms were below the limit of detection at all sites except HPX N187. Fucosylation, the subject of this study, was limited to singly fucosylated glycoforms which is in stark contrast to our previously reported analysis of haptoglobin, in the same population, where multiply fucosylated glycoforms with up to six fucoses per glycan were detectable in liver disease.16 Nevertheless, the tendency toward enhanced fucosylation in liver disease groups was clearly detected (Table S-3, Supporting Information).
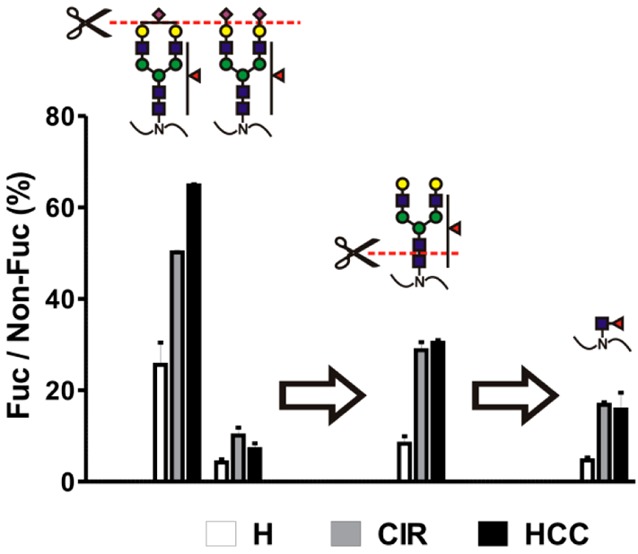
This is even better visualized by comparison of the ratio of corresponding fucosylated to nonfucosylated glycoforms (Figure 1). Approximation of quantities by the XIC signal intensity is reasonable because under the conditions of our study fucose has minimal effect on chromatographic retention time and ionization efficiency (Figure S-2, Supporting Information). This relative quantification allows one to compare the structure-specific fucosylation changes without the need for an internal standard as described for haptoglobin51 and α-2 macroglobulin.30 Our analysis confirms a clear trend toward enhanced fucosylation in liver disease but not a clear difference between cirrhosis and HCC. Interestingly, while fully sialylated structures dominated undersialylated glycoforms in both HPX and CFH (Table S-3, Supporting Information), undersialylated structures are fucosylated to a greater degree (Figure 1A, N187, compare triantennary A3G3S2 and A3G3S3; Figure 1A, N453, and Figure 1B, N911 and N1029, compare biantennary A2G2S1 and A2G2S2).
Figure 1.
Fucosylation of sialylated glycopetides in pooled samples of healthy controls (H), cirrhosis (CIR), and HCC patients. Relative abundance of each fucosylated glycoform, quantified as area of precursor ion XIC peak, is presented as a percent of its nonfucosylated counterpart. (A) Hemopexin; (B) CFH. Glycan structures are indicated above each group of corresponding bars representing three patient groups; the position of the glycosylation site in the protein sequence is shown below. Results are shown as mean ± SD; ∗, P < 0.05 vs H; #, P < 0.05 HCC vs CIR.
