

Abstract
A key enzyme in aerobic metabolism is cytochrome c oxidase (CcO), which catalyzes the reduction of molecular oxygen to water in the mitochondrial and bacterial membranes. Substrate electrons and protons are taken up from different sides of the membrane and protons are pumped across the membrane, thereby generating an electrochemical gradient. The well-studied A-type CcO uses two different entry channels for protons: the D-channel for all pumped and two consumed protons, and the K-channel for the other two consumed protons. In contrast, the B-type CcO uses only a single proton input channel for all consumed and pumped protons. It has the same location as the A-type K-channel (and thus is named the K-channel analog) without sharing any significant sequence homology. In this study, we performed molecular-dynamics simulations and electrostatic calculations to characterize the K-channel analog in terms of its energetic requirements and functionalities. The function of Glu-15B as a proton sink at the channel entrance is demonstrated by its rotational movement out of the channel when it is deprotonated and by its high pKA value when it points inside the channel. Tyr-244 in the middle of the channel is identified as the valve that ensures unidirectional proton transfer, as it moves inside the hydrogen-bond gap of the K-channel analog only while being deprotonated. The electrostatic energy landscape was calculated for all proton-transfer steps in the K-channel analog, which functions via proton-hole transfer. Overall, the K-channel analog has a very stable geometry without large energy barriers.
Introduction
A key element of aerobic metabolism is the enzyme cytochrome c oxidase (CcO), which catalyzes the reduction of molecular oxygen to water. Four electrons and four protons are transferred onto a single oxygen molecule in a stepwise reaction. The integral membrane protein CcO utilizes the resultant energy to pump protons across the membrane, thereby establishing an electrochemical gradient.
The CcO family may be divided into A, B, and C types. The well-studied A-type CcO is most abundant and constitutively expressed in mitochondria and several bacteria (1,2), whereas B- and C-type CcO are expressed in some specialized bacteria under low-oxygen conditions (3). Whereas the C-type CcO is more distant to the others (4), the A and B types share higher sequence similarity and have almost identical compositions of cofactors used for electron transport and enzymatic reaction (5,6). At the positively charged P side of the membrane, CcO takes up electrons from soluble cytochrome c. They are transported via the bimetallic copper A center (CuA) to heme a (A type) or heme b (B type), and finally to the binuclear center (BNC), where the chemical reaction takes place. The BNC is composed of heme a3 and the copper B center (CuB), and has a nearly identical three-dimensional structure in both A- and B-type CcO (5,6). The proton-pumping efficiency differs between the A- and B-type CcO (1 H+pump/e− and 0.5 H+pump/e−, respectively) (7), although it was speculated that the B-type pump efficiency may be underestimated due to experimental limitations (8). The intermediate states of the catalytic cycle in A-type CcO, which have been investigated intensively (2,9), may differ from those in the B type (10). Also, the proton input pathways of A- and B-type CcO differ from each other. All protons are taken up from the negatively charged N side of the membrane and can be differentiated into chemical protons that are consumed in the reaction and pumped protons, which are pumped across the membrane. In the A-type CcO, the protons are conducted via two different pathways (1,2): 1), the D-channel leading from aspartate (Asp-132, R. sphaeroides numbering) at the entrance to glutamate (Glu-286) situated between the hemes; and 2), the K-channel containing lysine (Lys-362) and terminating at tyrosine (Tyr-288), which is covalently bound to His-284 ligating CuB. For the A-type CcO, it is well established that all pumped protons are taken up via the D-channel, and that two chemical protons enter via the D-channel and two enter via the K-channel. In contrast, the B-type CcO was shown to transport all chemical and pumped protons via the same input channel (11), which is located at a similar position as the K-channel of the A-type CcO (6) without sharing significant sequence homology. This K-channel analog in B-type CcO (Fig. 1) involves a hydrogen bond (H-bond) chain of threonines, tyrosines, and serine, and a water molecule with only one 4.9 Å gap in between Thr-312 and Ser-309 (Fig. 1). Only glutamate and tyrosine at the entrance and terminus, respectively, are conserved among the K-channels of A- and B-type CcO.
Figure 1.
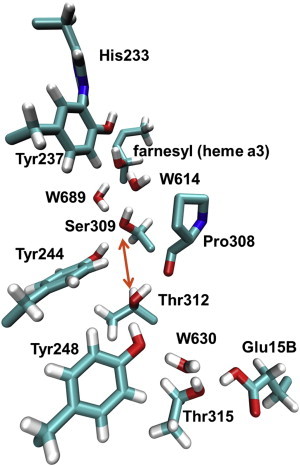
Proton wire in the K-channel analog in CcO of T. thermophilus. The channel runs from Glu-15B to Tyr-237, with one gap of 4.9 Å in the H-bond chain between Thr-312 and Ser-309 (indicated by an orange arrow). For proton transport, a proton hole is presumably transported from Tyr-237 to Glu-15B via farnesyl, W614, Ser-309, Tyr-244, Thr-312, Tyr-248, W630, and Thr-315. Coordinates were taken from crystal structure PDB 3S8F (6) and hydrogen atoms were added with CHARMM (19). In addition to the channel residues, Pro-308 is depicted (without hydrogens) because its backbone CO group accepts an H-bond from Thr-312, and His-233 is depicted because it is covalently bound to Tyr-237. For clarity, the farnesyl chain of heme a3 is shown only partially. To see this figure in color, go online.
For the A-type CcO, the properties of the D-channel for collecting and gating protons have been discussed extensively (12–14), and recently we also found a gating element in the K-channel (15). In the B-type CcO, these functionalities must be accomplished by a single proton input channel that ensures rapid unidirectional proton flow and translocation of pumped protons.
