

Abstract
Epidemiological and clinical data point to a close association between chronic hepatitis B virus infection or chronic hepatitis C virus infection and development of hepatocellular carcinoma (HCC). HCC develops over several decades and is associated with fibrosis. This sequence suggests that persistent viral infection and chronic inflammation can synergistically induce liver fibrosis and hepatocarcinogenesis. The transforming growth factor-β (TGF-β) signaling pathway plays a pivotal role in diverse cellular processes and contributes to hepatic fibro-carcinogenesis under inflammatory microenvironments during chronic liver diseases. The biological activities of TGF-β are initiated by the binding of the ligand to TGF-β receptors, which phosphorylate Smad proteins. TGF-β type I receptor activates Smad3 to create COOH-terminally phosphorylated Smad3 (pSmad3C), while pro-inflammatory cytokine-activated kinases phosphorylates Smad3 to create the linker phosphorylated Smad3 (pSmad3L). During chronic liver disease progression, virus components, together with pro-inflammatory cytokines and somatic mutations, convert the Smad3 signal from tumor-suppressive pSmad3C to fibro-carcinogenic pSmad3L pathways, accelerating liver fibrosis and increasing the risk of HCC. The understanding of Smad3 phosphorylation profiles may provide new opportunities for effective chemoprevention and personalized therapy for patients with hepatitis virus-related HCC in the future.
Keywords: Chronic viral hepatitis, Transforming growth factor-β, Smad3, Phosphorylation, Fibro-carcinogenesis, Hepatocellular carcinoma
Core tip: Chronic hepatitis B and C infections are major causes of cirrhosis and hepatocellular carcinoma (HCC). Most patients with persistent viral infection remain asymptomatic, while some patients have poor prognosis and develop HCC. Therefore, identifying persons at high-HCC risk among chronic hepatitis patients is crucial for preventing HCC. Analyses of domain-specific phosphorylation of Smad3 in liver specimens can be helpful to understand the stages of diseases and might represent a marker to predict HCC development.
INTRODUCTION
The incidence of hepatocellular carcinoma (HCC) is increasing, and it is expected that the rates of the HCC development will continue to rise for the foreseeable future. Epidemiological evidence clearly indicates that approximately 80% of HCC depend on chronically hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, worldwide. The remaining 20% of patients are complicated, with their liver cancer being caused by various factors such as excessive alcohol intake, fatty liver, hemochromatosis and metabolic syndrome[1]. In the natural progress of the viral hepatitis, there is persistence of a long latency of HBV and HCV infection; however, most of the patients with HBV and HCV are asymptomatic or have slight symptoms without progression[2,3]. In the livers of some patients, the progress of various stages with active inflammation and fibrosis, and eventually cirrhosis, is seen. Cirrhosis precedes liver-related complications, including HCC. It usually takes 20 years or more to lead to cirrhosis from viral infection. Subsequently, HCC often develops over another 10 years[4]. Therefore, the liver carcinogenic process related to HBV or HCV infection tends to be insidious, most likely requiring multiple sequential genetic alterations and complex interactions between the virus, the host and the environment.
In the last few decades, research has provided significant insights into the transforming growth factor (TGF)-β signal transduction network, which regulates many biological processes. In a normal system, TGF-β inhibits the physiological activity of epithelial cell growth, having a tumor suppressive function. At later stages, however, TGF-β can promote cancer progression[5-7]. As normal epithelial cells progress toward cancer cells, the oncogenic potential of TGF-β in malignant cells rises, accompanied by selective reduction of tumor-suppressive activity[6]. In chronic hepatitis, the cytostatic effect of TGF-β for hepatocytes attenuates as liver disease progresses from cirrhosis to HCC under persistent inflammatory microenvironments[8]. Under pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α) and interleukin-β (IL-β), TGF-β promotes extracellular matrix (ECM) accumulation, while the cell proliferative pro-inflammatory cytokine signal has a contradictory relationship with cytostatic signal through TGF-β[8,9]. By these regulatory actions, hepatic TGF-β signaling with pro-inflammatory cytokines can promote fibro-carcinogenesis.
The TGF-β signal is transmitted mainly by a transcription factor called Smad. Smads proteins are comprise Mad-homology (MH) 1 and MH2 domains, and a linker between them[10]. Activated type I receptors (TβRI) phosphorylate the C-terminus of Smad2 and Smad3 directly to form pSmad2C and pSmad3C[11]. Mitogenic signals are generated by the phosphorylation of their linker regions instead of C-terminal phosphorylation[12-18]. After formation of hetero-oligomers with Smad4, a common partner, Smad2 and Smad3 translocate from the cytoplasm to the nucleus, leading to regulated transcription of target genes[19].
Mechanisms linking fibrosis and HCC remain largely unsettled. Considering that the roles of phospho-Smads are greatly influenced by differential phosphorylation, a better understanding of mechanisms of phospho-Smads signaling should help to design approaches to diagnosis, prevention and treatment of hepatitis virus-related hepato-carcinogenesis. In this review, we summarize Smad3 phosphorylation profiles and consider how Smad3 affects human fibro-carcinogenesis at the molecular level.
