

Abstract
Aims: Chronic granulomatous disease (CGD) is a primary immunodeficiency caused by mutations in the phagocyte reactive oxygen species (ROS)–producing NOX2 enzyme complex and characterized by recurrent infections associated with hyperinflammatory and autoimmune manifestations. A translational, comparative analysis of CGD patients and the corresponding ROS-deficient Ncf1m1J mutated mouse model was performed to reveal the molecular pathways operating in NOX2 complex deficient inflammation. Results: A prominent type I interferon (IFN) response signature that was accompanied by elevated autoantibody levels was identified in both mice and humans lacking functional NOX2 complex. To further underline the systemic lupus erythematosus (SLE)-related autoimmune process, we show that naïve Ncf1m1J mutated mice, similar to SLE patients, suffer from inflammatory kidney disease with IgG and C3 deposits in the glomeruli. Expression analysis of germ-free Ncf1m1J mutated mice reproduced the type I IFN signature, enabling us to conclude that the upregulated signaling pathway is of endogenous origin. Innovation: Our findings link the previously unexplained connection between ROS deficiency and increased susceptibility to autoimmunity by the discovery that activation of IFN signaling is a major pathway downstream of a deficient NOX2 complex in both mice and humans. Conclusion: We conclude that the lack of phagocyte-derived oxidative burst is associated with spontaneous autoimmunity and linked with type I IFN signature in both mice and humans. Antioxid. Redox Signal. 21, 2231–2245.
Introduction
Chronic Granulomatous Disease (CGD) patients are deficient in phagocyte oxidative burst, as they carry mutations in the critical subunits of the phagocyte NADPH oxidase 2 (NOX2) complex. While the hallmarks of CGD are recurrent life-threatening infections caused by catalase-positive bacteria and fungi (22), the disease is also characterized by hyperinflammatory and autoimmune manifestations without pathogen involvement. Inflammatory bowel disease (IBD) is the most common noninfectious inflammatory manifestation among CGD patients (36).
CGD patients are also known to have increased risk of developing systemic lupus erythematosus (SLE) (3, 12, 16, 19, 35, 56, 59, 63, 64). Anti-phospholipid syndrome, IgA neurophathy (16), Kawasaki disease (66), and polyarthritis (33) are other autoimmune manifestations reported to occur in CGD patients. In addition, to highlight the relationship between autoimmunity and NOX2 complex function, several recent genetic studies have provided us with data linking polymorphisms in the NOX2 complex components to autoimmune diseases (13, 27, 40, 43, 44, 48, 49, 61, 62).
Innovation.
The immunological pathways contributing to the hyperinflammatory phenotype that is associated with the loss of NOX2 complex function remain unknown. Here, we identified a prominent type I interferon (IFN) signature in both mice and chronic granulomatous disease (CGD) patients, both of which lack functional NOX2 complex. The observed type I IFN signature was associated with autoimmune manifestations involving spontaneous formation of inflammation-associated antibodies in both species and inflammatory kidney deposits in mice. Importantly, the autoimmune process was not triggered by pathogen-induced immune stimulation, as germ-free mice reproduced the same gene expression pattern as initially discovered in CGD patients and mice housed in a conventional environment.
The mechanism causing these noninfectious, inflammatory manifestations remains incompletely described. Similar to humans, mice with functional deficiency in the NOX2 complex develop more severe bacterial and fungal infections and exhibit less efficient pathogen killing (26, 39). Furthermore, even in rodents, this deficiency in phagocyte reactive oxygen species (ROS) production leads to enhanced autoimmune arthritis (24, 42), lupus (11), and, in addition, exacerbated innate immunity-mediated inflammation in ears (55), lungs (57), and joints (65).
We performed a genome-wide hypothesis-free gene expression analysis to find out the pathogenic mechanism driving inflammation in the CGD patients and in its Ncf1m1J mutated mouse model. We describe a predominant STAT1 (downstream type I interferon, IFN) signature in ROS-deficient humans and mice that is associated with elevated autoantibody titers in both species.
Results
CGD patients have impaired oxidative burst on phagocytes and on B cells
A cohort of 7 children with CGD along with their healthy family members (13 healthy controls and 4 heterozygous X-linked CGD carriers) was recruited (Table 1). All CGD patients were confirmed to have impaired oxidative burst in granulocytes, monocytes, and B cells when compared with healthy controls after PMA stimulation (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars).
Table 1.
Demographic and Clinical Data of the Chronic Granulomatous Disease Patients and Their Healthy Relatives
| Family | Individual | Mutation | Age | Age at 1st symptoms | CRP (mg/L) | Clinical history | Therapy |
|---|---|---|---|---|---|---|---|
| 1 | CGD (m) | CYBA | 16 | 9 months | 7.0 | Perianal abscess/Anal fissure, osteomyelitis, lymphadenitis cervical, colitis (Crohn-like) | Cotrimoxazol, itraconazol |
| 1 | Brother | None | 17 | n.a | <1.0 | No inf. | No med. |
| 1 | Cousin (m) | None | 20 | n.a | 1.1 | No inf. | No med. |
| 1 | Cousin (m) | None | 14 | n.a | <1.0 | No inf. | No med. |
| 1 | Father | None | Adult | n.a | <1.0 | No inf. | No med. |
| 1 | Mother | None | Adult | n.a | 10.8 | No inf. | No med. |
| 2 | CGD (f) | Unknown | 8 | 4 years | 1.9 | Nasal furuncolosis - Staphylococcus aureus, gengivoestomatitis/caries, oral candidiasis, palato enantema, aphthae | Cotrimoxazol, |
| 2 | CGD (m) | Unknown | 5 | 8 months | 1.2 | Urinary tract infection, gengivoestomatitis/caries, palato enantema, aphthae | Cotrimoxazol, itraconazol |
| 2 | Mother | None | Adult | n.a | <1.0 | No inf. | No med. |
| 2 | Father | None | Adult | n.a | 1.4 | No inf. | No med. |
| 3 | CGD (f) | NCF1 | 10 | 5 years | 1.5 | Oral ulcers; Burkolderia cepacia causing cervical lymph node and splenic abscesses; visceral Leishmaniasis leading to hepatosplenomegaly and pancytopenia | Cotrimoxazol, itraconazol s.c. IFN-γ (age 5–9) |
| 3 | Brother | None | 15 | n.a | <1.0 | No inf. | No med. |
| 3 | Mother | None | Adult | n.a | 1.4 | No inf. | No med. |
| 4 | CGD (m) | CYBB | 4 | 1 month | 14.4 | Pneumonia, cervical lymphadenitis | Cotrimoxazol, itraconazol |
| 4 | Sister | Carrier | 8 | n.a | <1.0 | No inf. | No med. |
| 4 | Mother | Carrier | Adult | n.a | 3.3 | No inf. | No med. |
| 4 | Father | None | Adult | n.a | <1.0 | No inf. | No med. |
| 4 | Cousin (m) | None | 8 | n.a | <1.0 | No inf. | No med. |
| 5 | CGD (m) | CYBB | 5 | 3 weeks | 2.7 | Perianal abscess, intra-abdominal infections, enlarged, lymph nodes, sepsis, pneumonia, lymphadenitis, granulomas in colon | Itraconazol, sulfatrimetoprim, ASA, penicillin, immunoglobulin infusions |
| 5 | Sister | Carrier | 7 | n.a | <1.0 | No inf. | No med. |
| 5 | Mother | Carrier | Adult | n.a | 3.3 | No inf. | No med. |
| 6 | CGD (m) | Unknown | 4 | 22.7 | Colitis | Prednisolon | |
| 6 | Brother | None | 17 | n.a | 1.1 | No inf. | No med. |
| 6 | Mother | None | Adult | n.a | 3.9 | No inf. | No med. |
n.a., not applicable; (m), male; (f), female; No inf., no infections of chronic inflammation; No med., no regular intake of medication that could interfere with the immune system.
