

Abstract
Isobaric tags for relative and absolute quantitation (iTRAQ) is a prominent mass spectrometry technology for protein identification and quantification that is capable of analyzing multiple samples in a single experiment. Frequently, iTRAQ experiments are carried out using an aliquot from a pool of all samples, or “masterpool”, in one of the channels as a reference sample standard to estimate protein relative abundances in the biological samples and to combine abundance estimates from multiple experiments. In this manuscript, we show that using a masterpool is counterproductive. We obtain more precise estimates of protein relative abundance by using the available biological data instead of the masterpool and do not need to occupy a channel that could otherwise be used for another biological sample. In addition, we introduce a simple statistical method to associate proteomic data from multiple iTRAQ experiments with a numeric response and show that this approach is more powerful than the conventionally employed masterpool-based approach. We illustrate our methods using data from four replicate iTRAQ experiments on aliquots of the same pool of plasma samples and from a 406-sample project designed to identify plasma proteins that covary with nutrient concentrations in chronically undernourished children from South Asia.
Keywords: Mass spectrometry, iTRAQ, statistical analysis, experimental design
INTRODUCTION
A common reference standard is frequently employed in quantitative proteomic studies to quantify differences in protein expression between individual samples. The standard sample is typically a pool of an equal amount from each sample in the study and can be referred to as the “masterpool”. If the exact protein concentrations in the masterpool were known, then in mass tags techniques such as isobaric tags for relative and absolute quantitation iTRAQ 1–5 which compares up to eight samples in a single experiment, the masterpool protein concentrations could be used to compare protein abundances among the biological samples within the experiment and also among experiments. Estimates of protein relative abundance could be calculated with reference to the same standard, and the estimates could simply be combined across experiments. However, the exact masterpool protein concentrations are typically unknown. Because the relative protein concentrations in the masterpool have to be estimated from the experimental data, they are subject to variability. Although typically ignored, this variability in the masterpool protein concentrations affects the precision of the relative abundance estimates within an experiment6 and raises the question how to address this source of unwanted variability among experiments.
In multiplexing iTRAQ experiments, differences in a protein’s expression among samples is determined from comparing intensities of the eight different iTRAQ reporter ions, representing the eight different samples, in the MS/MS peptide fragmentation spectra.7 When using masterpools, the reporter ion intensities of the biological samples are divided by the reporter ion intensity from the masterpool sample for each peptide spectrum used for quantification. The estimate of relative abundance of a particular protein is typically calculated as the median of the reporter ion ratios from all peptide spectra belonging to the protein, as implemented, for example, in the Proteome Discoverer software (Thermo Scientific). Corrections for differences in the amounts of material loaded in the channels or differences due to sample processing can also be implemented: dividing each protein relative abundance estimate by the channel median relative abundance (the median of all relative protein abundances within a channel) normalizes all channels to have median one. Equivalently, on the logarithmic scale, subtracting the channel median log2 relative abundance from each log2 relative abundance estimate normalizes all channels to have median zero. The fact that all estimates stemming from the reference standard channel are also subject to experimental noise can result in highly variable estimates when ratios are calculated. Denoting the reporter ion intensity of a biological sample by ŶS and the reporter ion intensity of the reference sample by ŶR, the variability in the ratio of these intensities can be approximated via a first-order Taylor expansion,8 yielding
| (1) |
This term indicates that the variability in reporter ion intensity ratios is particularly susceptible to the variability in the estimates in the denominator (i.e., the reference channel). Thus, using less variable references, such as the mean or median of the reporter ion intensities observed in the biological samples, can provide more precise estimates of protein relative abundances.
In this manuscript, we are mainly concerned with how proteomic measurements covary with a numeric response, specifically, plasma micronutrient concentrations. We introduce a simple and scalable statistical method to associate quantitative proteomic data from multiple iTRAQ experiments with numeric outcomes, but also discuss that case-control data can easily be analyzed in this framework assuming proper experimental design. We show that our method is more powerful than conventional approaches that rely on masterpool or standard reference samples. The method yields more precise estimates of protein relative abundance in a single iTRAQ experiment and improves inference when multiple iTRAQ experiments are performed. We illustrate the performance of our methods using data from four replicate iTRAQ experiments on aliquots of a masterpool sample and from a 406-sample project designed to identify plasma proteins that covary with nutrient concentrations in chronically undernourished children from South Asia.
MATERIALS AND METHODS
Experimental Parameters
Eight-plex stable isotope masstags (iTRAQ) were employed to identify and quantify plasma proteins of 406 Nepalese children by high-throughput tandem mass spectrometry. Isotopically resolved masses in MS and MS/MS spectra were extracted with and without deconvolution using Thermo Scientific Xtract software. Both data sets were searched against the RefSeq 40 database using Mascot (Matrix Science) through Proteome Discoverer software (v1.3, Thermo Scientific), producing two mascot scores for each fragmentation spectrum. To avoid redundancies in peptide identification, only the highest of these two scores was used to identify the peptide and determine if the peptide identification passed the 5% false discovery rate filter, based on a concatenated decoy database search. To avoid redundancies in quantitation, only the reporter ions from the spectrum matched to peptide identifications with the highest Mascot score before or after deconvolution by the Xtract function were used for quantitation of reporter ion intensities. Only spectra with reporter ion intensities observed in all eight channels were used for peptide and protein quantitation. More details are given in the section Experimental Parameters in the Supporting Information.
Sample Description
The data stem from 1000 early-school-aged children, sampled from a micronutrient-deficient population typical of the Terai of rural Nepal.9 Parental consent was obtained prior to conducting child assessment procedures. The original field trial was registered with ClinicalTrials.gov (NCT00115271).10 The study protocol was reviewed and approved by institutional review boards at the Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA, and the Institute of Medicine, Tribhuvan University, Kathmandu, Nepal. A total of 20 nutrient indicators (including vitamins A, B6, B12, copper, iron, and zinc) and indicators of inflammation (including the C-reactive protein) are the outcomes of interest, measured for all 1000 children. To date, proteomic measurements are available for individual samples of 406 children and masterpools consisting of an equal amount of plasma from all 1000 children. A total of 58 iTRAQ experiments were run, each with seven biological samples and one masterpool aliquot randomly allocated to the eight channels, defined by isobaric tags, using a randomized block design (see the section Experimental Design in the Supporting Information). To assess technical variability, four “masterpool iTRAQ experiments” were carried out, each consisting of eight aliquots (technical replicates) of the plasma masterpool.
Metrics for Comparison
A logarithmic transformation of the reporter ion intensities is commonly employed because systematic effects and variance components are usually assumed to be additive on this scale.11,12 Thus, our calculations are carried out on the log2 scale in general, but for ease of interpretation, the findings are reported on the fold change scale where possible. Comparing different approaches for estimation of relative protein abundance in the four experiments with technical replicates of the masterpool, we know that, in truth, all protein abundances should be the same, that is, the relative abundances should all be equal to 1 (the log2 relative abundances should all be equal to 0). Thus, we assess the accuracy and precision by calculating root mean squared and median absolute fold changes. Specifically, for a set X1 …, Xn of fold changes, we calculate
| (2) |
and
| (3) |
to account for fold changes in both directions and to quantify “typical” deviations from the expected value of 1. In the data from the 58 iTRAQ experiments using plasma from 406 children, we also expect biological variability among the individual samples for any particular protein. For these samples, we use the concordance correlation coefficient to measure the agreement in relative abundance estimates between two approaches. The concordance correlation between two random variables X and Y is defined as
| (4) |
Unlike the Pearson correlation coefficient, it is not invariant to changes in location and scale and assesses the actual agreement between X and Y, rather then their correlation alone.