TGF-β AND JNK SIGNALING INVOLVEMENT IN CHRONIC LIVER DISEASE PROGRESSION
TGF-β appears highly important in hepatic fibro-carcinogenesis in patients with chronic liver diseases. TGF-β participates in many cell functions, such as tissue and organ development, cell proliferation, differentiation, cell survival, and the control of the apoptosis[20]. TGF-β, the most potent hepatic pro-fibrogenic cytokine, is deeply involved in chronic liver diseases, especially liver cirrhosis and HCC[21], and is produced mostly by activated mesenchymal cells during chronic liver damage[22]. TGF-β plays an important role in epithelial-mesenchymal transition during fibrogenesis[23]. HCC usually occurs in cirrhotic livers where more TGF-β existed in comparison with healthy livers, which suggests that TGF-β has a role in pro-oncogenesis[24]. TGF-β regulates a large number of genetic expressions in conjunction with carcinogenesis[25].
Another important factor influencing HCC development in chronic HBV or HCV infection is c-Jun-N-terminal kinase (JNK) activity[26,27]. JNK is a mitogen-activated protein kinase (MAPK) family member that is activated by diverse stimuli, including cytokines such as IL-1β and TNF-α. These cytokines are transcriptionally activated in the injured liver. Upon activation, JNK induces multiple biological events through several transcription factors and transcription-independent control of effector molecules[28].
Smads mediate the intracellular TGF-β signal as transcription factors. Eight kinds of Smads are known in mammals, and they are classified as signal-specific Smad2/3, common type Smad4 and inhibitory type Smad7 by their functions[29]. An SXS motif in the C-terminus of Smad3 is phosphorylated by TβRI upon TGF-β binding, leading to activation of Smad3 signaling (Figure 1A)[10,29]. On the other hand, signal-specific Smad2/3 at the linker lesion is phosphorylated directly by extracellular signal-regulated kinase (ERK), which is representative of the downstream molecules of the Ras cascade, such as JNK, p38 MAPK, cyclin-dependent kinase (CDK), glycogen synthase kinase 3-β, Ca2+-calmodulin-dependent protein kinase II, and G protein-coupled receptor kinase-2 (Figure 1B)[12-17,30-35]. As a substitute for pro-inflammatory cytokines and TGF-β, TGF-β activated kinase (TAK) 1 plays an important role in the signal reaction in the cell through the non-Smad cascade. TAK1 mediates activation of JNK and p38 MAPK signaling via mitogen-activated kinase (MKK) 4/7 and MKK3/6[36,37]. In addition, JNK and p38 MAPK interfere with TGF-β signal by pro-inflammatory cytokines through their regulation of many physiological functions[38].
Figure 1.
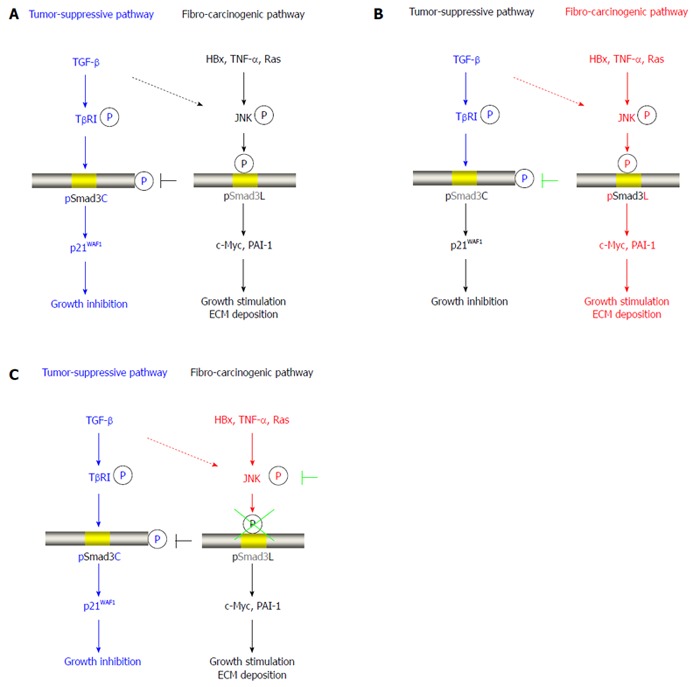
Reversibility of phospho-Smad3 signaling between tumor suppression and fibro-carcinogenesis. A: Additional treatment of transforming growth factor-β (TGF-β) activates TGF-β receptor type I (TβRI), further leading to direct phosphorylation of Smad3C, which inhibits normally hepatocytic growth by upregulating p21WAF1 transcription; B: Mitogens drastically alter phospho-Smad3 signaling via the c-Jun N-terminal kinase (JNK) pathway, increasing nuclear fibro-carcinogenic pSmad3L activity while shutting down TGF-β-dependent cytostatic pSmad3C. Although the TGF-β signal weakly phosphorylates Smad3L in normal hepatocytes (dotted line), hepatitis viral components, including HBx, pro-inflammatory cytokines including tumor necrosis factor-α (TNF-α), and somatic mutations such as Ras, additively transmit a fibro-carcinogenic signal through the JNK-dependent pSmad3L pathway to participate in hepatocytic growth and ECM deposition, possibly by stimulating transcription of c-Myc and PAI-1 genes. Linker phosphorylation of Smad3 indirectly prevents COOH-tail phosphorylation, pSmad3C-mediated p21WAF1 transcription and cytostatic function; C: Either various JNK inhibitors or a Smad3 mutation causing lack of JNK phosphorylation sites in the linker region can eliminate fibro-carcinogenic pSmad3L signaling, restoring or maintaining the tumor-suppressive pSmad3C signaling characteristic of mature hepatocytes.