Gene expression profiling revealed significant changes in gene expression in the blood samples of CGD patients
Whole blood samples were subjected to genome-wide gene expression analysis using Illumina HumanHT-12 v3 Expression BeadChip technology. Of all the differentially expressed transcripts between CGD patients and healthy controls, the majority (58%, n=182) was upregulated. The technical performance of the microarray was verified by quantitative real-time polymerase chain reaction (Q-RT-PCR) analysis for the 10 most significantly (i.e., lowest p-value) differentially up- and downregulated (n=10+10) transcripts. All selected transcripts that were upregulated in CGD patients were successfully reproduced (Supplementary Table S1). The downregulated genes could not be statistically significantly reproduced by Q-RT-PCR, although 90% of the tested downregulated genes reproduced the trend that was observed in array analysis.
Principal component analysis of CGD data Supplementary Fig. S2A) as well as mouse blood (Supplementary Fig. S2B) and spleen data (Supplementary Fig. S2C) showed that the observed gene expression differences did not alter the global gene expression profile in the studied samples. Different genotypes did not clearly cluster together, but were randomly spatially scattered.
Hypothesis-free Ingenuity pathway analysis revealed 27 significantly differentially regulated canonical pathways between CGD patients and the healthy controls (Supplementary Table S2). The top three most significantly affected pathways were as follows: B-cell development, role of pattern recognition receptors in recognition of bacteria and viruses, and IFN signaling. Thirteen out of the 20 most significant pathways had a direct immunological connection and the functions of these pathways covered IFN signaling, lymphocyte (LC) function, and inflammation.
The transcription of type I IFN-responsive genes was upregulated in CGD patients
All differentially regulated transcripts that fulfilled the inclusion criteria (adjusted p-value<0.05 and fold change (FC)>1.5) were manually curated into IFN-regulated (IRG), LC-related and inflammatory (INFL) transcripts. By definition, IRGs are genes that are expressed after exposure to IFNs and they can be divided into two main categories: Type I (induced by IFN-α and IFN-β) and type II (induced by IFN-γ) IRGs (45).
Most (53%) of the upregulated transcripts in CGD samples were IRGs (Fig. 1A). A prototype IFN-α-responsive gene, IFI27, was the most powerfully upregulated gene in the CGD patient cohort (FC=28.9) (Fig. 1B and Supplementary Table S3) (58). Another type I IRG, IFI44L, was upregulated in the CGD patient samples with FC 8.9 (47). OAS1, OAS2, OASL, and MX1 are additional type I IRGs that were more than 2.5-fold upregulated in CGD patients (4). All of the top 10 transcripts with the largest FC between patients and controls displayed an intermediate phenotype in X-linked CGD carriers (Fig. 1B).
FIG. 1.

Chronic granulomatous disease patients display a type I interferon signature. (A) All significantly differentially regulated transcripts (adjusted p-value<0.05 and FC>1.5) that were upregulated in chronic granulomatous disease (CGD) patients were curated into interferon-regulated (IRG), lymphocyte-related (LC), and inflammatory (INFL) transcripts. The numbers and percentages of genes in each category are presented. (B) Gene expression analysis results describing the ten significantly differentially regulated genes (corrected p-value<0.05) with the largest fold change between CGD patients and healthy controls are presented. CGD patients n=7, x-linked carriers n=4, healthy controls n=13. Benjamini–Hochberg corrected p-values for the correlation between the gene expression and phenotype are given. Boxplots present the minimum, maximum, median, and the 25% and 75% quartiles for each gene; the angular dashed line illustrates the slope of the correlation.
Importantly, we did not observe any differences in the expression levels of genes coding for IFN cytokines (data not shown), but all reported changes in gene expression took place in cellular processes downstream of IFN receptor signaling.
CGD patients had alterations in the peripheral blood B-cell populations, which translated into changes in the gene expression profile
B LC-related transcripts (22%) formed the second distinguishable group of upregulated genes in the CGD patients. CD79A, CD79B, and CD19, all of which were intimately connected with B-cell receptor signaling, as well as VPREB3, CD72, and CD38, regulators of B-cell maturation (46), were more than two-fold upregulated in the CGD samples. Furthermore, flow cytometry analysis revealed that the mean fluorescence intensity of CD38 expression on the B cells from CGD patients (95.9±26.5) was significantly (p=0.0021) higher than in the controls (25.3±6.4). The carriers displayed an intermediate phenotype (33.8±15.8). The observed B LC signature could be explained by the increased number and higher percentage of circulating CD19+ B cells in the CGD patients when compared with the healthy controls (Fig. 2A, B). In addition, CGD patients had more naïve IgD+CD27− B cells, and less memory B cells (both nonswitched IgD+CD27+ and switched IgD−CD27+) than the control group. The analysis of T- and NK-cell populations yielded no differences between the patients and healthy controls, while the CD11b+CD14+CD16− classical monocytes were significantly lower in the CGD blood (Fig. 2C, D).
FIG. 2.
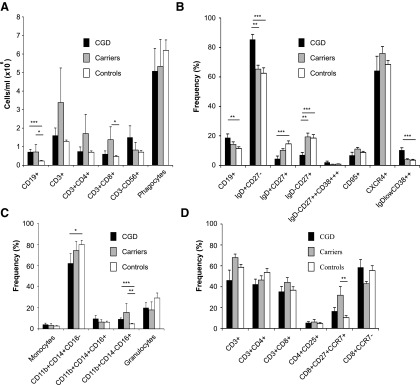
Chronic granulomatous disease patients have alterations in their B-lymphocyte populations. (A) Total cell counts per ml of blood of the main leukocyte populations. Total CD4+ T cells and total CD8+ T cells were calculated within total CD3+ cells. CD11b++SSChiFCShi are total phagocytes. (B) The frequency of total CD19+ B cells was calculated within total lymphocytes. The frequencies of the different B-cell subsets were calculated within total CD19+ B cells. (C) Frequency of total CD11b+ monocytes and CD11b++SSChi granulocytes were calculated within total leukocytes. Frequencies of monocyte subsets were calculated within CD11b+ cells. (D) The frequency of CD3+ T cells was calculated within total lymphocytes. The frequency of the CD4+, CD8+, and CD4+CD8+ cell subsets was calculated within total CD3+ T cells. The frequency of the CD4+ subsets was calculated within total CD4+ T cells, and that of the CD8+ subsets was calculated within the total CD8+ T cells. Bars represent average frequencies,±SEM, Kruskall–Wallis test with Bonferroni–Dunn post hoc analysis. *Represents p<0.05, **represents p<0.01, and ***represents p<0.001 between the indicated groups.
The downregulated genes in CGD patients gave no specific information about immunological pathways, which is most likely explained by compromised gene expression data quality. None of the tested downregulated genes could be statistically significantly reproduced by Q-RT-PCR (Supplementary Table S1).
The downstream type I IFN signature also predominated in the Ncf1 mutated mouse
Similar gene expression analysis as was done for the CGD patients was performed using its Ncf1m1J mutated mouse model. We identified a larger number of differentially regulated genes in the spleen samples (n=36) than in the blood samples (n=17), and the majority (n=22 and n=11, respectively) of the differentially expressed genes were upregulated in the Ncf1m1J mouse. (5)
The most significantly differentially expressed genes (adjusted p-value<0.05 and FC>1.5) in the mouse samples were manually curated into IRG, LC-related, and inflammatory (INFL) transcripts. Most (n=6, 55%) of the manually curated genes in mouse blood samples were IRGs (Fig. 3A and Supplementary Table S4).
FIG. 3.

