

Abstract
Mapping protein–protein interactions is essential to fully characterize the biological function of a protein and improve our understanding of diseases. Affinity purification coupled to mass spectrometry (AP-MS) using selective antibodies against a target protein has been commonly applied to study protein complexes. However, one major limitation is a lack of specificity as a substantial part of the proposed binders is due to nonspecific interactions. Here, we describe an innovative immuno-competitive capture mass spectrometry (ICC-MS) method to allow systematic investigation of protein–protein interactions. ICC-MS markedly increases the specificity of classical immunoprecipitation (IP) by introducing a competition step between free and capturing antibody prior to IP. Instead of comparing only one experimental sample with a control, the methodology generates a 12-concentration antibody competition profile. Label-free quantitation followed by a robust statistical analysis of the data is then used to extract the cellular interactome of a protein of interest and to filter out background proteins. We applied this new approach to specifically map the interactome of hepatitis C virus (HCV) nonstructural protein 5A (NS5A) in a cellular HCV replication system and uncovered eight new NS5A-interacting protein candidates along with two previously validated binding partners. Follow-up biological validation experiments revealed that large tumor suppressor homolog 1 and 2 (LATS1 and LATS2, respectively), two closely related human protein kinases, are novel host kinases responsible for NS5A phosphorylation at a highly conserved position required for optimal HCV genome replication. These results are the first illustration of the value of ICC-MS for the analysis of endogenous protein complexes to identify biologically relevant protein–protein interactions with high specificity.
The exploration of a protein's interactome in a given biological system is often critical to understand its function. Since the introduction of yeast two-hybrid experiments, alternative methods to explore protein–protein interactions have emerged (1–3). In particular, the combination of affinity-purification with mass spectrometry (AP-MS)1 (4) has shown great promise for the identification of protein complexes directly in mammalian cell lines (5). This approach typically involves capturing the protein of interest either through an epitope tag or using a selective antibody. The main challenge with AP-MS is to discern bona fide interactors from highly abundant cellular proteins e.g. cytoskeletal or ribosomal proteins that bind nonspecifically to the affinity matrix (6). This can be partially addressed by including a negative control, such as IP with an antibody of the same isotype against an irrelevant protein or using samples where the target protein is absent (4). More recently, the introduction of quantitative MS (7–9), involving either isotope labeling or label-free strategies (for a review see (9, 10)), have led to a better distinction between true and false-positive interactions. While most of the recent efforts to reduce false positive rates have concentrated on refining data analysis (11), very few attempts have been made to improve the selectivity at the IP step (12). Consequently, classical quantitative side-by-side comparison of a sample with its control (wild type versus knockout cell lysates or capturing antibody versus control isotype) still suffers from the fact that the control sample is not identical to the probed one and both samples can lead to the association of different nonspecific binders.
In this study, we present an innovative approach, termed immuno-competitive capture MS (ICC-MS), which involves a competition step between free and bound antibody in the same cellular extract and quantitation using label-free MS. Instead of comparing only one IP with a control, the methodology generates a 12-concentration antibody competition profile. Combined with a robust statistical analysis of the quantified MS signals, the cellular endogenous interactome of a protein of interest can be extracted out of the background of hundreds of proteins. We used this new approach to specifically map the interactome of the HCV NS5A protein, an essential viral regulatory protein for both genome replication and modulation of the host environment (13). Proteins interacting with NS5A have been previously identified using yeast two-hybrid (14) or classical co-expression and co-immunoprecipitation methods (15). In this study, we use a human hepatocyte-derived cellular model of HCV genome replication and uncover eight new NS5A-interacting protein candidates in addition to other well-known partners. In particular, we highlight LATS1 and LATS2, two closely related human serine/threonine protein kinases, and demonstrate that they are new host kinases responsible for NS5A phosphorylation and optimal HCV replication.
EXPERIMENTAL PROCEDURES
Cell Culture and Lysates Preparation
As previously described (16), we maintained Huh7 cells, stably expressing Con-1 genotype 1b replicon and the renilla luciferase reporter gene (Huh7 2209–23) (17) in DMEM (with GlutaMAX) containing antibiotics (500 μg/ml penicillin-streptomycin, 500 μg/ml G418, all Invitrogen, Carlsbad, CA) and 10% fetal bovine serum (Sigma, St. Louis, MO). Harvested replicon cells were resuspended in lysis buffer containing 50 mm Tris pH 7.5, 150 mm NaCl, 0.5% (v/v) Nonidet P-40, and protease and phosphatase inhibitor tablets (Complete and PhosStop, Roche Applied Science, Penzberg, Germany). After 15 min on ice, lysates were cleared by centrifugation at 1500 × g for 10 min at 4 °C, and protein concentration was estimated using the bicinchoninic acid protein assay kit (Pierce Rockford, IL).
Immunoprecipitation, Competition Experiment, and in-gel digestion
We used anti-NS5A clone 7-D4 for Western blot detection (Thermo Fischer Scientific, Waltham, MA) and anti-NS5A clone H26 (epitope between residues 228 and 278) for IP and competition experiments (Abcam, Cambridge, UK). The antibody was desalted on Zeba columns (Pierce) following manufacturer's instructions and then coupled to Affi-Gel 10 agarose beads (BioRad, Hercules, CA) for 3 h at 4 °C. Unreacted binding sites were blocked by the addition of 0.2 m ethanolamine, beads washed four times with cold PBS, and stored at 4 °C. Beads (20 μl) were incubated overnight with cell lysate (260 μg total protein per condition) at 4 °C, washed four times with lysis buffer, and eluted with SDS sample buffer.
For competition experiments, we pre-incubated lysates with increasing amounts of free anti-NS5A antibody (0, 0.06, 0.1, 0.17, 0.29, 0.5, 0.85, 1.45, 2.46, 4.19, 7.12, and 12.1 μg) for 3 h (100 μl of final volume) before treating them with immobilized anti-NS5A resin for 3 h (all at 4 °C). Eluates were separated on a 4–20% Tris-Gly SDS-PAGE by migrating the gel half its length, and proteins stained with colloidal blue. Four bands spanning from 20 to 150 kDa (arbitrarily named 20, 40, 80, and 140 kDa, respectively) were cut and in-gel trypsin digested following standard procedures.