Analysis of Desialylated Glycopeptides in Pooled Samples
Sialylated glycopeptides have lower ionization efficiencies in positive ionization mode, and the degree of sialylation contributes to microheterogeneity at each glycosite which leads, ultimately, to lower sensitivity of detection of fucosylated glycoforms we are interested in. In order to enhance sensitivity, we desialylated samples from the above analysis with nonspecific neuraminidase cleaving all sialic acids with the α(2-3,6,8) linkage as described previously.36 Under these conditions, we have detected quantifiable amounts of singly fucosylated triantennary glycans at three additional peptides, N64 and N453 of HPX and N911 of CFH (Table S-4, Supporting Information). Relative quantification of the corresponding glycoforms shows increased fucosylation in liver disease compared to healthy controls (Figure 2). We observe that the triantennary glycoforms have a substantially higher proportion of fucosylated structures compared to their biantennary counterparts. In healthy controls, the ratio of fucosylated to nonfucosylated biantennary glycoforms ranges from 2% to 13%, while the ratio of triantennary glycoforms ranges from 15% to 200% with a large increase at each individual glycosite (Figure 2). The largest difference was detected at N64 of hemopexin with A2G2F1 (6%) compared to A3G3F1 (200%), but the observation holds true for every site (Figure 2). In addition, we were able to detect doubly fucosylated triantennary and singly fucosylated tetra-antennary glycan structures at the N187 site of hemopexin (Figure 2A); these structures were reported to be associated with HCC in a study of detached glycans.11 The sum of intensities of the above two glycans (A3G3F2 + A4G4F1) divided by the intensity of A2G2 was used to distinguish HCC from cirrhosis. We did not find changes in this ratio (0.0142 ± 0.011 for cirrhosis, 0.0133 ± 0.0045 for HCC; see Table S-4, Supporting Information). This is possibly related to the difference between the studied populations or the difference in the quantification of detached glycans and glycopeptides. Since we did not analyze structures attached to doubly glycosylated hemopexin peptide N*GTGHGN*STHHGPEYMR, our analysis may be missing potentially informative glycans attached to this site, but it is also possible that other glycoproteins contributed to the total pool of glycans in the above study11 because hemin-affinity purification was the only purification step performed which is in our hands insufficient to achieve hemopexin purity (Figure S-1, Supporting Information). In addition, the HCC group in the reported study had significantly increased levels of C-reactive protein over the cirrhosis group11 which might be a factor contributing to these discrepancies. Overall, the tendency toward enhanced fucosylation is apparent in liver disease compared to healthy subjects, but we did not observe significant HCC-specific changes.
Figure 2.
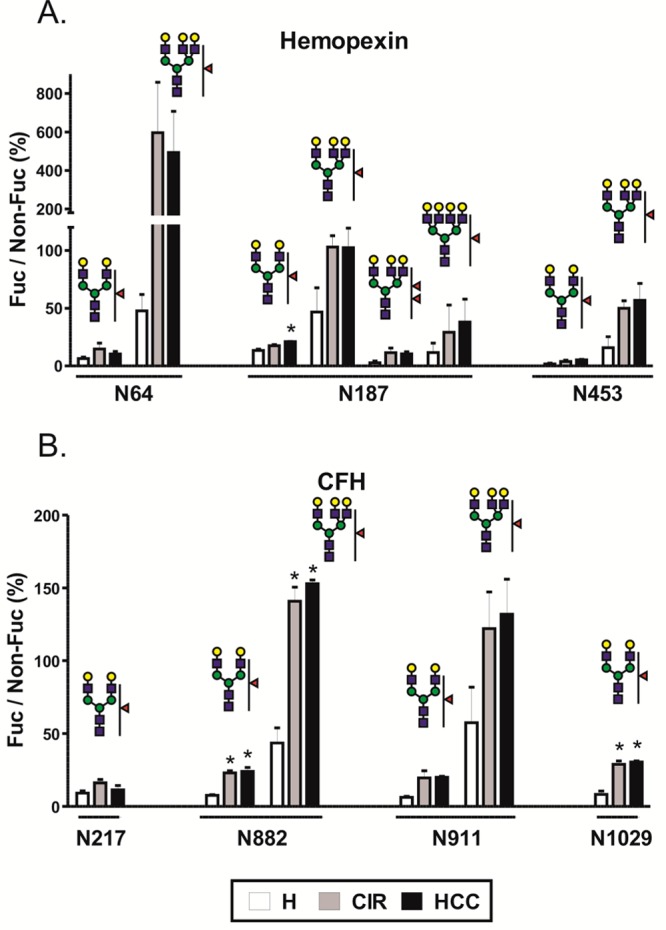
Fucosylation of desialylated glycopetides in pooled samples of healthy controls (H), cirrhosis (CIR), and HCC patients. Relative abundance of each fucosylated glycoform, quantified as area of precursor ion XIC peak, is presented as a percent of its nonfucosylated counterpart. (A) Hemopexin; (B) CFH. Glycan structures are indicated above each group of corresponding bars representing three patient groups; the position of the glycosylation site in the protein sequence is shown below. Results are shown as mean ± SD; ∗, P < 0.05 vs H.