ROS-deficient Ncf1m1J mutated mice share the type I IFN response. (A) All significantly differentially regulated transcripts (adjusted p-value<0.05 and FC>1.5) that were upregulated in ROS-deficient BQ.Ncf1m1J mice were curated into interferon-regulated (IRG), lymphocyte-related (LC), and inflammatory (INFL) transcripts. The percentages and the numbers of genes in each category are presented. BQ.Ncf1m1J n=12 and in BQ n=12. (B) STAT1 expression in wild-type (BQ) and Ncf1 mutated (BQ.Ncf1m1J) mice was analyzed in two independent pooled experiments by flow cytometry. Heparinized blood was subjected to red blood cell lysis and stained for STAT1 expression. The normalized GeoMean values (BQ signal set as 100) of the STAT1-specific signal are reported. Mean±SEM, n=11–12, two-tailed unpaired Student's t-test. (C) Heparinized blood was suspended into IMDM (Gibco) supplemented with 10% FCS and heparin. The cells were stimulated with recombinant mouse IFN-β (1000 U/ml; GenWay Biotech) at +37°C for 10 min and stained with pSTAT1-PE (pY701) antibody to assess the levels of activated STAT1. Results were calculated as the GeoMean for PE fluorescence, mean±SEM, n=6, two-tailed Mann–Whitney U test. The relative frequencies of CD3+, CD3+CD8+, CD3+CD4+ T lymphocytes, and B220+ B lymphocytes were analyzed in the (D) blood and (E) spleen samples from BQ.Ncf1m1J and BQ mice. Phagocytes were divided into CD11b+Gr-1HI granulocytes and CD11b+Gr-1− monocytes (and CD11c+ dendritic cells in spleen samples). Mean±SEM, n=16, two-tailed unpaired Student's t-test. ***p<0.001, **p<0.01, *p<0.05, ns, not significant.
The expression of Stat1, a transcription factor that regulates IFN signaling, was upregulated (FC 1.6) in the Ncf1m1J mice. The transcription of Stat1 has been shown to be more potently upregulated by type I IFNs (IFN-α and IFN-β), although IFN-γ also stimulates Stat1 expression (4). The transcriptional difference in Stat1 expression was confirmed at the protein level by intracellular flow cytometry analysis (Fig. 3B). After IFN-β treatment, there was significantly more phosphorylated (Signal transducer and activator of transcription 1) STAT1 in the NCF1-deficient cells than in the wild-type cells, thus reflecting the observed difference in the total STAT1 level (Fig. 3C).
Irgm and Iigp2 (also known as Irgm2) were other significantly upregulated genes in the blood samples collected from the naïve Ncf1m1J mice. Since the promoter regions of these genes contain γ-IFN activation sites (GAS) and IFN-stimulated response elements, they can be switched on by both type I and II IFNs. Similarly, the transcription of Ifitm3 (9), Gvin1 (29), and Gbp2 (15) was upregulated in the Ncf1m1J mutated mice. Furthermore, Oasl2, a relatively sparsely studied family member of the type I IFN-responsive OAS proteins (51), was upregulated in the Ncf1m1J mouse.
Similar to CGD patients, no significant differences were observed in the mouse with regard to the gene expression levels of IFNs (alphas, beta, zeta, epsilon, and kappa) (data not shown) or serum levels of IFN-α.
Expression of inflammation-related genes in the Ncf1m1j mutated mouse
Flow cytometry analysis of the main leukocyte populations could not reveal any significant differences in the granulocyte and macrophage populations in either blood or spleen samples. (Fig. 3D, E). Lactotransferrin (coded by Ltf) and proteinase 3 (coded by Prtn3) are proteins localized in the neutrophil granules that were significantly upregulated in the Ncf1m1J mutated mice when compared with the wild-type mice. Additional proinflammatory transcripts that contributed to the inflammatory signature observed in the Ncf1m1J mouse (Fig. 3A) include Ctsg, Lcn2, and Mpo.
Ncf1 was the only significantly downregulated gene in the blood samples collected from the Ncf1m1J mice. The complete blockage of induced oxidative burst in the Ncf1m1J mouse is due to a splice site point mutation that abolishes functional NCF1 expression (23, 24, 52). This explains the emergence of Ncf1 among the top downregulated genes in both blood and spleen samples (Supplementary Table S4).
The Ncf1m1J mice had reduced numbers of splenic CD3+CD27+ T cells (Supplementary Fig. S3), which explains the emergence of CD27 among downregulated genes. Similarly, the reduced CD8 expression could be assigned for the reduced CD3+CD8+ LC population observed in the spleen samples (Fig. 3E).
Blood B LC populations in the Ncf1m1J mice presented a similar subset distribution as in CGD patients, with a significantly reduced B-cell memory compartment (Fig. 4) and no alterations in the frequency of cells expressing activation (CD69), apoptosis (CD95), or homing (CXCR4) surface markers. However, no reduction in the total B-cell compartment was observed, which explains the lack of transcriptional B-cell signature in the Ncf1m1J mice.
FIG. 4.

ROS deficient Ncf1m1J mutated mice have a reduced total memory B-cell compartment. Flow cytometry analysis of heparinized peripheral blood from the Ncf1m1J mutated and BQ wild-type mice was performed to determine B-cell subset frequencies. The frequency of total B cells (CD19+) was calculated within the total lymphocyte gate based on FSCxSSC. Total memory B cells (CD19+CD23+CD21+IgM+/−IgD−) and naïve B cells (CD19+IgM+IgD++) were calculated within total B cells. B cells expressing activation marker CD69 (CD19+CD69+), homing receptor (CD19+CXCR4+), or the apoptosis-associated Fas receptor (CD19CD95+) were calculated within total B cells. Mean±SEM, n=6/group, two-way Mann–Whitney test.
CGD patients and the Ncf1m1J mutated mice had elevated levels of lupus-associated autoantibodies
Since the type I IFN response is known to be associated with autoimmune diseases, in particular SLE, we systematically screened all subjects enrolled in the study for the presence of antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibodies (ANCA), anti-cyclic citrullinated peptide (CCP) antibodies, anti-saccharomyces cerevisiae antibodies (ASCA), and the rheumatoid factor (RF).
Inflammation-associated antibodies were detected in all CGD patients. We detected IBD-associated ASCA (either IgG or both IgG and IgA isotypes) in six patients (Fig. 5A, B). SLE-associated anti-double-stranded DNA (dsDNA), anti-Th/To ribonucleoprotein (Th/To), and/or vasculitis-associated anti-bactericidal permeability increasing protein (anti-BPI) antibodies were detected in the three of the ASCA-positive patients. A single patient was positive for RF. Clinically, significant levels of anti-CCP autoantibodies could not be detected in any of the patients. However, the anti-CCP titers correlated with the genotype, with CGD patients displaying higher anti-CCP titers than the healthy controls (Fig. 5C). Among the CYBB-mutation carriers, only one had borderline positivity for RF; whereas the remaining carriers were negative for all the tested autoantibodies. Low but noteworthy levels of autoantibodies (ASCA, RF, and ANA) were present in six of the healthy noncarrier family members.
FIG. 5.

Anti-Saccharomyces cerevisiae antibodies (ASCA) and anti-cyclic citrullinated protein antibodies in chronic granulomatous disease patients. (A) IgG and (B) IgA ASCA were analyzed from serum samples from all subjects enrolled in the study. (C) Similarly, the titers of anti-cyclic citrullinated protein (CCP) antibodies were measured. Bar indicates the mean of each group, Kruskall–Wallis test followed by Bonferroni post hoc test. CGD patients n=7, x-linked carriers n=4, and healthy controls n=13. *Represents p<0.05, **represents p<0.01, and ***represents p<0.001 between the indicated groups.
The Ncf1m1J mutation does not induce spontaneous formation of lupus-associated autoantibodies on C57BL/10.Q/rhd genetic background. Therefore, we backcrossed the mutation to Balb/c mice that are known to be susceptible to pristane-induced lupus (53).
Significantly elevated levels of anti-dsDNA and anti-Sm/ribonucleoprotein (Sm/RNP) antibodies were found in Balb/c.Ncf1m1J mice (Fig. 6A, B). The observed increase in anti-histone autoantibodies did not reach statistical significance (Fig. 6C). Likewise, naïve Balb/c.Ncf1m1J mice aged between 8 and 16 weeks showed significant deposits of IgG (only micrograph shown) and complement factor C3 (Fig. 6D, E) within their kidney glomeruli. Thus, the observed type I IFN response is likely to be associated with lupus-related autoimmunity in both CGD patients and its Ncf1m1J mutated mouse model.
FIG. 6.