Estimable Parameters and Statistical Inference
Consider one particular protein, and let μ be the population mean for the log2 abundance (log2 absolute concentration) of this protein. If we select a random subset of eight subjects from this population, the average concentration in this sample will differ from the population mean, with a magnitude that depends on the biological variability across subjects in this population (i.e., a random effect). For this sample, denote Δ to be the shift from the population mean μ . In other words, the true sample mean is μ + Δ, and we assume that Δ ~ N(0, σΔ2). Further, let δ = (δ1, …, δ8) be the vector that indicates the deviation for each subject from this sample mean. Note that this implies Σk δk = 0. Thus, the true absolute protein abundance for subject k ∈ {1, …, 8} is ak = μ + Δ + δk. However, for each spectrum, s ∈ {1, …, S} of log2 reporter ion intensities, we observe only Ysk = Δ s + δk + εsk. Here, Δs is the average log2 ion intensity across all eight channels (the outcome of a random process, but considered fixed for any particular experiment), and we assume that ε ~ N(0, σ2) is random Gaussian noise. This implies that the absolute protein abundances ak are not estimable, as the observed data do not contain information about either μ or Δ.
Denote the average of the eight log2 reporter ion intensities for spectrum s as Ȳs = ∑k Ysk/8, with E[ȲS] = Δs (since ∑k δk= 0). Thus, for the “de-meaned” log2 reporter ion intensities Zsk = Ysk−Ȳs, we have E[Zsk] = E[Ysk] − E[ȲS] = δk. This implies that the relative abundances δk are estimable in each iTRAQ experiment. Also note that strictly speaking, the errors εsk − ε̄s for the demeaned reporter ion intensities Zsk are not independent (because they sum to 0), but instead can be described by an exchangeable variance-covariance matrix for the error term.
The question arises how relative abundance estimates from multiple iTRAQ runs can be combined. Assume that for each iTRAQ experiment, r ∈ {1, …, R}, we have estimates for δr = (δr1, …, δr8). The true log2 protein abundance for subject k in experiment r is given by ark = μ + Δ r + δrk; however, the among experiment variance component Δ r is not estimable from the proteomic data, so simply augmenting the δr across experiments as a surrogate for absolute abundance fails to take the variance component Δ r into account. Note that randomization of subjects to experiments helps to avoid systematic biases but does not eliminate the random mean shift Δ r expected among experiments. In other words, randomization cannot eliminate the variance component due to differences in mean abundances among experiments and thus cannot avoid a reduction in power to detect the association with an outcome of interest, unless Δ r is accounted for in the statistical inference.
Since Δ r cannot be estimated and eliminated using the proteomic data alone, the question arises whether the information in the outcome data (in our example, the micronutrient concentrations) can help to account for this unwanted variance component. Assume that in truth we have E[Nrk] = β0 + β1 × ark, where Nrk is a numeric outcome for the sample in experiment r channel k, and ark is the corresponding log2 absolute protein abundances. Substituting μ + Δ r + δrk for ark, we can rewrite this equation as
| (5) |
where r indicates the experiment and k the channel. Thus, eq 5 represents a linear mixed effects model with intercept γ0 = β0 + β1 μ , a random shift Br ~ N(0, (β1σΔ)2) that accounts for differences in means seen among experiments, and the slope β1 as the actual parameter of interest, assessing the relationship between protein and nutrient concentrations.
The statistical significance for this protein–nutrient association can be derived by testing the hypothesis β1 = 0. Note that the normality assumption for the random intercept is not very strong: if the responses are normally distributed, then so are the means of the samples chosen for an iTRAQ experiment, implying normality of the random effect. Also note that if more than one protein is linearly related to the nutrient concentration, the above equation can be extended to
| (6) |
Thus, even though we have multiple proteins, the resulting linear mixed effects model still has only one random effect, B͂r, that jointly summarizes the among experiment differences.
Estimation of Relative Protein Abundances
For a single iTRAQ experiment, denote Ysij = (Ysij1, …, Ysij8) to be the log2 reporter ion intensities for spectrum s belonging to peptide j of protein i, and let Zsij = (Zsij1,…, Zsij8) be the mean or median-polished log2 reporter ion intensities; that is, the residuals of the log2 reporter ion intensities after subtracting the sample mean or sample median from each of the eight log2 intensities. The estimates of relative abundance of a particular protein can be calculated, for example, as the mean or the median of the derived values from all reporter ion intensity spectra belonging to this protein. Alternatively, an approach that incorporates the hierarchical structure of the data can be employed. That is, the information from all spectra belonging to the same peptide is combined first (via means or medians), and then these values are combined by a mean or median to derive the log2 relative protein abundances. A weighted summary can also be used in case a strong inverse relationship exists between the ion counts across all channels and the variance in the respective expression ratios;6 however, we do not see that phenomenon in our data (see the section Variance Heterogeneity in the Supporting Information). Corrections for differences in amounts of material loaded in the channels or sample processing should be considered by subtracting the channel mean or median from the relative abundance estimate, normalizing all channels to have mean or median 0 (Figure 1).
Figure 1.
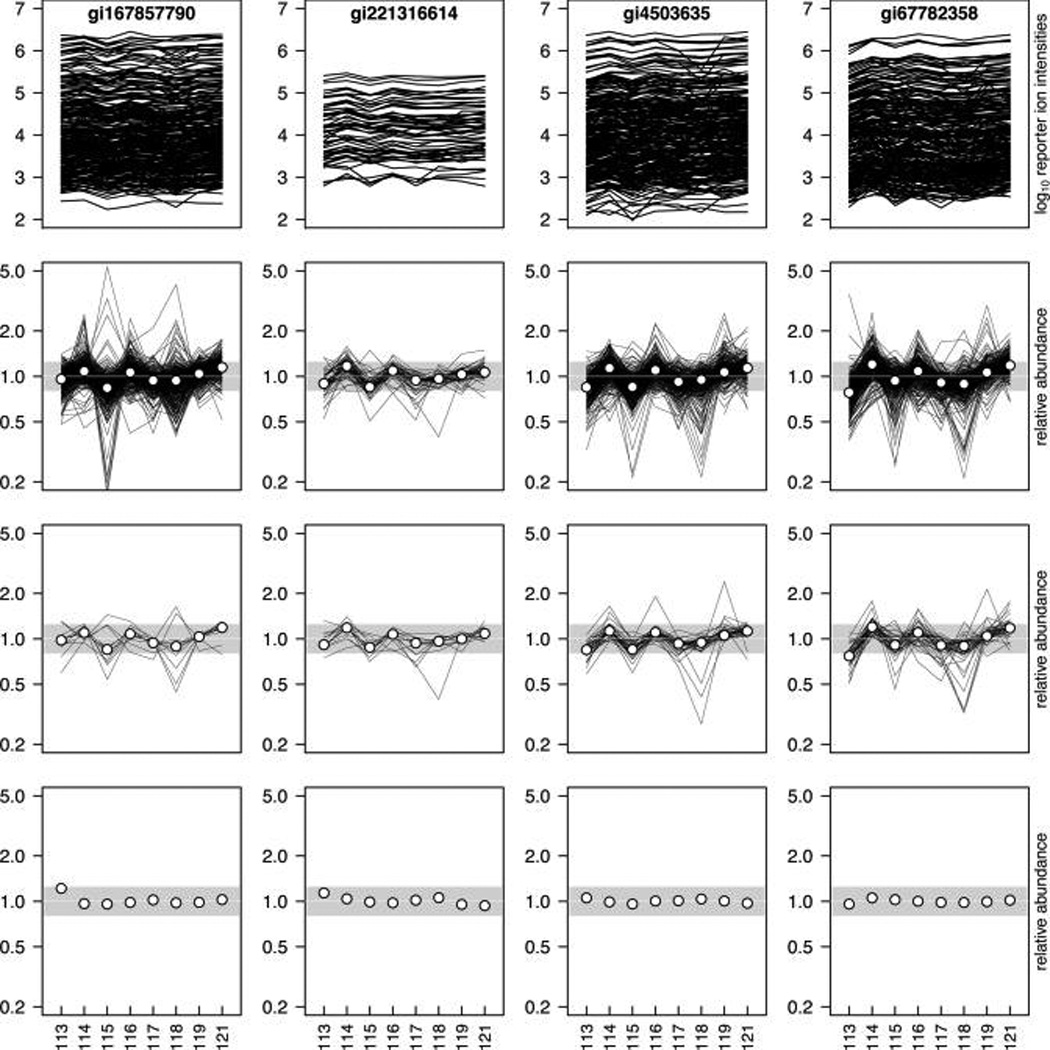
Quantification of relative peptide and protein abundances in an experiment with eight technical replicates of pooled plasma samples. The normalization steps are shown for four randomly selected proteins (accession numbers gil678S7790, gi221316614, gi4S0363S, and gi677823S8, in columns 1–4). Shown are the absolute reporter ion intensities on the log10 scale for all spectra detected for the respective proteins (first row), the same data after removal of the spectrum median (second row), the relative peptide abundances calculated as the median of the above row 2 values for each channel and peptide (third row), and the relative protein abundances after removing the loading effect (fourth row). The gray regions (rows 2–4) represent 25% fold changes in relative abundances. Since the same samples were loaded in the eight channels, all relative protein abundances should be equal to 1.