ANTAGONISTIC SMAD3 PHOSPHO-ISOFORM SIGNALING: TUMOR-SUPPRESSIVE (CYTOSTATIC) PSMAD3C VS ARCINOGENIC (MITOGENIC) PSMAD3L
Recent reports suggested that each step of the Smad3 signal transmission, including its ability to form a complex with Smad4, the local existence in the cell, binding to target gene promoter and the resolution, are controlled by phosphorylation[29]. Activation of TβRI with TGF-β binding and Ras-related kinase, including JNK and CDK, differentially phosphorylate Smad3 to form pSmad3C and the pSmad3L. These two domain-specific phosphorylation forms have different actions. Phosphorylation of Smad3C activates a cytostatic signal, while phosphorylation at the linker region upregulates the mitogenic signal. Notably, as pSmad3L is produced during fibro-carcinogenesis, leading to structure changes, phosphorylation of Smad3C is inhibited by pro-fibrogenic and pro-tumorigenic non-Smad pathways[39].
In normal epithelial cells, TGF-β regulates the growth and proliferation of cells. In particular, after pSmad3C shifts into the nucleus, cell proliferation stops by activation of p15INK4B and p21WAF gene transcription or suppression of the c-Myc gene, resulting in apoptosis with inhibition of Bcl-2 expression (Figure 1A)[40-43].
Traditionally, the cytostatic effects of TGF-β have been thought to oppose mitogenic signaling in normal cells, while in cancer cells, potent mitogenic actions of certain oncogenes can overwhelm the anti-mitogenic capacity of TGF-β signaling. Virus components, including the HBx protein; inflammatory cytokines such as IL-1β and TNF-α; growth factors acting through a tyrosine kinase type receptor, including hepatocyte growth factor and platelet-derived growth factor (PDGF); and Ras mutations additively upregulate phosphorylation of Smad3L by activated JNK[16,44,45]. After pSmad3L moves into the nucleus, nuclear pSmad3L induces the growth of hepatocytes and ECM deposition by upregulating c-Myc and PAI-1 transcription. Importantly, phosphorylation of Smad3 at the linker region inhibits C-terminal phosphorylation induced by TβRI[13,16,32,45,46]. When Smad3L phosphorylation is promoted by the activation of mitogenic signaling, phosphorylation of pSmad3C is suppressed indirectly, possibly by the cytostatic system (Figure 1B)[13,16].
A principal finding is the shift between JNK/pSmad3L and TRβI/pSmad3C signaling, representing a delicate balance between carcinogenesis and tumor-suppression (Figure 1C). Specifically blocking phosphorylation of Smad3 at the linker legion using Smad3 mutants lacking linker phosphorylation sites or inhibition of JNK activity, can recover to the pathway going through the tumor-suppressive pSmad3C pathway[16,45,46]. Therefore, we suggest that the cytostatic TβRI/pSmad3C signal has a contradictory relationship with the mitogenic JNK/pSmad3L signal.
MODULATION OF SMAD3 SIGNALING FAVORS CARCINOGENESIS
TGF-β activates cytostatic and apoptotic processes to maintain homeostasis in normal epithelial cells. In the human intestine, apoptosis induced by the TGF-β/pSmad3C pathway, which is essential for normal homeostasis, acts to inhibit human colorectal cancer cell proliferation. Similarly, hepatocytes show a balance between proliferation and differentiation during liver regeneration. Proliferation of normal hepatocytes to compensate for partial hepatectomy or diffuse liver injury is constructed by the interaction of polypeptide cytokines and growth factors.
In quiescent hepatocytes, Smad3C phosphorylation via the activin type I receptor (ActRI) persists[47]. During acute liver damage, mitogenic pSmad3L signaling induced by TNF-α becomes more dominant than pSmad3C signaling. As decreases in TNF-α and pSmad3L allow increased sensitivity to phosphorylation at Smad3C by TβRI, hepatocytic proliferation ceases. Such a competitive mode of hepatocytic Smad3 signaling is a protective reaction, which may prevent malignant transformation from normal cells. In this manner, hepatocytes with non-proliferative pSmad3C signaling may be destined to undergo apoptotic cell death[48].
On the other hand, constitutive pSmad3L (Ser-213)/c-Myc signaling accelerates cancer development[49]. Hepatocytes keep their cell proliferative potential by Smad3 linker phosphorylation and inhibition of anti-apoptotic action by Smad3C, which make an environment in which mutant genes are easily accumulated. This step mainly contributes to progression of hepatocarcinogenesis.
FIBRO-CARCINOGENESIS IN HUMAN CHRONIC LIVER DISEASES: RECIPROCAL CHANGES IN PSMAD3L AND PSMAD3C PATHWAYS
Around 80% to 90% of HCCs arise as a complication of long-standing symptomatic cirrhosis[50]. Cirrhosis is seen in 20% to 30% of patients with persistent HCV infection, several decades after viral infection. As fibrosis progresses, the risks of the HCC increase, in particular, HCC develops at an annual rate of 1% to 7% in HCV-infected patients with cirrhosis[51], and 0.02% to 3.7% in HBV-infected patients with cirrhosis[52,53]. These epidemiological findings indicate that hepato-carcinogenesis in chronic viral hepatitis is a sequential step from chronic liver injury, through cirrhosis to HCC, with fibrosis being pivotal at the pre-neoplastic stage.