ROS-deficient naïve Balb/c.Ncf1m1J mutated mice spontaneously produce lupus-associated autoantibodies and develop inflammatory kidney inflammation. (A) Anti-dsDNA, (B) anti-Sm/RNP, and (C) anti-histone autoantibodies were measured in sera of wild-type and Ncf1m1J mutated Balb/c mice using ELISA. Horizontal lines represent the mean medians. Two-tailed Mann–Whitney U test, *p<0.05, and **p<0.01, ns, not significant, n=47 in Balb/c.Ncf1m1J and n=21 in Balb/c mice. (D) Representative immunofluorescence images of glomeruli from wild-type and Ncf1m1J mutated mice stained with FITC-conjugated antibodies to mouse IgG and complement factor C3. Glomeruli of Ncf1m1J mutated mice revealed moderate granular IgG deposits, mostly in mesangial areas, and a marked mesangial pattern of complement factor C3-deposition. In contrast, glomeruli from wild-type mice showed only minimal depositions of IgG or C3 in the glomeruli. Microscope; Nikon Eclipse 80i, Nikon Plan Apo 20×0.75 objective, Camera: Nikon DS-QiMc, Original magnification×200, DAKO Fluorescent Mounting Medium, scale bar 50 μm. (E) Glomerular deposition of IgG and C3 was analyzed using a semiquantitative scoring system. 0=no deposits, 1=mild mesangial deposition, 2=marked mesangial deposition, 3=mesangial and slight capillary deposition, 4=intense mesangial and mesangocapillary deposition. Horizontal lines represent the medians. n=5 in Balb/c.Ncf1m1J and n=6 in Balb/c mice. Horizontal lines represent the medians, two-tailed Mann–Whitney U test, *p<0.05, ns, not significant.
Germ-free Ncf1m1J mice confirmed the endogenous origin of the type I IFN signature
Even though all CGD patients and all experimental mice used in the study were free from clinically detectable infections, all of them were surrounded by and interacting with normal flora. So far, the role of microbial products as inducers of granuloma and as autoimmunity inducing factors has not been conclusively elucidated.
The type I IFN response is evoked to combat viral infections, and it can be induced by either viral or bacterial epitopes (37). The mice used in the study were clinically fully healthy and according to institutional health monitoring, they were free from all investigated FELASA-listed pathogens. However, the monitoring revealed the presence of anti-mouse norovirus (MNV) antibodies. MNV is a nonpathogenic virus commonly occurring in animal colonies.
All BQ wild type (100%) and six out of the eight tested (75%) Ncf1m1J mutated mice were positive for anti-MNV antibodies. The antibody titers in the Ncf1m1J mutated mice were significantly lower than in the BQ wild-type mice (Fig. 7A).
FIG. 7.
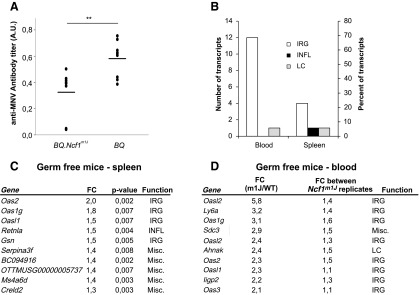
The type I IFN signature was confirmed in naïve germ-free BQ.Ncf1m1J mutated mice. (A) Anti-mouse norovirus antibodies in mouse sera were analyzed by fluorometric immunoassay. Results are presented as arbitrary units (A.U.). Mean±SEM, in BQ.Ncf1m1J n=8 and in BQ n=9, two-tailed unpaired Student's t-test, mean values±SEM are presented, **p<0.01. Spleen and blood samples from germ-free mice were subjected to genome-wide gene expression analysis. (B) All significantly differentially regulated transcripts (p-value<0.05 and FC>1.3) that were upregulated in the spleen samples collected Ncf1m1J mutated were curated into interferon-regulated (IRG), lymphocyte-related (LC), and inflammatory (INFL) transcripts. A similar analysis was performed for the transcripts yielding the largest possible difference between the genotypes but the smallest possible difference between the Ncf1m1J mutated replicates. The numbers and percentages of genes in each category are presented. In spleen samples, n=6 in both groups, in blood samples BQ.Ncf1m1J n=2, and in BQ n=1. (C) Ten significantly differentially regulated transcripts with the largest difference (FC) between the genotypes in spleen samples and (D) ten transcripts with the largest difference between the genotypes but the smallest difference between the Ncf1m1J mutated replicates are presented with functional curation into the groups mentioned earlier (IRG, LC, and INFL).
To analyze the role of environmental microbes as inducers of the transcriptional type I IFN signature, we transferred the mice to a germ-free facility. Spleen samples from the germ-free mice were of excellent quality, but due to the challenging sampling environment, we could only analyze blood samples from one wild-type mouse and from two Ncf1m1J mice.
Type I IRGs dominated the lists of most significantly differentially regulated genes in the germ-free mice (Fig. 7B). In the spleen, four out of the five transcripts with largest transcriptional upregulation in the Ncf1m1J mouse were classical type I IRGs (Fig. 7C). Similarly, in blood samples, 12 out of the 17 transcripts with largest transcriptional upregulation in the Ncf1m1J mutated mouse were predominantly type I IRGs (Fig. 7D). Oas1g, Oas2, and Oasl1 are the type I IRGs that were upregulated in both tissues. Furthermore, the transcription of Oasl2, Oas3, and Iigp2 was upregulated in the germ-free blood samples from the Ncf1m1J mice.
Interestingly, no signs of type II IFN response or granulocytic inflammation were seen in the germ-free samples. Taken together, these results show that the upregulated type I IFN signature in the ROS-deficient mouse is not dependent on microbial antigens but is of endogenous origin.
Discussion
Comparative translational genome-wide gene expression analysis revealed a pronounced type I IFN signature in both mice and humans in the absence of functional phagocyte ROS production. The classically SLE-associated type I IFN response was accompanied by signs of developing autoimmunity in both species. All CGD patients who were included in the study were tested positive for inflammation-associated antibodies or autoantibodies, and elevated levels of anti-dsDNA anti-Sm/RNP and anti-histone autoantibodies along with signs of inflammatory kidney disease were detected in the ROS-deficient Balb/c.Ncf1m1J mice.
Type I IFN signature is a well-established finding in autoimmune diseases such as SLE (6, 7) and Sjögren's syndrome. An activated type I IFN signature was also recently shown to precede seroconversion and to exist throughout the development of type I diabetes (28). We are the first to describe the induction of a type I IFN signature in CGD patients and in the corresponding Ncf1m1J mutated mouse model. The observed type I IFN signature was not associated with alterations in the transcription levels of IFN cytokines, but was completely dominated by transcripts whose expression is upregulated after IFN receptor activation-inducible Jak-Stat signaling cascade.
The connection between SLE and CGD is not novel, as already in the early 1970s two carriers of the x-linked form of CGD were reported to exhibit discoid lupus (54). The augmented SLE incidence in the x-linked CGD carriers with partially impaired ROS production has also been extensively validated (10).
CGD patients have been previously shown to have lower numbers of T cells (21) and reduced peripheral blood memory T- and memory B-cell compartments (8, 20). Here, we reproduce those findings of a reduced B-cell memory compartment in the CGD patients, and we show for the first time that a similar reduction is present in Ncf1m1J mice. The reduced memory B-cell compartment, however, has been shown not to affect the humoral immunologic memory (38). However, we also report, for the first time, an increase in the frequency of circulating type 1 transitional B cells in CGD patients. These cells link B-cell development in the bone marrow and spleen, and have been reported to be increased in the blood of SLE patients (60), which further stresses an SLE-like signature in CGD patients. In the presence of IFN-α, B cells increase their surface expression of CD38 (18), which is involved in cell adhesion (17) and apoptosis prevention (67). Therefore, we hypothesize that the higher expression of CD38 found on CGD B cells could be a consequence of the marked type I IFN signature.
Autoantibodies are the hallmark of SLE, but the prevalence of autoantibody positivity in CGD patients has not been systematically addressed earlier. There is a report describing a CGD patient with juvenile arthritis that was associated with elevated RF (33), and another CGD patient with SLE has been reported as ANA and anti-dsDNA antibody positive (35). Furthermore, a study of several cases of CGD with autoimmune manifestations reported the presence of autoantibodies in many, but not in all CGD patients (16). Even though ASCA are primarily associated with IBD, increased ASCA levels have also been measured from patients with SLE (14). Our systematic autoantibody analysis revealed that all CGD patients enrolled in the study were positive for either SLE-associated ANA or ANCA or for ASCA. Furthermore, the genotype correlated significantly with anti-CCP antibody titers, with CGD patients displaying higher anti-CCP levels than the controls.