LC-MS/MS Analyses
We analyzed each sample in duplicate with a nanoflow Easy-nLC system (Proxeon, Odense, Denmark) connected to an LTQ-Orbitrap Velos (Thermo Fisher Scientific). Peptides were concentrated on an AQUA C18 trap (100 μm × 10 mm, Phenomenex, Torrance, CA) before being separated on a ReproSil-Pur C18-AQ (75 μm × 200 mm, 3 μm particle size, 120 Å, Dr. Maisch GmbH, Ammerbuch-Entringen Germany) analytical column using a 50 min gradient of 0–35% acetonitrile (with 0.6% acetic acid) at 250 nl/min. Acquisitions were cycled between full scan at 60,000 resolution (at m/z 400) in the Orbitrap and 10 data-dependent collision-induced dissociation scans in the ion trap. Full-scan MS was recalibrated on the fly using the polycyclodimethylsiloxane at m/z 445.120084 as a lock mass (18). Ions were selected only with assigned charge states >1+ and then excluded for 30 s. MS raw files were converted into .dta files using Extract-MSn (version 1.0.0.8) and data searched using Sequest (version 27.0, revision 12, both Thermo Fisher Scientific). A database consisting of the human part of the SwissProt (June 2009, 34,275 entries, including splice variants) augmented with the five viral nonstructural proteins from the Con1 replicon NS3, NS4A, NS4B, NS5A, and NS5B was used. Searching parameters were trypsin (full) as an enzyme, one missed-cleavage and a mass tolerance of 10 ppm, and 1.0 Da for precursor and fragment ions, respectively. Oxidized methionines (+15.9949 Da), phosphorylated serine, threonine, and tyrosines (+79.9663 Da) were set as differential, while carbamidomethylated cysteines (+57.0215 Da) were set as static modifications. The spectral false discovery rate (specFDR) was restricted to 2.5% or 1.0% by performing a target-decoy search using a concatenated decoy database (19). Processing and analysis, as described below, was performed with the 2.5% specFDR dataset, and the final protein-binding partners were verified with the 1.0% specFDR search. Data from the in vitro phosphorylation experiment were processed using Proteome Discoverer (version 1.2) with the same parameters mentioned above and selected extracted ion chromatograms (EICs) generated with XCalibur's Qual Browser (Thermo Fisher Scientific).
Data Processing and Analysis
The data analysis was performed as described previously with some modifications (20). MS data from each of the four SDS gel bands were processed independently using RefinerMS (version 5.2.6., Genedata Basel, Switzerland) and peptide EICs were estimated as measure of peptide quantitation. Samples from each antibody concentration and each gel band were normalized, respectively, using the quantile–quantile approach (21, 22). The peaks found in RefinerMS were assigned to the sequence information via their scan identifiers; peaks with no identification or with ambiguous identification were removed and quantitative information was log2-transformed (23). In order to control quality, the log2-transformed quantitation data were subjected to principal component analysis. Scores of the first four principal components were correlated with process parameters (antibody concentration, gel lane, LC-MS run number). Mahalanobis distance outliers were estimated based on the scores of the first two principle components; samples outside the 99% confidence interval were considered as outliers and removed from further analysis. Relative protein quantitations were estimated by summarizing peptide EICs into relative abundance of their corresponding protein quantitation group. This was achieved by using the median polish procedure (24), in analogy to the robust multichip analysis method applied in the microarray field (25). In order to detect dose–response relationships, the concepts of contrast tests (26, 27) combined with a moderated linear model (available from the Limma package (21) and already applied to label-free quantitative MS data similar to ours (22)) were used and resulted in a series of T-statistic values for each protein quantitation group. A maximum T-statistic from each series was determined per protein quantitation group. p values were obtained based on the algorithm for step-down max(T) adjusted p values, with 1000 permutations (28), which originates from Westfall and Young (29). The latter was modified with respect to the one-sidedness of the statistic (T instead of T). An adjusted p value of ≤ 5% was chosen as a threshold to distinguish specific from nonspecific binders. Data computations and visualizations were performed with R (version 2.10.1; http://www.r-project.org/).
In Vitro NS5A Phosphorylation
Recombinant NS5A Δ32 (BK strain, residues 33–447) was expressed in Escherichia coli and isolated from the soluble fraction as reported (30). The purified protein (2 μg) was premixed with active human LATS2 (residues 480–1088, SignalChem, Richmond, Canada 500 ng) in kinase activity buffer (12.5 mm β-glycerol phosphate, 25 mm MgCl2, 5 mm EGTA, 2 mm EDTA, 0.25 mm DTT in 25 mm MOPS pH 7.2). The reaction was initiated by the addition of 1 mm ATP (15 μl reaction volume), kept at 30 °C for 0, 20 and 120 min, and stopped by the addition of SDS sample buffer. Proteins were separated by SDS-PAGE and stained with Pro-Q Diamond and SYPRO Ruby (Invitrogen). The same experiment was repeated with 400 ng of purified protein and 400 ng LATS2 for 0, 20, 40, 60, and 80 min and processed for LC-MS analysis as described above.
Transient Replicon Assay
Transient wild type replicon Con1 (expressing firefly luciferase reporter gene) (16) was used to introduce the Ser71Ala mutation in the NS5A region using Quick Change site-directed mutagenesis kit following manufacturer's instructions (Stratagene, La Jolla, CA). The introduced mutation was confirmed by DNA sequencing. Lunet Huh7 cells (4 × 106) were transfected with 10 μg of in vitro transcribed HCV transient subgenomic replicon RNAs (16) (WT or NS5A mutant). The normalized replication efficiency of the NS5A Ser71Ala mutant was determined as the firefly luciferase signal at 96 h post-transfection divided by the signal at 4 h. The replication capacity of the NS5A Ser71Ala mutant replicons was expressed as its normalized replication efficiency compared with that of the WT replicon set at a value of 100%. Average and standard deviation were calculated from four independent experiments.
siRNA Knockdown Experiments
All siRNAs used in the study were obtained from Integrated DNA Technologies, Inc., Coralville, IA, Transfection of siRNA in the HCV replicon cells was performed using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer's protocol. Briefly, 5000 replicon cells, seeded in a 96-well plate 24 h pre-siRNA transfection, were transfected in duplicate with 1 pmol of siRNA. The level of knockdown for each siRNA used in the study was evaluated by real-time PCR (17). Briefly, total cellular RNA was extracted with RNeasy (Qiagen, Venlo, Netherlands) according to manufacturer's instructions. Reverse transcription was carried out using the Taqman RT reagents (Applied Biosystems, Foster City, CA). To quantitate the level of HCV replicon, cDNA was amplified using Taqman Universal PCR mix (PE Biosystems, Norwalk, CT) with a set of primers and probe complementary to a region of the NPTII gene (fluorogenic probe labeled with FAM (6-carboxyfluorescein), 5′-TCC TGC CGA GAA AGT ATC CAT CAT GGC T-3′; forward primer, 5′-GCT GCT ATT GGG CGA AGT G-3′; reverse primer, GCC GCC GCA TTG CA; all obtained from Integrated DNA Technologies, Inc.). A second set of primers and probe complementary to the β-actin gene (Applied Biosystems) was used to quantitate the β-actin mRNA level as the endogenous control. For the quantitation of PI4KA, LATS1 and LATS2, sets of primers and probes (Applied Biosystems) were used according to the manufacturer's instructions. Level of knockdown for each target gene was normalized to the one obtained using negative control siRNAs. Replicon renilla luciferase signal was read 72 h post-siRNA transfection using the Renilla Luciferase Assay System (Promega, Fitchburg, WI, USA) following the manufacturer's protocol. The signal for each gene-specific siRNA experiment was normalized to the one obtained using negative control siRNAs. Average and standard deviation were calculated from 8 to 12 independent experiments.