In this study, we use molecular-dynamics (MD) simulations and electrostatic energy computations to explore the energy landscape of proton transfer via the K-channel analog in the B-type CcO. We highlight the key functionalities of specific molecular geometries and offer an explanation for how the channel may ensure proton transport that is rapid as well as unidirectional.
Materials and Methods
Preparing and performing MD simulations of CcO
The coordinates of subunits I, II, and IIa of CcO from Thermus thermophilus were taken from the Protein Data Bank (16) (PDB ID: 3S8F (5)), embedded in a lipid bilayer of phosphatidylcholines modeled with the plug-in of VMD (17), and solvated in a TIP3P (18) water box applying periodic boundary conditions (dimensions: 100 Å × 100 Å × 107.8 Å). We employed the CHARMM22 force field (19), CHARMM36 extension for lipids (20), and in-house-determined parameters for the cofactors (13). The same setup of MD simulation was used previously for the A-type CcO (13). The MD simulations were performed with the software NAMD (21) using a 2 fs time step with SHAKE to fix the bond lengths involving hydrogen atoms and Langevin dynamics with friction constant β = 1 ps−1 at 300 K temperature. The MD simulations were performed with a flexible cell size and constant ratio of 1:1 for the x and y dimensions to stabilize the membrane, which was placed in the x-y plane. All MD simulations (listed in Table 1) exhibited stable geometries for 100 ns, with root mean-square deviation (RMSD) values for the backbone atoms essentially below 1.2 Å (Fig. S1 in the Supporting Material) and all-atom RMSD values essentially below 1.5 Å (Fig. S2).
Table 1.
MD simulations performed in this work
| MD simulation | Redox and protonation state of BNC | Specific protonation in channel |
|---|---|---|
| 1 | Fe(IV)=O/CuB(II)-OH/Tyr-237-O− | all channel residues protonated (except Tyr-237) |
| 2 | Fe(IV)=O/CuB(II)-OH/Tyr-237-OH | all channel residues protonated |
| 3 | Fe(IV)=O/CuB(II)-OH/Tyr-237-O− | deprotonated Tyr-244 |
| 4 | Fe(IV)=O/CuB(II)-OH/Tyr-237-OH | deprotonated Tyr-244 |
| 5 | Fe(IV)=O/CuB(II)-OH/Tyr-237-O− | deprotonated Glu-15B |
| 6 | Fe(IV)=O/CuB(II)-OH/Tyr-237-OH | deprotonated Glu-15B |
Atomic partial charges of CcO cofactors
We calculated the atomic partial charges of the cofactors as described by Woelke et al. (13), using the quantum-chemical program Jaguar v.7.7 (Schrödinger, LLC, New York, NY) and the B3LYP DFT functional with the LACVP∗∗ basis set. First, the cofactor geometries were optimized quantum chemically. All hydrogen atoms were geometry optimized without constraints, whereas the coordinates of nonhydrogen atoms were optimized with respect to bond lengths and bond angles, keeping the corresponding torsion angles fixed. We then computed the electrostatic potentials in the vicinity of the cofactors based on the electronic wave functions and charges of the nuclei, using the same procedure we employed for geometry optimization. We determined the atomic partial charges from the electrostatic potentials of the cofactors by employing a two-stage restraint-electrostatic-potential (RESP) (22,23) procedure. The only difference between our approach and the procedure described by Woelke et al. (13) is that to compute the atomic partial charges of the CuB center, we included Tyr-237 covalently bound to His-233. The coordinates and atomic partial charges are given in Table S2.
Computation of pKA values and electrostatic energies
We investigated the protonation states in equilibrium of all CcO residues based on preliminary pKA computations (24,25) using just a single structure with optimized hydrogen atom positions based on the crystal structure from T. thermophilus with the highest resolution of 1.8 Å (6). The histidine protonations (involving 18 His) are given in Table S1. All other titratable groups (Asp, Glu, Arg, and Lys) are charged, with the following exceptions: the charge-neutral Glu-15B at the entrance of the K-channel analog (discussed in the next section) and the charge-neutral Glu-203, Glu-131B, and Asp-372, which might be involved in proton pumping, but were not analyzed in this study because they are far from the K-channel analog.
To calculate the energy landscape of the proton transfer pathway via the K-channel analog, we used the crystal structure PDB 3S8F (6) embedded in a membrane as described above. The BNC ligands were modeled as described in Woelke et al. (13). We added hydrogen atoms and adjusted the geometry of OH groups of water and side chains (tyrosine and serine) to the different protonation states that potentially can occur during proton transport in the K-channel analog. This modeling also involved adjusting the Tyr-244 side chain for the Tyr-244–Thr-312 switch mechanism (explained in the Results and Discussion section), followed by constrained energy minimization of the modeled region. The pKA values were calculated using karlsberg+ (24,25). Crystal water molecules were removed (except for the three crystal water molecules inside the K-channel analog (W614, W630, and W689), which were kept) and a dielectric continuum with ε = 80 was placed in the resulting cavities. Lipid molecules of the membrane with atoms within a 20 Å sphere around the center of the K-channel analog were kept. All other lipids were deleted. We tested this procedure and found it to be appropriate (15). A dielectric continuum with ε = 80 was placed outside of the volume of all atoms (from protein, lipids, and water molecules) that were kept explicitly, whereas inside the volume of explicit atoms, ε = 4. The moderate value of the dielectric constant of ε = 4 inside the volume of explicit atoms accounts implicitly for a limited variation of atom positions, which is not considered explicitly when only the crystal structure is used to evaluate electrostatic energies. Since the electrostatic continuum implicitly accounts for conformational variability, the computed electrostatic energies also contain entropy contributions from the protein and solvent.
