

Background: ASH2L is a component of MLL complexes conferring H3K4 trimethylation.
Results: Increased ASH2L expression induces ERα expression in breast cancers. ASH2L controls estrogen receptor (ER) α expression by directly potentiating GATA3 transcriptional activity.
Conclusion: ASH2L enhances ERα expression as a coactivator of GATA3 in breast cancers.
Significance: Amplification of the ASH2L gene may contribute to breast cancer development by increasing ERα expression.
Keywords: Breast Cancer, Estrogen Receptor, GATA Transcription Factor, Gene Regulation, Oncogene
Abstract
ASH2L is a component of MLL complexes that confer H3K4 trimethylation. The ASH2L gene is located at 8q11–12, which is often amplified in breast cancers. We found that increased ASH2L expression, which can result from gene amplification, is often correlated with increased ERα expression in both breast cancer cell lines and primary breast cancers. Forced expression of ASH2L induced ERα expression in mammary epithelial cells, whereas depletion of ASH2L suppressed ERα expression in breast cancer cells. To understand the mechanism by which ASH2L regulates ERα expression, we identified GATA3 as the binding protein of ASH2L. ASH2L was shown to potentiate the transcriptional activity of GATA3. ASH2L was recruited to the enhancer of the ERα gene through GATA3 to promote ERα transcription. This study established that ASH2L enhances ERα expression as a coactivator of GATA3 in breast cancers.
Introduction
Estrogens are indispensable for the development of mammary glands and involved in the growth and development of breast cancers (1). During puberty, ductal growth is largely directed by estrogen (2). During early pregnancy, estrogen and progesterone are responsible for further ductal branching and extensive lobulo-alveolar proliferation (3). Exposure to excessive estrogen increases the risk of breast cancers, whereas estrogen antagonist tamoxifen decreases the incidence of breast cancers (4).
Estrogens exert their cellular effects through estrogen receptor (ER)2 that is a member of the nuclear receptor superfamily (5). Although there are two estrogen receptors (ERα and ERβ) in human, it is ERα which plays a major role in breast normal and cancer development (6, 7). ER modulates target gene transcription by directly binding to a short DNA sequence, termed the estrogen response element located in the promoter regions of the target genes. Estrogen receptor also modulates gene expression by communicating with other transcription factors such as AP-1 and Sp1 (8, 9). About 70% of breast cancers express ERα. About 70–80% of ER positive breast cancers are responsive to anti-estrogen therapy (10). However, these tumors gradually become resistant to anti-estrogen agents after several months of anti-estrogen therapy (11). The emergence of anti-estrogen-resistant cancer cells poses a major impediment to the successful treatment of breast cancers.
Expression of the ERα gene is under complicated control with at least seven promoters (12). Analysis of ERα promoters identified AP2C as an important transcription factor controlling ER expression (13). Other important transcription factors involved in ERα expression include FOXM1, FOXO1, GATA3, and NR2E3 (14–17). In addition to transcriptional regulation, ERα is also regulated through mRNA stability and protein stability (18). The ERα protein level is suppressed by the treatment of estrogen in normal breast tissues and breast cancers (19). ERα expression has been shown to be up-regulated in breast cancers but the underlying mechanisms remain elusive (20).
Histone H3K4 trimethylation is often associated with actively transcribed genes (21, 22). MLL complexes have histone methyltransferase activity, which methylates Lys-4 on histone H3 (23, 24). ASH2L is a component of MLL complexes, which include one of seven Set/MLL proteins (SETA, SETB, MLL1 to 5), ASH2L, RBBP5, WDR5, HDPY-30, and others (25–27). Different MLL complexes have different components besides the core proteins. For example, the MLL3/4 complex contains UTX and can be associated with NCOA6 (25, 26, 28). UTX is a histone demethylase that demethylates trimethylated Lys-27 on histone H3 (29, 30). NCOA6 (also known as PRIP, NRC, and AIB3) is a nuclear receptor coactivator (31–35). In addition to the MLL complex, ASH2L is also present in another complex, which consists of ASH2L, RBBP5, and WDR5 without SET/MLL proteins (36, 37). It was shown that this complex also carries methyltransferase activity. ASH2L possesses histone methyltransferase activity when it forms a complex with a 26-amino acid short peptide from RBBP5 (37). ASH2L can also bind DNA directly using a forkhead-like helix-wing-helix domain located on its N-terminal region (38, 39). ASH2L has been shown to promote the transformation of primary fibroblast cells (40). ASH2L interacts with TBX1, AP2G, and MEF2 and is involved in their transcriptional activation (41–43).
Here we report that overexpression of ASH2L due to gene amplification induces expression of the ERα gene in breast cancers. In an effort to understand the mechanisms by which ASH2L activates the ERα gene, we identified GATA3 as an ASH2L interacting protein. We found that ASH2L enhances the transcriptional activity of GATA3, indicating that ASH2L is a coactivator of GATA3. Further studies established that ASH2L promotes ERα expression through its enhancer harboring GATA3 binding sites.
MATERIALS AND METHODS
Plasmids and Antibodies
pCMV-sport6-GATA3 and pCMV-ASH2L are IMAGE clones from Open Biosystem (Huntsville, AL). GATA3-TK-Luc reporter vector was generated by inserting an oligonucleotide carrying three GATA3 binding sites into the SalI/HindIII sites of TK-Luc. pGBKT7-ASH2L was generated by inserting ASH2L cDNA into the BamHI site of pGBKT7. Anti-ASH2L was obtained from Bethyl Laboratories (Montgomery, TX). Anti-ER and anti-GATA3 were purchased from Abcam (Cambridge, MA).
Establishment of Mammary Epithelial Cell Line
The inguinal glands were collected from wild type mice. Mammary epithelial cells were separated from stromal cells by collagenase digestion and Percoll gradient centrifugation (44). 5 × 106 of 293 FT packaging cells were transfected with 15 μg of lentiviral vector for P53DD (Addgene) along with packaging vectors through the calcium phosphate precipitation method. Forty-eight hours after the transfection, viral supernatant was added to 5 × 105 mammary epithelial cells with 8 μg/ml of Polybrene and incubated for 5 h. Immortalized cells appeared 2 weeks later.
RT-PCR and PCR
Total RNAs were prepared by the TRizol (Invitrogen) method. RT-PCR was performed with SuperScript one-step RT-PCR kit from Invitrogen. Real-time RT-PCR and PCR were performed on ABI 7300 (Applied Biosystems) using SYBR Green Supermix (Applied Biosystems) according to the manufacturer's protocol. The β-actin locus was used for the internal reference copy number to determine the ASH2L copy number.