During progression of liver injury, hepatic stellate cells (HSCs) undergo a complex transformation or activation process, losing lipid droplets, retaining retinoids and changing into myofibroblast (MFB)-like cells, which progress ECM accumulation[54]. This step is the beginning of fibrosis. If this process is repeated, the liver shifts to irreversible cirrhosis. Activation of HSCs promotes fibrogenesis and control intracellular signaling networks by proliferative PDGF[55,56] and fibrogenic TGF-β cytokines[57]. Moreover, our in vivo model indicated that the pSmad3C-mediated signal decreased, while the pSmad3L pathway predominated during transdifferentiation in culture[31]. In chronic hepatitis C, α-smooth muscle actin (SMA)-positive MFB in the portal area is strongly affected by the pSmad3L signal rather than the pSmad3C signal[44]. Increase of α-SMA in HSC creates scar-forming MFB, leading to liver fibrosis[58]. Similar to MFB, pSmad3L is predominantly located in hepatocytic nuclei in portal tracts, in sharp contrast to pSmad3C[44]. Kupffer cells in the portal tract produce and release TGF-β and a variety of pro-inflammatory cytokines, which provoke JNK activation[59,60]. According to these findings, JNK activated pro-inflammatory cytokines are able to transform Smad3 into pSmad3L in both hepatocytes and MFB during the course of chronic hepatitis.
Our studies compared pSmad3L with pSmad3C distribution in biopsy specimens from patients chronically infected with HBV or HCV. These results demonstrated reciprocal changes in pSmad3L and pSmad3C pathways during hepato-carcinogenesis, with pSmad3L being seen predominantly in hepatocytes with progress of liver disease (Figure 2)[44,45]. In contrast, pSmad3C staining decreased in hepatocytes during fibrosis progression (Figure 2)[44,45]. Furthermore, the JNK/pSmad3L-mediated pathway in hepatocytes and activated HSC involves hepatic fibrosis during a long cancerous process. This is a principal mechanism in the development of liver fibrosis toward HCC in both HBV- and HCV-related liver diseases.
Figure 2.
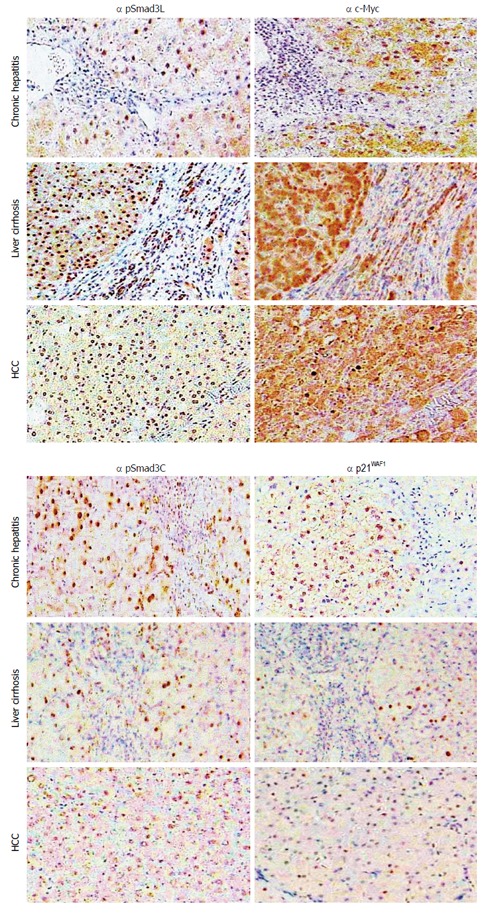
Hepatic fibro-carcinogenesis: reciprocal change in linker phosphorylated and COOH-terminally phosphorylated Smad3 pathways. The linker phosphorylated Smad3 (pSmad3L)/c-Myc pathway shows increasing prominence in hepatocytes, as hepatitis C virus (HCV)-infected liver progresses from chronic hepatitis, through cirrhosis to hepatocellular carcinoma (HCC). In contrast to the intense staining for pSmad3L and c-Myc, the COOH-terminally phosphorylated Smad3 (pSmad3C)/p21WAF1 pathway staining decreases in hepatocytes as liver disease progresses toward HCC.
PROMOTION OF HEPATO-CARCINOGENESIS BY CHRONIC VIRAL INFECTION TOGETHER WITH INFLAMMATION IN THE HUMAN LIVER
A number of pathogenic mechanisms have been proposed for HCC associated with chronic inflammatory diseases, including chronic viral hepatitis B and C, alcoholic hepatitis, non-alcoholic fatty liver disease, autoimmune hepatitis, primary biliary cirrhosis, hemochromatosis, Wilson’s disease and others. Although cell division is very rare in normal hepatocytes, in viral chronic liver disease, the hepatocyte turnover rate is significantly accelerated under repeated hepatocytic necrosis and regeneration. Cell proliferation fundamentally does not have a function to transform cells, but cell proliferation is necessary in the process of liver carcinogenesis[61]. By accelerating hepatocyte turnover, the chronic inflammatory liver is exposed to the risk of cell progression into a fully malignant phenotype, acting as a potent tumor promoter. Moreover, stepwise accumulation of mutations in cancer-related genes and molecular alterations participate in human carcinogenesis[62]. Eventually, somatic mutations in hepatocytes stimulate hepatocytic growth, and HCC occurs as a result of malignant cell transformation.