RESULTS
ICC-MS Principle
To improve the specificity of classical AP-MS, we devised a label-free strategy in which a pre-incubation step of the cell lysates with a concentration range of free antibody is introduced prior to IP with the same antibody immobilized on agarose beads (Fig. 1). The bound proteins can then be eluted from the beads, and digested by trypsin for further analysis by nanoflow liquid chromatography-tandem mass spectrometry. After protein identification by database search and adequate quality control, the MS data are normalized and peptide EICs are translated into relative protein abundances. Increasing concentrations of competing free antibody in the samples result in fewer amounts of target proteins captured from the mixture by the immobilized antibody. Therefore, by plotting the relative abundance of the target protein from the bound fraction versus the increasing amounts of free target-specific antibody, a concentration-dependent displacement profile can be generated. We hypothesized that any protein specifically associated with the target protein in a complex would behave similarly, and we applied a statistical model (20, 22, 27) to analyze each protein's displacement profile. Compared with classical binary comparison, this gradual increase of free antibody concentrations provides additional support for efficient elimination of nonspecific binders and isolation of the true interactors.
Fig. 1.
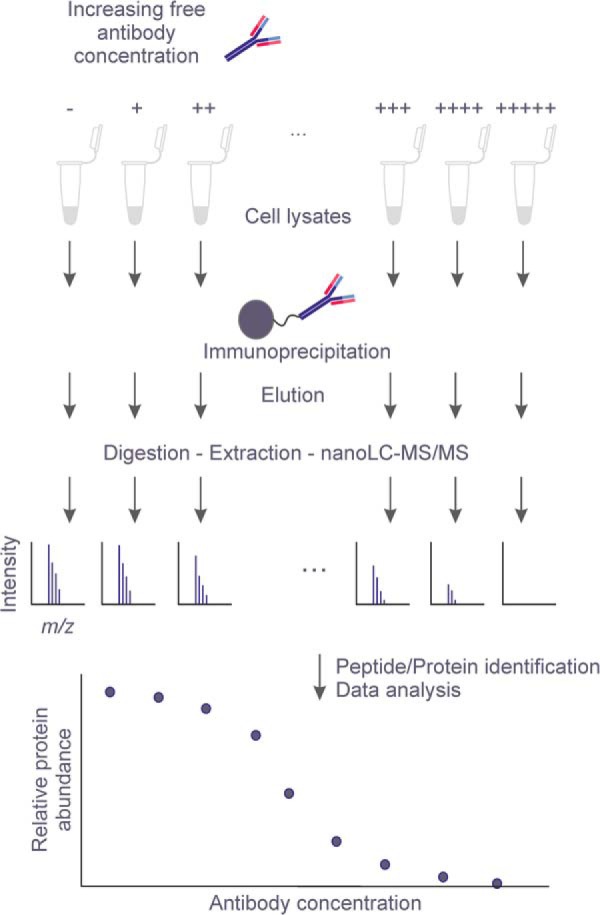
Overview of the label-free quantitative immuno-competitive capture mass spectrometry (ICC-MS) experimental procedure for the detection of protein interactors. Cell lysates are first pre-incubated with increasing concentrations of free antibody and then individually immunoprecipitated using the same antibody immobilized on agarose beads. After discarding the unbound fractions and performing several washing steps, the eluted fractions are separated by SDS-PAGE, proteins digested with trypsin, and extracted peptides analyzed by LC-MS/MS on an Orbitrap. Proteins are quantified on the basis of their corresponding peptide EICs and protein signals are subjected to a statistical analysis in order to derive the concentration-dependent signal decrease of the specific interactors.
Identification of HCV NS5A Interactors Using the ICC-MS Approach in Con1-Huh7 Cells
We applied this new strategy to the analysis of the NS5A interactome in an HCV subgenomic replicon system, a stable Con1 (genotype 1b) replicon cell line derived from human hepatoma Huh7 cells (Con1-Huh7). This in vitro model expresses the nonstructural HCV proteins NS3/NS4A, NS4B, NS5A, and NS5B at a physiologically relevant level and encompasses all steps involved in viral RNA replication (16). We first used Con1-Huh7 cell lysate to confirm by immunoblot that the NS5A-specific antibody cross-linked to the agarose beads was able to selectively and efficiently pull down NS5A (supplemental Fig. S1). We further observed that a pre-incubation of the lysate with 100 μg/ml of NS5A-specific antibody resulted in a near-complete disappearance of the signal corresponding to the immuno-precipitated NS5A protein (supplemental Fig. S2). These results confirmed the specific capture of NS5A by the immobilized antibody and efficient competition by the free antibody.
To generate well-resolved displacement profiles, the Con1-Huh7 cell lysate was divided into 12 equal aliquots that were then pre-incubated with 12 increasing concentrations of free antibody to NS5A with an equal dilution factor of 1.7. As initially postulated, we observed a dose-dependent inhibition of NS5A protein capture by the immobilized antibody (Fig. 2A). The largest drop of signal occurred between 1.7 and 2.9 μg/ml of free NS5A antibody and only minimal signal was observed at the highest concentrations. We analyzed the same samples by nanoLC-MS to detect NS5A co-immunoprecipitated proteins. Out of the entire sample set, we initially identified and quantified a total of 769 proteins (i.e. protein groups representing 1116 individual proteins and splice variants) in the NS5A-specific antibody bound fraction (supplemental Table S1). By adopting a robust statistical approach analyzing dose–response relationship in the entire dataset, the target protein NS5A appeared as the best hit (adjusted p value of 6.7 × 10−6) with a displacement curve showing a low technical variance for replicate analyses and an excellent correlation with the immunoblot signals (Fig. 2B, supplemental Table S2). As before, the largest drop of the NS5A signal occurred between 1.7 and 2.9 μg/ml. This result is validating our ICC-MS approach for the target NS5A and is confirming that the label-free quantitative MS is a reliable readout.
Fig. 2.