The antibody findings support the notion of developing autoimmunity in CGD patients. SLE patients are known to develop disease-associated anti-SSA antibodies even years before the disease becomes clinically apparent (2) and similarly, autoantibodies are reported to precede and predict the development of rheumatoid arthritis (30, 31). Furthermore, ASCA and ANCA are reported to predict the development of IBD (25).
The Ncf1m1J mouse with a point mutation in the activator subunit of the NOX2 complex displayed a similar type IFN signature as was first identified in CGD patients. Furthermore, the mouse model also exhibited clear signs of developing autoimmunity with elevated autoantibody titers and histologically evident inflammatory kidney pathology on Balb/c background. Importantly, all the autoimmune manifestations reported in the mouse were identified in naïve state. The same mouse model has previously been shown to develop enhanced experimental autoimmune arthritis and encephalomyelitis (24, 42) and lupus on MRL.RAS(lpr) background (11). Furthermore, the Ncf1m1J mouse has also been reported to spontaneously develop clinically evident erosive arthritis postpartum and (24) and with older age (32).
Furthermore, since the life expectancy of CGD patients is increasing, the development of overt autoimmune diseases has become a therapeutic challenge for CGD (34). Therefore, early detection of autoantibodies in CGD patients might provide an efficient means for the prophylaxis of autoimmunity, in particular SLE and IBD.
In SLE, autoantibodies are reported to show cross-reactivity with viral and bacterial epitopes,46 suggesting that environmental priming may participate in the disease progression. In addition, compromised bacterial and fungal defense expose the ROS-deficient mice and CGD patients to prolonged contact with pathogenic microbial antigens. To study the potential role of environmental priming in the development of type I IFN response, we collected blood and spleen samples from germ-free mice. The type I IFN response was replicated in the germ-free samples, while no type II IFN response-related or phagocyte-derived inflammatory transcripts were upregulated in the germ-free Ncf1m1J mice. Thus, we conclude that the type I IFN signature was induced by an endogenous process and was not triggered by microbiological stimulation.
Materials and Methods
Patient samples
Blood was collected from seven children with CGD [according to the International Union of Immunological Societies classification of primary immunodeficiencies (1)], from eight age-matched healthy relatives, and from nine adult healthy relatives. Identification of the mutations in the NOX2 components was done by either flow cytometry or Western blot using antibodies specific for gp91phox, p22phox, p47phox, and p67phox as previously described (50). Four of the healthy controls were x-linked CGD carriers (Table 1). Gene expression samples (2 ml) were collected into RNA PaxGene Blood RNA tubes (Becton Dickinson, Franklin Lakes, NJ), and heparinized samples (3 ml) were used in flow-cytometry analysis. At the time of collection, the CGD patients had no observed undergoing infections, no clinically manifested inflammatory processes and were not under IFN-γ therapy. The healthy relatives had no clinical signs of infection or inflammation, did not take any immune-suppressant drugs, nor had a history of chronic infections or inflammation. At the time of blood collection, patients were taking the WHO/IUIS-approved prophylactic medication for CGD as indicated in Table 1. However, since these are antibiotics and antifungal drugs, their influence as immune-modulators was considered negligible.
Flow cytometry on human samples
White blood cells from whole blood were stained with fluorochrome-conjugated monoclonal antibodies against CD19, CD27, CD38, CD3, CD4, CD8, CCR7/CD197, CXCR4/CD184, CD25, CD11b, CD16, CD14 (Ebioscience), CD56 (Biolegend), IgD, and CD95 (BD Biosciences). Data were acquired on an FACSCalibur (BD Biosciences), and analyzed with FlowJo Software v7.6 (Tree Star, Inc.). Statistical analyses were performed by using the Kruskal–Wallis one-way analysis of variance with Bonferroni–Dunn post hoc analysis; p-values<0.05 were considered statistically significant.
PHAGOBURST™ kit (Glycotope Biotechnology) was used to measure the oxidative burst of peripheral blood phagocytes from all subjects. Data were acquired using Coulter Epics XL-MCL (Beckman Coulter, Inc.), and analyzed with FlowJo Software v7.6.
Autoantibody analysis in CGD patients
All subjects were screened for ANA, ANCA, anti-CCP antibodies, ASCA, and the RF. ANA were quantified by direct immunofluorescence on Hep2 cells (ANA HEp-2 cells IFA kit; Menarini Diagnostics). When ANA titers were positive (>1/160) with a speckled or homogenous pattern, the presence of anti-DsDNA, anti-SM, anti-SSA, anti-SSB, and anti-RNP antibodies was analyzed by Fluoro-Immuno-Enzymatic assay (FEIA); whenever there were 46 nuclear dots, the presence of anti-centromer antibodies was detected by FEIA; and for ANA, positive titers with nucleolar pattern anti-Slc-70 antibodies were assessed by FEIA. The presence of anti-RNA polymerase I and III, anti-PM-SCL and anti-KU antibodies was assessed in anti-Slc-70-positive samples. Serum ANCA was detected by direct immunofluorescence on leukocytes. Subjects displaying positive titer (>1/20) of P- and/or C-ANCA, the presence of anti-PR3, anti-myeloperoxidase, anti-catepsin-G, anti-BPI, anti-lactoferrin, and anti-elastase were assessed by immunoblot or FEIA. X-ANCA-positive sera were not further evaluated. The serological titer of IgM anti-IgG RF was determined by immunoturbidimetry. The presence of anti-CCP1 antibodies was measured by FEIA. Serum titers of IgG+ or IgA+ ASCA antibodies were quantified by ELISA. Statistical analyses of ASCA IgG, ASCA IgA, and anti-CCP antibody levels between the different experimental groups were performed by using the Kruskal–Wallis one-way analysis of variance with Bonferroni–Dunn post hoc analysis; p-values<0.05 were considered statistically significant.
Mice
Age- and sex-matched 8–12 week-old, specific pathogen-free C57BL/10.Q/rhd mice with (BQ.Ncf1m1J) and without (BQ) the m1J mutation in the Ncf1 gene (24) were used. The mutation was backcrossed for more than ten generations onto BQ and Balb/c genetic background, and the BQ.Ncf1m1J mice were ascertained to be genetically clean even in the linked fragment. The germfree BQ and BQ.Ncf1m1J mice were produced at the Core Facility for Germ-free Research, Karolinska Institutet, Stockholm, Sweden.
Mouse sample collection and flow cytometry
After hypotonic lysis of the red blood cells and Fc block (CD16/CD32, 2.4G2), blood and spleen leukocytes cells were stained with rat anti-mouse monoclonal antibodies anti: CD3e-FITC (145-2C11), CD4-AF647 (RM4-5), CD8a-PerCpCy5.5 (53–6.7), B220-Pacific Blue (RA3-6B2), CD11c-APC (HL3) Gr-1-FITC (RB6-8C5), CD11b-APC (M1/70) (BD Biosciences), and CD11b-PE (M1/70) (eBioscience). For STAT1 phosphorylation analysis, the cells were suspended into Lyse&Fix solution (BD Biosciences), permeabilized with 90% methanol (30 min), and stained with STAT1-PE (1/Stat1), IgG1-PE isotype control (X40), or pSTAT1-PE (pY701) (4a) antibody (BD Biosciences). Samples were acquired using BD LSR II operated by Diva software (BD Biosciences), and the data were analyzed using Flowing Software (Cell Imaging Core, Turku Centre for Biotechnology). Identification of blood B-cell subsets was done using rat anti-mouse monoclonal antibodies anti: IgD-FITC (11-26c2a), CXCR4-AF647 (TG12/CXCR4), CD69-PE (H1.2F3), CD21/CD35-FITC (7E9), CD23-PE (B3B4) (Biolegend), IgM-APC (III41), CD95-APC (15A7), and CD19 PerCpCy5.5 (eBio 1D3) (Ebioscience). Samples were analyzed on an FACSCalibur (BD Biosciences), and data were analyzed with FlowJo software v7.6.