The primers used were as follows: β-actin, 5′-CCATCTACGAGGGCTATGCT-3′ and 5′-GCAAGTTAGGTTTTGTCAAAGA-3′; ASH2L, 5′-TGCCATCACAGTGGGAATAC-3′ and 5′-GGTTGTCATAAGCAGGACCA-3′; ERα, 5′-GCACCCGCCGCCGCAGCTGT-3′ and 5′-GGCTTGGCCAAAGGTTGGCA-3′; pS2, 5′-GATCTGCGCCCTGGTCCT-3′ and 5′-GGAAAACCACAATTCTGT-3′; Cathepsin D, 5′-TGATGCAGCAGAAGCTGGTG-3′ and 5′-GAGTCTGTGCCACCCAGCA-3′. The ASH2L for gene amplification were: 5′-CTGACGTCTTGTATCACGTG-3′ and 5′-GCATCTTTGGGAGAACATTTG-3′ and β-actin for gene amplification: 5′-TGTCCACCTTCCAGCAGATG-3′ and 5′-CTGCGCAAGTTAGGTTTTGTC-3′.
Immunohistochemistry
Immunohistochemical analysis was performed on paraffin-embedded human normal breast or breast cancer tissues. Sections were deparaffinized and rehydrated. After the inactivation of endogenous peroxidase activity and antigen retrieval, sections were blocked with 10% normal bovine serum in phosphate-buffered saline, followed by sequential incubation at room temperature with anti-ASH2L antibodies for 3 h, biotinylated goat anti-rabbit IgG for 1 h, streptavidin-linked horseradish peroxidase for 30 min, and finally 3,3′-diaminobenzidine tetrahydrochloride solution for 4 min. Sections were counterstained with hematoxylin.
Generation of Stable Cell Lines Expressing Human ASH2L
The immortalized mammary cells were infected with lentiviruses expressing ASH2L. After selection with blasticidin (10 μg/ml), the individual clones were combined, and expanded. The cell extracts were subject to Western blot analysis.
Yeast Two-hybrid Screening
Yeast two-hybrid screening was performed using the matchmaker two-hybrid system kit as instructed by the manufacturer (Clontech). Briefly, the yeast strain HF7C was cotransformed with a human matchmaker mammary gland cDNA expression library and pGBKT7-ASH2L. The HF7C cells were selected by their growth in medium lacking histidine and the expression of β-galactosidase. Four days later, the clones that likely contained the ASH2L interacting protein emerged. These clones were confirmed by its expression of β-galactosidase.
GST Pull-down Assays
GST and GST fusion proteins were produced in Escherichia coli BL21 and bound to glutathione-Sepharose beads according to the manufacturer's instructions (Pharmacia). In vitro translation was performed using rabbit reticulocyte lysate (Promega) and labeled with [35S]methionine. 50 μl of GST fusion protein was incubated with 10 μl of in vitro translated proteins for 2 h in 500 μl of NETN (20 mm Tris-HCl, pH 7.5, 100 mm KCl, 0.7 mm EDTA, 0.05% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride). Bound proteins were washed 5 times with NETN, eluted by boiling for 2 min in 30 μl of protein loading buffer, separated by SDS-PAGE, and autoradiographed.
Immunoprecipitation
HEK293 cells were transfected with GATA3 and ASH2L expression vectors. The nuclear extract was immunoprecipitated by anti-GATA3 or control IgG in GST binding buffer (20 mm Tris-HCl, pH 7.9, 180 mm KCl, 0.2 mm EDTA, 0.05% Nonidet P-40, 0.5 mm PMSF, and 1 mm DTT). After extensive washing using the same buffer, the bound proteins were eluted by boiling in SDS-PAGE sample buffer, resolved by SDS-PAGE, transferred onto a nitrocellulose membrane, and subjected to Western blot analysis using anti-ASH2L.
shRNA Transfection
Five ASH2L shRNA constructs and one mock shRNA (pKLO vector) construct were purchased from Sigma. Lentiviruses were produced by transfecting 293FT cells with shRNA vectors along with packaging vector for 48 h. BT-483 cells were infected for 24 h with viral supernatant and selected for 6–8 days with 0.5 μg/ml of puromycin. The cells were then combined for further analysis.
Enhancer Analysis
The ERα enhancer 1 fragment and its mutant fragment with two GATA3 binding sites mutated were amplified from human genome DNA using forward primers, 5′-AGACATCCCGGGGATGGTCCTGATTATGG-3′ and 5′-AGACATCCCGGGGATGGTCCTCCCTATGGCCCCCTGAAATATGCT and the common reverse primer, 5′-AGCTAGAGTCGACGAAAATATTTAGAGATACGG-3′, digested with SmaI and SalI, and cloned into the luciferase report vector pGL3-promoter. Immortalized mammary epithelial cells or BT-483 cells (1 × 105) were plated in DMEM containing 10% fetal calf serum in six-well plates and cultured for 24 h before transfection. Transfections were performed using Lipofectamine 2000 reagent (Invitrogen) with ER-LUC (1.8 μg) and pCMV-β (0.2 μg) as an internal control for transfection efficiency. Cell extracts were prepared 36 h after transfection and assayed for luciferase and β-galactosidase activities.
Chromatin Immunoprecipitation (ChIP) and ChIP-qPCR Analyses
BT483 cells were maintained in DMEM containing 10% fetal calf serum. Cells were cross-linked with 1% formaldehyde for 10 min at room temperature. After cells were collected, chromatin immunoprecipitation was performed as described (45) using the appropriate antibodies. The primers used were: F1/R1, 5′-GATGGTCCTGATTATGGGAT-3′ and 5′-TATTTAGAGATACGGAAAAGG-3′. The primers were F2/R2, 5′-AAATTGTAGGAGAGCTCCGC-3′ and 5′-CCTCTAAGAAGTTTTTCTACATC-3′ for the upstream sequence. Purified DNA fragments were amplified and visualized in agarose gel electrophoresis for ChIP analysis. Real-time qPCR was performed as described above for ChIP-qPCR analysis. Each assay was repeated three times. Representative results were presented.
RESULTS
Overexpression of ASH2L Often Correlates to Increased ERα Expression
To identify genes that control the expression of ERα, we searched for genes whose expressions are highly correlated with ERα expression in the microarray data from a total of 51 human breast cancer cell lines (46). A close examination of the data revealed that high ERα mRNA levels were often associated with high ASH2L mRNA levels (supplemental Table 1S). To confirm this observation, we examined ERα and ASH2L mRNA and protein in four ER positive tumor cell lines. Real-time RT-PCR revealed that BT-483 and MDA-MB-134 expressed high ERα and ASH2L mRNA, whereas MCF-7 and T47D express relatively low ERα and ASH2L mRNA (Fig. 1A). Western blots showed that both the ERα and ASH2L proteins correspond to their mRNA level in these four cell lines (Fig. 1A). Both BT-483 and MDA-MB-134 carry amplification of 8p11–12 where the ASH2L gene is located (47, 48). As expected, we confirmed that the ASH2L gene is amplified in both BT-483 and MDA-MB-134 cells but not in MCF-7 and T47D cells (Fig. 1A), indicating that ASH2L overexpression in these two cancer cell lines are due to gene amplification. We examined 9 additional breast cancer cell lines for ERα and ASH2L expression by real-time RT-PCR. We found that ER negative cells also express ASH2L, indicating that ASH2L alone is not sufficient to activate ERα expression (Fig. 1A).
FIGURE 1.