An alternative approach is to employ linear mixed effects models for protein relative abundance estimation as well. For a single iTRAQ experiment, these estimates for the relative protein abundances can be derived from the protein-sample interaction term in a mixed effects model; for example,
| (7) |
where Z denotes the mean-polished reporter log2 ion intensities in channel k, for spectrum s from peptide j of protein i. C denotes the channel, Π denotes the protein, π denotes the peptide, and ε denotes the residuals of the observed data after accounting for the systematic effects. In the above model, Ck is the loading effect, a random effect that models systematic deviation of the log2 reporter ion intensities from zero in the respective channels. Since the average for each mean-polished log2 reporter ion intensities spectrum is 0, it is not necessary to specify the main effects for spectrum, peptide, and protein. The term {C:Π}ik is the sample by protein interaction, a fixed effect that determines the deviation (up or down regulation) of protein i in channel k. The term {C:π}kij is a random effect that models the variability of the relative peptide abundances (indexed by j) around the relative protein abundance of protein i in channel k; that is, potential differences between relative peptide abundances that contribute to the estimate of the relative protein abundance. Finally, the last variance component is the residual error εsijk assumed to have mean 0 and a constant variance.
When violations of model assumptions are a concern, simpler, more robust approaches can possibly yield better results. For example, if the assumption of a normally distributed random effect for peptide nested within protein does not hold, the above model could be simplified to
| (8) |
omitting the peptide random effect. Note that the correction for differences in loadings or sample processing could also be carried out after estimating the protein abundances by subtracting the mean or median from all protein abundances within each channel. In particular, omitting the term Ck in eq 8 simply yields the log2 reporter ion intensity average for each protein as the initial abundance estimate in each channel because only fixed effects are present.
In this manuscript, we consider a total of seven approaches to quantify protein relative abundances from a single iTRAQ experiment. In particular, we compare methods with and without masterpool normalization, linear models with and without random effects, and simple summaries of log-transformed reporter ion intensities. We also investigate whether means or medians in general perform better for data summaries. For all methods, we remove the loading effect after abundance estimation (a significant source of variability; see the section Loading Effects in the Supporting Information). We note that missing data patterns can substantially affect parameter estimates, such as the estimated loading effects, in particular when the missingness cannot be ignored. It is often assumed or observed that missingness is related to protein concentration (for example, due to instrument detection limits);13 however, we did not observe such patterns in our 8-plex iTRAQ data (manuscript in preparation).
As defined above, let Zsij = (Zsij1, …, Zsij8) denote the mean-polished log2 reporter ion intensities for spectrum s (that is, the residuals of the log2 reporter ion intensities after removing the sample mean across the eight channels), belonging to peptide j of protein i. In addition, let Z͂sij = (Zsij1, …, Z͂sij8) denote the median-polished reporter log ion intensities. Without loss of generality, assume that a masterpool has been run in channel 1. We compared the following approaches: (1) Masterpool-based normalization using means; that is, calculating Zsijk − Zsij1 for k ∈ 2,…, 8, and then averaging those numbers across all spectra s, for each protein i in each channel k, ignoring peptides. (2) Masterpool-based normalization using medians; that is, calculating Z͂sijk − Z͂sij1 for k ∈ 2, …, 8, and then calculating the median of those numbers across all spectra s, for each protein i in each channel k. (3) Linear mixed effects models as considered above, where we model the peptides nested within a protein as random effects. (4) Linear models as considered above, ignoring the hierarchical structure, averaging the demeaned log2 reporter ion intensities Zsijk across all spectra s, for each protein i in each channel k. (5) A “median sweep” ignoring peptides; that is, simply calculating the median of all Zsijk, across all spectra s, for each protein i in each channel k. (6) A “mean sweep” taking the hierarchical data structure into account; that is, calculating Zijk = means{Zsijk}, the average of all Zsijk across all spectra s, for each protein i and peptide j in each channel k, and then averaging the resulting means over the peptides; that is, calculating Zik = meanj{Zijk}. (7) A “median sweep” taking the hierarchical data structure into account, calculating Z͂ijk = medians{Z͂sijk} for each protein i and peptide j in each channel k, and then Z͂ik = medianj{Z͂ijk}.
Under the assumption that specific variance components are the same in each experiment (such as the peptide variability around the respective protein estimates, the model error variability, etc), more efficient estimates might be derived by estimating protein quantities simultaneously across multiple iTRAQ experiments. For example, protein abundances might be derived from the mixed effects model,
| (9) |
where Z denotes the mean-polished reporter log ion intensities in iTRAQ experiment r, channel k, for spectrum s from peptide j of protein i. Again, C denotes the channel, Π denotes the protein, π denotes the peptide, and ε denotes the residuals of the observed data after accounting for the systematic effects. As before, the term {C:Π}ikr denotes a sample by protein interaction, a fixed effect that determines the deviation (up or down regulation) of protein i in the sample loaded in channel k of iTRAQ experiment r. Note that omitting the term {C:π}kijr, the random effect that models the variability of the relative peptide abundances around the relative protein abundance, leads to a fixed effects model. Combining multiple iTRAQ experiments in this setting yields a design matrix with orthogonal components, thus resulting in the same protein relative abundance estimates as the ones derived separately from each single experiment. In this manuscript, we also delineate relative abundance estimates simultaneously from multiple iTRAQ experiments using mixed effects models and investigate whether the increase in computing complexity is warranted.
RESULTS
The relative abundance estimates for our data derived by simultaneously analyzing multiple iTRAQ experiments are virtually identical to those derived separately from each iTRAQ experiment. Using the 406 samples from 58 separate iTRAQ experiments (seven biological samples plus one masterpool, the latter being omitted for this part of the analysis), we compared linear models (ignoring the hierarchical data structure of peptides nested in proteins) and linear mixed effects model (using peptide as a random effect). For each of those two methods, we compared the results by deriving the relative abundance estimates jointly for the samples in all experiments, and derived separately for the samples in each experiment. The fixed effects linear model generates a design matrix with orthogonal entries when all experiments are assessed simultaneously, and thus yields estimates identical to those obtained from analyzing one experiment at a time. The results from both fixed effects approaches are included solely to demonstrate this feature. Using the concordance correlation (defined in eq 4), we get nearly identical results for all four approaches and, as expected, identical results for the fixed effects models whether the estimates are derived simultaneously or separately (Table 1). Thus, there is no evidence that the added complexity of introducing a random peptide effect is warranted for the estimation of relative protein abundances.
Table 1.
Quantiles of the Distributions of the Concordance Correlations, Defined in Eq 4, Comparing Four Different Approaches Based on Linear Models and Linear Mixed Effects Modelsa
| 0% | 25% | 50% | 75% | 100% | |
|---|---|---|---|---|---|
| LM vs LME | 0.841 | 0.966 | 0.983 | 0.992 | 0.999 |
| LM vs LMS | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| LM vs LMES | 0.852 | 0.967 | 0.985 | 0.993 | 0.999 |
| LME vs LMS | 0.841 | 0.966 | 0.983 | 0.992 | 0.999 |
| LME vs LMES | 0.986 | 0.998 | 0.999 | 1.000 | 1.000 |
| LMS vs LMES | 0.852 | 0.967 | 0.985 | 0.993 | 0.999 |
Proteins observed in all biological samples of the 58 experiments (406 samples total) and at least two peptides per protein were included (n = 92). Both linear models and linear mixed effects models were run separately for each experiment to generate protein relative abundance estimates (LM and LME, respectively) and simultaneously for all 58 experiments (LMS and LMES, respectively). The relative abundance estimates derived from the different methods are almost identical, and thus, there is no evidence that the added complexity of introducing a random effect for peptide is warranted.