Two-tailed Mann–Whitney U test was used to evaluate whether null hypothesis can be rejected when the groups did not have equal variance (pSTAT assay and B-cell subset quantification). Student's t-test was used with groups of equal variance (STAT1 expression and relative frequencies of different blood and spleen cells); p-values<0.05 were considered statistically significant.
Lupus-associated autoantibodies and renal pathology in mouse
For detection of autoantibodies in sera from 8- to 16 week-old naïve Balb/c.Ncf1m1J and BALB/c mice, microtiter plates were coated overnight with 50 μg/ml histone from calf thymus (Roche Diagnostics), 1 μg/ml Sm/RNP (GenWay Biotech), or precoated with 20 μg/ml poly-L-Lysine (Sigma-Aldrich) before coating with 20 μg/ml calf thymus DNA (Sigma-Aldrich), respectively. Sera were added at a 1:20 dilution in PBS containing 2% FCS. Bound IgG was detected with a horseradish peroxidase-conjugated goat anti-mouse IgG (Southern Biotech). Two-tailed Mann–Whitney U test, p-values<0.05 were considered statistically significant.
For assessing glomerular deposits, PBS-flushed kidneys were embedded in OTC Tissue-Tek (Sakura) compound, snap frozen, and stored at −80°C. Five mocrometer cryo-sections were fixed, permeabilized with acetone, and stained with FITC-labeled antibodies to complement factor C3 (Cedarlane) and IgG (BioLegend). Glomerular deposits were evaluated by two blinded individual scorers by immunofluorescence microscopy using a semiquantitative scoring system as previously described (41). Two-tailed Mann–Whitney U test, p-values<0.05 were considered statistically significant.
Genome-wide gene expression analysis
RNA preservation, isolation, and purification reagents are listed in Supplementary Table S5. RNA amplifications were performed using Illumina Total Prep RNA Amplification kit (Ambion); in vitro transcription and biotinylation were performed overnight. cRNA concentration measurement with Nanodrop ND-1000 was followed by Experion electrophoresis station (Biorad) quality check.
Sentrix BeadChip Array MouseRef-8 v2 (Illumina) and HumanHT-12 v3 Expression BeadChip (Illumina) hybridizations were detected with Cyanine3-streptavidine; the arrays were scanned with Bead Array Reader (Illumina); and the numerical results were extracted with Bead Studio v3.2.6 without normalization or background subtraction.
The microarray data were quantile normalized, log2 transformed, and filtered for absent calls (germ-free data) and differentially regulated genes were identified using linear modeling (LIMMA packages; Bioconductor). Hierarchical clustering divided Finish and Portuguese CGD samples into two separate groups, and the samples were treated as batches in subsequent data analysis. Neither mouse blood nor spleen samples revealed biased clustering. For all further analyses, false discovery rate (FDR, Benjamini–Hochberg procedure) adjusted p-values<0.05 were considered statistically significant. Blood samples from germ-free mice had compromised RNA quality, and the analysis was restricted to the good-quality samples (BQ.Ncf1m1J n=2, BQ n=1). Transcripts were ranked to yield a list of genes with highest possible FC between the genotypes but the smallest variation between the mutant replicates. The microarray data have been deposited in the NCBI gene expression omnibus (GEO) MIAME-compliant database with accession numbers GSE44969 (CGD data) and GSE45125 (mouse data).
The relationship between the genotype and the log2-transformed gene expression level was analyzed for all probes (n=48 802, no absent call filtering) by fitting the data into the linear regression model. The slope p-values were corrected for multiple testing by using the Benjamini–Hochberg procedure.
Ingenuity pathway analysis
The canonical pathways from transcripts with adjusted p-value p<0.05 were generated using the Ingenuity pathway analysis (IPA, Ingenuity Systems, www.ingenuity.com). The IPA software measured the significance of the association between the data set and the canonical pathway in two ways: (i) A ratio of the number of molecules from the data set that map to the pathway divided by the total number of molecules which map to the canonical pathway. (ii) Fisher's exact test was used to calculate a p-value determining the probability that the association between the genes in the dataset and the canonical pathway is explained by chance alone.
Manual curation of the most significantly differentially expressed genes
Selected transcripts passing inclusion criteria (adjusted p-value<0.05 and FC>1.5) were curated into IRG, LC-related, inflammatory (INFL), and miscellaneous (Misc.) transcripts by accessing Interferome.org and PubMed Gene (www.ncbi.nlm.nih.gov/gene) databases (June 2012).
Quantitative RT-PCR
The most significantly differentially regulated transcripts were confirmed by Q-RT-PCR. RNA was reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad) followed by Q-PCR amplification using TaqMan Universal Master Mix (Applied Biosystems) with primers and probes specified in Supplementary Table S1. PCR cycling was performed using Applied Biosystems 7900HT Fast Sequence Detection System. p-values<0.05, calculated by using Student's t-test, were considered statistically significant.
Anti-MNV antibodies
MNV antibody analysis was performed by multiplexed fluorometric immunoassay at Surrey diagnostics Ltd. Statistical significance was tested by using the two-tailed unpaired Student's t-test; p-values<0.05 were considered statistically significant.
Study approvals
All subjects were enrolled after signed written consent had been obtained. Parents provided consent for the participants younger than 18 years of age. The study was approved by the ethics committee of the Centro Hospitalar do Porto, and it was in accordance with the Helsinki Declaration governing clinical trials.
All mouse experiments were performed in accordance with EU guidelines with ethical permits from the national Animal Experiment Board (Eläinkoelautakunta, ELLA, Finland) under license numbers ESHL-2008-02873/Ym-23 and ESAVI-0000497/041003/2011 and in Erlangen, Germany, with permit number 54-2532.1-29/12.
Supplementary Material
Abbreviations Used
- ANA
antinuclear antibodies
- ANCA
anti-neutrophil cytoplasmic antibodies
- anti-BPI
anti-bactericidal permeability increasing protein
- anti-Sm/RNP
anti-Sm/ribonucleoprotein
- ASCA
anti-saccharomyces cerevisiae antibodies
- C3
complement factor 3
- CCP
cyclic citrullinated peptide
- CGD
chronic granulomatous disease
- dsDNA
double-stranded DNA
- FC
fold change
- GAS
γ-interferon activation sites
- IBD
inflammatory bowel disease
- IFN
interferon
- IgA
immunoglobulin A
- IgG
immunoglobulin G
- INFL
inflammation-related gene
- IRG
interferon-regulated gene
- LC
lymphocyte
- MNV
mouse norovirus
- NCF1
neutrophil cytosol factor 1
- NOX2
nicotinamide adenine dinucleotide phosphate-oxidase
- OAS
2′-5′-oligoadenylate synthetase
- PCA
principal component analysis
- PMA
phorbhol 12-myristate 13-acetate
- Q-RT-PCR
quantitative real-time polymerase chain reaction
- RF
rheumatoid factor
- ROS
reactive oxygen species
- SLE
systemic lupus erythematosus
Acknowledgments
The authors are thankful to all patients and their relatives who had collaborated in this study. Paulo Rodrigues-Santos is acknowledged for helping with flow cytometry phenotyping of the patient samples, and Harri Lähdesmäki is thanked for expert supervision of gene expression analysis. The authors also want to acknowledge the personnel at the Mikro Department of the Central Animal Laboratory of the University of Turku. Animal technicians Seija Lindqvist and Tiina Kyrölä are especially thanked for their expert care of the mice. The authors are also grateful to the team of Serviço de Imunologia, Centro Hospitalar do Porto, Otília Figueiras, Paula Carneiro, and Sílvia Lima for technical support.
This work was supported by Academy, Sigrid Juselius Foundation, the Swedish Research Council, and Academy of Finland Center of Excellence in Molecular Systems Immunology and Physiology Research, 2012–2017, Decision No. 250114. The research leading to these results has also received funding from the European Community's Framework programs under grant agreements Neurinox (Health-F2-2011-278611), Masterswitch (HEALTH-F2-2008-223404), and AUTOCURE (LSHM-CT-2005-018661) and the IMI program BeTheCure. T.K. has received funding from the Nordic Center of Excellence in Disease Genetics, and M.M.S-C was funded by the Marie Curie grant PERG-GA-2008-239422-BNOX. D.K. was supported by the ELAN-program of the University clinic Erlangen and Staedler Stiftung, and M. Hoffmann was funded by the Austrian Science Fund (FWF): J3102-B13.