A: a, the levels of ERα expression in breast cancer cell lines are correlated with the levels of ASH2L expression. The ERα and ASH2L mRNA (left panel) and protein (right panel) in MCF-7, T47D, BT483, and MDA-MB-134 cells were determined by real-time RT-PCR and Western blot, respectively. **, p < 0.01 (versus MCF-7 and T47D). b, copy number of the ASHL gene was determined in MCF-7, T47D, BT483, and MDA-MB-134 cells by real-time PCR. **, p < 0.01 (versus MCF-7 and T47D). c, ASH2L and ERα expression in additional breast cancer cell lines. Lanes 1, primary breast epithelial cells; 2, MCF-10A; 3, MDA-MB-361; 4, ZNF-75–1; 5, BT-20; 6, BT474; 7, SKBR3; 8, MDA-MB-231; 9, MDA-MB-157; 10, MDA-MB-175; 11, MDA-MB-134. B: a, real-time PCR analyses of ASH2L and ERα mRNA in human primary ER positive breast cancers. Lanes 1, human normal mammary epithelial cells served as a normal control; 2 to 33, primary breast cancers. **, p < 0.01 (versus other tumors). b, immunostaining of ASH2L protein. N, normal breast; T, breast cancer with increased expression of ASH2L protein (tumor number 4 from a is shown as a representative).
To find if ERα expression correlates to ASH2L expression in primary breast cancers, we examined ERα mRNA and ASH2L mRNA in a total of 32 cases of ER positive breast cancers. We found that eight cases expressed high levels of ASH2L mRNA. Seven of the eight cases also expressed high levels of ERα mRNA, whereas one case expressed a low level of ERα mRNA (Fig. 1B), confirming that increased ASH2L expression is associated with increased ERα expression in primary breast cancers. We also found that three breast tumors without high ASH2L expression express high levels of ERα, indicating that other factors can contribute to the high level expression of ERα. Immunostaining revealed that the ASH2L protein is heterogeneously expressed in the nuclei of normal mammary epithelial cells (Fig. 1B), whereas these breast cancers with high levels of ASH2L mRNA showed rather homogeneous ASH2L protein expression with increased intensity (Fig. 1B).
Increased Expression of ASH2L Induces ERα Expression
To demonstrate the involvement of ASH2L in regulation of the ERα gene, an immortalized mammary epithelial cell line was infected with lentivirus expressing ASH2L or control empty virus. Blasticidin-resistant clones were combined and analyzed for ASH2L and ERα expression. The overexpression of ASH2L was shown by real-time RT-PCR and Western blot (Fig. 2A). The cells overexpressing ASH2L were found to have much increased ERα mRNA and protein expression (Fig. 2A), indicating that ASH2L can induce the expression of ERα in mammary epithelial cells.
FIGURE 2.

A, ASH2L induces ERα expression in mammary epithelial cells. Immortalized mammary epithelial cells were infected with lentivirus expressing ASH2L or LacZ as a control. The ASH2L and ERα mRNA were examined by real-time RT-PCR, which were normalized to control β-actin mRNA levels (left panel). **, p < 0.01 (versus control). The ASH2L and ERα protein were examined by Western blot (right panel). B: a, depletion of ASH2L leads to deceased expression of ERα in human breast cancer cells. BT483 cells were infected with viruses expressing two different shRNAs targeting ASH2L (sh1, sh2) or control shRNA. Real-time RT-PCR was performed to determine ERα and ASH2L mRNA levels (left panel). **, p < 0.01 (versus control). The level of ERα and ASH2L protein were evaluated by Western blot (right panel). b, reduced expression of ERα target genes with the depletion of ASH2L expression. The cells were treated with 100 nm E2 or control vehicle for 24 h and collected for the preparation of total RNA. Real time RT-PCR was performed to determine cathepsin D (CTSD) and pS2 mRNA levels, which were normalized to internal control β-actin mRNA levels. **, p < 0.01 (versus control vehicle). ##, p < 0.01(versus control shRNA). c, depletion of ASH2L results in cell growth inhibition of BT483. ASH2L-depleted cells (sh1 and sh2) and cells expressing control shRNA (WT) were counted by trypan blue staining at different times after initial seeding of 2 × 104 cells with the treatment of 100 nm E2. The experiment was done independently three times, and the results were averaged with error bars representing S.D. **, p < 0.01 (versus control shRNA).
Depletion of ASH2L Inhibits ERα Expression with Reduced Expression of ERα Target Genes and Results in Cell Growth Inhibition of ER Positive Breast Cancers
To examine the effect of ASH2L on ERα expression in breast cancer, BT483 cells were infected with two lentiviruses expressing different shRNAs against ASH2L, each of which reduced the ASH2L transcript about 80% compared with controls (Fig. 2B). We found that cells with reduced ASH2L expression showed decreased ERα expression (Fig. 2B). We checked the expression of ER target genes pS2 and cathepsin D (CTSD) in response to estrogen. Accordingly, the expression of these genes in response to estrogen was decreased with the depletion of ASH2L (Fig. 2B). We further sought to determine whether estrogen-dependent growth of BT483 cells was affected by depletion of ASH2L. In comparison with control cells, the depletion of ASH2L significantly inhibited the estrogen-dependent growth of BT483 cells (Fig. 2B), suggesting that ASH2L is involved in estrogen-dependent growth of breast cancer cells.
ASH2L Expression Is Also Regulated at Its Protein Level in Breast Cancers
When we examined ASH2L expression in breast cancer cell lines, we found that the ASH2L mRNA level is slightly lower in T47D cells than that in another breast cancer cell line BT20 (Fig. 3A). However, the ASH2L protein level is much higher in T47D cells than that in BT20 cells (Fig. 3B). A pulse-chase experiment revealed that the ASH2L protein in T47D cells is more stable in BT20 cells (Fig. 3C), indicating that in addition to gene amplification, ASH2L expression is also regulated at its protein level.
FIGURE 3.