In comparison with other influences, chronic infection with HBV or HCV represents the most important risk factor for the development of HCC. Hepato-carcinogenesis in HBV and HCV infection has been studied extensively with necrosis and chronic inflammation, followed by fibrosis and cell proliferation, playing a role. Nevertheless, HCC only occurs in a small proportion of HBV and HCV carriers. The hepato-carcinogenic process involves the interplay between hepatitis viruses and host inflammatory responses; thus, both factors may contribute to the final malignant outcome, either individually or synergistically[63-67].
CHRONIC INFLAMMATION AND HEPATITIS VIRUS ALTER THE HEPATOCYTIC TGF-β SIGNALING
Hepatitis viruses and inflammation influence the fibro-carcinogenic TGF-β pathway, which leads to development of HCC. We investigated the correlation between hepatocytic pSmad3L positivity and plasma HBV DNA concentration in chronic hepatitis B patients, because HBV itself can upregulate phosphorylation of hepatocytic Smad3L[45]. pSmad3L is predominantly located in the nuclei of hepatocytes adjoining inflammatory cells in the portal tracts[44]. Furthermore, Smad3L phosphorylation showed strong correlation with inflammatory activity[44].
At the onset of HCC, chronic inflammation, cell death and persistence of viral infection in liver cells, play a central role. Hepatocytes with abundant pSmad3L and little pSmad3C can survive during the progress of chronic hepatitis, resulting in the accumulation of various mutations in sequences. These mutations are associated with genes in the Ras pathway[68], which activate phosphorylation of Smad3L constantly in pre-neoplastic hepatocytes, resulting in the eventual development of overt HCC[69]. These observations are consistent with the general hypothesis of the additive promotion of hepato-carcinogenesis by persistent hepatitis virus infection and chronic inflammation.
SMAD3 PHOSPHORYLATION FOR PREDICTING RISK OF HCC
Identification of high-risk patients among virus hepatitis carriers is of great importance. Well-planned treatment allocation and effective screening for HCC in chronic hepatitis patients are essential. Several factors have been identified as associated with higher risk of development of HCC, including male gender, advanced age[70], virological factors[71-73], and disease factors, including alanine aminotransferase concentrations and the presence of cirrhosis[70,74].
Our retrospective study using pSmad3L and pSmad3C in human liver have provide physiopathogenic insights. In the chronic hepatitis B patients, HCC develops from the group with high pSmad3L but low pSmad3C in their hepatocytes. By contrast, HCC does not occur from the group with high pSmad3C but little pSmad3L in their hepatocytes[45]. A similar tendency is observed in human HCV-related hepato-carcinogenesis[44]. Striking differences in cumulative HCC incidence have been observed between the two groups during 12 years of follow-up. These findings clarify the position of pSmad3C as a tumor suppressor, while pSmad3L is a tumor promoter during human hepato-carcinogenesis. In addition, according to the results of multivariate analysis, high pSmad3L and low pSmad3C in hepatocytes were each independent risk factors for liver carcinogenesis[45]. These observations suggested that the Smad3 phosphorylation profile has an excellent discriminating ability to triage chronic hepatitis patients for the most appropriate treatment.
CONCLUSION
Over the last decade, our understanding of the mechanism of hepato-carcinogenesis has deepened. HCC associated with chronic with HBV and HCV infections remains associated with poor prognosis. Nonetheless, our studies clearly demonstrated that as the stage of viral chronic liver disease progresses, fibrogenic and oncogenic pSmad3L signaling gradually increases in pre-neoplastic hepatocytes persistently affected by TGF-β, together with pro-inflammatory cytokines, leading to liver fibrosis and initiating HCC (Figure 3). The model focusing on phospho-Smad signaling that we proposed may be understood as a crucial molecular mechanism by which most HCC develops in the context of severe fibrosis[75]. Thus, understanding of Smad phospho-isoform signaling is important to understand the mechanisms underlying hepato-carcinogenesis.
Figure 3.
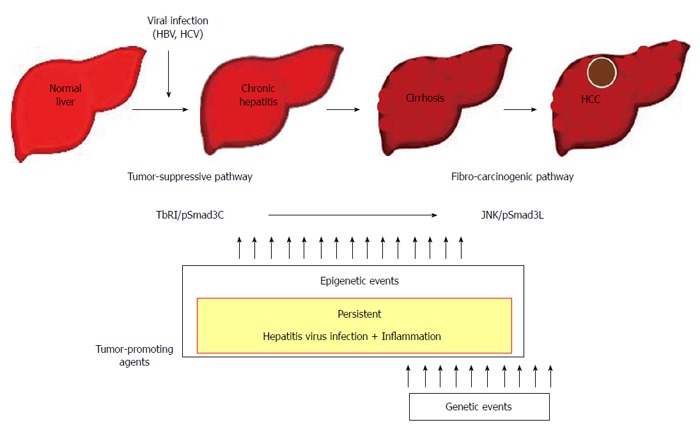
Contribution to fibro-carcinogenesis by tumor-promoting agents. Together, persistent hepatitis virus infection and chronic inflammation shift hepatocytic phospho-Smad3 signaling from the tumor-suppressive transforming growth factor-β (TGF-β) receptor type I (TβRI)/COOH-terminally phosphorylated Smad3 (pSmad3C) mode to the fibro-carcinogenic c-Jun N-terminal kinase (JNK)/linker phosphorylated Smad3 (pSmad3L) mode characteristic of median forebrain bundle, accelerating liver fibrosis while increasing risk of hepatocellular carcinoma (HCC). In the liver, persistent hepatitis virus infection and chronic inflammation contribute to fibro-carcinogenesis. In proportion to the severity of fibrosis, mitogenic genetic or epigenetic alterations can drive fibro-carcinogenesis via the pSmad3L pathway. Escaping the cytostatic action of pSmad3C is a critical step for progression to full malignancy in cancers, which must overcome multiple fail-safe genetic controls. HBV: Hepatitis B virus; HCV: Hepatitis C virus.