Author Disclosure Statement
The authors declare no competing financial interests except that Peter Olofsson and Rikard Holmdahl are cofounders of a small company, Redoxis AB, aiming at developing treatments using activators of the NOX2 complex.
References
- 1.Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, Etzioni A, Fischer A, Franco JL, Geha RS, Hammarstrom L, Nonoyama S, Notarangelo LD, Ochs HD, Puck JM, Roifman CM, Seger R, and Tang ML. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol 2: 54, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, and Harley JB. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med 349: 1526–1533, 2003 [DOI] [PubMed] [Google Scholar]
- 3.Badolato R, Notarangelo LD, Plebani A, and Roos D. Development of systemic lupus erythematosus in a young child affected with chronic granulomatous disease following withdrawal of treatment with interferon-gamma. Rheumatology (Oxford) 42: 804–805, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, and Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100: 2610–2615, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekpen C, Hunn JP, Rohde C, Parvanova I, Guethlein L, Dunn DM, Glowalla E, Leptin M, and Howard JC. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol 6: R92, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, and Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197: 711–723, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanco P, Palucka AK, Gill M, Pascual V, and Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science 294: 1540–1543, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Bleesing JJ, Souto-Carneiro MM, Savage WJ, Brown MR, Martinez C, Yavuz S, Brenner S, Siegel RM, Horwitz ME, Lipsky PE, Malech HL, and Fleisher TA. Patients with chronic granulomatous disease have a reduced peripheral blood memory B cell compartment. J Immunol 176: 7096–7103, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, and Elledge SJ. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139: 1243–1254, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cale CM, Morton L, and Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol 148: 79–84, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell AM, Kashgarian M, and Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med 4: 157ra141, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cobeta-Garcia JC, Domingo-Morera JA, Monteagudo-Saez I, and Lopez-Longo FJ. Autosomal chronic granulomatous disease and systemic lupus erythematosus with fatal outcome. Br J Rheumatol 37: 109–111, 1998 [DOI] [PubMed] [Google Scholar]
- 13.Cunninghame Graham DS, Morris DL, Bhangale TR, Criswell LA, Syvanen AC, Ronnblom L, Behrens TW, Graham RR, and Vyse TJ. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet 7: e1002341, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai H, Li Z, Zhang Y, Lv P, and Gao XM. Elevated levels of serum antibodies against Saccharomyces cerevisiae mannan in patients with systemic lupus erythematosus. Lupus 18: 1087–1090, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Degrandi D, Konermann C, Beuter-Gunia C, Kresse A, Wurthner J, Kurig S, Beer S, and Pfeffer K. Extensive characterization of IFN-induced GTPases mGBP1 to mGBP10 involved in host defense. J Immunol 179: 7729–7740, 2007 [DOI] [PubMed] [Google Scholar]
- 16.De Ravin SS, Naumann N, Cowen EW, Friend J, Hilligoss D, Marquesen M, Balow JE, Barron KS, Turner ML, Gallin JI, and Malech HL. Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol 122: 1097–1103, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dianzani U, Funaro A, DiFranco D, Garbarino G, Bragardo M, Redoglia V, Buonfiglio D, De Monte LB, Pileri A, and Malavasi F. Interaction between endothelium and CD4+CD45RA+ lymphocytes. Role of the human CD38 molecule. J Immunol 153: 952–959, 1994 [PubMed] [Google Scholar]
- 18.Galibert L, Burdin N, de Saint-Vis B, Garrone P, Van Kooten C, Banchereau J, and Rousset F. CD40 and B cell antigen receptor dual triggering of resting B lymphocytes turns on a partial germinal center phenotype. J Exp Med 183: 77–85, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez-Moyano E, Vera-Casano A, Moreno-Perez D, Sanz-Trelles A, and Crespo-Erchiga V. Lupus erythematosus-like lesions by voriconazole in an infant with chronic granulomatous disease. Pediatr Dermatol 27: 105–106, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Hasui M, Hattori K, Taniuchi S, Kohdera U, Nishikawa A, Kinoshita Y, and Kobayashi Y. Decreased CD4+CD29+ (memory T) cells in patients with chronic granulomatous disease. J Infect Dis 167: 983–985, 1993 [DOI] [PubMed] [Google Scholar]
- 21.Heltzer M, Jawad AF, Rae J, Curnutte JT, and Sullivan KE. Diminished T cell numbers in patients with chronic granulomatous disease. Clin Immunol 105: 273–278, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Holland SM. Chronic granulomatous disease. Clin Rev Allergy Immunol 38: 3–10, 2010 [DOI] [PubMed] [Google Scholar]
- 23.Huang CK, Zhan L, Hannigan MO, Ai Y, and Leto TL. P47(phox)-deficient NADPH oxidase defect in neutrophils of diabetic mouse strains, C57BL/6J-m db/db and db/+. J Leukoc Biol 67: 210–215, 2000 [DOI] [PubMed] [Google Scholar]
- 24.Hultqvist M, Olofsson P, Holmberg J, Backstrom BT, Tordsson J, and Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci U S A 101: 12646–12651, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Israeli E, Grotto I, Gilburd B, Balicer RD, Goldin E, Wiik A, and Shoenfeld Y. Anti-Saccharomyces cerevisiae and antineutrophil cytoplasmic antibodies as predictors of inflammatory bowel disease. Gut 54: 1232–1236, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson SH, Gallin JI, and Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med 182: 751–758, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, Quismorio FP, Jr., Reiff A, Myones BL, Kaufman KM, McCurdy D, Harley JB, Silverman E, Kimberly RP, Vyse TJ, Gaffney PM, Moser KL, Klein-Gitelman M, Wagner-Weiner L, Langefeld CD, Armstrong DL, and Zidovetzki R. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci U S A 109: E59–67, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kallionpaa H, Elo LL, Laajala E, Mykkanen J, Ricano-Ponce I, Vaarma M, Laajala TD, Hyoty H, Ilonen J, Veijola R, Simell T, Wijmenga C, Knip M, Lahdesmaki H, Simell O, and Lahesmaa R. Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes, 2014 [DOI] [PubMed] [Google Scholar]
- 29.Klamp T, Boehm U, Schenk D, Pfeffer K, and Howard JC. A giant GTPase, very large inducible GTPase-1, is inducible by IFNs. J Immunol 171: 1255–1265, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Koivula MK, Heliovaara M, Ramberg J, Knekt P, Rissanen H, Palosuo T, and Risteli J. Autoantibodies binding to citrullinated telopeptide of type II collagen and to cyclic citrullinated peptides predict synergistically the development of seropositive rheumatoid arthritis. Ann Rheum Dis 66: 1450–1455, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koivula MK, Heliovaara M, Rissanen H, Palosuo T, Knekt P, Immonen H, and Risteli J. Antibodies binding to citrullinated telopeptides of type I and type II collagens and to mutated citrullinated vimentin synergistically predict the development of seropositive rheumatoid arthritis. Ann Rheum Dis 71: 1666–1670, 2012 [DOI] [PubMed] [Google Scholar]
- 32.Lee K, Won HY, Bae MA, Hong JH, and Hwang ES. Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc Natl Acad Sci U S A 108: 9548–9553, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee BW. and Yap HK. Polyarthritis resembling juvenile rheumatoid arthritis in a girl with chronic granulomatous disease. Arthritis Rheum 37: 773–776, 1994 [DOI] [PubMed] [Google Scholar]
- 34.Leiding JW, Marciano BE, Zerbe CS, Deravin SS, Malech HL, and Holland SM. Diabetes, renal and cardiovascular disease in p47 phox-/- chronic granulomatous disease. J Clin Immunol 33: 725–730, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manzi S, Urbach AH, McCune AB, Altman HA, Kaplan SS, Medsger TA, Jr., and Ramsey-Goldman R. Systemic lupus erythematosus in a boy with chronic granulomatous disease: case report and review of the literature. Arthritis Rheum 34: 101–105, 1991 [DOI] [PubMed] [Google Scholar]