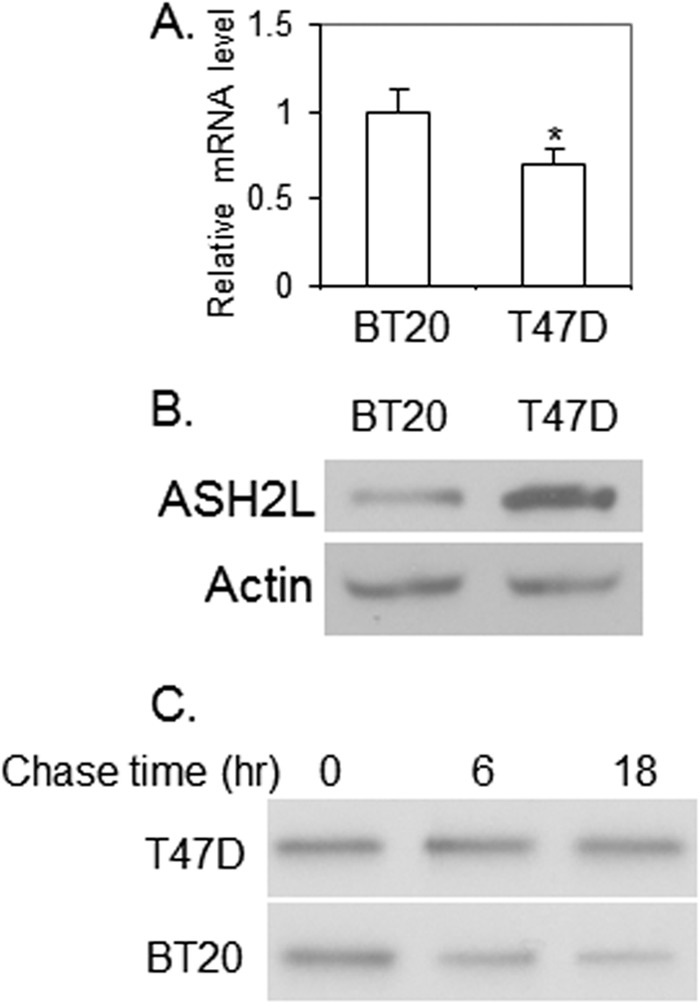
ASH2L expression is regulated at its protein level. A, ASH2L mRNA level in BT20 and T47D cells, as revealed by real-time RT-PCR. *, p < 0.05 (versus BT20). B, ASH2L protein level in BT20 and T47D cells, as examined by Western blot. C, ASH2L protein is more stable in T47D cells than in BT20 cells, as revealed by pulse-chase analysis. 35S-Labeled ASH2L was precipitated by anti-ASH2L at several time points following the labeling.
Identification of GATA3 as an ASH2L Interacting Protein
As ASH2L is a component of MLL/Set complexes, it is likely that ASH2L is recruited to the ERα gene by a certain transcription factor to promote ERα expression. To identify such a transcription factor, we performed yeast two-hybrid screening using ASH2L as the bait and a human mammary cDNA library as the prey. One of the interacting proteins isolated is GATA3, which has been shown to regulate ERα expression (16). The pGBKT7-ASH2L vector allowed yeast growth when co-transformed with pACT2-GATA3 vector but not with control pACT2 vector (Fig. 4A), indicating that ASH2L indeed interacts with GATA3 in yeast. GST pull-down assay was performed to further demonstrate the interaction between GATA3 and ASH2L. In the GST pull-down assay, 35S-labeled GATA3 specifically interacted with the GST-ASH2L fusion protein (Fig. 4B). The interaction domain was localized to the 100-amino acid carboxyl-terminal fragment of GATA3 (Fig. 4B).
FIGURE 4.

A, ASH2L interacts with GATA3, as revealed by a yeast two-hybrid system. Co-expression of ASH2L fused to the Gal4 DNA binding domain and GATA3 fused to the Gal4 activation domain but not either of the two proteins alone allowed the growth of yeast. The interaction between P53 and Large T antigen served as a positive control. B, interaction of ASH2L with GATA3 in vitro. Immobilized GST and GST-ASH2L proteins were incubated with 3S-labeled full-length or partial GATA3 proteins. Bound proteins were eluted, separated by SDS-PAGE, and autoradiographed. AD, activation domain; DBD, DNA binding domain; CTD, COOH-terminal domain. C, interaction of GATA3 with ASH2L in vivo. Plasmid expressing GATA3 was co-transfected with plasmid expressing ASH2L into HEK293 cells. The whole cell extract was prepared and used for immunoprecipitation assay with anti-GATA3 or control serum. The precipitates were analyzed by Western blot with anti-ASH2L antibody. D, ASH2L potentiated GATA3 transcriptional activity. CV-1 cells were transfected with 1.5 μg of GATA3-TK-LUC or TK-LUC, 20 ng of PCMV-GATA3, 20 ng of PCMV-LacZ, and different amounts of PCMV-ASH2L as indicated or PCMV control vector. The luciferase activities were determined. The activity obtained on transfection of GATA3-TK-LUC without exogenous ASH2L was taken as 1. ##, p < 0.01 (versus the control with no GATA3). **, p < 0.01 (versus the control with no ASH2L).
GATA3 Interacts with ASH2L in Vivo
To detect the interaction between GATA3 and ASH2L in vivo, plasmid expressing ASH2L was co-transfected with plasmid expressing GATA3 into 293T cells. The whole cell extract was prepared and used for an immunoprecipitation assay with anti-GATA3 antibody. Later the immunoprecipitated samples were analyzed by Western blot with anti-ASH2L. Signals corresponding to the size of ASH2L protein were only detected in the samples precipitated with anti-GATA3, whereas there was no signal in the samples precipitated with control serum (Fig. 4C), indicating that ASH2L binds to GATA3 in vivo.
ASH2L Potentiates the Transcriptional Activity of GATA3
To investigate the effects of ASH2L on the transcriptional activity of GATA3, we conducted a transient transfection assay with overexpressed ASH2L. As expected, GATA3 enhanced the expression of luciferase controlled by the GATA3 binding element (Fig. 4D). Overexpression of ASH2L leads to about a 2.5-fold increase in the transcription of the reporter gene mediated by GATA3 in a dose-dependent manner (Fig. 4D). The result strongly suggested that ASH2L played an important role in determining the transcriptional activity of GATA3.
ASH2L Potentiates the Activity of ERα Enhancer
The ERα gene is under control of at least seven alternative promoters and two enhancers (Fig. 5A). GATA3 has been shown to be recruited to enhancer 1 (16). To gain insight into the mechanism by which ASH2L regulates the transcription of ERα in breast cancers, the enhancer 1 fragment was cloned into a vector carrying a luciferase reporter gene and tested in ASH2L-depleted or control BT483 cells by transfection assays. Unlike the control reporter gene, the enhancer-directed expression of luciferase was decreased in ASH2L-depleted cells (Fig. 5B). The enhancer-directed expression of luciferase was also decreased in GATA3-depleted cells (Fig. 5C), suggesting that GATA3 is indeed involved in the regulation of ERα through this enhancer. To further confirm that ASH2L acts on GATA3 to regulate ERα gene expression, we transfected the ERα enhancer directed reporter gene, ASH2L expression vector, and GATA3 expression vector into immortalized mammary epithelial cells. GATA3 increased the ERα enhancer activity. ASH2L further potentiated the GATA3-mediated transcriptional activity (Fig. 5D). To confirm the role of GATA3 in mediating activation of ERα enhancer by ASH2L, the two GATA3 binding motifs in ERα enhancer were mutated. As expected, both GATA3 and ASH2L had a minimal effect on the activity of the mutated ERα enhancer (Fig. 5D).
FIGURE 5.