Analysis of Core Fucosylation in Pooled Samples
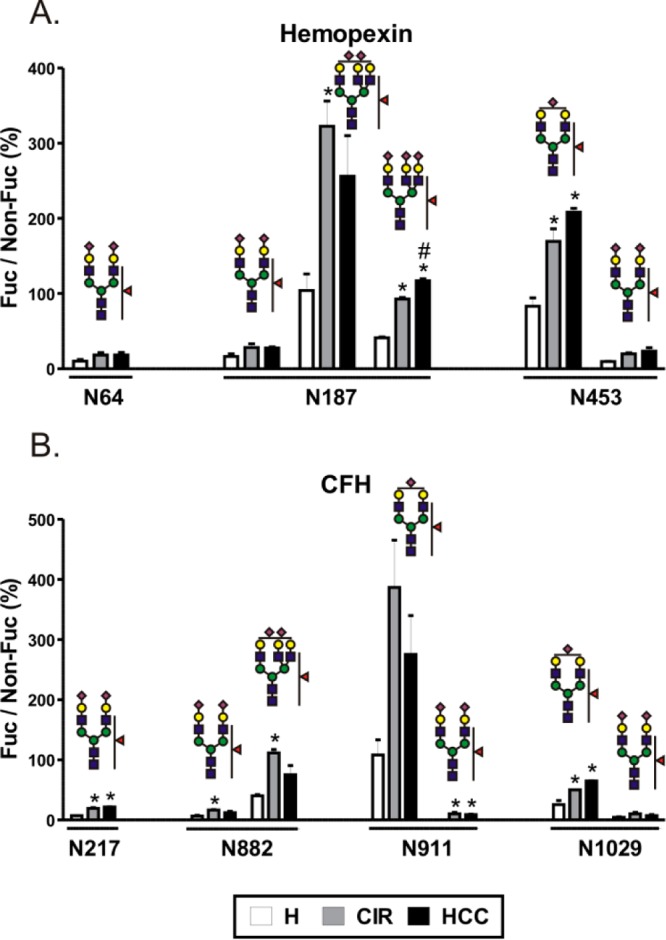
The preceding analysis does not allow unequivocal differentiation of core α(1–6) linkage from outer arm fucosylation; these modifications are carried out by a different set of enzymes and are important to distinguish in the liver disease context.11,19,52 To quantify core fucosylation, we treated glycopeptides with a combination of endoglycosidase F2 and F3. This cleaves complex bi- and triantennary glycans leaving the innermost N-linked GlcNAc with or without core fucose attached to the peptide. We have utilized both enzymes in excess and did not observe any residual uncleaved glycopeptides after overnight digest. The proportion of core fucosylated peptides, expressed as a percent of the nonfucosylated form, is presented in Figure 3. In the case of HPX, core fucose was identified on all three singly glycosylated peptides. The percentage of core fucose in healthy subjects was below 5% on all sites; a tendency toward an increased ratio of fucosylated structures was observed in liver disease, but this increase was minor (Figure 3A). In contrast, core fucosylation of CFH was clearly elevated in the liver disease groups at N217 and N1029 positions (Figure 3B), but a difference between cirrhosis and HCC groups was not observed. We did not detect any core fucosylation at N882 and N911 sites of CFH although the nonfucosylated glycopeptides were readily detected. This indicates that the changes in overall fucosylation at these sites (Figures 1, 2, and 4) are associated with alterations in outer arm fucosylation.
Figure 3.
Core fucosylation in pooled samples of healthy controls (H), cirrhosis (CIR), and HCC patients. Core fucosylation of (A) hemopexin and (B) CFH was analyzed following endoglycosidase F2/F3 treatment. Relative abundance of each fucosylated glycoform, quantified as area of precursor ion XIC peak, is presented as a percent of its nonfucosylated counterpart. The position of the glycosylation site in the protein sequence is shown below the corresponding group of bars representing three patient groups. Results are shown as mean ± SD; ∗, P < 0.05 vs H; ND, nondetectable.
