

Abstract
Allostery is the process by which biological macromolecules (mostly proteins) transmit the effect of binding at one site to another, often distal, functional site, allowing for regulation of activity. Recent experimental observations demonstrating that allostery can be facilitated by dynamic and intrinsically disordered proteins have resulted in a new paradigm for understanding allosteric mechanisms, which focuses on the conformational ensemble and the statistical nature of the interactions responsible for the transmission of information. Analysis of allosteric ensembles reveals a rich spectrum of regulatory strategies, as well as a framework to unify the description of allosteric mechanisms from different systems.
Allostery, first articulated more than 50 years ago1–3, has remained a central focus in biology, owing to the fact that a quantitative description of allostery is fundamental to an understanding of most processes beyond the molecular level, such as cellular signalling4 and disease5. Allostery has even been referred to as the ‘second secret of life’6,7. In spite of its importance, allosteric mechanisms in most instances remain a biophysical enigma, eluding a general, quantifiable and predictive atomic description7,8.
Through the lens of history, it is clear that allosteric concepts and models have evolved over decades as the experimental technologies have improved. The first allosteric proteins studied, such as haemoglobin9, were extensively structured and thus amenable to high-resolution X-ray crystallography. Models of allosteric mechanisms based on such studies were understandably greatly influenced by the static structural images10,11. However, as more experimental structural, dynamic and thermodynamic approaches have been developed, descriptions for allostery have necessarily become more complex. The largely qualitative, static images of end point protein structures have been replaced by more quantitative, dynamic views of allostery, which, unlike static structural models, are more difficult to visualize. Here the historically dominant paradigm of structure-based allostery11–14 is briefly reviewed and placed in the context of new discoveries, revealing that allostery can also be associated with changes in dynamics and large-scale conformational disorder15–18. Indeed, more recent discoveries emphasize how nature uses the full spectrum of conformational heterogeneity19 (for example, rigid body movement20–22, folded yet dynamic structure4,23–27, and intrinsic disorder28–30) to facilitate allostery. These recent discoveries challenge traditional understanding of allostery and provide the foundations for developing a framework that unifies descriptions of allostery in structured, dynamic and disordered systems.
From structures to ensembles
The first statement of allostery31, arising from the classic experiments of Changeux2, is that two distinct sites within one protein, each binding different ligands, could nonetheless interact despite being ‘non-overlapping’ (that is, presumably distant from each other) in the molecular structure. This ‘action at a distance’ phenomenon was difficult to interpret in the absence of structural information or site-specific biochemical data.
For decades, the two dominant models for allostery were the ‘sequential’, or KNF (Koshland–Nemethy–Filmer), model32 and the ‘symmetric’, or MWC (Monod–Wyman–Changeux), model3. Both proposals were influential and agreed on the importance of conformational change between two well-defined structural end states33. Whereas the MWC model posited the existence of two pre-existing quaternary states (tensed (T) and relaxed (R)), whose equilibrium was shifted upon binding ligand3, the KNF model was based on the appealing, general notion of the inherent flexibility of proteins32 and ‘induced-fit’ of a binding site in response to ligand34,35. Importantly, both models were successful in describing allostery31–36.
However, it is well known that both the MWC and KNF models are phenomenological33, and consequently, do not provide insight into how the structure facilitates allosteric communication between sites. It was the birth of structural biology and the development of the influential stereo-chemical model by Perutz10,13 that first addressed this issue and set the course for future allosteric studies. According to this model, which was built upon by many others33,37,38, allostery could be understood in terms of structural changes that could be gleaned through inspection of the high-resolution structure. This structural view of allosteric mechanism has been the reigning paradigm ever since39–43, with some approaches even positing the existence of conserved allosteric pathways that presumably mediate the transmission of information between sites. In fact, such pathways are even evident in haemoglobin44 (Fig. 1a).
Figure 1. Structure-based views of allostery.