ASH2L promotes the enhancer activity of the ERα gene through GATA3. A, the diagram of ERα regulatory elements. The six promoters are indicated from A to F along with two enhancers (EN1 and EN2). Enhancer 1 (EN1) was used for luciferase activity and ChIP assays. B, depletion of ASH2L decreased the activity of ERα enhancer. ASH2L-depleted cells (sh2 as described above) or wild type cells expressing control shRNA (control) were transfected with control luciferase reporter (pGL-Luc) or the luciferase reporter directed by ERα enhancer (pGL-ER-Luc) and β-gal expression vector. Luciferase activity was normalized to β-gal activity. **, p < 0.01 (versus control reporter). ##, p < 0.01 (versus control shRNA). C, depletion of GATA3 decreased activity of the ERα enhancer. BT483 cells were infected with viruses expressing two different shRNAs targeting GATA3 (sh1, sh2) or control shRNA (NS). Real-time RT-PCR was performed to determine GATA3 mRNA levels (left panel). GATA3-depleted cells or wild type cells expressing control shRNA (control) were transfected and analyzed as described in B (right panel). ##, p < 0.01 (versus control shRNA); **, p < 0.01 (versus control reporter). D, ASH2L activated ERα enhancer through GATA3. Immortalized mammary epithelial cells were transfected with pGL-ER-Luc or mutant pGL-ER-Luc reporters, β-gal expression vector, plus ASH2L expression vector and GATA3 expression vector as indicated. ##, p < 0.01 (versus control); **, p < 0.01 (versus normal reporter).
ASH2L Is Recruited to the Enhancer of ERα
To investigate if ASH2L is recruited to the ERα enhancer in the nuclei of living cells, we performed a ChIP assay. The antibodies against ASH2L or GATA3 could efficiently precipitate the enhancer of the ERα gene but could not pull down the fragment upstream of the enhancer (Fig. 6A). The depletion of GATA3 decreased the recruitment of GATA3 and ASH2L to the enhancer, whereas the depletion of ASH2L abolished ASH2L binding but retained GATA3 binding at the enhancer, confirming the specificity of the antibodies and indicating the dependence of ASH2L on GATA3 for its binding at the enhancer (Fig. 6A). WDR5 is another component of MLL complexes. ChIP assays with anti-WDR5 revealed the enriched WDR5 binding to the ERα enhancer but not to the fragment upstream of the enhancer in wild type cells. Compared with wild type cells, a substantial reduced amplification was seen in ASH2L- or GATA3-depleted cells (Fig. 6A). These results suggested that the complex is recruited to the ERα enhancer in breast cancer cells through ASH2L. As ASH2L is a component of protein complexes carrying histone methyltransferase activity, we performed chromatin immunoprecipitation to find if the recruitment of ASH2L alters the methylated status of histones in ERα enhancer. We observed a marked increase of H3K4 trimethylation in the ERα enhancer in wild type BT483 cells compared with ASH2L or GATA3-depleted BT483 cells (Fig. 6A). Sequential ChIP was performed to find if ASH2L binds to GATA3 on the ERα enhancer in breast cancer cells. The eluted chromatin precipitated by anti-GATA3 was subjected to a second immunoprecipitation with anti-ASH2L or control IgG. There was specific enrichment of DNA corresponding to the ERα enhancer with anti-ASH2L compared with IgG controls, whereas no enrichment of DNA upstream of ERα enhancer was observed (Fig. 6B), indicating that ASH2L and GATA3 form a complex on ERα enhancer.
FIGURE 6.
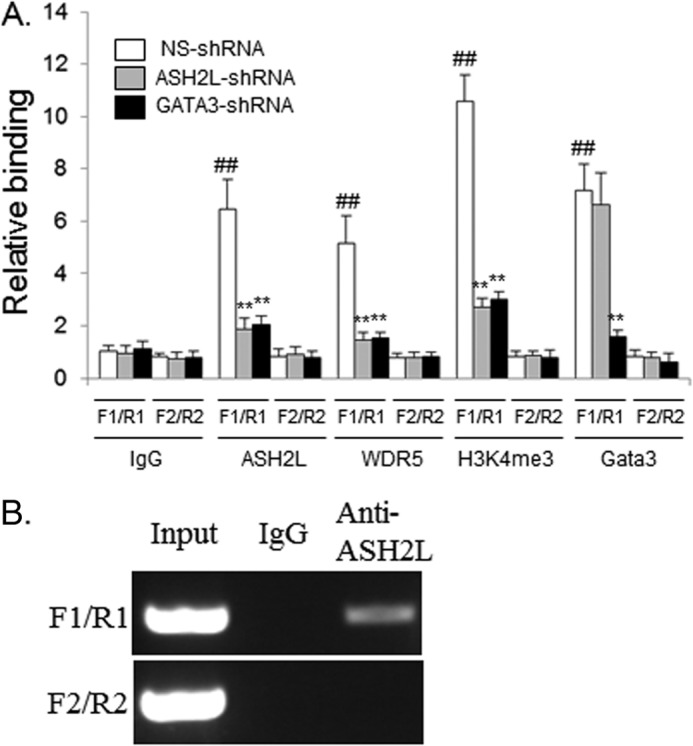
A, ASH2L is recruited to the ERα enhancer and enhances H3K4-trimethylation at the ERα enhancer in breast cancers. One pair of primers for the human ERα enhancer region (F1/R1) and another pair of primers ∼2 kb upstream of the ERα enhancer (F2/R2) were used in ChIP-qPCR assays. ChIP was performed with BT483 cells expressing nonspecific shRNA (NS), ASH2L shRNA (sh2 as described above), or GATA3 shRNA (sh2 as described above), and anti-ASH2L, anti-H3K4me3, anti-WDR5, anti-GATA3, or control IgG. ##, p < 0.01 (versus IgG control); **, p < 0.01 (versus control shRNA). B, ASH2L and GATA3 protein form complexes on the ERα enhancer, as revealed by sequential ChIP assays. The first-step ChIP was performed with BT483 cells and anti-GATA3. The second-step ChIP was carried out with the eluate of the initial ChIP and anti-ASH2L or IgG. PCR were performed using primer sets F1/R1 and F2/R2 as described above.
DISCUSSION
ERα signaling is critical for normal breast and breast cancer development. ERα expression is up-regulated at the mRNA level in breast cancers (20). The mechanisms by which ERα expression is regulated in normal breast and up-regulated in breast cancers are not fully understood. In this study, we demonstrated that ASH2L enhances ERα expression. We found that ASH2L interacts with GATA3 and potentiates GATA3 transcriptional activity. ASH2L was shown to be recruited to the enhancer of the ERα gene through GATA3 to promote ERα transcription. This study established that ASH2L enhances ERα expression through acting as a coactivator of GATA3.
We found that the high expression of ERα is often correlated with increased ASH2L expression in both breast cancer cell lines and primary breast cancers. However, we also observed that one of the primary tumors with high ASH2L expression expressed a low level of ERα mRNA. It is likely that other factors in addition to ASH2L are needed to enhance ERα expression.
ASH2L is the component of MLL2 and the ASCOM complex (28, 49). These two complexes are recruited to ERα through MLL2 and PRIP, respectively. Both complexes have been shown to promote ERα transcriptional activity. Although ASH2L does not directly interact with ERα, it may be required for the function of complexes. In addition to regulating ERα expression, it could also be required for ERα transcriptional activity. So the decreased expression of ERα target genes due to ASH2L depletion could result from both decreased ERα expression and activity.