For each of the titratable residues involved in proton translocation inside the channel (Tyr-237, farnesylhemea3, W614, Ser-309, Tyr-244, Thr-312, Tyr-248, W630, Thr-315, and Glu-15B), a structure was modeled for the corresponding deprotonated state. For each of these structures, the pKA values and thus the protonation energies of the corresponding residue were calculated with karlsberg+ (24,25). To account for structural changes due to the modeling, the electrostatic contribution to the conformational energy was added to the protonation energy. All of these energy terms were calculated by numerically solving the linearized Poisson-Boltzmann equation using the program APBS (26). To obtain reliable results, the finest grid spacing of 0.25 Å was used.
Role of electrostatic energy in characterizing proton transfer energetics
A precise evaluation of proton transfer energetics requires a consideration of the free energy, which is computationally difficult to assess. Usually, it requires MD simulations beyond the microsecond time regime and is subject to the uncertainties of the employed energy function, which for such long simulation times may drive the molecular system into unphysical conformational regimes. The free energy combines the influence of enthalpy and entropy. Whereas entropy is only indirectly accessible by the conformational variability, enthalpy relies on the interplay between kinetic and potential energies. Based on the second viral theorem (27), the average kinetic energy is proportional to the number of degrees of freedom of the molecular system and therefore does not change in a proton transfer process.
The enthalpy consists of bonded (covalent) and nonbonded (van der Waals (vdW) and Coulomb) energies. A critical part of the bonded energy is the contribution of the transferred proton that is covalently bound to different molecular groups before and after the transfer step. However, we showed that ab initio computations of pKA values using a combination of high-precision quantum-chemical and electrostatic energy evaluations yield an RMSD to corresponding measured values of 0.5 pH units (28). This agreement demonstrates that the thermodynamic averages of 1), nonelectrostatic quantum-chemical contributions to pKA values do not depend on the environment; 2), vdW interactions are practically identical before and after a change in protonation; and 3), covalent interactions inside other not directly covalently connected molecular groups (of the solute) that are spectators in the considered proton transfer step also do not change. Further support for these conclusions comes from extensive evaluations of pKA values of titratable groups in proteins based on the measured pKA values in solution (24). These pKA values are transformed by electrostatic energy computations to the corresponding protein environment with an RMSD to measured values of 1.1 pH units. Hence, it is justifiable to consider only electrostatic energies to characterize proton transfer processes, since they make the main contribution to the energetics of such processes.
Another reason to consider only the electrostatic energies is the fact that our computations are based on crystal structures. This may yield arbitrary contributions for covalent and vdW interactions, since these interactions are very sensitive to small variations of 0.1 Å in atomic coordinates for individual conformations, whereas electrostatic energies with their very mild 1/r distance dependence are relatively insensitive to such small changes. Although the electrostatic energies computed in this study refer to a single conformation, they implicitly consider many conformations by using a dielectric continuum for the solvent and the protein. Thus, they also account for entropy contributions as noted above.
Results and Discussion
Investigation of the K-channel analog
After oxygen splitting occurs, the first proton is likely taken up in the PR state (29), which is characterized by an access electron inside the BNC that is composed of Fe(IV)=O/Cu(II)-OH/Tyr-237-O−. The K-channel analog, as depicted in Fig. 1, starts with Glu-15B at the N side of the membrane and terminates at Tyr-237. As has been shown by quantum-chemical calculations (30), the proton wire in the channel is composed of several H-bonded OH groups that are protonated in the resting state and conduct protons via a proton-hole transfer mechanism. We performed MD simulations to investigate the conformational stability or mobility of residues in the K-channel analog in different protonation states. A 100 ns MD simulation was performed with all three subunits of CcO embedded in a phospholipid membrane, as described in Materials and Methods, with the BNC modeled in the PR state (Table 1, simulation No. 1). In the PR state (Table 1, No. 1), the K-channel analog exhibits a very stable geometry for the full MD simulation time, as evidenced, for example, by the low RMSD value of 0.8 Å of the K-channel analog residues (alignment on all backbone atoms and RMSD measurement on all atoms of channel residues; Fig. S3). All H-bonds from Glu-15B to the deprotonated Tyr-237 are pointing toward the BNC fluctuating around an oxygen-oxygen distance of 2.8 Å corresponding to ideal H-bond geometry (Fig. S4 A). Tyr-244 donates an H-bond to Ser-309, whereas Thr-312 donates its H-bond to the backbone oxygen of Pro-308 (see Fig. 1), thus leaving a gap of ∼5 Å in the H-bond chain, which is not bridged by a water molecule throughout the simulation (see further discussion below). The crystal water molecule W614 moves from downward of the Tyr-237 oxygen to upward, but remains H-bonded to the negatively charged Tyr-237 side chain. The other two crystal waters inside the K-channel analog, W630 and W689, remain at their positions during the whole MD simulation time of 100 ns. This rigid channel geometry may be essential for function in the high-temperature environment in which T. thermophilus resides.
The MD simulation with protonated Tyr-237 (Table 1, No. 2) exhibits a geometry similar to that observed in the simulation with deprotonated Tyr-237, but some H-bonds of the K-channel analog are slightly less stable (see Fig. S4 B). Tyr-237 loses its H-bond contact to the OH group of farnesyl from heme a3, and also the H-bond between Ser-309 and Tyr-244 is split, but bridged by a water molecule.