In our recent studies, HCV clearance at an early stage by anti-viral therapy restored hepatocytic TGF-β signaling from fibro-carcinogenesis toward the tumor-suppression[76]. Moreover, low pSmad3C and high pSmad3L positivity were significantly predictive of human HCC development[44,45]. We concluded that pSmad3L and pSmad3C can serve as useful markers to determine whether antiviral therapy will be effective against viral related HCC. Using an individual’s phospho-Smad3 markers, clinicians may be able to achieve more effective chemoprevention and individualized therapy for HBV- and HCV-infected patients.
Footnotes
P- Reviewer: Ferreira CN, Sipos F S- Editor: Gou SX L- Editor: Stewart G E- Editor: Zhang DN
References
- 1.Thomas MB, Zhu AX. Hepatocellular carcinoma: the need for progress. J Clin Oncol. 2005;23:2892–2899. doi: 10.1200/JCO.2005.03.196. [DOI] [PubMed] [Google Scholar]
- 2.Ganem D, Prince AM. Hepatitis B virus infection--natural history and clinical consequences. N Engl J Med. 2004;350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 3.Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med. 2000;132:296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- 4.Tong MJ, el-Farra NS, Reikes AR, Co RL. Clinical outcomes after transfusion-associated hepatitis C. N Engl J Med. 1995;332:1463–1466. doi: 10.1056/NEJM199506013322202. [DOI] [PubMed] [Google Scholar]
- 5.Moses HL, Yang EY, Pietenpol JA. TGF-beta stimulation and inhibition of cell proliferation: new mechanistic insights. Cell. 1990;63:245–247. doi: 10.1016/0092-8674(90)90155-8. [DOI] [PubMed] [Google Scholar]
- 6.Roberts AB, Sporn MB. The transforming growth factor-βs. In: Sporn MB, Roberts AB, editors. Peptide Growth Factors and Their Receptors. Berlin: Springer; 1990. pp. 419–472. [Google Scholar]
- 7.Bellam N, Pasche B. Tgf-beta signaling alterations and colon cancer. Cancer Treat Res. 2010;155:85–103. doi: 10.1007/978-1-4419-6033-7_5. [DOI] [PubMed] [Google Scholar]
- 8.Matsuzaki K. Smad phosphoisoform signals in acute and chronic liver injury: similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res. 2012;347:225–243. doi: 10.1007/s00441-011-1178-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 11.Wrana JL. Regulation of Smad activity. Cell. 2000;100:189–192. doi: 10.1016/s0092-8674(00)81556-1. [DOI] [PubMed] [Google Scholar]
- 12.Kretzschmar M, Doody J, Timokhina I, Massagué J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa M, et al. TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene. 2004;23:7416–7429. doi: 10.1038/sj.onc.1207981. [DOI] [PubMed] [Google Scholar]
- 14.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–231. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 15.Kamaraju AK, Roberts AB. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-beta-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J Biol Chem. 2005;280:1024–1036. doi: 10.1074/jbc.M403960200. [DOI] [PubMed] [Google Scholar]
- 16.Sekimoto G, Matsuzaki K, Yoshida K, Mori S, Murata M, Seki T, Matsui H, Fujisawa J, Okazaki K. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 2007;67:5090–5096. doi: 10.1158/0008-5472.CAN-06-4629. [DOI] [PubMed] [Google Scholar]
- 17.Matsuzaki K, Kitano C, Murata M, Sekimoto G, Yoshida K, Uemura Y, Seki T, Taketani S, Fujisawa J, Okazaki K. Smad2 and Smad3 phosphorylated at both linker and COOH-terminal regions transmit malignant TGF-beta signal in later stages of human colorectal cancer. Cancer Res. 2009;69:5321–5330. doi: 10.1158/0008-5472.CAN-08-4203. [DOI] [PubMed] [Google Scholar]
- 18.Matsuzaki K. Smad phospho-isoforms direct context-dependent TGF-β signaling. Cytokine Growth Factor Rev. 2013;24:385–399. doi: 10.1016/j.cytogfr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 20.Cohen MM. TGF beta/Smad signaling system and its pathologic correlates. Am J Med Genet A. 2003;116A:1–10. doi: 10.1002/ajmg.a.10750. [DOI] [PubMed] [Google Scholar]
- 21.Bissell DM. Chronic liver injury, TGF-beta, and cancer. Exp Mol Med. 2001;33:179–190. doi: 10.1038/emm.2001.31. [DOI] [PubMed] [Google Scholar]
- 22.Inagaki Y, Okazaki I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56:284–292. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, Piek E, Bottinger EP. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci USA. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bissell DM, Roulot D, George J. Transforming growth factor beta and the liver. Hepatology. 2001;34:859–867. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- 25.Massagué J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 27.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsutsumi T, Suzuki T, Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Matsuura Y, Kimura S, Koike K, Miyamura T. Alteration of intrahepatic cytokine expression and AP-1 activation in transgenic mice expressing hepatitis C virus core protein. Virology. 2002;304:415–424. doi: 10.1006/viro.2002.1702. [DOI] [PubMed] [Google Scholar]