GATA3 is a member of the GATA transcription factor family (51, 52). In addition to its function in T cell differentiation (53), GATA3 plays an essential role in maintaining the proliferation and differentiation of breast luminal cells (54, 55). GATA3 expression along with FOXA1 expression is associated with ER positive luminal breast cancers that tend to have a favorable prognosis (56, 57). The molecular mechanism by which GATA3 functions in mammary glands and ER positive breast cancer development remains to be fully defined. Point mutations on the GATA3 protein have been isolated in about 5% ER positive breast cancers (58, 59). Although the role of these mutations in ER positive cancer development remains elusive, it indicates that altered GATA3 signaling could contribute to ER positive breast cancer development. As a coactivator of GATA3, ASH2L could contribute to ER positive tumor development through regulating other GATA3 target genes. It remains to be determined how ASH2L overexpression affects the function of mutant GATA3 in breast cancers.
The increased ASH2L expression in the two breast cancer cell lines we tested are due to ASH2L gene amplification. In addition, our result also showed that ASH2L expression is regulated at the protein level. Immunostaining revealed that ASH2L expression is heterogeneous in normal breast epithelial cells, which could partially result from regulation of the protein level. The dysregulation of ASH2L at the protein level could also contribute to its abnormal expression in breast cancers.
The ASH2L gene is located on 8p11–12, which is amplified in 10 to 15% of human breast cancers (60–62). Amplification of 8p11–12 has been associated with estrogen receptor positive luminal breast cancers. The 8p11 amplification is complicated as it contains four amplicons, indicating the involvement of multiple genes from 8p11 amplification in breast cancers (47). So far, only ZNF703 has been identified as a candidate oncogene from 8p11–12 amplification (50, 63). The search for oncogenes in this region with the transformation assay using MCF-10A cells provided inconclusive results about the role of individual candidate genes (48). MCF-10A is a basal breast epithelial cell line. When ASH2L was overexpressed in MCF-10A cells, we found that ERα expression was not induced (data not shown), indicating the cell-type specific role of the ASH2L protein. So given that the amplification is often associated with ER positive tumors, ASH2L could be one of the genes from 8p11–12 amplicons contributing to the tumor development by inducing ERα expression.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant CA 88898 (to Y. J. Z).

This article contains supplemental Table S1.
- ERα
- estrogen receptor α
- ASH2L
- absent, small or homeotic 2-like protein
- GATA3
- GATA-binding protein 3
- FOXO1
- Forkhead box protein O1
- FOXM1
- Forkhead box protein M1
- RBBP5
- retinoblastoma-binding protein 5
- WDR5
- WD repeat-containing protein 5
- UTX
- ubiquitously transcribed tetratricopeptide repeat, X chromosome
- MLL
- mixed lineage leukemia protein.
REFERENCES
- 1. Hennighausen L., Robinson G. W. (1998) Think globally, act locally: the making of a mouse mammary gland. Genes Dev. 12, 449–455 [DOI] [PubMed] [Google Scholar]
- 2. Neville M. C., McFadden T. B., Forsyth I. (2002) Hormonal regulation of mammary differentiation and milk secretion. J. Mammary Gland Biol. Neoplasia 7, 49–66 [DOI] [PubMed] [Google Scholar]
- 3. Brisken C. (2002) Hormonal control of alveolar development and its implications for breast carcinogenesis. J. Mammary Gland Biol. Neoplasia 7, 39–48 [DOI] [PubMed] [Google Scholar]
- 4. Shoker B. S., Jarvis C., Clarke R. B., Anderson E., Hewlett J., Davies M. P., Sibson D. R., Sloane J. P. (1999) Estrogen receptor-positive proliferating cells in the normal and precancerous breast. Am. J. Pathol. 155, 1811–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mangelsdorf D. J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., Evans R. M. (1995) The nuclear receptor superfamily: the second decade. Cell 83, 835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mosselman S., Polman J., Dijkema R. (1996) ERβ: identification and characterization of a novel human estrogen receptor. FEBS Lett. 392, 49–53 [DOI] [PubMed] [Google Scholar]
- 7. Korach K. S., Couse J. F., Curtis S. W., Washburn T. F., Lindzey J., Kimbro K. S., Eddy E. M., Migliaccio S., Snedeker S. M., Lubahn D. B., Schomberg D. W., Smith E. P. (1996) Estrogen receptor gene disruption: molecular characterization and experimental and clinical phenotypes. Recent Prog. Horm. Res. 51, 159–186; discussion 186–158 [PubMed] [Google Scholar]
- 8. Porter W., Wang F., Wang W., Duan R., Safe S. (1996) Role of estrogen receptor/Sp1 complexes in estrogen-induced heat shock protein 27 gene expression. Mol. Endocrinol. 10, 1371–1378 [DOI] [PubMed] [Google Scholar]
- 9. Webb P., Lopez G. N., Uht R. M., Kushner P. J. (1995) Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol. Endocrinol. 9, 443–456 [DOI] [PubMed] [Google Scholar]
- 10. Early Breast Cancer Trialists' Collaborative Group. (1992) Systemic treatment of early breast cancer by hormonal, cytotoxic, or immune therapy: 133 randomised trials involving 31,000 recurrences and 24,000 deaths among 75,000 women. Lancet 339, 71–85 [PubMed] [Google Scholar]
- 11. Nass S. J., Davidson N. E. (1999) The biology of breast cancer. Hematol. Oncol. Clin. North Am. 13, 311–332 [DOI] [PubMed] [Google Scholar]
- 12. Kos M., Reid G., Denger S., Gannon F. (2001) Minireview: genomic organization of the human ERα gene promoter region. Mol. Endocrinol. 15, 2057–2063 [DOI] [PubMed] [Google Scholar]
- 13. McPherson L. A., Baichwal V. R., Weigel R. J. (1997) Identification of ERF-1 as a member of the AP2 transcription factor family. Proc. Natl. Acad. Sci. U.S.A. 94, 4342–4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo S., Sonenshein G. E. (2004) Forkhead box transcription factor FOXO3a regulates estrogen receptor α expression and is repressed by the Her-2/neu/phosphatidylinositol 3-kinase/Akt signaling pathway. Mol. Cell. Biol. 24, 8681–8690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Madureira P. A., Varshochi R., Constantinidou D., Francis R. E., Coombes R. C., Yao K. M., Lam E. W. (2006) The Forkhead box M1 protein regulates the transcription of the estrogen receptor α in breast cancer cells. J. Biol. Chem. 281, 25167–25176 [DOI] [PubMed] [Google Scholar]