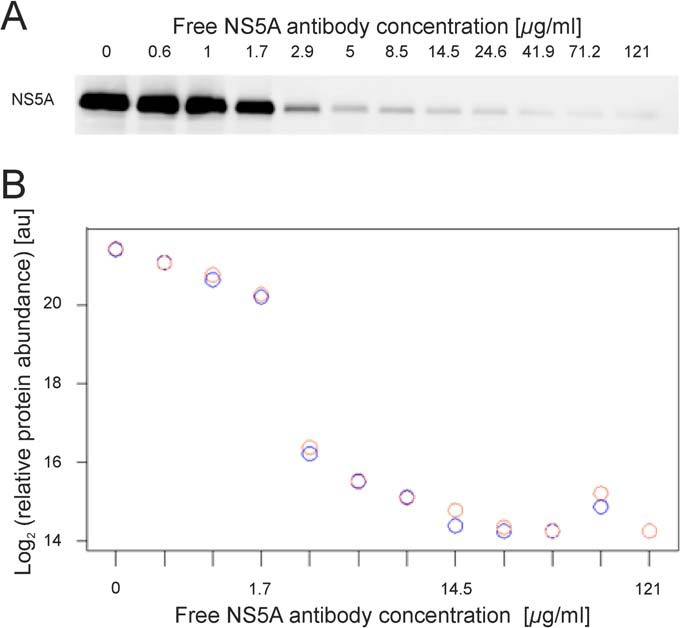
Concentration-dependent decrease of the NS5A capture by applying ICC-MS. A. NS5A capture from Con1-Huh7 cell lysates pre-incubated with 0, 0.6, 1, 1.7, 2.9, 5, 8.5, 14.5, 24.6, 41.9, 71.2, and 121 μg/ml of free NS5A antibody was probed by Western blot analysis. NS5A IP gradually decreased due to pre-incubation of the cell lysates with increasing concentration of the free form of the antibody. B. NS5A from each immunoprecipitate was identified and quantified by LC-MS. The relative abundance was plotted on the y axis and the free NS5A antibody concentrations on the x axis. Replicates are indicated by red and blue circles.
In addition to NS5A, the free-antibody competition strategy led to significant displacement of 10 host proteins (NP1L4, PKHG2, FBW1B, NP1L1, PI4KA, UBP19, VAPA, LATS1, LATS2, and PGAM5) with adjusted p values below 5% (Fig. 3). Two of them, PI4KA and VAPA, are already well-known NS5A interacting proteins from literature, validating our approach for specific binder identification (31, 32). All candidates proposed were verified by a 1% specFDR database search (supplemental Table S3). To validate that the 10 candidates mentioned above were pulled-down in a complex with NS5A and not through direct binding to the antibody, we used the H26 capture antibody for Western blot detection on Con1-Huh7 crude lysate (supplemental Fig. S3A). We confirmed that H26 antibody mainly detects NS5A with one extra weak band around 45 kDa. An additional Western blotting using the recombinant proteins of NP1L1 and NP1L4 (the only candidates with a molecular weight near 45 kDa) confirmed that there is no cross-reactivity of these two proteins with the capture antibody (supplemental Fig. S3B).
Fig. 3.
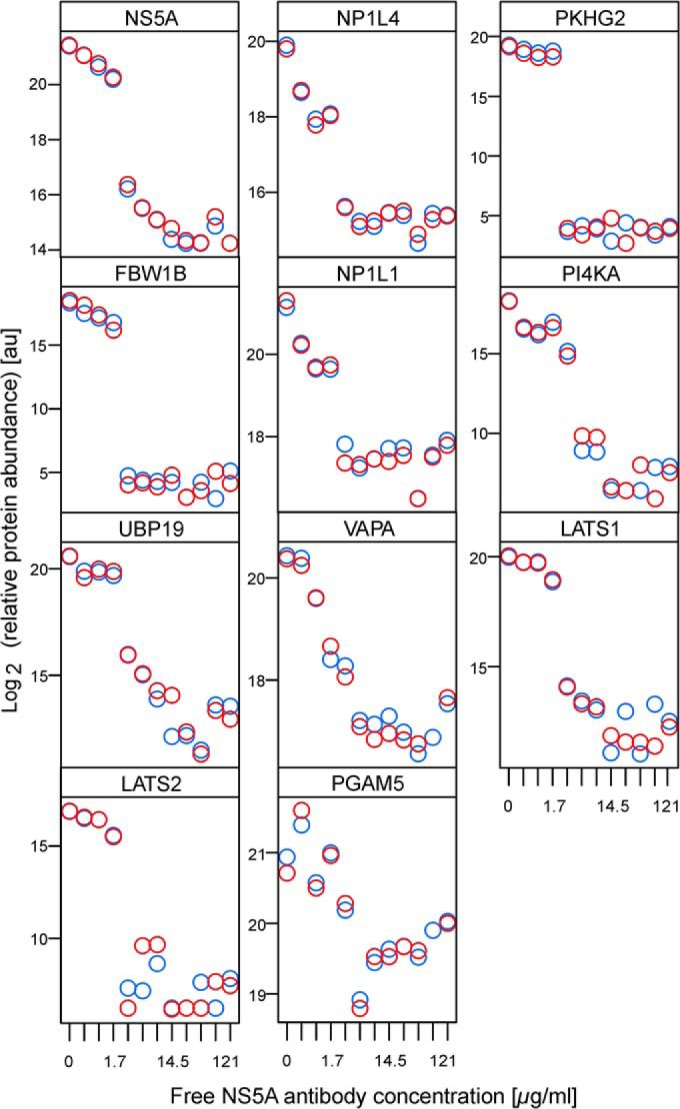
Concentration-dependent decreases of the 10 statistically significant NS5A interactors identified by ICC-MS. Candidates are ranked by increasing adjusted p value. Replicates are indicated by red and blue circles.
We also performed a mock immuno-capture in the Huh7 parental cell line that does not express NS5A. PKHG2, FBW1B, PI4KA, UBP19, NP1L4, LATS1, and LATS2 were absent from the bound fraction, therefore confirming that these proteins are binding NS5A and not directly the capture antibody. NP1L1, VAPA, and PGAM5 were identified in the bound fraction from Huh7 cell lysates, but their capture was independent of the concentration of free competing antibody (supplemental Fig. S4). Using uncoated beads to perform pull down from Con1-Huh7 lysates revealed that these three proteins may bind to the agarose beads at low level (supplemental Fig. S5). The fact that NP1L1 and VAPA are significantly enriched when using the H26-antibody-coupled beads and can only be displaced by the competing antibody in a dose-dependent manner in the presence of NS5A supports their candidacy as specific NS5A-interactors. On the contrary, all of the previous candidates except PGAM5 have over 80% signal reduction between the highest and the lowest concentration of competitor antibody (supplemental Table S2). We therefore performed transient transfection of tagged PGAM5 in Con1-Huh7 cells but could not detect NS5A after immunoprecipitation (supplemental Fig. S6), thereby confirming PGAM5 as a false positive.