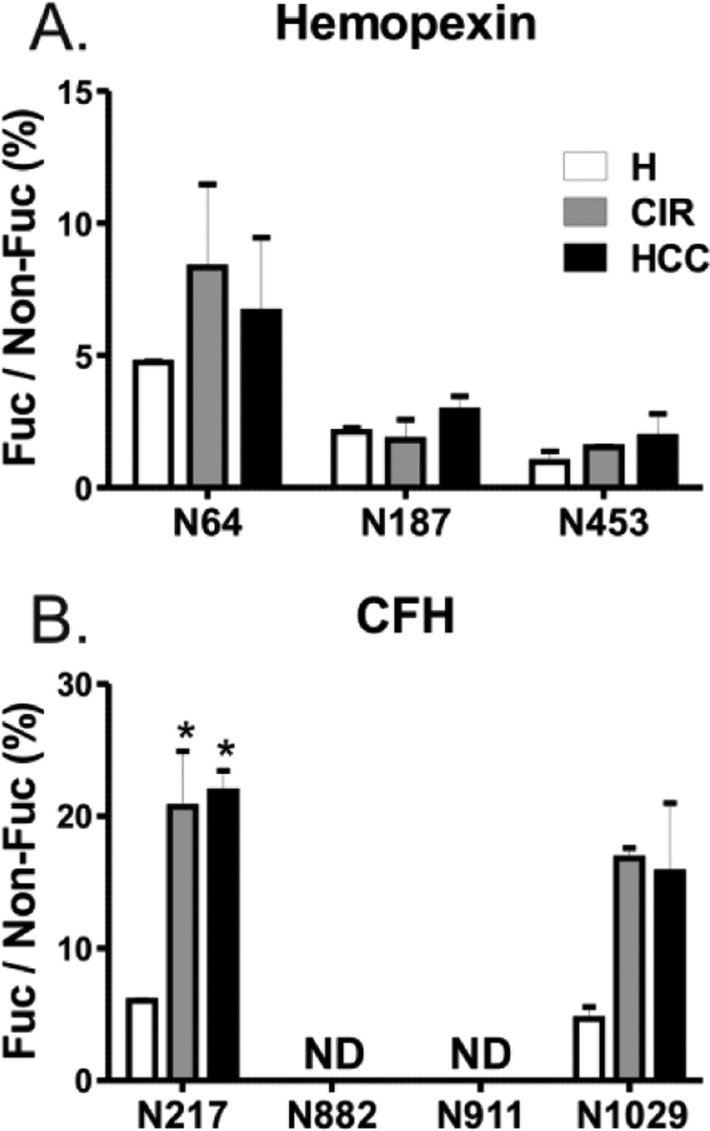
Figure 4.
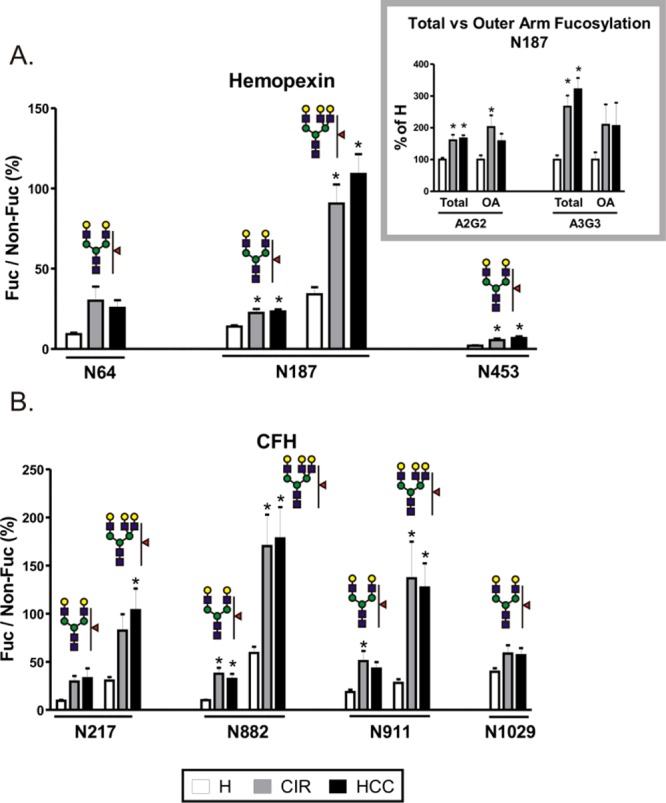
Fucosylation of desialylated glycopetides in individual samples of healthy controls (H), cirrhosis (CIR), and HCC patients. Tryptic glycopeptides of (A) hemopexin and (B) CFH from the heme-bound fraction of individual patient samples were quantified by LC-MS MRM. Data are expressed as a relative ratio of signal intensities of fucosylated glycopeptide to its nonfucosylated counterpart monitored as the 366 transition (Hex–HexNAc) and shown as percent of the nonfucosylated form. Glycan structures representing specific glycoforms are indicated above each group of corresponding bars; the position of the glycosylation site in the protein sequence is shown below. Inset. Comparison of total and outer arm fucosylation at the N187 site of hemopexin. Outer arm fucosylation was quantified as the 512 transition (Fuc–GlcNAc–Gal) of fucosylated precursor normalized to the 366 transition of its nonfucosylated counterpart; total fucosylation was quantified as above. The relative change in fucosylation in liver disease groups is shown as a percent of H. OA; outer arm fucosylation. Results are shown as mean ± SEM; ∗, P < 0.05 vs H.