The proton-hole sink: Glu-15B
The entrance of the K-channel analog consists of the glutamate residue Glu-15B, which is conserved among channels of A- and B-type CcO (5,6,31,32). We performed MD simulations with protonated (Table 1, No. 2) and deprotonated (Table 1, No. 6) Glu-15B, and found that the protonated Glu-15B forms a stable H-bond toward Thr-315 with an average distance of 2.8 Å (Fig. S4, A and B) over 100 ns (Fig. 2 A), whereas the deprotonated Glu-15B does not (Fig. S5, A and B). The deprotonated Glu-15B first accepts an H-bond from Thr-315 (Fig. 2 B), but loses this contact after 10.8 ns or 1.4 ns MD simulation with protonated Tyr-237 or deprotonated Tyr-237, respectively. The deprotonated Glu-15B swings out of the hydrophobic channel entrance toward bulk water, as has also been demonstrated by quantum-chemical calculations (30,33), and does not return in the deprotonated state (Fig. S5, A and B). As soon as Glu-15B loses contact to Thr-315, the H-bonds of the proton wire of the K-channel analog (see Fig. 1) turn inward, pointing with the hydrogen atoms toward the BNC (Fig. 2 C). After a >40 ns simulation with deprotonated Glu-15B, the water molecule W630 leaves the channel toward bulk water without returning (Fig. S5, A and B). This creates a gap between Thr-315 and Tyr-248, which slightly destabilizes the H-bond chain in the K-channel analog.
Figure 2.
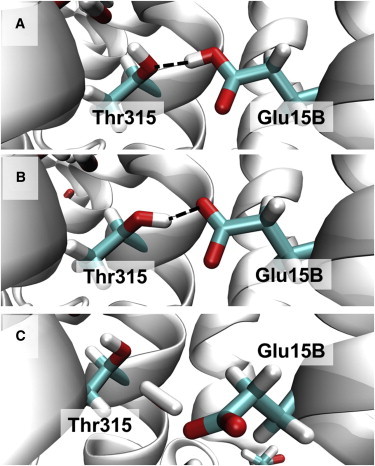
Different positions of Glu-15B depending on the protonation state. (A) The crystal structure (PDB ID: 3S8F (6)) with protonated Glu-15B donating an H-bond to Thr-315. The MD simulation with protonated Glu-15B remains close to this position. (B) After energy minimization, deprotonated Glu-15B accepts an H-bond from Thr-315. (C) Snapshot of the MD simulation after 15.0 ns. Glu-15B moves out of the K-channel analog toward bulk water and loses connectivity to Thr-315, which then forms an H-bond inward with the hydrogen atom pointing to the BNC and remains there for the rest of the 100 ns MD simulation. H-bonds are indicated by dashed lines. To see this figure in color, go online.
In the crystal structure PDB 3S8F (6), Glu-15B is pointing toward Thr-315 (see Fig. 1) and may either be protonated and donate an H-bond to Thr-315 or be deprotonated and accept an H-bond. Preliminary pKA computations based solely on the crystal structure yield a pKA value for Glu-15B of 6.8. However, in MD simulations, the deprotonated Glu-15B moves out of the K-channel analog toward bulk water and the protonated Glu-15B remains precisely at the crystal structure position. Thus, we conclude that Glu-15B is protonated in the 3S8F structure (6). Interestingly, the other crystal structures of CcO from T. thermophilus exhibit a Glu-15B position more distant from Thr-315 (3.41 Å in PDB 1EHK (34) and 3.64 Å in PDB 1XME (35)) and the mean B factor of the Glu-15B side chain atoms is 41.8 Å2 larger than the mean B factor (26.1 Å2) of the side-chain atoms of the other K-channel analog residues in the crystal structure 3S8F (6) (similar relations hold for the other crystal structures). This suggests that Glu-15B is more flexible and may occasionally move from its main conformation, where it is protonated and points inward to a second conformation that is deprotonated and points to bulk water.
In summary, Glu-15B of the K-channel analog is likely protonated and donates an H-bond to Thr-315 in the resting state of the channel, where CcO is ready to take up a new electron. However, Glu-15B may easily donate its proton, become deprotonated, and move out toward the channel entrance in bulk water, where it may transiently accept a new proton. This behavior of Glu-15B renders it an excellent sink for proton holes, promoting rapid proton uptake by CcO.
Tyr-244, the valve
In the crystal structure obtained by Tiefenbrunn et al. (6), the only gap in the H-bond chain of the K-channel analog is between Thr-312 and Ser-309, which are 4.9 Å apart (oxygen-oxygen distance; Fig. 1). This gap could be bridged either by Tyr-244 swinging toward Thr-312 or by one additional water molecule, as suggested previously (11). However, after carefully inspecting the latest crystal structure at 1.8 Å resolution, Tiefenbrunn et al. (6) did not observe an electron density that could correspond to a water molecule bridging this gap. Furthermore, the Tyr-244 position is virtually identical in all available crystal structures and has a low B factor throughout (6,34,35). Likewise, all of our modeling attempts to move Tyr-244 or place an additional water molecule inside this gap did not lead to a stable conformation in MD simulations (data not shown). Instead, we observed a switching mechanism of Tyr-244.
Whereas the protonated Tyr-244 remained at the crystal structure position during the whole 100 ns MD simulation (Fig. 3 A; Table 1, No. 2; H-bond distances in Fig. S6, A and B), the deprotonated Tyr-244 immediately moved between Ser-309 and Thr-312, accepting H-bonds from both of them (Fig. 3 B; Table 1, No. 3; H-bond distances in Fig. S6, A and B). This geometry was stable for only 0.3 ns, and then a water molecule that was placed inside the protein during solvation moved between Tyr-244 and Ser-309, such that they were still H-bond connected (Fig. and H-bond distances in Fig. S6, A and B). The other H-bond between Thr-312 and Tyr-244 remained stable for practically the whole 100 ns simulation, with an average distance of 2.8 Å (Fig. 3 C and H-bond distances in Fig. S6, A and B).