- 16. Eeckhoute J., Keeton E. K., Lupien M., Krum S. A., Carroll J. S., Brown M. (2007) Positive cross-regulatory loop ties GATA-3 to estrogen receptor α expression in breast cancer. Cancer Res. 67, 6477–6483 [DOI] [PubMed] [Google Scholar]
- 17. Park Y. Y., Kim K., Kim S. B., Hennessy B. T., Kim S. M., Park E. S., Lim J. Y., Li J., Lu Y., Gonzalez-Angulo A. M., Jeong W., Mills G. B., Safe S., Lee J. S. (2012) Reconstruction of nuclear receptor network reveals that NR2E3 is a novel upstream regulator of ESR1 in breast cancer. EMBO Mol. Med. 4, 52–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pinzone J. J., Stevenson H., Strobl J. S., Berg P. E. (2004) Molecular and cellular determinants of estrogen receptor α expression. Mol. Cell. Biol. 24, 4605–4612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nawaz Z., Lonard D. M., Dennis A. P., Smith C. L., O'Malley B. W. (1999) Proteasome-dependent degradation of the human estrogen receptor. Proc. Natl. Acad. Sci. U.S.A. 96, 1858–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clarke R. B., Anderson E., Howell A. (2004) Steroid receptors in human breast cancer. Trends Endocrinol. Metab. 15, 316–323 [DOI] [PubMed] [Google Scholar]
- 21. Martin C., Zhang Y. (2005) The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6, 838–849 [DOI] [PubMed] [Google Scholar]
- 22. Shilatifard A. (2008) Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 20, 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tenney K., Shilatifard A. (2005) A COMPASS in the voyage of defining the role of trithorax/MLL-containing complexes: linking leukemogensis to covalent modifications of chromatin. J. Cell. Biochem. 95, 429–436 [DOI] [PubMed] [Google Scholar]
- 24. Ansari K. I., Mandal S. S. (2010) Mixed lineage leukemia: roles in gene expression, hormone signaling and mRNA processing. FEBS J. 277, 1790–1804 [DOI] [PubMed] [Google Scholar]
- 25. Cho Y. W., Hong T., Hong S., Guo H., Yu H., Kim D., Guszczynski T., Dressler G. R., Copeland T. D., Kalkum M., Ge K. (2007) PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 282, 20395–20406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Issaeva I., Zonis Y., Rozovskaia T., Orlovsky K., Croce C. M., Nakamura T., Mazo A., Eisenbach L., Canaani E. (2007) Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol. Cell. Biol. 27, 1889–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miller T., Krogan N. J., Dover J., Erdjument-Bromage H., Tempst P., Johnston M., Greenblatt J. F., Shilatifard A. (2001) COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. U.S.A. 98, 12902–12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goo Y. H., Sohn Y. C., Kim D. H., Kim S. W., Kang M. J., Jung D. J., Kwak E., Barlev N. A., Berger S. L., Chow V. T., Roeder R. G., Azorsa D. O., Meltzer P. S., Suh P. G., Song E. J., Lee K. J., Lee Y. C., Lee J. W. (2003) Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol. Cell. Biol. 23, 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Agger K., Cloos P. A., Christensen J., Pasini D., Rose S., Rappsilber J., Issaeva I., Canaani E., Salcini A. E., Helin K. (2007) UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449, 731–734 [DOI] [PubMed] [Google Scholar]
- 30. Lan F., Bayliss P. E., Rinn J. L., Whetstine J. R., Wang J. K., Chen S., Iwase S., Alpatov R., Issaeva I., Canaani E., Roberts T. M., Chang H. Y., Shi Y. (2007) A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449, 689–694 [DOI] [PubMed] [Google Scholar]
- 31. Zhu Y., Kan L., Qi C., Kanwar Y. S., Yeldandi A. V., Rao M. S., Reddy J. K. (2000) Isolation and characterization of peroxisome proliferator-activated receptor (PPAR) interacting protein (PRIP) as a coactivator for PPAR. J. Biol. Chem. 275, 13510–13516 [DOI] [PubMed] [Google Scholar]
- 32. Mahajan M. A., Samuels H. H. (2000) A new family of nuclear receptor coregulators that integrate nuclear receptor signaling through CREB-binding protein. Mol. Cell. Biol. 20, 5048–5063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Caira F., Antonson P., Pelto-Huikko M., Treuter E., Gustafsson J. A. (2000) Cloning and characterization of RAP250, a novel nuclear receptor coactivator. J. Biol. Chem. 275, 5308–5317 [DOI] [PubMed] [Google Scholar]
- 34. Lee S. K., Anzick S. L., Choi J. E., Bubendorf L., Guan X. Y., Jung Y. K., Kallioniemi O. P., Kononen J., Trent J. M., Azorsa D., Jhun B. H., Cheong J. H., Lee Y. C., Meltzer P. S., Lee J. W. (1999) A nuclear factor, ASC-2, as a cancer-amplified transcriptional coactivator essential for ligand-dependent transactivation by nuclear receptors in vivo. J. Biol. Chem. 274, 34283–34293 [DOI] [PubMed] [Google Scholar]
- 35. Garapaty S., Xu C. F., Trojer P., Mahajan M. A., Neubert T. A., Samuels H. H. (2009) Identification and characterization of a novel nuclear protein complex involved in nuclear hormone receptor-mediated gene regulation. J. Biol. Chem. 284, 7542–7552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patel A., Vought V. E., Dharmarajan V., Cosgrove M. S. (2011) A novel non-SET domain multi-subunit methyltransferase required for sequential nucleosomal histone H3 methylation by the mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 286, 3359–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cao F., Chen Y., Cierpicki T., Liu Y., Basrur V., Lei M., Dou Y. (2010) An Ash2L/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 SET domain. PLoS One 5, e14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen Y., Wan B., Wang K. C., Cao F., Yang Y., Protacio A., Dou Y., Chang H. Y., Lei M. (2011) Crystal structure of the N-terminal region of human Ash2L shows a winged-helix motif involved in DNA binding. EMBO Rep. 12, 797–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sarvan S., Avdic V., Tremblay V., Chaturvedi C. P., Zhang P., Lanouette S., Blais A., Brunzelle J. S., Brand M., Couture J. F. (2011) Crystal structure of the trithorax group protein ASH2L reveals a forkhead-like DNA binding domain. Nat. Struct. Mol. Biol. 18, 857–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lüscher-Firzlaff J., Gawlista I., Vervoorts J., Kapelle K., Braunschweig T., Walsemann G., Rodgarkia-Schamberger C., Schuchlautz H., Dreschers S., Kremmer E., Lilischkis R., Cerni C., Wellmann A., Lüscher B. (2008) The human trithorax protein hASH2 functions as an oncoprotein. Cancer Res. 68, 749–758 [DOI] [PubMed] [Google Scholar]