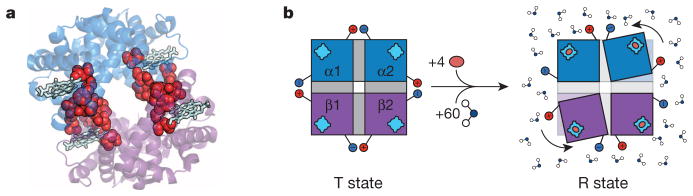
a, Ribbon diagram representation of tetrameric haemoglobin (PDB accession 1GZX) rendered in PyMol (Schrödinger). The proposed pathway responsible for the cooperative transition from tensed (T) to relaxed (R) is highlighted with red spheres and the haem groups are represented as light blue sticks44. b, Allosteric transition of tetrameric haemoglobin, as proposed by Perutz10,13. Tetrameric haemoglobin in the T state is depicted on the left with the two α-subunits (blue) and the two β-subunits (purple) each with their own haem group (light blue). Salt bridges, depicted as the red positive and blue negative charges, hold the molecule in the T conformation, and these salt bridges are released upon binding of oxygen (orange oval) in the transition to the R conformation (on the right) accompanied by a 15° turn of the subunits relative to each another. Also contributing to the equilibrium are 60 additional water molecules preferentially binding the R state45.
Nonetheless, the notion that something was amiss (or at least incomplete) with a purely structure-centric understanding of allostery had been recognized for decades. In a classic thermodynamic study of haemoglobin, Parsegian and colleagues45 found that the T-to-R transition in haemoglobin involved the binding of approximately 60 additional water molecules (Fig. 1b), and noted “… it is doubtful that the R state would even exist without the stabilization promoted by hydration of the extra surface.” In effect, the entire molecule must have a role in mediating allostery. Contrasting the different ‘images’ of allostery conveyed by Fig. 1 reveals the challenge in quantitatively describing mechanism. Clearly, an understanding of allostery requires not only knowledge of the concerted structural changes between two binding sites, but also the factors that render an allosteric protein ‘poised to undergo’ the structural change.
The Parsegian quote itself hints at the realization (well-appreciated by the authors of the early models) that allosteric transitions involve multiple states, whose relative populations must be tunable over a range of physiological conditions. Nonetheless, the fact that initial structures were found for the T and R forms of haemoglobin supported the notion that allostery as a general phenomenon might be understood, perhaps in all systems, if only the structures of the allosteric states could be determined. To that end, extensive molecular dynamics33,46,47 and coarse-grained network simulation20,48 (for those proteins in which rigid body motions could be proposed for the allosteric transition) further suggested that ‘allosteric mechanisms reliant primarily upon observable changes in the ensemble-average structure are common and may be dominant’20. Although the discovery of an alternate ‘relaxed’ structure of haemoglobin by Arnone and co-workers49 laid to rest the simple two-state view of allostery, at least for haemoglobin, not fully appreciated was the range of possible regulatory strategies that could be used if the full spectrum of binding and conformational states of proteins were to be harnessed.
In 1984 Cooper and Dryden touched on one such possibility when proposing that allostery could be manifested from changes in the width of a conformational distribution, raising the spectre of significant entropic contributions to allostery50. These authors rigorously demonstrated, using a statistical thermodynamic formalism, that changes in the frequency and amplitude of thermal fluctuations in a protein upon ligand binding could result in cooperative energies on the order of a few kcal per mol without perturbing the average structure. Importantly, such a situation would be difficult to reconcile by simple inspection of the end-state structures, explaining why experimental examples had escaped detection until recently. The work of Cooper and Dryden has proven prescient33. The term ‘dynamic allostery’ has even found its way into the modern lexicon, ostensibly to describe the role of entropy in the thermodynamics of allostery, although unfortunately it is often conflated with motions between relevant ensembles. However, with the development of NMR techniques to study allosteric protein systems at a site-resolved level, both aspects of the role of motion in transitions between functional states are becoming illuminated51–55.