The striking difference in core fucosylation of CFH at different glycosites substantiates the need for site specific analysis. Maturation of glycans, including core fucosylation, takes place in the Golgi compartment and is believed to occur on fully folded proteins.53 It has been therefore proposed that core fucosylation depends on solvent accessibility at the site of fucosylation.29 The two sites without core fucose have rather low predicted accessibility;29 thus, a low level of core fucosylation would be expected. Conversely, we clearly detected core fucose at N1029 which has even lower predicted accessibility. In addition, core fucose was not detected at N1095 (data not shown) which has the highest predicted solvent accessibility of all the above sites. This indicates that site accessibility, although potentially an important factor overall, does not explain the differences of core fucosylation in the case of CFH.
Besides the peptides shown in Figure 3, we also analyzed core fucosylation at peptide WDPEVN*CSMAQIQLCPPPPQIPNSHN*MTTTLNYR doubly glycosylated at positions N802 and N822 (relative abundance of the fucosylated glycoform 13.040 ± 0.949, 57.510 ± 2.003, and 48.251 ± 9.127 for healthy, cirrhosis, and HCC, respectively, expressed as percent of nonfucosylated counterpart). Interestingly, either both sites or neither of them were core fucosylated; we did not find any peptide fucosylated at only one of the two positions. The amount of this doubly fucosylated peptide increases 4-fold in liver disease but to the same degree in the cirrhosis and HCC groups.
MRM Analysis of Desialylated Glycopeptides in Individual Samples
To validate the results of our analysis of pooled samples, we employed an MRM workflow to quantify site specific glycoforms in individual patient samples. CFH and HPX were partially purified by hemin-affinity (as for pooled samples) and further enriched on the C18 SPE cartridge using conditions based on RP-HPLC separation. Elution with 40% acetonitrile was sufficient to elute both proteins and eliminate more hydrophobic protein contaminants. In order to eliminate the potential variations in binding capacity, we used aliquots of the same batch of hemin–agarose beads for all samples. The enriched hemin-bound fraction was digested by trypsin, and HPX and CFH glycopeptides were desialylated with neuraminidase prior to MRM analysis. Of the monitored MRM transitions (Table S-5, Supporting Information), glycopeptide precursor → 366.1 (Hex–HexNAc) transition, the most intense transition, was used for quantitative comparisons. We present a comparison of relative abundances of the fucosylated form normalized to the intensity of its nonfucosylated counterpart in Figure 4 (n = 10 per group). The dot plot version of Figure 4 showing the distribution of individual values is provided in the Supporting Information (Figure S-3), and quantitative data for each patient are shown in Table S-6 (Supporting Information). Due to high signal-to-noise, we could not reliably quantify A3G3F1 at N64, A3G3F2 and A4G4F1 at N187, and A3G3F1 at N453; quantitative data obtained on the remaining structures for both HPX and CFH closely resemble those obtained by XIC-based quantification on pooled samples (Figure 2) and validate the results.
The transition glycopeptide precursor → 512.2 (Fuc–GlcNAc–Gal) was used to determine the presence of outer arm fucosylation. We could quantify the 512 transition at the two core fucose-lacking glycosites of CFH (N882 and N911) and at the N187 site of hemopexin (inset in Figure 4A). The intensity of the 512 transition of the fucosylated glycopeptide was normalized to the intensity of the 366 transition of its nonfucosylated counterpart. For total fucosylation, the 366 transition was used for both fucosylated and nonfucosylated form. Because of differences in intensities between the two transitions, data are presented as a percent of healthy controls. Outer arm fucose was identified on both biantennary (A2G2) and triantennary (A3G3) glycans, and the level of fucosylation was increased to the same extent in both liver disease groups. The comparison with core fucosylation at the same site (Figure 3A, N187), which is not significantly altered in disease, allows us to conclude that the liver disease-related increase in fucosylation at this site is due to enhanced outer arm fucosylation. For other sites with detected core fucosylation (Figure 3, N64 and N453 of HPX and N217 and N1029 of CFH), the intensity of the 512 transition was indistinguishable from noise, indicating that core fucose is the major contributor to the fucosylation.