LATS2 Kinase Phosphorylates NS5A at Ser71 in Vitro
Among the identified candidate interacting partners, LATS1 and LATS2, two members of the nuclear Dbf2-related (NDR) family of AGC kinases, were of particular interest (33, 34). These two Ser/Thr kinases bind and phosphorylate the specific substrate consensus sequence His-X-Arg/His/Lys-X-X-Ser/Thr (35, 36). This motif is present in domain 1 of NS5A (His65 to Ser71) and is highly conserved across all HCV viral genotypes (supplemental Table S4). By performing an immunoblot against LATS1 and LATS2 on the different elution fractions, we confirmed the MS observation that both kinases specifically bind to NS5A (supplemental Fig. S7A and B). Interactions between NS5A and LATS kinases were further validated by immunoprecipitating LATS1 and LATS2 (supplemental Fig. S7C). To test whether NS5A is indeed a substrate of the LATS kinases, we performed in vitro phosphorylation using purified recombinant NS5A derived from E. coli (NS5AΔ32, a truncated version of NS5A spanning residues 33–447) with catalytically active recombinant LATS2. The level of phosphorylated NS5A significantly increased upon incubation with the kinase, suggesting that NS5A is a bona fide substrate of LATS2 (Fig. 4A and B).
Fig. 4.
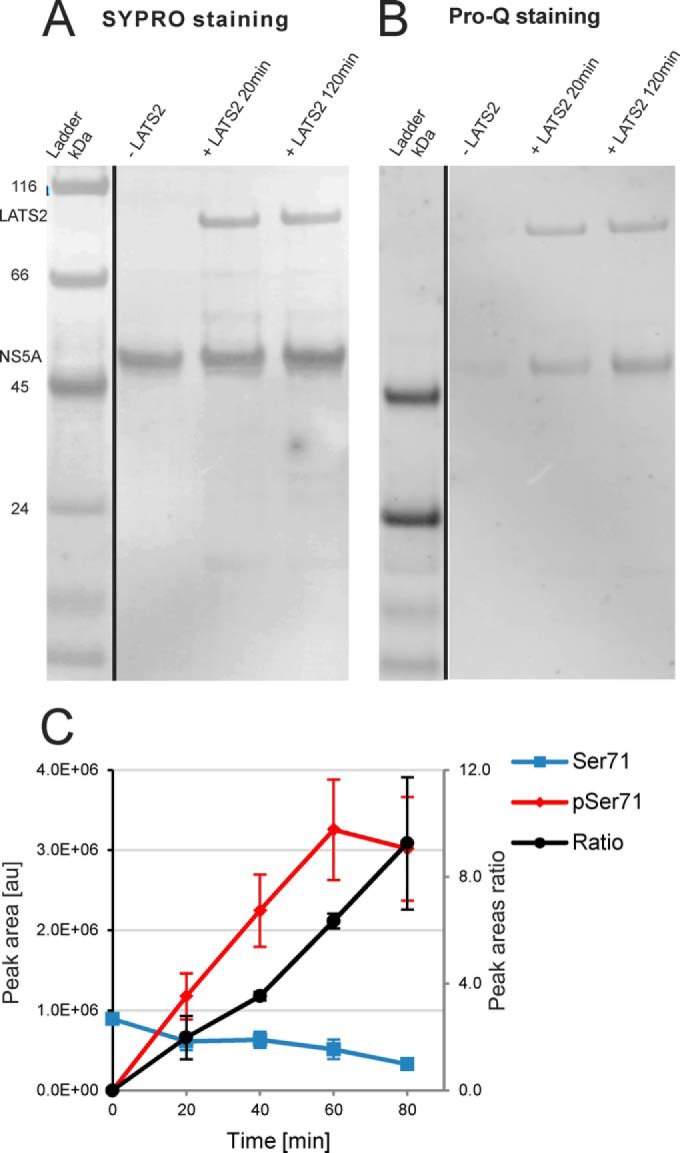
In vitro phosphorylation of NS5A by LATS2. A. Total protein (SYPRO) and B. phosphoprotein (Pro-Q Diamond) staining of the SDS-PAGE separated reaction mixtures. We used NS5A Δ32 (residues 33–447) recombinant form of the protein detected at ca. 50 kDa. We clearly observed phosphorylation of NS5A after 20 min treatment with the kinase. C. Relative quantitation of Ser71 phosphorylation by LC-MS. Ratios between phosphorylated and un-phosphorylated NGSMRIVGPK peptide EICs are represented. Error bars, S.E. (n = 2).
To define the NS5A phosphorylation sites targeted by LATS2, we repeated the same experiment and analyzed the samples by LC-MS. NS5A was detected with 32 unique peptides corresponding to 55% sequence coverage. The phosphopeptide 69NGpSMRIVGPK78 was clearly identified and the phosphorylation site unambiguously localized to Ser71 (supplemental Fig. S8). We performed semiquantitative assessment of the phosphorylation stoichiometry by comparing the peak areas (37) of both the unmodified and the modified peptide (Fig. 4C, supplemental Table S5). The kinetic analysis shows that the phosphorylated 69NGSMRIVGPK78 is approximately nine times more abundant than its unmodified counterpart after 80 min of reaction and clearly supports LATS2-mediated NS5A Ser71 phosphorylation. We identified four additional phosphopeptides, but as their respective phosphorylation stoichiometry ratios were considerably smaller (in the range of 0.02) and also did not contain the sequence motif recognized by the kinase, they were not considered as LATS2 specific (data not shown).
Ser71Ala Mutation Reduces HCV Replication Capacity
Phosphorylation of NS5A (38) is known to modulate its protein interactions, as well as to affect HCV RNA replication (39, 40). To investigate the role of NS5A phosphorylation at residue 71 in HCV RNA replication, a point mutation (Ser-to-Ala) at this position was engineered by site-directed mutagenesis in the wild type transient genotype 1b Con1 replicon. Replicon RNAs were transiently transfected into Lunet Huh7 cells, and their replication levels were measured as described previously (41). The Ser71Ala mutant replicon showed a replication capacity of 14% compared with the wild type replicon replication, suggesting that prevention of phosphorylation at residue 71 of NS5A significantly impairs viral replication (Fig. 5).
Fig. 5.

Replication capacity of mutant transient replicon Con1-NS5A-Ser71Ala. The replication capacity of the mutant transient replicon Con1-NS5A-Ser71Ala was expressed as its normalized replication efficiency compared with that of the reference strain Con1 that is set at a value of 100%. Mean and standard deviation were obtained from four independent experiments.
LATS1 and 2 Silencing Inhibits HCV Genome Replication
siRNA molecules targeting the LATS1 and LATS2 genes were used to evaluate the potential role of the LATS kinases in the HCV replication cycle. As positive controls, the Con1 HCV replicon sequence (located in the NS4B gene) (42) and the PI4KA gene (43), known to be important for the HCV replication, were targeted by specific siRNAs. The knockdown level for each siRNA used in the study was first evaluated by real-time PCR (Fig. 6A). Con1, PI4K, and LATS2 siRNAs reduced the mRNA transcription level of their targets by 91%, 91%, and 80%, respectively; whereas LATS1 siRNA reduced the LATS1 mRNA level by 51%.
Fig. 6.