Figure 3.
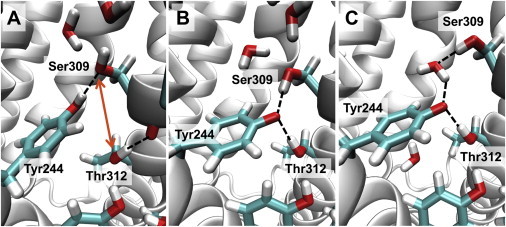
Different positions of Tyr-244 depending on its protonation state. (A) The crystal structure (PDB ID: 3S8F (6)) with protonated Tyr-244. The 4.9 Å gap between Thr-312 and Ser-309 is indicated by an orange arrow. During the MD simulation with Tyr-244 protonated (Table 1, No. 2), it remains close to the position in the crystal structure. (B) Snapshot of the MD simulation with deprotonated Tyr-244 (Table 1, No. 4) after 50 ps. The deprotonated Tyr-244 moves into the Thr-312-Ser-309 gap immediately. H-bonds are indicated by dashed lines. (C) Snapshot of the MD simulation with deprotonated Tyr-244 (Table 1, No. 4) after 17.6 ns. One water molecule moves in between Ser-309 and Tyr-244.
In conclusion, the gap between Ser-309 and Thr-312 is only bridged by Tyr-244 after its deprotonation. This behavior renders the Tyr-244–Thr-312 pair an optimal switch, ensuring unidirectional proton transport. In the resting state of the channel, where CcO is ready to take up a new electron, Tyr-244 is only H-bonded with the upstream Ser-309 close to the BNC, whereas Thr-312 donates an H-bond to the Pro-308 backbone oxygen atom, which has no hydrogen atom that could potentially leak toward the channel entrance. Only after Tyr-244 has donated its proton toward the BNC does the Tyr-244–Thr-312 switch make the K-channel analog proton conducting again. The deprotonated Tyr-244 may quickly obtain a new proton from Glu-15B at the channel entrance, which converts the protonation of the K-channel analog back to the resting state of the channel. Thus, the K-channel analog does not function solely via a simple Grotthuss proton conductor consisting of a single wire of connected H-bonds (36), as suggested previously (11,30); rather, it is a two-wire Grotthuss conductor interrupted by Tyr-244, which if deprotonated makes a conformational change to connect the two wires transiently.
We also calculated the so-called action pKA of Tyr-244, assuming that a (nonequilibrium) proton hole is localized at Tyr-244. Such action pKA values describe the local electrostatic energies within the reaction sequence of proton-hole transfer inside the K-channel analog. The alternative pKA of Tyr-244 in equilibrium with solvent pH is not useful for our question of interest because it depends on the changes in protonation pattern of all other titratable residues and the corresponding conformational change of the protein. However, these changes do not reflect the physiological conditions of the proton-transfer reaction. The action pKA for Tyr-244 is 7.0 in a conformation corresponding to the crystal structure involving only one H-bond with Ser-309. However, this value drops to 2.8 with the conformational change of Tyr-244 accepting two H-bonds from Ser-309 and Thr-312. This clearly demonstrates that the deprotonated Tyr-244 needs to be stabilized by an additional H-bond from Thr-312, whereas the protonated Tyr-244 is in a more stable conformation when it donates only one H-bond to Ser-309, as observed in the crystal structure.
The Tyr-244–Thr-312 switch is supported by experimental data. The Thr312Val mutant was shown to have more severe effects on CcO function than other channel mutants, such as Tyr248Phe and Thr315Val, which strongly reduced or even abolished enzymatic activity in experiments (11,37,38). In a study by Smirnova et al. (39), the Thr312Val and Tyr244Phe mutants of CcO were able to take up the first proton after oxygen splitting, but the efficiency of the second proton uptake was significantly reduced. The authors concluded that these mutations interfere with structural changes that are rate limiting for the second proton transfer (39). In this study, we have demonstrated that the suggested structural change is connected to the ability of Tyr-244 to move and fulfill its switching function in the K-channel analog, thereby controlling proton conductance.
Tyr-237: the branching point
The K-channel analog terminates at Tyr-237, which is covalently bound to one of the CuB-ligating histidines and is conserved in all types of CcO (4–6). It takes part in the enzymatic reaction of CcO by donating a proton as well as an electron in the oxygen-splitting reaction, thus forming a tyrosyl radical. In the catalytic cycle, this Tyr-237 radical receives a new electron and a new proton in separate steps. In this study, we investigated the PR state, in which Tyr-237 has received an electron but not yet a proton. Interestingly, the F state could not be observed spectroscopically in the B-type CcO (29,40), perhaps because in the B-type CcO, the Tyr-237 receives the first chemical proton (41) instead of the CuB-ligating hydroxyl as in A-type CcO. The state after this proton is received in the B-type CcO may be the F state or an intermediate before F-state formation.
We also performed MD simulations with protonated (Table 1, No. 2) and deprotonated (Table 1, No. 1) Tyr-237 (Fig. 4). In both MD simulations, the H-bond network of the K-channel analog is very stable. For deprotonated Tyr-237 (Table 1, No. 1), all H-bonds of the K-channel analog point inward to the BNC, whereas the crystal water W614 moves slightly above Tyr-237 but remains H-bonded with it (H-bond distances in Fig. S4 A). For the protonated Tyr-237 (Table 1, No. 2), the H-bonds of the proton wire in the upper part of the channel point down toward the N side, but in the lower part of the channel from Thr-312 to Glu-15B, the H-bonds still point inward to the BNC (H-bond distances in Fig. S4 B). This underlines the function of the Tyr-244–Thr-312 switch, because this geometry is not prone to proton leakage from Tyr-237 to the N side of the K-channel analog.