The same holds when we compare the relative abundance estimates in the four all-masterpool 8-plex iTRAQ experiments. Since there is no biological variability in these 32 samples and, thus, the relative abundance estimate for each protein in each sample should be equal to 1, in truth we report the absolute median fold change (defined in eq 3) for each sample. In most instances, the results are virtually identical, and in the samples for which a modest difference among methods can be seen, the simplest model based on fixed effects without peptide random effects actually achieves the best results (Figure 2). Thus, for the proteomic data in our hands, there does not appear to be a benefit of using more computationally intensive methods. The simplest method, ignoring the hierarchical structure of peptides nested in proteins and estimating relative abundances one experiment at a time, yields results at least as good as the mixed effects models, and is much faster to run. Moreover, the data can be analyzed sequentially as iTRAQ experiments are carried out in the laboratory, without the need to refit previous samples, an additional attractive feature. Therefore, only abundance estimates derived from one experiment at a time are considered in the remainder of the manuscript.
Figure 2.
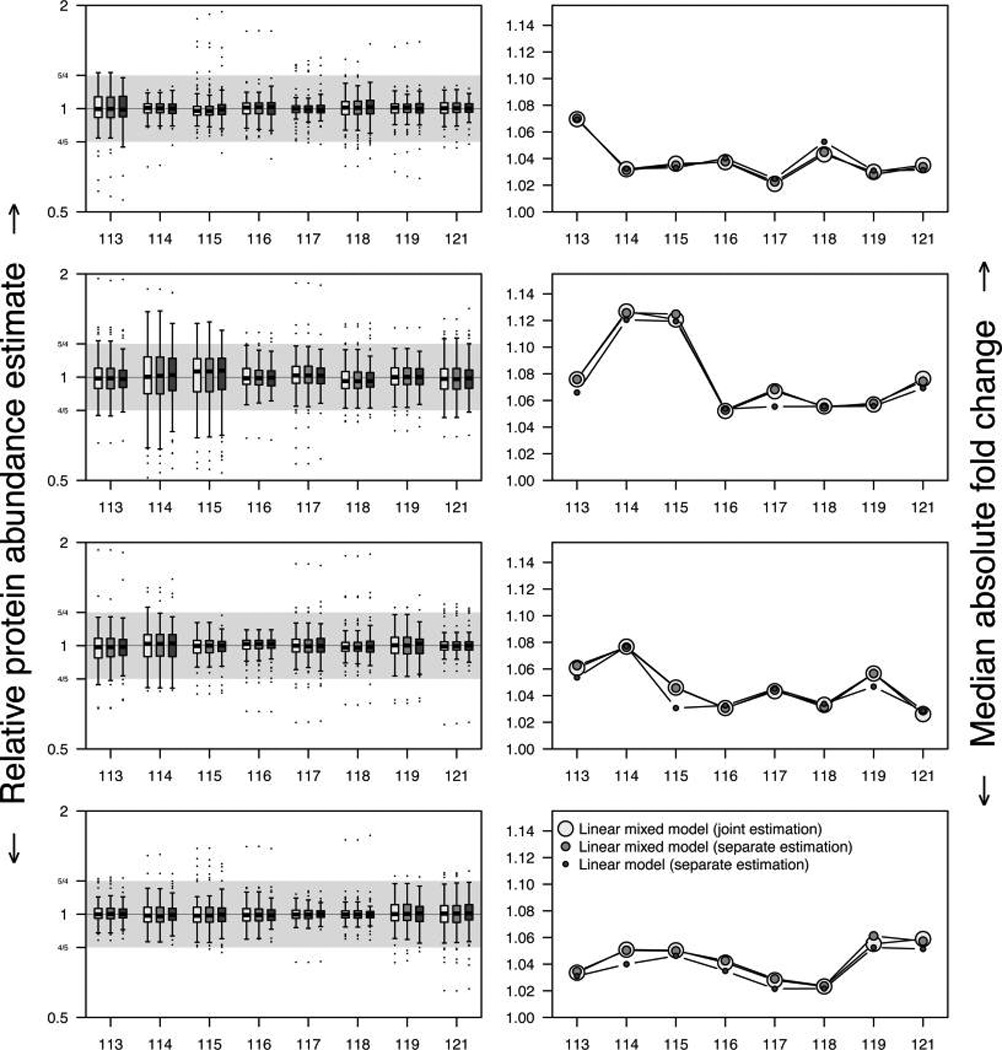
Protein relative abundance estimates (left column) derived from four all-masterpool 8-plex iTRAQ experiments (top to bottom). Proteins observed in all four experiments with two or more peptides were considered (n = 130). Boxplots of the relative abundance estimates are shown for the eight channels in each of the four experiments, using linear mixed effects models estimating all relative abundances simultaneously (white), linear mixed effects models estimating all relative abundances separately for each experiment (light gray), and standard linear models estimating all relative abundances separately for each experiment (dark gray). The median absolute fold change (right column) represents a typical value for the deviation from 0 on the log2 scale, mapped back to the original fold change scale. A value of 1 represents no variability, 1.1 indicates that typical deviation is 10%. The gray regions in the left panels represent 25% fold changes in relative abundances.
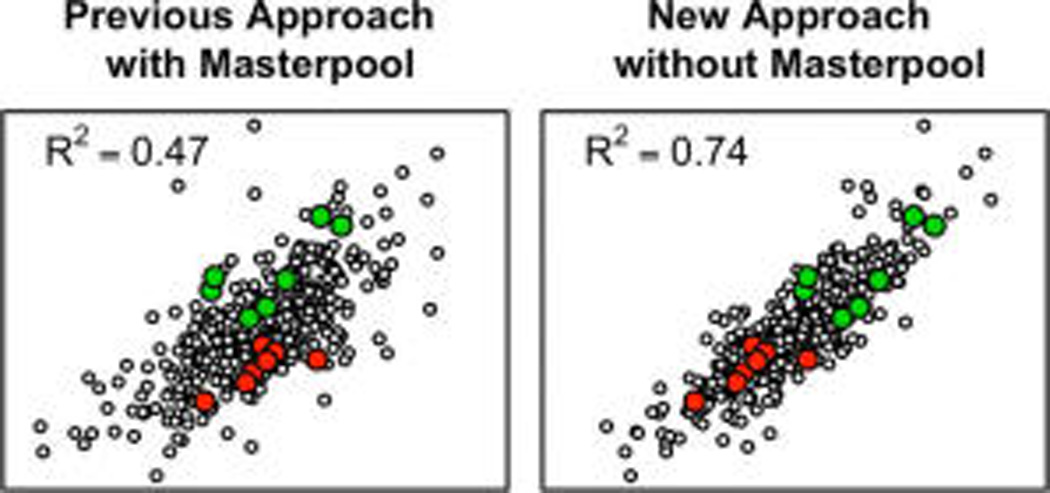
We show next that properly accounting for among-experiments variability can substantially boost the power to detect protein-nutrient associations and can improve the overall assessment of these relationships (as compared with ignoring the among-experiments variability or using the masterpool for abundance estimation and comparison of estimates among experiments). For example, using the “median sweep” without employing the masterpool (Figure 1), the estimated relative abundances of the retinol-binding protein 4 derived from 58 8-plex iTRAQ experiments with seven individual samples and one masterpool each (406 individual samples total) explain 50% of the variability in measured plasma retinol concentrations when the data across experiments are simply augmented, but 74% of that variability can be explained when the among-experiments variance component Br in eq 5 is estimated and accounted for (Figure 3). In this example, allowing a random intercept accounts for 42% of the total variability (the sum of the among-experiments variability and the residual variability). Using the masterpools to estimate protein abundances in all experiments yields an R2 of only 0.47. The observed improvement is a trend that holds in general, shown here for the 15 most significant associations of mass-spectrometry-based protein abundances with plasma retinol (vitamin A), the C-reactive protein measured in plasma as an indicator of inflammation, and the α1-acid glycoprotein: the random intercepts model (eq 5) always yields a higher coefficient of determination (Figure 4 and Table 2).