- 36.Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O'Brien S, Hilligoss DM, Malech HL, Gallin JI, and Holland SM. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics 114: 462–468, 2004 [DOI] [PubMed] [Google Scholar]
- 37.McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, and James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med 11: 85–89, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Moir S, De Ravin SS, Santich BH, Kim JY, Posada JG, Ho J, Buckner CM, Wang W, Kardava L, Garofalo M, Marciano BE, Manischewitz J, King LR, Khurana S, Chun TW, Golding H, Fauci AS, and Malech HL. Humans with chronic granulomatous disease maintain humoral immunologic memory despite low frequencies of circulating memory B cells. Blood 120: 4850–4858, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, and Dinauer MC. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med 185: 207–218, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muise AM, Xu W, Guo CH, Walters TD, Wolters VM, Fattouh R, Lam GY, Hu P, Murchie R, Sherlock M, Gana JC, Neopics , Russell RK, Glogauer M, Duerr RH, Cho JH, Lees CW, Satsangi J, Wilson DC, Paterson AD, Griffiths AM, Silverberg MS, and Brumell JH. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut 61: 1028–1035, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, Wiethe C, Winkler TH, Kalden JR, Manz RA, and Voll RE. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med 14: 748–755, 2008 [DOI] [PubMed] [Google Scholar]
- 42.Olofsson P, Holmberg J, Tordsson J, Lu S, Akerstrom B, and Holmdahl R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet 33: 25–32, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Olsson LM, Lindqvist AK, Kallberg H, Padyukov L, Burkhardt H, Alfredsson L, Klareskog L, and Holmdahl R. A case-control study of rheumatoid arthritis identifies an associated single nucleotide polymorphism in the NCF4 gene, supporting a role for the NADPH-oxidase complex in autoimmunity. Arthritis Res Ther 9: R98, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olsson LM, Nerstedt A, Lindqvist AK, Johansson SC, Medstrand P, Olofsson P, and Holmdahl R. Copy number variation of the gene NCF1 is associated with rheumatoid arthritis. Antioxid Redox Signal 16: 71–78, 2012 [DOI] [PubMed] [Google Scholar]
- 45.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5: 375–386, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Rangel R, McKeller MR, Sims-Mourtada JC, Kashi C, Cain K, Wieder ED, Molldrem JJ, Pham LV, Ford RJ, Yotnda P, Guret C, Frances V, and Martinez-Valdez H. Assembly of the kappa preB receptor requires a V kappa-like protein encoded by a germline transcript. J Biol Chem 280: 17807–17814, 2005 [DOI] [PubMed] [Google Scholar]
- 47.Raterman HG, Vosslamber S, de Ridder S, Nurmohamed MT, Lems WF, Boers M, van de Wiel M, Dijkmans BA, Verweij CL, and Voskuyl AE. The interferon type I signature towards prediction of non-response to rituximab in rheumatoid arthritis patients. Arthritis Res Ther 14: R95, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, Griffiths AM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL, Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, Cho JH, Daly MJ, and Brant SR. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39: 596–604, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, and Merriman TR. Confirmation of association of IRGM and NCF4 with ileal Crohn's disease in a population-based cohort. Genes Immun 9: 561–565, 2008 [DOI] [PubMed] [Google Scholar]
- 50.Roos D. and de Boer M. Molecular diagnosis of chronic granulomatous disease. Clin Exp Immunol 175: 139–149, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rutherford MN, Kumar A, Nissim A, Chebath J, and Williams BR. The murine 2-5A synthetase locus: three distinct transcripts from two linked genes. Nucleic Acids Res 19: 1917–1924, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sareila O, Jaakkola N, Olofsson P, Kelkka T, and Holmdahl R. Identification of a region in p47phox/NCF1 crucial for phagocytic NADPH oxidase (NOX2) activation. J Leukoc Biol 93: 427–435, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Satoh M. and Reeves WH. Induction of lupus-associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. J Exp Med 180: 2341–2346, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schaller J. Illness resembling lupus erythematosus in mothers of boys with chronic granulomatous disease. Ann Intern Med 76: 747–750, 1972 [DOI] [PubMed] [Google Scholar]
- 55.Schappi M, Deffert C, Fiette L, Gavazzi G, Herrmann F, Belli D, and Krause KH. Branched fungal beta-glucan causes hyperinflammation and necrosis in phagocyte NADPH oxidase-deficient mice. J Pathol 214: 434–444, 2008 [DOI] [PubMed] [Google Scholar]
- 56.Schmitt CP, Scharer K, Waldherr R, Seger RA, and Debatin KM. Glomerulonephritis associated with chronic granulomatous disease and systemic lupus erythematosus. Nephrol Dial Transplant 10: 891–895, 1995 [PubMed] [Google Scholar]
- 57.Segal BH, Davidson BA, Hutson AD, Russo TA, Holm BA, Mullan B, Habitzruther M, Holland SM, and Knight PR, 3rd. Acid aspiration-induced lung inflammation and injury are exacerbated in NADPH oxidase-deficient mice. Am J Physiol Lung Cell Mol Physiol 292: L760–L768, 2007 [DOI] [PubMed] [Google Scholar]
- 58.Sellebjerg F, Krakauer M, Hesse D, Ryder LP, Alsing I, Jensen PE, Koch-Henriksen N, Svejgaard A, and Soelberg Sorensen P. Identification of new sensitive biomarkers for the in vivo response to interferon-beta treatment in multiple sclerosis using DNA-array evaluation. Eur J Neurol 16: 1291–1298, 2009 [DOI] [PubMed] [Google Scholar]
- 59.Sillevis Smitt JH, Bos JD, Weening RS, and Krieg SR. Discoid lupus erythematosus-like skin changes in patients with autosomal recessive chronic granulomatous disease. Arch Dermatol 126: 1656–1658, 1990 [DOI] [PubMed] [Google Scholar]
- 60.Sims GP, Ettinger R, Shirota Y, Yarboro CH, Illei GG, and Lipsky PE. Identification and characterization of circulating human transitional B cells. Blood 105: 4390–4398, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sironi M, Guerini FR, Agliardi C, Biasin M, Cagliani R, Fumagalli M, Caputo D, Cassinotti A, Ardizzone S, Zanzottera M, Bolognesi E, Riva S, Kanari Y, Miyazawa M, and Clerici M. An evolutionary analysis of RAC2 identifies haplotypes associated with human autoimmune diseases. Mol Biol Evol 28: 3319–3329, 2011 [DOI] [PubMed] [Google Scholar]
- 62.Somasundaram R, Deuring JJ, van der Woude CJ, Peppelenbosch MP, and Fuhler GM. Linking risk conferring mutations in NCF4 to functional consequences in Crohn's disease. Gut 61: 1097; author reply 109 7–1098, 2012 [DOI] [PubMed] [Google Scholar]
- 63.Stalder JF, Dreno B, Bureau B, and Hakim J. Discoid lupus erythematosus-like lesions in an autosomal form of chronic granulomatous disease. Br J Dermatol 114: 251–254, 1986 [DOI] [PubMed] [Google Scholar]
- 64.Strate M, Brandrup F, and Wang P. Discoid lupus erythematosus-like skin lesions in a patient with autosomal recessive chronic granulomatous disease. Clin Genet 30: 184–190, 1986 [DOI] [PubMed] [Google Scholar]
- 65.van de Loo FA, Bennink MB, Arntz OJ, Smeets RL, Lubberts E, Joosten LA, van Lent PL, Coenen-de Roo CJ, Cuzzocrea S, Segal BH, Holland SM, and van den Berg WB. Deficiency of NADPH oxidase components p47phox and gp91phox caused granulomatous synovitis and increased connective tissue destruction in experimental arthritis models. Am J Pathol 163: 1525–1537, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamazaki-Nakashimada MA, Ramirez-Vargas N, and De Rubens-Figueroa J. Chronic granulomatous disease associated with atypical Kawasaki disease. Pediatr Cardiol 29: 169–171, 2008 [DOI] [PubMed] [Google Scholar]
- 67.Zupo S, Rugari E, Dono M, Taborelli G, Malavasi F, and Ferrarini M. CD38 signaling by agonistic monoclonal antibody prevents apoptosis of human germinal center B cells. Eur J Immunol 24: 1218–1222, 1994 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.