Figure 4.
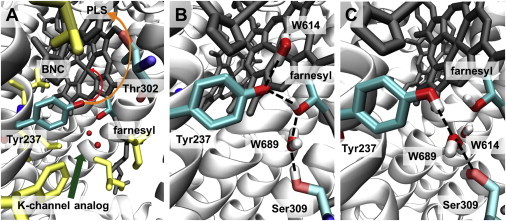
Different positions of Tyr-237 depending on its protonation state. (A) The crystal structure (PDB ID: 3S8F (6)) with the environment of Tyr-237. Tyr-237 at the end of the K-channel analog (green arrow) is close to or even part of the BNC (black stick model) and at a 5.0 Å distance from the OH group of farnesyl from heme a3. The putative PLS is above the BNC, with a few hydrophilic residues (e.g., Thr-302) in between. Hydrophobic residues around Tyr-237 are shown in yellow. The putative pathways of the chemical (red) and pumped (orange) protons above Tyr-237 are indicated by arrows, but the exact locations of the proton pathways are not known. (B) Typical snapshot of MD simulation with deprotonated Tyr-237 (Table 1, No. 1) at 47.0 ns. The deprotonated Tyr-237 remains at the crystal structure position with a stable H-bond from farnesyl of heme a3 (O-O distance 2.6 Å). The crystal water W614 moves above Tyr-237 and forms a stable H-bond (O-O distance 2.8 Å). H-bonds are indicated by dashed lines. Nonpolar hydrogens are not shown. (C) Typical snapshot of MD simulation with protonated Tyr-237 (Table 1, No. 2) at 64.0 ns. The H-bond between Tyr-237 and farnesyl of heme a3 is lost, and the water molecule W614 bridges Tyr-237 and Ser-309 (the O-O distances of Tyr-237 and Ser-309 with W614 are 2.8 Å and 2.6 Å, respectively).
The oxygen atom of Tyr-237 is located at a distance of 5.0 Å from the oxygen ligand of heme a3 (6) (Fig. 4) and may donate chemical protons to this oxygen, probably via one or two water molecules (a suggested pathway is indicated in Fig. 4), which are not present in the crystal structure and thus may occupy their positions only transiently. Tyr-237 is the residue that terminates the proton input channel at the BNC. Since there is only one proton input channel in B-type CcO, it is suggestive that also the pumped protons are transferred via Tyr-237. No alternative pathway for proton transfer from the K-channel analog to the putative proton loading site (PLS) is visible in the CcO crystal structure (Fig. 4). Between Tyr-237 and the putative PLS, which was proposed to be around the propionates of heme a3 (42,43), the only titratable groups are the CuB ligands and Thr-302 (Fig. 4). The surroundings of Tyr-237 involve several hydrophobic residues, which are not suitable for proton transfer. Therefore, Tyr-237 may likely be the branching point at which the routes of chemical and pumped protons diverge (suggested pathways are indicated in Fig. 4). A function analog to Tyr-237 is accomplished by Glu-286 in the D-channel of A-type CcO (2) (R. sphaeroides numbering).
The approximate location of the pathways for chemical and pumped protons beyond Tyr-237 is indicated in Fig. 4. However, the mechanism underlying how chemical and pumped protons are differentiated beyond Tyr-237 needs further investigation. Proton movement is controlled by electrons entering the BNC and, most likely, chemical protons are dragged from Tyr-237 into the BNC by a low pKA of the oxygen ligand at heme a3 and pumped protons are dragged by a low pKA of the PLS. From the crystal structure with modeled hydrogens, we computed a pKA of 12.8 for Tyr-237 in the PR state based on electrostatic calculations. However, as Tyr-237 is covalently bound to the CuB-ligating histidine, its pKA will be strongly influenced by the redox-state of CuB and refinement of the pKA computation using quantum-mechanical calculations will be needed.
The actual protonation state of Tyr-237 in the crystal structure (PDB ID: 3S8F (6)) cannot be determined with certainty. Still, two arguments point to a protonated Tyr-237: 1), the average geometry from MD simulation with a protonated Tyr-237 deviates the least from the crystal structure geometry, and even crystal water molecules around Tyr-237 remain at their position; and 2), if Tiefenbrunn et al.’s (6) conclusion about a peroxide in the BNC is correct, Tyr-237 would be in the state right before donating a proton.
Proton transfer energy landscape
To demonstrate how the K-channel analog might operate, we calculated the electrostatic contribution of the energy differences of all steps of proton transport using our software karlsberg+ (24,25). However, these energy barriers are only approximate because we used the crystal structure (6) for our calculations and adjusted only the BNC ligands to the corresponding redox state with split dioxygen (see Table 1), added hydrogen atoms, made small geometric adjustments of the OH groups for the different protonation states, and modeled the conformational change of Tyr-244 for the Thr-312–Tyr-244 switch. The proton-transfer sequence starts in the PR state with a deprotonated Tyr-237 and all other residues involved in K-channel proton transport in their neutral-charge state. As can be seen in Fig. 5, almost all proton-transfer steps exhibit a favorable electrostatic energy relative to the proton hole at the starting point at Tyr-237. The most unfavorable energy is observed for the crystal water W630 forming a hydroxyl ion, which is due to the high general pKA value of water in solution (pKA = 15.75). However, another reason for the large activation barrier could be the uncertainty of the oxygen atom position of W630, which is taken from the CcO crystal structure and may differ under physiological conditions in the proton-transfer reaction.