Figure 3.
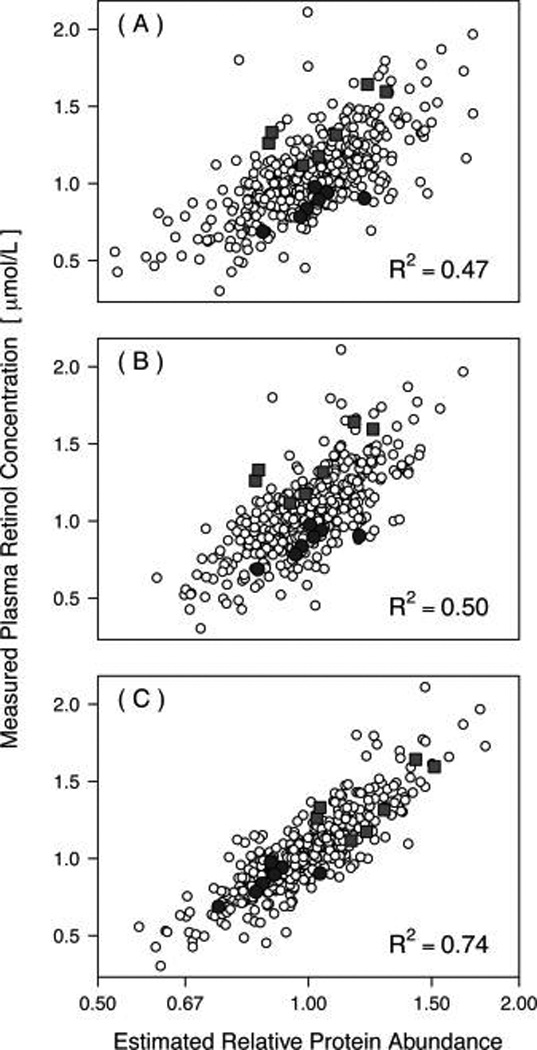
Estimates of relative abundances of retinol-binding protein 4 (RBP4) derived from 58 8-plex iTRAQ experiments (seven individual samples and one masterpool each, 406 biological samples total) versus measured plasma retinol concentration. Relative abundance estimates from different experiments were combined using the masterpool (A), the median sweep augmenting the estimates from each experiment (B), and the linear mixed effects model (eq 5), using the median sweep-derived relative abundance estimates (C). Highlighted are two experiments, one with high (lighter gray) and one with low (darker gray) average plasma retinol concentration.
Figure 4.
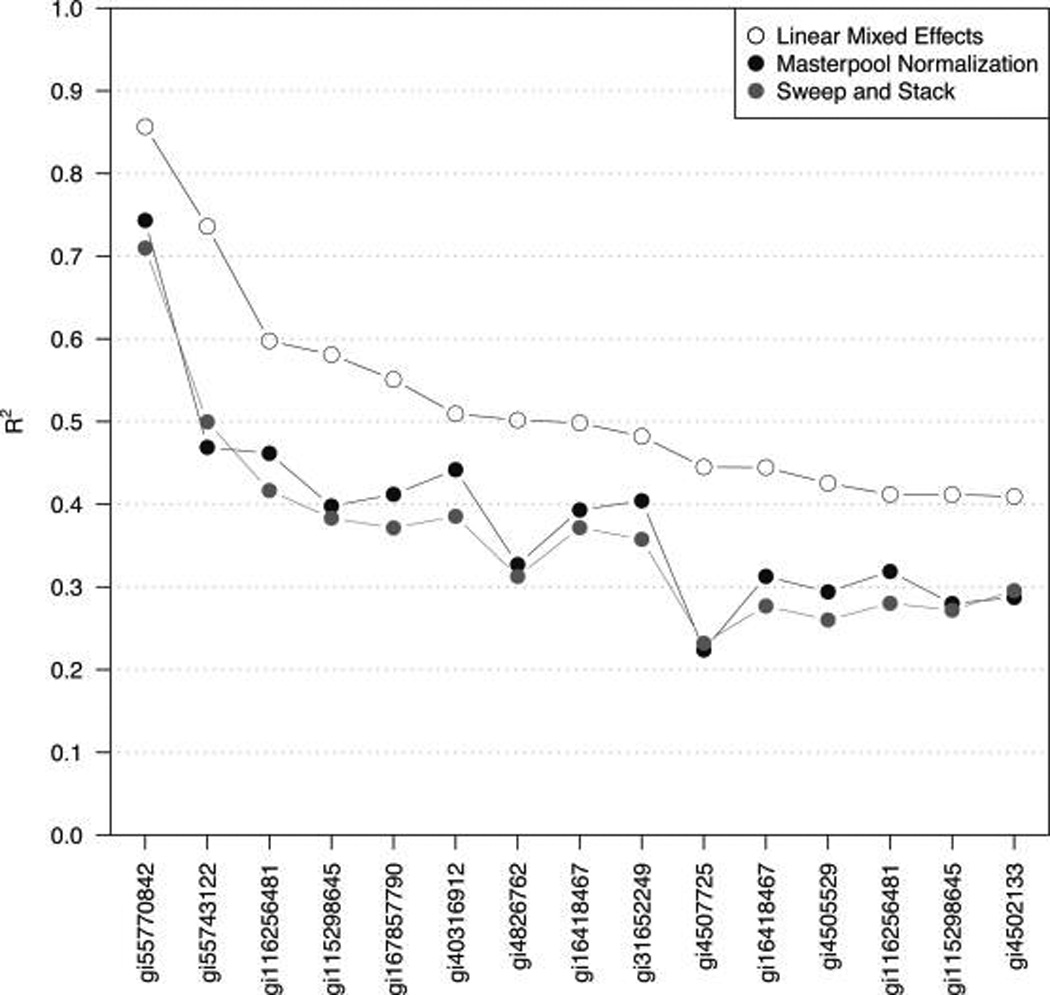
The strongest associations (R2, y-axis) of proteins (accession numbers on the x-axis) with nutrients or indicators of acute phase response observed in our experiments (Table 2). Estimates of relative protein abundance from different experiments were combined using the linear mixed effects model (eq 5), the masterpool normalization, and the median sweep, augmenting (stacking) the estimates from each experiment. The association of RBP4 (accession number gi55743122) and plasma retinol is shown in detail in Figure 3.
Table 2.
The Strongest Associations of Proteins and Nutrients (or indicators of acute phase response) Observed in Our Experimentsa
| N/I | accession | protein | n | ME | MP | SS |
|---|---|---|---|---|---|---|
| Crp | gi55770842 | C-reactive protein | 357 | 0.86 | 0.74 | 0.71 |
| ret | gi55743122 | retinol-binding protein 4 | 406 | 0.74 | 0.47 | 0.50 |
| agP | gi116256481 | TNFAIP3-interacting protein 1 | 301 | 0.60 | 0.46 | 0.42 |
| agP | gi115298645 | mitogen-activated protein kinase kinase kinase | 259 | 0.58 | 0.40 | 0.38 |
| agP | gi167857790 | orosomucoid 1 | 406 | 0.55 | 0.41 | 0.37 |
| crp | gi40316912 | serum amyloid A protein preproprotein | 399 | 0.51 | 0.44 | 0.39 |
| agP | gi4826762 | haptoglobin isoform 1 preproprotein | 266 | 0.50 | 0.33 | 0.31 |
| crp | gi16418467 | leucine-rich α-2-glycoprotein 1 | 406 | 0.50 | 0.39 | 0.37 |
| crp | gi31652249 | lipopolysaccharide-binding protein | 406 | 0.48 | 0.40 | 0.36 |
| ret | gi4507725 | transthyretin | 406 | 0.45 | 0.22 | 0.23 |
| agP | gi16418467 | leucine-rich α-2-glycoprotein 1 | 406 | 0.44 | 0.31 | 0.28 |
| agP | gi4505529 | orosomucoid 2 | 406 | 0.43 | 0.29 | 0.26 |
| crp | gi116256481 | TNFAIP3-interacting protein 1 | 301 | 0.41 | 0.32 | 0.28 |
| crp | gi115298645 | mitogen-activated protein kinase kinase kinase | 259 | 0.41 | 0.28 | 0.27 |
| crp | gi4502133 | serum amyloid P component | 406 | 0.41 | 0.29 | 0.30 |
The nutrients/indicators (N/I) are the C-reactive protein (crp), plasma retinol (ret), and the α(1)-acid glycoprotein (agp). The statistical inference is based on three different approaches. SS: estimates of relative protein abundance from different experiments were combined using the median sweep (Figure 1), and the estimates from each experiment were simply stacked, ignoring among-experiments variability. MP: the masterpool approach, using a reference sample for relative abundance estimation, and combining estimates among experiments. ME: using linear mixed effects models (eq 5) to account for among-experiments variability, with median sweep-derived relative abundance estimates. The total number of observed protein–outcome pairs (n) ranged from 259 (37 experiments with 7 biological samples each) to 406 (observed in all samples of the 58 experiments).