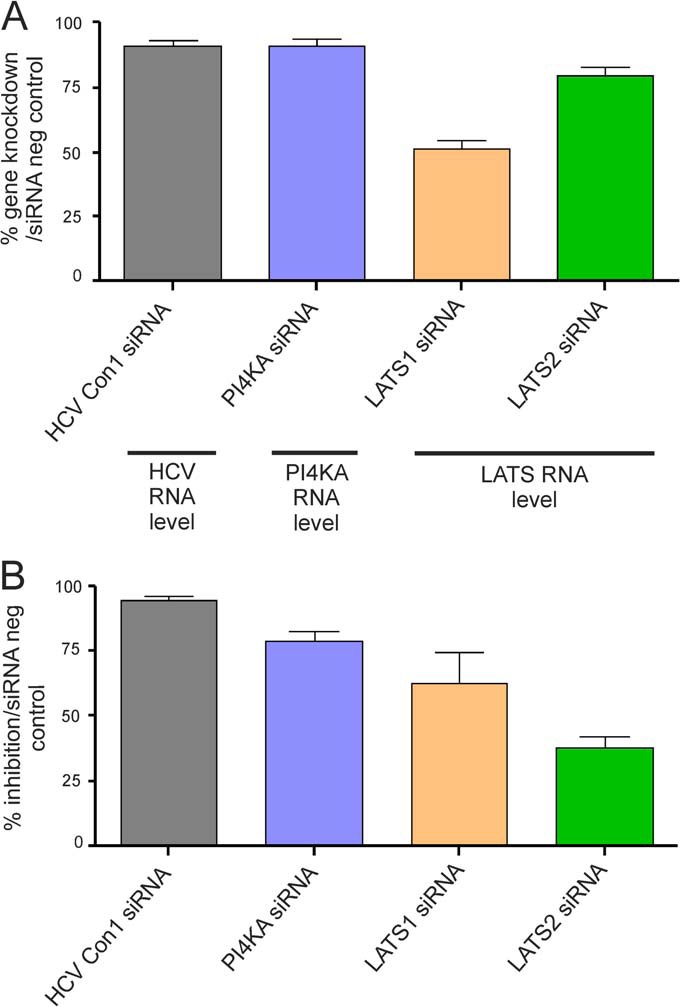
Effect of siRNA knockdown on HCV replicon replication. A. Gene knockdown after Con1, PI4KA, LATS1, or LATS2 siRNA transfection. Knockdown was evaluated by real-time PCR and normalized to negative control siRNAs. Mean and standard deviation were calculated from five to nine independent experiments. B. Inhibition of HCV replicon renilla luciferase signal by siRNA targeting either Con1, PI4KA, LATS1, or LATS2. The renilla luciferase signal for each gene-specific siRNA experiment was normalized to the one obtained using negative control siRNA (siRNA Neg Control). Mean and standard deviation were calculated from 8 to 12 independent experiments.
The effect of siRNA knockdown on HCV replication was evaluated by measuring the HCV replicon renilla luciferase signal. As previously described, the siRNAs targeting either Con1 or PI4KA efficiently decreased the renilla luciferase signal by 94% and 79% respectively, as compared with the siRNA negative control (Fig. 6B) (40, 41). The siRNAs targeting LATS1 and the siRNAs targeting LATS2 inhibited the signal by 63% and 38%, respectively (Fig. 6B), suggesting a role of LATS1 and LATS2 kinases in the HCV life cycle, which is likely through phosphorylation of NS5A.
DISCUSSION
Understanding the biological function of a protein often relies on the identification of its interactome in a cellular context. The ICC-MS approach described in this study can generate highly specific protein–protein interaction maps. We show the first application of this new strategy by elucidating the HCV NS5A interactome in human hepatoma cells. We demonstrated that the free antibody can efficiently compete with its immobilized counterpart for the capture of NS5A. The extent of displacement correlates with the MS signals in a dose-dependent manner in accordance with conventional immunoblotting techniques. This approach revealed 10 specific dose-dependent profiles out of the 769 proteins initially identified in the elution fractions, with an adjusted p value ≤ 0.05. The invalidation of PGAM5 showed that the percentage of signal reduction should also be taken into account. Moreover, all candidates meeting these two selection criteria (an adjusted p value ≤ 0.05 and a percentage of signal reduction ≥ 80%) should be subjected to biological validation in order to be considered as functional NS5A interactors. Because functional validation could be time consuming and in some cases technically challenging, our goal of developing the ICC-MS approach was to provide a manageably short list of proteins, which have a high chance to be validated biologically. In our study, four candidates have been shown to be biologically relevant NS5A-interacting proteins. Phosphatidylinositol 4-kinase III alpha (PI4KA) and 33 kDa vesicle-associated membrane protein-associated protein A (VAPA), are well-known NS5A-interacting proteins, validating the approach. PI4KA has been previously described as being activated by NS5A through direct interaction and is required for optimal HCV replication (31). VAPA has been shown to bind NS5A and to colocalize with the viral protein in the membrane lipid raft, thereby contributing to the formation of the HCV replication complex (32).
LATS1 and LATS2 kinases are identified for the first time as potential NS5A-interacting proteins. Although NS5A phosphorylation at various sites has been reported previously, it was often under conditions of NS5A overexpression while viral genome replication was absent (for a review see (38)). In addition, the upstream host kinase(s) responsible for phosphorylating these sites remain largely unknown (except casein kinase I and II (44–47)). In our follow-up studies, we demonstrated that LATS2 can phosphorylate NS5A at residue Ser71 in vitro. Furthermore, we showed that preventing the phosphorylation at this position by site-directed mutagenesis impairs viral genome replication in cells. This is consistent with the observation that siRNA knockdown of LATS1/2 in replicon cells reduces viral replication in this system. Altogether, our results strongly suggest that LATS kinases play an important role in the regulation of NS5A function through site-specific post-translational modification and that phosphorylation of Ser/Thr71 is essential for optimal viral genome replication. Whereas further investigations are needed to fully define the respective roles of each kinase, LATS1 and LATS2 could be potential new host targets for developing HCV therapeutics.
Further examination, e.g. by siRNA knockdown, would be needed to validate the other newly identified NS5A-interacting proteins found by ICC-MS (NP1L4, PKHG2, FBW1B, NP1L1, and UBP19). Their respective biological relevance to the function of NS5A is a priori less obvious than LATS kinases but could eventually lead to a better understanding of the various roles that HCV NS5A plays during the viral life cycle.