Figure 5.
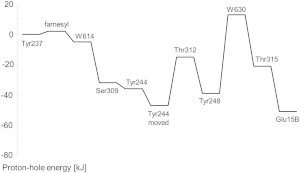
Electrostatic energy landscape of the proton hole inside the K-channel analog. Electrostatic energies for all deprotonation steps were calculated using the software karlsberg+ (24,25) as described in Materials and Methods. Transport of a proton hole along the K-channel analog starts at Tyr-237, which is deprotonated and donates its proton to the BNC. The proton hole subsequently moves along a linear chain of deprotonatable residues until it reaches Glu-15B. After deprotonation of Tyr-244, this residue moves inside the gap between Ser-309 and Thr-312. The electrostatic energies before and after this conformational change are depicted.
Our results demonstrate the feasibility of the Tyr-244–Thr-312 switch, since deprotonation of Tyr-244 is energetically favorable when it accepts one H-bond from Ser-309, but becomes even more favorable when Tyr-244 changes its conformation to form H-bonds with both Ser-309 and Thr-312 (Fig. 5).
In the last step in proton-hole conduction in the K-channel analog, Glu-15B donates its proton with the largest favorable energy. This may ensure that no negative charge accumulates inside the proton channel that could lead to proton leakage through the channel. The deprotonated Glu-15B then swings out toward bulk water and receives a new proton to fulfill its function to serve as proton-hole sink at the channel entrance.
Conclusions
In this study, we analyzed the properties of the K-channel analog in CcO of T. thermophilus. We demonstrated that proton transport via the K-channel analog works smoothly, with no oversized energy barriers, using a proton-hole transport mechanism. We identified two important functionalities: 1), Glu-15B at the channel entrance is a proton-hole sink; and 2), unidirectional proton flow is accomplished by the Tyr-244–Thr-312 switch, where deprotonated Tyr-244 moves inside the gap between Thr-312 and Ser-309. These findings may explain the outcome of several mutational studies and may inform the setup for new experiments. Furthermore, enhanced knowledge about the K-channel analog will provide an important basis for investigating the aberrant pumping properties of B-type as compared with A-type CcO (44).
Acknowledgments
The authors thank Dr. Joachim Heberle for fruitful discussions.
This work was supported by the Deutsche Forschungsgemeinschaft within the framework of Collaborative Research Center 1078 (project C2) and the research training group Computational System Biology.
Supporting Material
References
- 1.Brzezinski P., Gennis R.B. Cytochrome c oxidase: exciting progress and remaining mysteries. J. Bioenerg. Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaila V.R., Verkhovsky M.I., Wikström M. Proton-coupled electron transfer in cytochrome oxidase. Chem. Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 3.Han H., Hemp J., Gennis R.B. Adaptation of aerobic respiration to low O2 environments. Proc. Natl. Acad. Sci. USA. 2011;108:14109–14114. doi: 10.1073/pnas.1018958108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buschmann S., Warkentin E., Michel H. The structure of cbb3 cytochrome oxidase provides insights into proton pumping. Science. 2010;329:327–330. doi: 10.1126/science.1187303. [DOI] [PubMed] [Google Scholar]
- 5.Qin L., Hiser C., Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tiefenbrunn T., Liu W., Cherezov V. High resolution structure of the ba3 cytochrome c oxidase from Thermus thermophilus in a lipidic environment. PLoS ONE. 2011;6:e22348. doi: 10.1371/journal.pone.0022348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Ballmoos C., Adelroth P., Brzezinski P. Proton transfer in ba(3) cytochrome c oxidase from Thermus thermophilus. Biochim. Biophys. Acta. 2012;1817:650–657. doi: 10.1016/j.bbabio.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 8.Rauhamäki V., Wikström M. The causes of reduced proton-pumping efficiency in type B and C respiratory heme-copper oxidases, and in some mutated variants of type A. Biochim. Biophys. Acta. 2014;1837:999–1003. doi: 10.1016/j.bbabio.2014.02.020. [DOI] [PubMed] [Google Scholar]
- 9.Siegbahn P.E., Blomberg M.R. Quantum chemical studies of proton-coupled electron transfer in metalloenzymes. Chem. Rev. 2010;110:7040–7061. doi: 10.1021/cr100070p. [DOI] [PubMed] [Google Scholar]
- 10.Siletsky S.A., Belevich I., Verkhovsky M.I. Time-resolved OH—>EH transition of the aberrant ba3 oxidase from Thermus thermophilus. Biochim. Biophys. Acta. 2009;1787:201–205. doi: 10.1016/j.bbabio.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 11.Chang H.Y., Hemp J., Gennis R.B. The cytochrome ba3 oxygen reductase from Thermus thermophilus uses a single input channel for proton delivery to the active site and for proton pumping. Proc. Natl. Acad. Sci. USA. 2009;106:16169–16173. doi: 10.1073/pnas.0905264106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaila V.R., Verkhovsky M.I., Wikström M. Glutamic acid 242 is a valve in the proton pump of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 2008;105:6255–6259. doi: 10.1073/pnas.0800770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woelke A.L., Galstyan G., Knapp E.W. Exploring the possible role of Glu286 in CcO by electrostatic energy computations combined with molecular dynamics. J. Phys. Chem. B. 2013;117:12432–12441. doi: 10.1021/jp407250d. [DOI] [PubMed] [Google Scholar]