We stress that the assessment of statistical significance of a protein-nutrient relationship is based on the test statistic for the slope parameter β1 in eq 5, whereas the improvement in R2 is mostly due to explaining the among-experiments variability. An increase in overall nutrient variability explained, however, does translate into a decrease in the standard error of the test statistics and, thus, an improvement in power. In the Supporting Information (section Expected Improvement in Power), we present a simulation study and give a more in-depth discussion of what improvements in power one can expect.
The question remains which of the approaches (1–7) defined in the Methods section yields the best results for the relative abundance estimation in single iTRAQ experiments. When investigating the quality of the respective relative abundance estimates using the four iTRAQ experiments, each with eight technical replicates of the masterpool, we find that methods (3–7) perform roughly equally well, clearly outperforming the two approaches for abundance estimation based on a reference sample. In these four masterpool experiments, the median absolute fold change is always the lowest for the median sweeps (5) and (7), and the methods using masterpool normalization appear substantially more variable than all others (Figure 5, top). This observation is even more pronounced when the root mean squared error (translated into a fold change) is considered (Figure 5, bottom).
Figure 5.
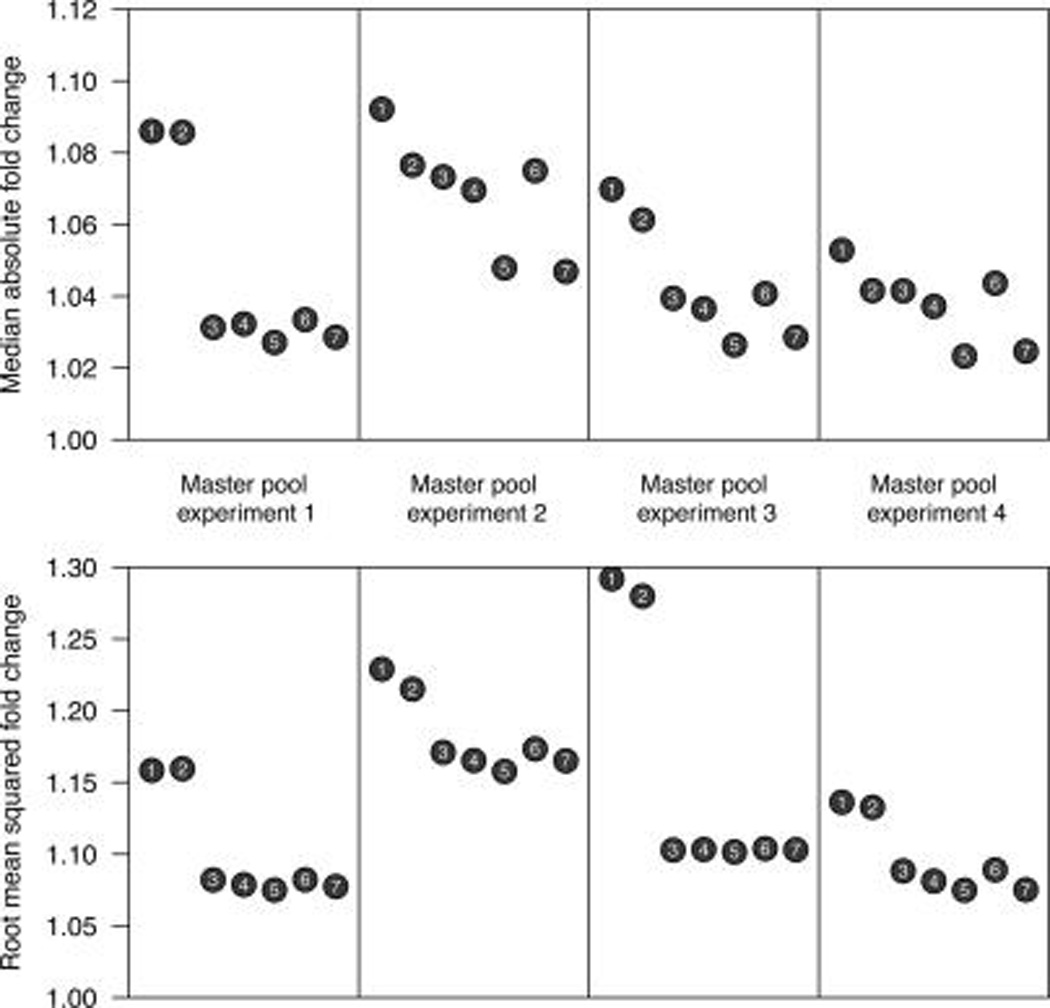
The median absolute fold changes (top) and the root mean-squared fold changes (bottom) for seven different relative abundance estimating procedures, defined in the Methods section. Proteins observed in all four experiments with two or more peptides were included in the assessment (n = 130), and results are shown for each procedure and each iTRAQ experiment, calculating summary statistics across all proteins and channels. The masterpool-based procedures 1 and 2 clearly fare the worst. In addition, median-based procedures, for example, 5 and 7, appear to perform better than the respective mean-based procedures, for example, 4 and 6.
When samples with biological variability and their associations with nutrient concentrations are considered, the median sweep ignoring peptides (5) again is arguably the best approach, typically producing among the highest coefficients of determination of all approaches considered (Figure 6). Interestingly, the other arguably superior method for samples with biological variability is the median-based masterpool approach (2), which seems to contradict the findings from the masterpool experiments (Figure 5). This apparent paradox can be explained by the fact that the random intercept models used to investigate protein–nutrient associations can account for some or all of the extra variability introduced by the masterpool-based normalization. The only difference between the median-based masterpool approach (2) and the median sweep ignoring peptides (5) is the denominator for calculating ion intensity ratios from the reporter ion intensity spectrum. For both approaches, the numerator is the observed ion intensity of the biological sample. For the masterpool approach, this intensity is divided by the intensity observed in the designated masterpool channel, which leads to higher variability than in the median sweep, where the denominator is simply the median of the ion intensities across all channels with biological samples (Figure 5). However, as long as the intensities in all biological samples are divided by the same number (in fact, divided by any number), the mixed effects models can account for this difference by shifting the random intercepts accordingly. Since the median sweep (5) produces the best or among the best results for all approaches considered when proteins are associated with the outcome (Figure 6) or null (Figure 5), we recommend this approach for use in practice.
Figure 6.