- 41. Stoller J. Z., Huang L., Tan C. C., Huang F., Zhou D. D., Yang J., Gelb B. D., Epstein J. A. (2010) Ash2l interacts with Tbx1 and is required during early embryogenesis. Exp. Biol. Med. 235, 569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rampalli S., Li L., Mak E., Ge K., Brand M., Tapscott S. J., Dilworth F. J. (2007) p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat. Struct. Mol. Biol. 14, 1150–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tan C. C., Sindhu K. V., Li S., Nishio H., Stoller J. Z., Oishi K., Puttreddy S., Lee T. J., Epstein J. A., Walsh M. J., Gelb B. D. (2008) Transcription factor Ap2δ associates with Ash2l and ALR, a trithorax family histone methyltransferase, to activate Hoxc8 transcription. Proc. Natl. Acad. Sci. U.S.A. 105, 7472–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang J., Richards J., Guzman R., Imagawa W., Nandi S. (1980) Sustained growth in primary culture of normal mammary epithelial cells embedded in collagen gels. Proc. Natl. Acad. Sci. U.S.A. 77, 2088–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shang Y., Hu X., DiRenzo J., Lazar M. A., Brown M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852 [DOI] [PubMed] [Google Scholar]
- 46. Neve R. M., Chin K., Fridlyand J., Yeh J., Baehner F. L., Fevr T., Clark L., Bayani N., Coppe J. P., Tong F., Speed T., Spellman P. T., DeVries S., Lapuk A., Wang N. J., Kuo W. L., Stilwell J. L., Pinkel D., Albertson D. G., Waldman F. M., McCormick F., Dickson R. B., Johnson M. D., Lippman M., Ethier S., Gazdar A., Gray J. W. (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gelsi-Boyer V., Orsetti B., Cervera N., Finetti P., Sircoulomb F., Rougé C., Lasorsa L., Letessier A., Ginestier C., Monville F., Esteyriès S., Adélaïde J., Esterni B., Henry C., Ethier S. P., Bibeau F., Mozziconacci M. J., Charafe-Jauffret E., Jacquemier J., Bertucci F., Birnbaum D., Theillet C., Chaffanet M. (2005) Comprehensive profiling of 8p11–12 amplification in breast cancer. Mol. Cancer Res. 3, 655–667 [DOI] [PubMed] [Google Scholar]
- 48. Yang Z. Q., Streicher K. L., Ray M. E., Abrams J., Ethier S. P. (2006) Multiple interacting oncogenes on the 8p11-p12 amplicon in human breast cancer. Cancer Res. 66, 11632–11643 [DOI] [PubMed] [Google Scholar]
- 49. Mo R., Rao S. M., Zhu Y. J. (2006) Identification of the MLL2 complex as a coactivator for estrogen receptor α. J. Biol. Chem. 281, 15714–15720 [DOI] [PubMed] [Google Scholar]
- 50. Sircoulomb F., Nicolas N., Ferrari A., Finetti P., Bekhouche I., Rousselet E., Lonigro A., Adélaïde J., Baudelet E., Esteyriès S., Wicinski J., Audebert S., Charafe-Jauffret E., Jacquemier J., Lopez M., Borg J. P., Sotiriou C., Popovici C., Bertucci F., Birnbaum D., Chaffanet M., Ginestier C. (2011) ZNF703 gene amplification at 8p12 specifies luminal B breast cancer. EMBO Mol. Med. 3, 153–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bresnick E. H., Martowicz M. L., Pal S., Johnson K. D. (2005) Developmental control via GATA factor interplay at chromatin domains. J. Cell. Physiol. 205, 1–9 [DOI] [PubMed] [Google Scholar]
- 52. Viger R. S., Guittot S. M., Anttonen M., Wilson D. B., Heikinheimo M. (2008) Role of the GATA family of transcription factors in endocrine development, function, and disease. Mol. Endocrinol. 22, 781–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hosoya T., Maillard I., Engel J. D. (2010) From the cradle to the grave: activities of GATA-3 throughout T-cell development and differentiation. Immunol. Rev. 238, 110–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kouros-Mehr H., Kim J. W., Bechis S. K., Werb Z. (2008) GATA-3 and the regulation of the mammary luminal cell fate. Curr. Opin. Cell Biol. 20, 164–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Asselin-Labat M. L., Sutherland K. D., Barker H., Thomas R., Shackleton M., Forrest N. C., Hartley L., Robb L., Grosveld F. G., van der Wees J., Lindeman G. J., Visvader J. E. (2007) Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell. Biol. 9, 201–209 [DOI] [PubMed] [Google Scholar]
- 56. Fang S. H., Chen Y., Weigel R. J. (2009) GATA-3 as a marker of hormone response in breast cancer. J. Surg. Res. 157, 290–295 [DOI] [PubMed] [Google Scholar]
- 57. Lacroix M., Leclercq G. (2004) About GATA3, HNF3A, and XBP1, three genes co-expressed with the oestrogen receptor-alpha gene (ESR1) in breast cancer. Mol. Cell. Endocrinol. 219, 1–7 [DOI] [PubMed] [Google Scholar]
- 58. Usary J., Llaca V., Karaca G., Presswala S., Karaca M., He X., Langerød A., Kåresen R., Oh D. S., Dressler L. G., Lønning P. E., Strausberg R. L., Chanock S., Børresen-Dale A. L., Perou C. M. (2004) Mutation of GATA3 in human breast tumors. Oncogene 23, 7669–7678 [DOI] [PubMed] [Google Scholar]
- 59. Arnold J. M., Choong D. Y., Thompson E. R., kConFab, Waddell N., Lindeman G. J., Visvader J. E., Campbell I. G., Chenevix-Trench G. (2010) Frequent somatic mutations of GATA3 in non-BRCA1/BRCA2 familial breast tumors, but not in BRCA1-, BRCA2- or sporadic breast tumors. Breast Cancer Res. Treat. 119, 491–496 [DOI] [PubMed] [Google Scholar]
- 60. Ray M. E., Yang Z. Q., Albertson D., Kleer C. G., Washburn J. G., Macoska J. A., Ethier S. P. (2004) Genomic and expression analysis of the 8p11–12 amplicon in human breast cancer cell lines. Cancer Res. 64, 40–47 [DOI] [PubMed] [Google Scholar]
- 61. Garcia M. J., Pole J. C., Chin S. F., Teschendorff A., Naderi A., Ozdag H., Vias M., Kranjac T., Subkhankulova T., Paish C., Ellis I., Brenton J. D., Edwards P. A., Caldas C. (2005) A 1 Mb minimal amplicon at 8p11–12 in breast cancer identifies new candidate oncogenes. Oncogene 24, 5235–5245 [DOI] [PubMed] [Google Scholar]
- 62. Adélaïde J., Chaffanet M., Imbert A., Allione F., Geneix J., Popovici C., van Alewijk D., Trapman J., Zeillinger R., Børresen-Dale A. L., Lidereau R., Birnbaum D., Pébusque M. J. (1998) Chromosome region 8p11-p21: refined mapping and molecular alterations in breast cancer. Genes Chromosomes Cancer 22, 186–199 [DOI] [PubMed] [Google Scholar]
- 63. Holland D. G., Burleigh A., Git A., Goldgraben M. A., Perez-Mancera P. A., Chin S. F., Hurtado A., Bruna A., Ali H. R., Greenwood W., Dunning M. J., Samarajiwa S., Menon S., Rueda O. M., Lynch A. G., McKinney S., Ellis I. O., Eaves C. J., Carroll J. S., Curtis C., Aparicio S., Caldas C. (2011) ZNF703 is a common Luminal B breast cancer oncogene that differentially regulates luminal and basal progenitors in human mammary epithelium. EMBO Mol. Med. 3, 167–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.