- 14.Yang S., Cui Q. Glu-286 rotation and water wire reorientation are unlikely the gating elements for proton pumping in cytochrome C oxidase. Biophys. J. 2011;101:61–69. doi: 10.1016/j.bpj.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woelke A.L., Galstyan G., Knapp E.W. Lysine 362 in Cytochrome c Oxidase Regulates Opening of the K-Channel via Changes in pKA and Conformation. Biochim. Biophys. Acta. 2014 doi: 10.1016/j.bbabio.2014.08.003. http://www.sciencedirect.com/science/article/pii/S0005272814005647 [DOI] [PubMed] [Google Scholar]
- 16.Berman H.M., Westbrook J., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 18.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 19.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bayly C.I., Cieplak P., Cornell W., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—the RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 23.Cornell W.D., Cieplak P., Bayly C.I., Kollman P.A. Application of RESP charges to calculate conformational energies, hydrogen-bond energies, and free-energies of solvation. J. Am. Chem. Soc. 1993;115:9620–9631. [Google Scholar]
- 24.Kieseritzky G., Knapp E.W. Optimizing pKa computation in proteins with pH adapted conformations. Proteins. 2008;71:1335–1348. doi: 10.1002/prot.21820. [DOI] [PubMed] [Google Scholar]
- 25.Rabenstein B., Knapp E.W. Calculated pH-dependent population and protonation of carbon-monoxy-myoglobin conformers. Biophys. J. 2001;80:1141–1150. doi: 10.1016/S0006-3495(01)76091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker N.A., Sept D., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirschfelder J.O., McClure F.T., Weeks I.F. Second virial coefficients and the forces between complex molecules. J. Chem. Phys. 1942;10:201. [Google Scholar]
- 28.Schmidt am Busch M., Knapp E.W. Accurate pKa determination for a heterogeneous group of organic molecules. ChemPhysChem. 2004;5:1513–1522. doi: 10.1002/cphc.200400171. [DOI] [PubMed] [Google Scholar]
- 29.Smirnova I.A., Zaslavsky D., Brzezinski P. Electron and proton transfer in the ba(3) oxidase from Thermus thermophilus. J. Bioenerg. Biomembr. 2008;40:281–287. doi: 10.1007/s10863-008-9157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noodleman L., Han Du W.G., Walker R.C. Linking chemical electron-proton transfer to proton pumping in cytochrome c oxidase: broken-symmetry DFT exploration of intermediates along the catalytic reaction pathway of the iron-copper dinuclear complex. Inorg. Chem. 2014;53:6458–6472. doi: 10.1021/ic500363h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostermeier C., Harrenga A., Michel H. Structure at 2.7 A resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody FV fragment. Proc. Natl. Acad. Sci. USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshikawa S., Shinzawa-Itoh K., Tsukihara T. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 33.Fee J.A., Case D.A., Noodleman L. Toward a chemical mechanism of proton pumping by the B-type cytochrome c oxidases: application of density functional theory to cytochrome ba3 of Thermus thermophilus. J. Am. Chem. Soc. 2008;130:15002–15021. doi: 10.1021/ja803112w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soulimane T., Buse G., Than M.E. Structure and mechanism of the aberrant ba(3)-cytochrome c oxidase from thermus thermophilus. EMBO J. 2000;19:1766–1776. doi: 10.1093/emboj/19.8.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hunsicker-Wang L.M., Pacoma R.L., Stout C.D. A novel cryoprotection scheme for enhancing the diffraction of crystals of recombinant cytochrome ba3 oxidase from Thermus thermophilus. Acta Crystallogr. D Biol. Crystallogr. 2005;61:340–343. doi: 10.1107/S0907444904033906. [DOI] [PubMed] [Google Scholar]
- 36.Agmon N. The Grotthuss mechanism. Chem. Phys. Lett. 1995;244:456–462. [Google Scholar]
- 37.Chang H.Y., Choi S.K., Gennis R.B. Exploring the proton pump and exit pathway for pumped protons in cytochrome ba3 from Thermus thermophilus. Proc. Natl. Acad. Sci. USA. 2012;109:5259–5264. doi: 10.1073/pnas.1107345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smirnova I., Reimann J., Adelroth P. Functional role of Thr-312 and Thr-315 in the proton-transfer pathway in ba3 Cytochrome c oxidase from Thermus thermophilus. Biochemistry. 2010;49:7033–7039. doi: 10.1021/bi100749p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smirnova I., Chang H.Y., Brzezinski P. Single mutations that redirect internal proton transfer in the ba3 oxidase from Thermus thermophilus. Biochemistry. 2013;52:7022–7030. doi: 10.1021/bi4008726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.von Ballmoos C., Gennis R.B., Brzezinski P. Kinetic design of the respiratory oxidases. Proc. Natl. Acad. Sci. USA. 2011;108:11057–11062. doi: 10.1073/pnas.1104103108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siletsky S.A., Belevich I., Verkhovsky M.I. Time-resolved single-turnover of ba3 oxidase from Thermus thermophilus. Biochim. Biophys. Acta. 2007;1767:1383–1392. doi: 10.1016/j.bbabio.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Blomberg M.R., Siegbahn P.E. The mechanism for proton pumping in cytochrome c oxidase from an electrostatic and quantum chemical perspective. Biochim. Biophys. Acta. 2012;1817:495–505. doi: 10.1016/j.bbabio.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 43.Kaila V.R., Sharma V., Wikström M. The identity of the transient proton loading site of the proton-pumping mechanism of cytochrome c oxidase. Biochim. Biophys. Acta. 2011;1807:80–84. doi: 10.1016/j.bbabio.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 44.Lee H.J., Reimann J., Adelroth P. Functional proton transfer pathways in the heme-copper oxidase superfamily. Biochim. Biophys. Acta. 2012;1817:537–544. doi: 10.1016/j.bbabio.2011.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.