Conclusions
Our glycosidase assisted workflow documents efficient analysis of site specific fucosylated glycoforms by LC-MSMS in pooled samples followed by simplified LC-MS-MRM quantification in a larger set of individual samples. The results document significant increases in fucosylation of HPX and CFH in the context of liver disease. Sialidase improves the sensitivity of detection of fucosylated glycopeptides. Digestion with endoglycosidase F2/F3 enables site-specific quantitative analysis of core fucosylation. Using the glycosidase-assisted approach, we have identified striking differences in core fucosylation among different glycosites of CFH as well as a higher degree of microheterogeneity at the N187 glycosite of HPX. The combination of glycosidase assisted analyses and specific CID fragmentation shows that the percent of core fucosylation is site-specific and higher in CFH than in HPX. The lack of changes differentiating HCC from cirrhosis in samples matched on liver damage indicates that increased fucosylation of hemopexin and CFH reflects the extent of damage of liver tissue rather than malignant transformation. This is further supported by correlation analysis in individual samples of liver disease patients showing positive association between the degree of fucosylation and MELD score and negative correlation with serum albumin (Figure S-4, Supporting Information). We present correlation analysis for site N217 of CFH, but the observation holds true for the remaining glycosites on both glycoproteins. These observations demonstrate the advantages of site-specific glycopeptide analysis and efficiency of the glycosidase assisted LC-MS-MRM workflow in quantitative comparisons of protein glycosylation.
Acknowledgments
This work was supported by National Institutes of Health Grants UO1 CA168926, UO1 CA171146, and RO1 CA135069 (to R.G.) and CCSG Grant P30 CA51008 (to Lombardi Comprehensive Cancer Center supporting the Proteomics and Metabolomics Shared Resource). P.P. was supported by AMVIS Czech Republic-U.S. exchange program LH13051 and by Grant Agency of the Czech Republic (Grant P206/12/0503), Charles University (ProjectUNCE_204025/2012).
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# J.B. and M.S. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lis H.; Sharon N. Eur. J. Biochem. 1993, 218, 1–27. [DOI] [PubMed] [Google Scholar]
- Varki A. Glycobiology 1993, 3, 97–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen K. W.; Tiemeyer M.; Nairn A. V. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold J. N.; Saldova R.; Hamid U. M.; Rudd P. M. Proteomics 2008, 8, 3284–3293. [DOI] [PubMed] [Google Scholar]
- Peracaula R.; Sarrats A.; Rudd P. M. Proteomics Clin. Appl. 2010, 4, 426–431. [DOI] [PubMed] [Google Scholar]
- Chandler K.; Goldman R. Mol. Cell. Proteomics 2013, 12, 836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomme B.; Francque S.; Trepo E.; Libbrecht L.; Vanderschaeghe D.; Verrijken A.; Pattyn P.; Nieuwenhove Y. V.; Putte D. V.; Geerts A.; Colle I.; Delanghe J.; Moreno C.; Gaal L. V.; Callewaert N.; Vlierberghe H. V. Dig. Liver Dis. 2012, 44, 315–322. [DOI] [PubMed] [Google Scholar]
- Comunale M. A.; Lowman M.; Long R. E.; Krakover J.; Philip R.; Seeholzer S.; Evans A. A.; Hann H. W.; Block T. M.; Mehta A. S. J. Proteome Res. 2006, 5, 308–315. [DOI] [PubMed] [Google Scholar]