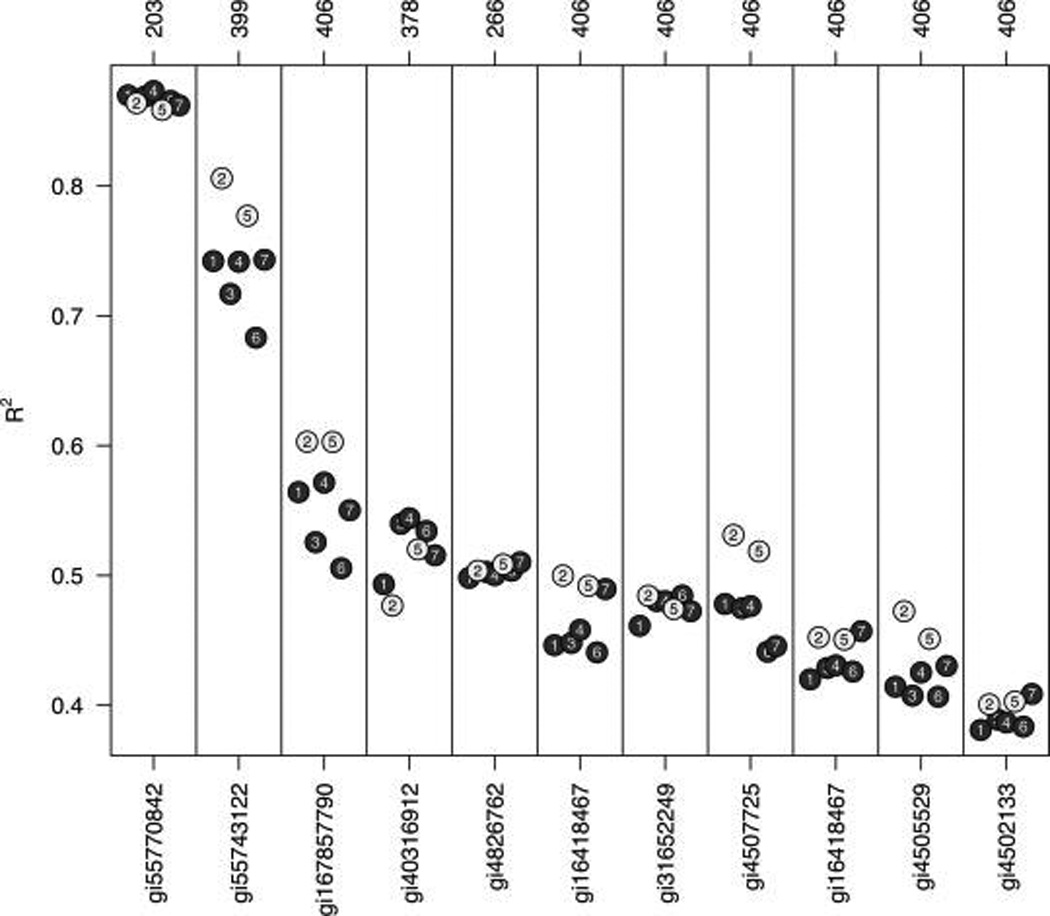
The associations (R2, y-axis) of proteins (accession numbers on the x-axis) with nutrients or indicators of acute phase response observed in our experiments (a subset of Table 2). Proteins observed in at least half of the samples (203, corresponding to 29 complete experiments with seven biological samples) with two or more peptides were included in the assessment (n = 92). The R2 values were derived from the random intercept model (eq 5), based on seven different procedures to estimate protein relative abundances (defined in the Methods section). Highlighted are the results for the median-based master pool approach (2) and the median sweep ignoring peptides (5), performing the best in this assessment.
DISCUSSION
In this manuscript, we show that using a masterpool, a commonly employed strategy in iTRAQ experiments, is not optimal in several regards. In a single iTRAQ experiment, more precise estimates of protein relative abundances can be obtained by using a summary of the biological data as the reference. The data from multiple iTRAQ experiments can then be analyzed, allowing for an experiment-specific random effect, which results in improved inference compared with the masterpool-based approach. And obviously, using a masterpool occupies a channel that otherwise could be used for an additional biological sample.
We note that in addition to iTRAQ, our new method is also applicable to other isobaric labeling methods with reporter fragment ions, such as tandem mass tag (TMT) reagents; however, our approach does not alleviate the often observed “shrinkage to the mean” effects, that is, the fact that the estimated and reported fold changes are closer to unity than the actual existing abundance ratios in the samples.6 Our method is based on the observed reporter ion intensities, which already are subject to these shrinkage effects, and thus, our method also does not yield unbiased estimates for relative abundances (see section Estimation Bias in the Supporting Information for an example).
Our approach was specifically developed to investigate associations between protein abundances and a numeric outcome of interest, and thus, other types of studies based on multiple experiments, such as chaining of experiments to track protein abundances and protein changes over time, are typically outside this framework. However, we note that in case-control studies, the inference is rather simple (and a masterpool would be of no use at all). If case and control samples are used in the same iTRAQ experiment, the difference in the respective sample means is an unbiased estimate for the true difference in log2 abundance, and its significance can be assessed by calculating the standard error of that difference. For a single iTRAQ experiment, this is simply a two-sample t test, and for multiple iTRAQ experiments, the test statistics could be weighted using inverse variances. It might be the case that the simultaneous analysis of multiple proteins in case-control settings could improve by pooling variances and, thus, derive more precise estimates for the standard errors, assuming that the variance components for these proteins are indeed the same, and that these protein sets were known a priori.
Alternative approaches, such as linear models that do not necessarily include a masterpool in the experiment, have also been proposed in the literature for the estimation of relative protein abundances. These methods also allow for statistical inference on multiple experiments simultaneously and attempt to describe the existing sources of variability and the biological and technical factors 14–18 in a single model. For case-control settings, Oberg et al.11 and Hill et al.12 extended a mixed effects approach originally used for gene expression studies 19,20 and proposed to use an analysis of variance (ANOVA) approach with mixed effects for proteomic data. Specifically, the authors write
| (10) |
where yi,j(i),c,q,s,l is the log-transformed reporter ion intensity corresponding to protein i, peptide j, condition or treatment group c, experiment q, tag or channel l, and spectrum s. In the above, p denotes the protein effect (a fixed effect); f denotes a peptide nested within a protein, a random effect,21 and r denotes potential differences in spectra intensities comparing case and control samples. The inference for the interaction between protein and group status (denoted as ri,c in eq 10) is of particular interest because it quantifies differences in protein abundances, comparing cases and controls (i.e., it answers which proteins are differentially expressed). The terms v and q allow adjustment for potential differences due to channel effects, loading, mixing, and sample handling. In the above, hi,j(i),c,q,s,l is the remaining experimental error, not explained by the linear model terms.
The mixed effects model above (eq 10) is specifically tailored to case-control settings or data stemming from experiments with more than two conditions (defining outcome groups) to carry out a corresponding analysis of variance. In principle, these models could also be extended into more general regression settings, such as ours; however, one drawback of this approach and presumably a reason for their somewhat limited use in the proteomics literature is that it can be computationally very difficult to fit these mixed effects models. A very large number of parameters need to be estimated numerically in these models (in contrast to balanced gene expression studies, in which closed-form solutions and exact null distributions are often available), and the design matrix is such that no orthogonal components exist that would allow for separate estimation of sets of parameters. In proteomic experiments, the data are typically unbalanced and contain missing values,21–23 adding significantly to the computational complexity. Oberg et al.11 and Schwacke et al.24. discuss these challenges and provide some practical advice for possible implementations of such ANOVA models for a limited number of complex samples. Needless to say, finding the global optimum for such a high dimensional likelihood function would be a daunting task with 58 iTRAQ experiments.
The main motivation for a comprehensive analysis, assessing all proteins across all samples and experiments with a single analysis, is that more precise estimates of protein abundance and better inference can be derived when the underlying model assumptions are met. These assumptions are fairly strong and are subject to debate in the proteomics literature,12,21 similar to the discussions in other fields, such as studies of gene expression.25–27 For the model in eq 7, we would have to assume that the loading effects are normally distributed with a constant variance for all channels (a reasonable assumption from a statistical perspective); that the peptide effects are normally distributed with a constant variance for all proteins across all channels (a less reasonable assumption); and that the residual error has a constant variability across all spectra, peptides, proteins, and channels (a fairly unrealistic assumption; see the section Model Assumptions in the Supporting Information). It is not obvious how such model violations affect abundance estimates and inference and if a highly complex single analysis might do more damage than good. Allowing for a random peptide effect did not improve the relative abundance estimates or the statistical inference for our data. Virtually identical results were obtained when the data were analyzed one experiment at a time, compared with a simultaneous analysis of all experiments. Ignoring the hierarchical structure of peptide nested in protein and estimating relative abundances one experiment at a time yields results as good as or better than the mixed effects models and is much faster to execute. This approach also allows for data to be analyzed as iTRAQ experiments are carried out sequentially in the laboratory, without the need to refit previous samples, which makes this approach even more attractive.