- 29.Derynck R, Miyazono K. The TGF-β signaling. New York: Cold Spring Harbor Laboratory Press; 2008. [Google Scholar]
- 30.Wicks SJ, Lui S, Abdel-Wahab N, Mason RM, Chantry A. Inactivation of smad-transforming growth factor beta signaling by Ca(2+)-calmodulin-dependent protein kinase II. Mol Cell Biol. 2000;20:8103–8111. doi: 10.1128/mcb.20.21.8103-8111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki Y, et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38:879–889. doi: 10.1053/jhep.2003.50384. [DOI] [PubMed] [Google Scholar]
- 32.Ho J, Cocolakis E, Dumas VM, Posner BI, Laporte SA, Lebrun JJ. The G protein-coupled receptor kinase-2 is a TGFbeta-inducible antagonist of TGFbeta signal transduction. EMBO J. 2005;24:3247–3258. doi: 10.1038/sj.emboj.7600794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu F. Smad3 phosphorylation by cyclin-dependent kinases. Cytokine Growth Factor Rev. 2006;17:9–17. doi: 10.1016/j.cytogfr.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 34.Millet C, Yamashita M, Heller M, Yu LR, Veenstra TD, Zhang YE. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J Biol Chem. 2009;284:19808–19816. doi: 10.1074/jbc.M109.016667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alarcón C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A, Miller AN, Manova-Todorova K, Macias MJ, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139:757–769. doi: 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 37.Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee KY, Bussey C, Steckel M, Tanaka N, et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol. 2007;9:1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- 40.Frederick JP, Liberati NT, Waddell DS, Shi Y, Wang XF. Transforming growth factor beta-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol Cell Biol. 2004;24:2546–2559. doi: 10.1128/MCB.24.6.2546-2559.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 42.Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang YA, Zhang GM, Feigenbaum L, Zhang YE. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of Bcl-2. Cancer Cell. 2006;9:445–457. doi: 10.1016/j.ccr.2006.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, Kaibori M, Kamiyama Y, Nishizawa M, Fujisawa J, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007;46:48–57. doi: 10.1002/hep.21672. [DOI] [PubMed] [Google Scholar]
- 45.Murata M, Matsuzaki K, Yoshida K, Sekimoto G, Tahashi Y, Mori S, Uemura Y, Sakaida N, Fujisawa J, Seki T, et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology. 2009;49:1203–1217. doi: 10.1002/hep.22765. [DOI] [PubMed] [Google Scholar]
- 46.Nagata H, Hatano E, Tada M, Murata M, Kitamura K, Asechi H, Narita M, Yanagida A, Tamaki N, Yagi S, et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor- suppression in rat hepatocellular carcinoma. Hepatology. 2009;49:1944–1953. doi: 10.1002/hep.22860. [DOI] [PubMed] [Google Scholar]
- 47.Date M, Matsuzaki K, Matsushita M, Tahashi Y, Sakitani K, Inoue K. Differential regulation of activin A for hepatocyte growth and fibronectin synthesis in rat liver injury. J Hepatol. 2000;32:251–260. doi: 10.1016/s0168-8278(00)80070-7. [DOI] [PubMed] [Google Scholar]
- 48.Matsuzaki K. Smad3 phosphoisoform-mediated signaling during sporadic human colorectal carcinogenesis. Histol Histopathol. 2006;21:645–662. doi: 10.14670/HH-21.645. [DOI] [PubMed] [Google Scholar]
- 49.Kawamata S, Matsuzaki K, Murata M, Seki T, Matsuoka K, Iwao Y, Hibi T, Okazaki K. Oncogenic Smad3 signaling induced by chronic inflammation is an early event in ulcerative colitis-associated carcinogenesis. Inflamm Bowel Dis. 2011;17:683–695. doi: 10.1002/ibd.21395. [DOI] [PubMed] [Google Scholar]
- 50.Caldwell S, Park SH. The epidemiology of hepatocellular cancer: from the perspectives of public health problem to tumor biology. J Gastroenterol. 2009;44 Suppl 19:96–101. doi: 10.1007/s00535-008-2258-6. [DOI] [PubMed] [Google Scholar]
- 51.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 52.Manno M, Cammà C, Schepis F, Bassi F, Gelmini R, Giannini F, Miselli F, Grottola A, Ferretti I, Vecchi C, et al. Natural history of chronic HBV carriers in northern Italy: morbidity and mortality after 30 years. Gastroenterology. 2004;127:756–763. doi: 10.1053/j.gastro.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 53.Beasley RP. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer. 1988;61:1942–1956. doi: 10.1002/1097-0142(19880515)61:10<1942::aid-cncr2820611003>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 54.Okuno M, Kojima S, Akita K, Matsushima-Nishiwaki R, Adachi S, Sano T, Takano Y, Takai K, Obora A, Yasuda I, et al. Retinoids in liver fibrosis and cancer. Front Biosci. 2002;7:d204–d218. doi: 10.2741/A775. [DOI] [PubMed] [Google Scholar]