- Comunale M. A.; Wang M.; Hafner J.; Krakover J.; Rodemich L.; Kopenhaver B.; Long R. E.; Junaidi O.; Bisceglie A. M.; Block T. M.; Mehta A. S. J. Proteome Res. 2009, 8, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comunale M. A.; Rodemich-Betesh L.; Hafner J.; Wang M.; Norton P.; Di Bisceglie A. M.; Block T.; Mehta A. PLoS One 2010, 5, e12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debruyne E. N.; Vanderschaeghe D.; Van V. H.; Vanhecke A.; Callewaert N.; Delanghe J. R. Clin. Chem. 2010, 56, 823–831. [DOI] [PubMed] [Google Scholar]
- Liao J.; Zhang R.; Qian H.; Cao L.; Zhang Y.; Xu W.; Li J.; Wu M.; Yin Z. Biochem. Biophys. Res. Commun. 2012, 420, 308–314. [DOI] [PubMed] [Google Scholar]
- Liu X. E.; Desmyter L.; Gao C. F.; Laroy W.; Dewaele S.; Vanhooren V.; Wang L.; Zhuang H.; Callewaert N.; Libert C.; Contreras R.; Chen C. Hepatology 2007, 46, 1426–1435. [DOI] [PubMed] [Google Scholar]
- Moriya S.; Morimoto M.; Numata K.; Nozaki A.; Shimoyama Y.; Kondo M.; Nakano M.; Maeda S.; Tanaka K. Anticancer Res. 2013, 33, 997–1001. [PubMed] [Google Scholar]
- Naitoh A.; Aoyagi Y.; Asakura H. J. Gastroenterol. Hepatol. 1999, 14, 436–445. [DOI] [PubMed] [Google Scholar]
- Pompach P.; Brnakova Z.; Sanda M.; Wu J.; Edwards N.; Goldman R. Mol. Cell. Proteomics 2013, 12, 1281–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschaeghe D.; Laroy W.; Sablon E.; Halfon P.; Van H. A.; Delanghe J.; Callewaert N. Mol. Cell. Proteomics 2009, 8, 986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Long R. E.; Comunale M. A.; Junaidi O.; Marrero J.; Di Bisceglie A. M.; Block T. M.; Mehta A. S. Cancer Epidemiol., Biomarkers Prev. 2009, 18, 1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Lin Z.; Wu J.; Yin H.; Dai J.; Feng Z.; Marrero J.; Lubman D. M. J. Proteome. Res. 2014, 13, 2986–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefaniuk P.; Cianciara J.; Wiercinska-Drapalo A. World J. Gastroenterol. 2010, 16, 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T.; Forgues M.; Wang W.; Kim J. W.; Ye Q.; Jia H.; Budhu A.; Zanetti K. A.; Chen Y.; Qin L. X.; Tang Z. Y.; Wang X. W. Cancer Res. 2008, 68, 1451–1461. [DOI] [PubMed] [Google Scholar]
- Goldman R.; Ressom H. W.; Varghese R. S.; Goldman L.; Bascug G.; Loffredo C. A.; Abdel-Hamid M.; Gouda I.; Ezzat S.; Kyselova Z.; Mechref Y.; Novotny M. V. Clin. Cancer Res. 2009, 15, 1808–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isailovic D.; Kurulugama R. T.; Plasencia M. D.; Stokes S. T.; Kyselova Z.; Goldman R.; Mechref Y.; Novotny M. V.; Clemmer D. E. J. Proteome. Res. 2008, 7, 1109–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A.; Michalski J. C.; Morelle W. Proteomics Clin. Appl. 2010, 4, 372–378. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Varghese R. S.; Bekesova S.; Loffredo C. A.; Hamid M. A.; Kyselova Z.; Mechref Y.; Novotny M. V.; Goldman R.; Ressom H. W. J. Proteome Res. 2010, 9, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekesova S.; Kosti O.; Chandler K. B.; Wu J.; Madej H. L.; Brown K. C.; Simonyan V.; Goldman R. J. Proteomics 2012, 75, 2216–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson P. J.; Poon T. C.; Hjelm N. M.; Ho C. S.; Ho S. K.; Welby C.; Stevenson D.; Patel T.; Parekh R.; Townsend R. R. Br. J. Cancer 1999, 81, 1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano M.; Nakagawa T.; Ito T.; Kitada T.; Hijioka T.; Kasahara A.; Tajiri M.; Wada Y.; Taniguchi N.; Miyoshi E. Int. J. Cancer 2008, 122, 2301–2309. [DOI] [PubMed] [Google Scholar]
- Thaysen-Andersen M.; Packer N. H. Glycobiology 2012, 22, 1440–1452. [DOI] [PubMed] [Google Scholar]