This first application of the ICC-MS approach highlighted several advantages for determining the interactome of a protein of interest. It has exquisite specificity enabling the identification of the true interactors and not nonspecific, highly abundant proteins. The approach results in a handful of potential interactors rather than hundreds of candidates and so time and cost for validation can be significantly reduced. Unlike a simple binary comparison method, the ICC-MS approach does not exclude a protein from the analysis even if it shows some level binding to the agarose beads. VAPA is a good example of a validated NS5A interactor that would have been excluded from further validation if the candidate selection was solely based on absence from the agarose bead proteome. Moreover, ICC-MS is technically straightforward and broadly applicable. It does not require protein tagging or silencing, nor sample labeling, and can be directly applied to endogenous proteins from cell lines and even tissues. The number of antibody concentrations as well as the number of replicates can be fine-tuned for each particular experiment. A successful ICC-MS, like other standard pull-down experiments, relies on antibody availability and specificity. The outcome of this profiling exercise may vary depending on the particular epitope of the antibody. Using multiple antibodies directed against different epitopes on the target protein will likely increase the coverage of the full interactome. It also should be noted that fast, transient interactions or low abundant interacting proteins might not be detected with the current approach. Therefore, it is critical to estimate a priori the yield of the immunocapture to ensure a proper enrichment of the target protein by the immobilized antibody.
In conclusion, we believe that the combination of immuno-competition and quantitative mass spectrometry is a powerful tool for studying biologically relevant protein–protein interactions. Furthermore, we expect that a broad application of this method together with additional available approaches will help to improve the accuracy and specificity of current protein–protein interaction databases.
Acknowledgments
We would like to thank Michel Petrovic for his support during data processing. We also would like to thank Hassan Javanbakht and Han Ma for critical suggestions and discussion, as well as Corinne Ploix, Norman Mazer, and Hans Bitter for their thorough reading of the manuscript.
Footnotes
Author contributions: H.M., J.G., and A.A. designed research; H.M., J.G., S.G., S.L., and S.S. performed research; H.M., J.G., J.L., S.L., and M.T. analyzed data; H.M. and J.G. wrote the paper; H.M. first author; J.G. cofirst author; J.L., S.L., and M.T. revised the manuscript; L.G., I.N., H.L., K.K., and A.A. provided support and revised the manuscript.
 This article contains supplemental material.
This article contains supplemental material.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental Figs. S1–S8 and Tables S1–S5.
1 The abbreviations used are:
- AP
- Affinity purification
- EICs
- Extracted ion chromatograms
- HCV
- Hepatitis C virus
- ICC-MS
- Immuno-competitive capture mass spectrometry
- IP
- Immunoprecipitation
- LATS
- Large tumor suppressor
- NDR
- Nuclear Dbf2-related family
- NS5A
- Nonstructural protein 5A
- PCA
- Principal component analysis
- RMA
- Robust multichip analysis
- specFDR
- Spectral false discovery rate.
REFERENCES
- 1. Braun P., Gingras A. C. (2012) History of protein–protein interactions: From egg-white to complex networks. Proteomics. 12, 1478–1498 [DOI] [PubMed] [Google Scholar]
- 2. Berggard T., Linse S., James P. (2007) Methods for the detection and analysis of protein–protein interactions. Proteomics. 7, 2833–2842 [DOI] [PubMed] [Google Scholar]
- 3. Köcher T., Superti-Furga G. (2007) Mass spectrometry-based functional proteomics: from molecular machines to protein networks. Nat. Methods. 4, 807–815 [DOI] [PubMed] [Google Scholar]
- 4. Dunham W. H., Mullin M., Gingras A. C. (2012) Affinity-purification coupled to mass spectrometry: Basic principles and strategies. Proteomics. 12, 1576–1590 [DOI] [PubMed] [Google Scholar]
- 5. Gingras A. C., Gstaiger M., Raught B., Aebersold R. (2007) Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8, 645–654 [DOI] [PubMed] [Google Scholar]
- 6. Trinkle-Mulcahy L., Boulon S., Lam Y. W., Urcia R., Boisvert F. M., Vandermoere F., Morrice N. A., Swift S., Rothbauer U., Leonhardt H., Lamond A. (2008) Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 183, 223–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paul F. E., Hosp F., Selbach M. (2011) Analyzing protein–protein interactions by quantitative mass spectrometry. Methods. 54, 387–395 [DOI] [PubMed] [Google Scholar]
- 8. Vermeulen M., Hubner N. C., Mann M. (2008) High confidence determination of specific protein–protein interactions using quantitative mass spectrometry. Curr. Opin. Biotechnol. 19, 331–337 [DOI] [PubMed] [Google Scholar]
- 9. Trinkle-Mulcahy L. (2012) Resolving protein interactions and complexes by affinity purification followed by label-based quantitative mass spectrometry. Proteomics. 12, 1623–1638 [DOI] [PubMed] [Google Scholar]
- 10. Pardo M., Choudhary J. S. (2012) Assignment of protein interactions from affinity purification/mass spectrometry data. J. Proteome Res. 11, 1462–1474 [DOI] [PubMed] [Google Scholar]
- 11. Nesvizhskii A. I. (2012) Computational and informatics strategies for identification of specific protein interaction partners in affinity purification mass spectrometry experiments. Proteomics. 12, 1639–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rinner O., Mueller L. N., Hubalek M., Muller M., Gstaiger M., Aebersold R. (2007) An integrated mass spectrometric and computational framework for the analysis of protein interaction networks. Nat. Biotechnol. 25, 345–352 [DOI] [PubMed] [Google Scholar]
- 13. He Y., Staschke K. A., Tan S. L. (2006) HCV NS5A: A Multifunctional Regulator of Cellular Pathways and Virus Replication in Hepatitis C Viruses: Genomes and Molecular Biology, pp. 267–292, Horizon Bioscience, Norfolk, UK: [PubMed] [Google Scholar]
- 14. Ahn J., Chung K. S., Kim D. U., Won M., Kim L., Kim K. S., Nam M., Choi S. J., Kim H. C., Yoon M., Chae S. K., Hoe K. L. (2004) Systematic identification of hepatocellular proteins interacting with NS5A of the hepatitis C virus. J. Biochem. Mol. Biol. 37, 741–748 [DOI] [PubMed] [Google Scholar]
- 15. Macdonald A., Harris M. (2004) Hepatitis C virus NS5A: Tales of a promiscuous protein. J. Gen. Virol. 85, 2485–2502 [DOI] [PubMed] [Google Scholar]
- 16. Lohmann V., Hoffmann S., Herian U., Penin F., Bartenschlager R. (2003) Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77, 3007–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klumpp K., Leveque V., Le Pogam S., Ma H., Jiang W. R., Kang H., Granycome C., Singer M., Laxton C., Hang J. Q., Sarma K., Smith D. B., Heindl D., Hobbs C. J., Merrett J. H., Symons J., Cammack N., Martin J. A., Devos R., Najera I. (2006) The novel nucleoside analog R1479 (4′-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J. Biol. Chem. 281, 3793–3799 [DOI] [PubMed] [Google Scholar]