Supplementary Material
ACKNOWLEDGMENTS
The nutriproteomics study was supported through the “Assessment of Micronutrient Status by Nutriproteomics” grant (GH 5241) from the Bill and Melinda Gates Foundation. The original intervention trial was supported by the US Agency for International Development, Washington, D.C. through Cooperative Agreements No. HRN-A-00-97-00015-00 and GHS-A-00-03-00019-00 with the Johns Hopkins University and by Grant GH614 from the Bill and Melinda Gates Foundation. Additional assistance was received from the Sight and Life Research Institute, Baltimore, MD. The cohort follow-up field study in Nepal from which plasma samples were obtained was supported through the “Global Control of Micronutrient Deficiency” grant (GH 614) from the Bill and Melinda Gates Foundation, Seattle, WA. Additional support was provided from a CTSA grant to the Johns Hopkins Medical Institutions. We also thank Laurent Gatto and Bernard Delanghe for helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental details and results. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive sobaric tagging reagents. Mol. Cell. Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal K, Choe LH, Lee KH. Quantitative analysis of protein expression using amine-specific isobaric tags in Escherichia coli cells expressing rhsA elements. Proteomics. 2005;5(9):2297–2308. doi: 10.1002/pmic.200401231. [DOI] [PubMed] [Google Scholar]
- 3.Lau KW, Jones AR, Swainston N, Siepen JA, Hubbard SJ. Capture and analysis of quantitative proteomic data. Proteomics. 2007;7(16):2787–2799. doi: 10.1002/pmic.200700127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiese S, Reidegeld KA, Meyer HE, Warscheid B. Protein labeling by iTRAQ: a new tool for quantitative mass spectrometry in proteome research. Proteomics. 2007;7(3):340–350. doi: 10.1002/pmic.200600422. [DOI] [PubMed] [Google Scholar]
- 5.Pierce A, Unwin RD, Evans CA, Griffiths S, Carney L, Zhang L, Jaworska E, Lee C-F, Blinco D, Okoniewski MJ, Miller CJ, Bitton DA, Spooncer E, Whetton AD. Eight-channel iTRAQ enables comparison of the activity of six leukemogenic tyrosine kinases. Mol. Cell. Proteomics. 2008;7(5):853–863. doi: 10.1074/mcp.M700251-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Karp NA, Huber W, Sadowski PG, Charles PD, Hester SV, Lilley KS. Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics. 2010;9(9):1885–1897. doi: 10.1074/mcp.M900628-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zieske L. R A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J. Exp. Bot. 2006;57(7):1501–1508. doi: 10.1093/jxb/erj168. [DOI] [PubMed] [Google Scholar]
- 8.Ruczinski I, Plaxco KW. Some recommendations for the practitioner to improve the precision of experimentally determined protein folding rates and phi values. Proteins. 2009;74(2):461–474. doi: 10.1002/prot.22155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stewart CP, Christian P, Schulze KJ, Leclerq SC, West KP, Khatry SK. Antenatal micronutrient supplementation reduces metabolic syndrome in 6- to 8-year-old children in rural Nepal. J. Nutr. 2009;139(8):1575–1581. doi: 10.3945/jn.109.106666. [DOI] [PubMed] [Google Scholar]
- 10.Christian P, Khatry SK, Katz J, Pradhan EK, LeClerq SC, Shrestha SR, Adhikari RK, Sommer A, West KP. Effects of alternative maternal micronutrient supplements on low birth weight in rural Nepal: double blind randomised community trial. BMJ (Br. Med. J.) 2003;326(7389):571. doi: 10.1136/bmj.326.7389.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oberg AL, Mahoney DW, Eckel-Passow JE, Malone CJ, Wolfinger RD, Hill EG, Cooper LT, Onuma OK, Spiro C, Therneau TM, Bergen HR. Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J. Proteome Res. 2008;7(1):225–233. doi: 10.1021/pr700734f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill EG, Schwacke JH, Comte-Walters S, Slate EH, Oberg AL, Eckel-Passow JE, Therneau TM, Schey KL. A statistical model for iTRAQ data analysis. J. Proteome Res. 2008;7(8):3091–3101. doi: 10.1021/pr070520u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang P, Tang H, Zhang H, Whiteaker J, Paulovich AG, Mcintosh M. Normalization regarding non-random missing values in high-throughput mass spectrometry data. Pac. Symp. Biocomput. 2006:315–326. 2006. [PubMed] [Google Scholar]
- 14.Urfer W, Grzegorczyk M, Jung K. Statistics for proteomics: a review of tools for analyzing experimental data. Proteomics. 2006;6(Suppl 2):48–55. doi: 10.1002/pmic.200600554. [DOI] [PubMed] [Google Scholar]
- 15.Gan CS, Chong PK, Pham TK, Wright PC. Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ) J. Proteome Res. 2007;6(2):821–827. doi: 10.1021/pr060474i. [DOI] [PubMed] [Google Scholar]
- 16.Prakash A, Piening B, Whiteaker J, Zhang H, Shaffer SA, Martin D, Hohmann L, Cooke K, Olson JM, Hansen S, Flory MR, Lee H, Watts J, Goodlett DR, Aebersold R, Paulovich A, Schwikowski B. Assessing bias in experiment design for large scale mass spectrometry-based quantitative proteomics. Mol. Cell. Proteomics. 2007;6(10):1741–1748. doi: 10.1074/mcp.M600470-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Vitek O. Getting started in computational mass spectrometry-based proteomics. PLoS Comput. Biol. 2009;5(5):e1000366. doi: 10.1371/journal.pcbi.1000366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaell L, Vitek O. Computational mass spectrometry-based proteomics. PLoS Comput. Biol. 2011;7(12):e1002277. doi: 10.1371/journal.pcbi.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. J. Comput. Biol. 2000;7(6):819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- 20.Kerr MK, Churchill GA. Experimental design for gene expression microarrays. Biostatistics. 2001;2(2):183–201. doi: 10.1093/biostatistics/2.2.183. [DOI] [PubMed] [Google Scholar]
- 21.Keshamouni VG, Michailidis G, Grasso CS, Anthwal S, Strahler JR, Walker A, Arenberg DA, Reddy RC, Akulapalli S, Thannickal VJ, Standiford TJ, Andrews PG, Omenn GS. Differential protein expression profiling by iTRAQ-2DLC-MS/MS of lung cancer cells undergoing epithelial-mesenchymal transition reveals a migratory/invasive phenotype. J. Proteome Res. 2006;5(5):1143–1154. doi: 10.1021/pr050455t. [DOI] [PubMed] [Google Scholar]
- 22.Liu H, Sadygov RG, Yates JR. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004;76(14):4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 23.Chong PK, Gan CS, Pham TK, Wright PC. Isobaric tags for relative and absolute quantitation (iTRAQ) reproducibility: Implication of multiple injections. J. Proteome Res. 2006;5(5):1232–1240. doi: 10.1021/pr060018u. [DOI] [PubMed] [Google Scholar]
- 24.Schwacke JH, Hill EG, Krug EL, Comte-Walters S, Schey KL. iQuantitator: a tool for protein expression inference using iTRAQ. BMC Bioinf. 2009;10:342. doi: 10.1186/1471-2105-10-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 2001;98(9):5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolfinger RD, Gibson G, Wolfinger ED, Bennett L, Hamadeh H, Bushel P, Atshari C, Paules RS. Assessing gene significance from cDNA microarray expression data via mixed models. J. Comput. Biol. 2001;8(6):625–637. doi: 10.1089/106652701753307520. [DOI] [PubMed] [Google Scholar]
- 27.Jain N, Thatte J, Braciale T, Ley K, O’Connell M, Lee JK. Local-pooled-error test for identifying differentially expressed genes with a small number of replicated microarrays. Bioinformatics. 2003;19(15):1945–1951. doi: 10.1093/bioinformatics/btg264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.