- Lin Z.; Yin H.; Lo A.; Ruffin M. T.; Anderson M. A.; Simeone D. M.; Lubman D. M. Electrophoresis 2013, 35, 2108–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumada T.; Nakano S.; Takeda I.; Kiriyama S.; Sone Y.; Hayashi K.; Katoh H.; Endoh T.; Sassa T.; Satomura S. J. Hepatol. 1999, 30, 125–130. [DOI] [PubMed] [Google Scholar]
- Sanda M.; Pompach P.; Benicky J.; Goldman R. Electrophoresis 2013, 34, 2342–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel D.; Flugge U. I. Anal. Biochem. 1984, 138, 141–143. [DOI] [PubMed] [Google Scholar]
- Chandler K. B.; Pompach P.; Goldman R.; Edwards N. J. Proteome Res. 2013, 12, 3652–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompach P.; Chandler K. B.; Lan R.; Edwards N.; Goldman R. J. Proteome Res. 2012, 11, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda M.; Pompach P.; Brnakova Z.; Wu J.; Makambi K.; Goldman R. Mol. Cell. Proteomics 2013, 12, 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Winter J. C. F.Pract. Assess. Res. Eval. 2013, 18 (10); Available online: http://pareonline.net/getvn.asp?v=18&n=10. [Google Scholar]
- Morota K.; Nakagawa M.; Sekiya R.; Hemken P. M.; Sokoll L. J.; Elliott D.; Chan D. W.; Dowell B. L. Clin. Chem. Lab Med. 2011, 49, 711–718. [DOI] [PubMed] [Google Scholar]
- Whaley K.; Ruddy S. J. Exp. Med. 1976, 144, 1147–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajona D.; Castano Z.; Garayoa M.; Zudaire E.; Pajares M. J.; Martinez A.; Cuttitta F.; Montuenga L. M.; Pio R. Cancer Res. 2004, 64, 6310–6318. [DOI] [PubMed] [Google Scholar]
- Junnikkala S.; Jokiranta T. S.; Friese M. A.; Jarva H.; Zipfel P. F.; Meri S. J. Immunol. 2000, 164, 6075–6081. [DOI] [PubMed] [Google Scholar]
- Junnikkala S.; Hakulinen J.; Jarva H.; Manuelian T.; Bjorge L.; Butzow R.; Zipfel P. F.; Meri S. Br. J. Cancer 2002, 87, 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczek E.; Rzepko R.; Nowis D.; Legat M.; Golab J.; Glab M.; Gorlewicz A.; Konopacki F.; Mazurkiewicz M.; Sladowski D.; Gornicka B.; Wasiutynski A.; Wilczynski G. M. Int. J. Cancer 2008, 122, 2030–2037. [DOI] [PubMed] [Google Scholar]
- Muller-Eberhard U. Methods Enzymol. 1988, 163, 536–565. [DOI] [PubMed] [Google Scholar]
- Delanghe J. R.; Langlois M. R. Clin. Chim. Acta 2001, 312, 13–23. [DOI] [PubMed] [Google Scholar]
- Ferreira V. P.; Pangburn M. K.; Cortes C. Mol. Immunol. 2010, 47, 2187–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenaille F.; Le M. M.; Groseil C.; Ramon C.; Riande S.; Siret L.; Bihoreau N. Glycobiology 2007, 17, 932–944. [DOI] [PubMed] [Google Scholar]
- Jia W.; Lu Z.; Fu Y.; Wang H. P.; Wang L. H.; Chi H.; Yuan Z. F.; Zheng Z. B.; Song L. N.; Han H. H.; Liang Y. M.; Wang J. L.; Cai Y.; Zhang Y. K.; Deng Y. L.; Ying W. T.; He S. M.; Qian X. H. Mol. Cell. Proteomics 2009, 8, 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Cao J.; He Y.; Qiao L.; Xu C.; Lu H.; Yang P. J. Proteome. Res. 2010, 9, 227–236. [DOI] [PubMed] [Google Scholar]
- Royle L.; Campbell M. P.; Radcliffe C. M.; White D. M.; Harvey D. J.; Abrahams J. L.; Kim Y. G.; Henry G. W.; Shadick N. A.; Weinblatt M. E.; Lee D. M.; Rudd P. M.; Dwek R. A. Anal. Biochem. 2008, 376, 1–12. [DOI] [PubMed] [Google Scholar]
- Lee H. J.; Cha H. J.; Lim J. S.; Lee S. H.; Song S. Y.; Kim H.; Hancock W. S.; Yoo J. S.; Paik Y. K. J. Proteome. Res. 2014, 13, 2328–2338. [DOI] [PubMed] [Google Scholar]
- Tanabe K.; Deguchi A.; Higashi M.; Usuki H.; Suzuki Y.; Uchimura Y.; Kuriyama S.; Ikenaka K. Biochem. Biophys. Res. Commun. 2008, 374, 219–225. [DOI] [PubMed] [Google Scholar]
- Parodi A. J. Annu. Rev. Biochem. 2000, 69, 69–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.