- 18. Olsen J. V., de Godoy L. M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics. 4, 2010–2021 [DOI] [PubMed] [Google Scholar]
- 19. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 20. Augustin A., Lamerz J., Meistermann H., Golling S., Scheiblich S., Hermann J. C., Duchateau-Nguyen G., Tzouros M., Avila D. W., Langen H., Essioux L., Klughammer B. (2013) Quantitative chemical proteomics profiling differentiates erlotinib from gefitinib in EGFR wild-type non-small cell lung carcinoma cell lines. Mol. Cancer Therapeut. Ahead of print 10.1158/1535–7163.MCT-1112–0880; [DOI] [PubMed] [Google Scholar]
- 21. Smyth G. K. (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 [DOI] [PubMed] [Google Scholar]
- 22. Wu Z., Doondeea J. B., Gholami A. M., Janning M. C., Lemeer S., Kramer K., Eccles S. A., Gollin S. M., Grenman R., Walch A., Feller S. M., Kuster B. (2011) Quantitative chemical proteomics reveals new potential drug targets in head and neck cancer. Mol. Cell. Proteomics. 10, M111.011635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oberg A. L., Mahoney D. W., Eckel-Passow J. E., Malone C. J., Wolfinger R. D., Hill E. G., Cooper L. T., Onuma O. K., Spiro C., Therneau T. M., Bergen H. R., 3rd. (2008) Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J. Proteome Res. 7, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emerson J. D., Hoaglin D. C. (1983) Analysis of two-tables by median in Understanding Robust and Exploratory Data Analysis, pp. 166–206, John Wiley & Sons Inc., New York [Google Scholar]
- 25. Irizarry R. A., Hobbs B., Collin F., Beazer-Barclay Y. D., Antonellis K. J., Scherf U., Speed T. P. (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 4, 249–264 [DOI] [PubMed] [Google Scholar]
- 26. Stewart W. H., Ruberg S. J. (2000) Detecting dose response with contrasts. Stat. Med. 19, 913–921 [DOI] [PubMed] [Google Scholar]
- 27. Bretz F., Hothorn L. A. (2001) Testing dose-response relationships with a priori unknown, possibly nonmonotone shapes. J. Biopharm. Stat. 11, 193–207 [DOI] [PubMed] [Google Scholar]
- 28. Ge Y., Dudoit S., Speed T. P. (2003) Resampling-based multiple testing for microarray data hypothesis. Test. 12, 1–44 [Google Scholar]
- 29. Westfall P. H., Young S. S. (1993) Resampling-Based Multiple Testing: Examples and Methods for p-Value Adjustment, John Wiley & Sons Inc., New York [Google Scholar]
- 30. Huang L., Sineva E. V., Hargittai M. R., Sharma S. D., Suthar M., Raney K. D., Cameron C. E. (2004) Purification and characterization of hepatitis C virus non-structural protein 5A expressed in Escherichia coli. Protein Expres. Purif. 37, 144–153 [DOI] [PubMed] [Google Scholar]
- 31. Reiss S., Rebhan I., Backes P., Romero-Brey I., Erfle H., Matula P., Kaderali L., Poenisch M., Blankenburg H., Hiet M. S., Longerich T., Diehl S., Ramirez F., Balla T., Rohr K., Kaul A., Buhler S., Pepperkok R., Lengauer T., Albrecht M., Eils R., Schirmacher P., Lohmann V., Bartenschlager R. (2011) Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 9, 32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao L., Aizaki H., He J. W., Lai M. M. (2004) Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 78, 3480–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pearce L. R., Komander D., Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 34. Visser S., Yang X. (2010) LATS tumor suppressor: A new governor of cellular homeostasis. Cell Cycle. 9, 3892–3903 [DOI] [PubMed] [Google Scholar]
- 35. Zhao B., Wei X., Li W., Udan R. S., Yang Q., Kim J., Xie J., Ikenoue T., Yu J., Li L., Zheng P., Ye K., Chinnaiyan A., Halder G., Lai Z. C., Guan K. L. (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lei Q. Y., Zhang H., Zhao B., Zha Z. Y., Bai F., Pei X. H., Zhao S., Xiong Y., Guan K. L. (2008) TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell. Biol. 28, 2426–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Steen H., Jebanathirajah J. A., Springer M., Kirschner M. W. (2005) Stable isotope-free relative and absolute quantitation of protein phosphorylation stoichiometry by MS. Proc. Natl. Acad. Sci. U.S.A. 102, 3948–3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanji Y., Kaneko T., Satoh S., Shimotohno K. (1995) Phosphorylation of hepatitis C virus-encoded nonstructural protein NS5A. J. Virol. 69, 3980–3986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Evans M. J., Rice C. M., Goff S. P. (2004) Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. U.S.A. 101, 13038–13043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang Y., Staschke K., De Francesco R., Tan S. L. (2007) Phosphorylation of hepatitis C virus NS5A nonstructural protein: A new paradigm for phosphorylation-dependent viral RNA replication? Virology. 364, 1–9 [DOI] [PubMed] [Google Scholar]
- 41. Le Pogam S., Jiang W. R., Leveque V., Rajyaguru S., Ma H., Kang H., Jiang S., Singer M., Ali S., Klumpp K., Smith D., Symons J., Cammack N., Najera I. (2006) In vitro selected Con1 subgenomic replicons resistant to 2′-C-methyl-cytidine or to R1479 show lack of cross resistance. Virology. 351, 349–359 [DOI] [PubMed] [Google Scholar]
- 42. Randall G., Grakoui A., Rice C. M. (2003) Clearance of replicating hepatitis C virus replicon RNAs in cell culture by small interfering RNAs. Proc. Natl. Acad. Sci. U.S.A. 100, 235–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Berger K. L., Cooper J. D., Heaton N. S., Yoon R., Oakland T. E., Jordan T. X., Mateu G., Grakoui A., Randall G. (2009) Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. U.S.A. 106, 7577–7582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang L., Hwang J., Sharma S. D., Hargittai M. R., Chen Y., Arnold J. J., Raney K. D., Cameron C. E. (2005) Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J. Biol. Chem. 280, 36417–36428 [DOI] [PubMed] [Google Scholar]
- 45. Kim J., Lee D., Choe J. (1999) Hepatitis C virus NS5A protein is phosphorylated by casein kinase II. Biochem. Biophys. Res. Commun. 257, 777–781 [DOI] [PubMed] [Google Scholar]
- 46. Dal Pero F., Di Maira G., Marin O., Bortoletto G., Pinna L. A., Alberti A., Ruzzene M., Gerotto M. (2007) Heterogeneity of CK2 phosphorylation sites in the NS5A protein of different hepatitis C virus genotypes. J. Hepatol. 47, 768–776 [DOI] [PubMed] [Google Scholar]
- 47. Quintavalle M., Sambucini S., Summa V., Orsatti L., Talamo F., De Francesco R., Neddermann P. (2007) Hepatitis C virus NS5A is a direct substrate of casein kinase I-alpha, a cellular kinase identified by inhibitor affinity chromatography using specific NS5A hyperphosphorylation inhibitors. J. Biol. Chem. 282, 5536–5544 [DOI] [PubMed] [Google Scholar]