- 55.Friedman SL, Arthur MJ. Activation of cultured rat hepatic lipocytes by Kupffer cell conditioned medium. Direct enhancement of matrix synthesis and stimulation of cell proliferation via induction of platelet-derived growth factor receptors. J Clin Invest. 1989;84:1780–1785. doi: 10.1172/JCI114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinzani M, Milani S, Herbst H, DeFranco R, Grappone C, Gentilini A, Caligiuri A, Pellegrini G, Ngo DV, Romanelli RG, et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am J Pathol. 1996;148:785–800. [PMC free article] [PubMed] [Google Scholar]
- 57.Tahashi Y, Matsuzaki K, Date M, Yoshida K, Furukawa F, Sugano Y, Matsushita M, Himeno Y, Inagaki Y, Inoue K. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology. 2002;35:49–61. doi: 10.1053/jhep.2002.30083. [DOI] [PubMed] [Google Scholar]
- 58.Pinzani M, Macias-Barragan J. Update on the pathophysiology of liver fibrosis. Expert Rev Gastroenterol Hepatol. 2010;4:459–472. doi: 10.1586/egh.10.47. [DOI] [PubMed] [Google Scholar]
- 59.Yoshida K, Matsuzaki K, Mori S, Tahashi Y, Yamagata H, Furukawa F, Seki T, Nishizawa M, Fujisawa J, Okazaki K. Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol. 2005;166:1029–1039. doi: 10.1016/s0002-9440(10)62324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, Colmenero J, Bataller R, Schwabe RF. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138:347–359. doi: 10.1053/j.gastro.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Webber EM, Wu JC, Wang L, Merlino G, Fausto N. Overexpression of transforming growth factor-alpha causes liver enlargement and increased hepatocyte proliferation in transgenic mice. Am J Pathol. 1994;145:398–408. [PMC free article] [PubMed] [Google Scholar]
- 62.Nishida N, Nishimura T, Ito T, Komeda T, Fukuda Y, Nakao K. Chromosomal instability and human hepatocarcinogenesis. Histol Histopathol. 2003;18:897–909. doi: 10.14670/HH-18.897. [DOI] [PubMed] [Google Scholar]
- 63.Tsai JF, Jeng JE, Ho MS, Chang WY, Hsieh MY, Lin ZY, Tsai JH. Effect of hepatitis C and B virus infection on risk of hepatocellular carcinoma: a prospective study. Br J Cancer. 1997;76:968–974. doi: 10.1038/bjc.1997.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hassan MM, Hwang LY, Hatten CJ, Swaim M, Li D, Abbruzzese JL, Beasley P, Patt YZ. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206–1213. doi: 10.1053/jhep.2002.36780. [DOI] [PubMed] [Google Scholar]
- 65.Park SZ, Nagorney DM, Czaja AJ. Hepatocellular carcinoma in autoimmune hepatitis. Dig Dis Sci. 2000;45:1944–1948. doi: 10.1023/a:1005638500236. [DOI] [PubMed] [Google Scholar]
- 66.Shibuya A, Tanaka K, Miyakawa H, Shibata M, Takatori M, Sekiyama K, Hashimoto N, Amaki S, Komatsu T, Morizane T. Hepatocellular carcinoma and survival in patients with primary biliary cirrhosis. Hepatology. 2002;35:1172–1178. doi: 10.1053/jhep.2002.33157. [DOI] [PubMed] [Google Scholar]
- 67.But DY, Lai CL, Yuen MF. Natural history of hepatitis-related hepatocellular carcinoma. World J Gastroenterol. 2008;14:1652–1656. doi: 10.3748/wjg.14.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuzaki K. Smad phosphoisoform signaling specificity: the right place at the right time. Carcinogenesis. 2011;32:1578–1588. doi: 10.1093/carcin/bgr172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sherman M. Hepatocellular carcinoma: epidemiology, risk factors, and screening. Semin Liver Dis. 2005;25:143–154. doi: 10.1055/s-2005-871194. [DOI] [PubMed] [Google Scholar]
- 71.Yang HI, Lu SN, Liaw YF, You SL, Sun CA, Wang LY, Hsiao CK, Chen PJ, Chen DS, Chen CJ. Hepatitis B e antigen and the risk of hepatocellular carcinoma. N Engl J Med. 2002;347:168–174. doi: 10.1056/NEJMoa013215. [DOI] [PubMed] [Google Scholar]
- 72.Chan HL, Hui AY, Wong ML, Tse AM, Hung LC, Wong VW, Sung JJ. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut. 2004;53:1494–1498. doi: 10.1136/gut.2003.033324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen CJ, Yang HI, Su J, Jen CL, You SL, Lu SN, Huang GT, Iloeje UH. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA. 2006;295:65–73. doi: 10.1001/jama.295.1.65. [DOI] [PubMed] [Google Scholar]
- 74.Wai CT, Greenson JK, Fontana RJ, Kalbfleisch JD, Marrero JA, Conjeevaram HS, Lok AS. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology. 2003;38:518–526. doi: 10.1053/jhep.2003.50346. [DOI] [PubMed] [Google Scholar]
- 75.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 76.Yamaguchi T, Matsuzaki K, Inokuchi R, Kawamura R, Yoshida K, Murata M, Fujisawa J, Fukushima N, Sata M, Kage M, et al. Phosphorylated Smad2 and Smad3 signaling: Shifting between tumor suppression and fibro-carcinogenesis in chronic hepatitis C. Hepatol Res. 2013;43:1327–1342. doi: 10.1111/hepr.12082. [DOI] [PubMed] [Google Scholar]