One of the most surprising observations over the past half-decade has been the appearance of allostery in intrinsically disordered proteins (IDPs), systems that are defined by a lack of fixed structure, exhibiting instead extreme conformational fluctuations56–60. Clearly both conformational dynamics of folded structures and large-scale disorder are important for allostery61, but quantitatively understanding this phenomenon remains a challenge. It is not obvious how the phenomenon of allostery can be understood and described in terms that can do equal justice to both highly structured and highly disordered systems.
Below, we use several recently studied allosteric systems, spanning the continuum of structure/dynamics classification space (Fig. 2), to show how changes in dynamics and disorder are phenomenologically associated with allostery. Initially highlighted are the challenges associated with reconciling allostery in terms of classic structural or deterministic models. However, when allosteric proteins are viewed as ensembles of states, an entirely new palette of expected behaviour is revealed. Importantly, it becomes clear that taxonomic schemes segregating allosteric proteins by amount and/or type of dynamics that change during an allosteric transition may actually obscure similarities between mechanisms that are only phenomenologically different (Box 1). The ensemble nature of allostery revealed through analysis of the full dynamic spectrum of allosteric systems permits a rich repertoire of regulatory strategies and illuminates key organizing principles for describing allostery.
Figure 2. The dynamic continuum of allosteric phenomena.

Schematic representation of allosteric systems with increasing dynamics, disorder or fluctuations on the vertical axis.
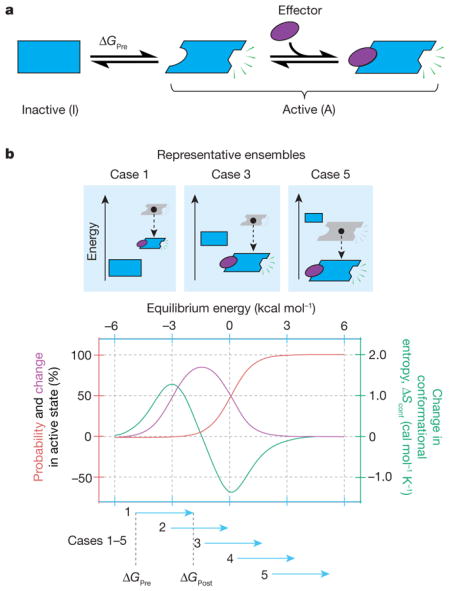
BOX 1. Tunable sensitivity in allosteric ensembles.
Tuning the sensitivity of the ensemble to an allosteric effector. a, Two-state allosteric protein wherein the effector ligand binds to and stabilizes the active state. This equilibrium (ΔGPre) can be shifted (ΔGPost) by adding effector. b, Shown is the response of the system to an allosteric effector that increases the stability of the active state by 3 kcal mol−1. The observed response is dependent on where the equilibrium is poised before activation. When the inactive state is too dominant (b, top, case 1), activation (that is, adding effector) produces a modest change in activity and an increase in the conformational entropy, Sconf (= −RΣPilnPi, where Piis the probability of the ith state and the summation is over all states in the ensemble). When the active state is already dominant (case 5), activation again produces a modest effect, but in this case the conformational entropy decreases. Under conditions in which both states are populated (case 3), the change in activity is maximal, but the change in conformational entropy is near zero. This is depicted quantitatively in the graph in the bottom of b: the probability of being active (red), the change in activity (purple) and the change in conformational entropy (green) as a function of ΔGPre. Importantly, the free energy before (ΔGPre) and after (ΔGPost) addition of effector ligand determines how the ensemble is poised, which in turn determines both the degree of activity change and whether conformational entropy (as determined from a positions-specific probe of dynamics) will increase, decrease or remain constant upon activation.
The dynamic continuum of allostery
Arguably the most significant experimental advance in the analysis of allostery has been the development of NMR spectroscopy, which provides information about protein structure, dynamics and thermodynamics16,53,54. Although the high-resolution structure of a protein provides exquisite insight into the enthalpic contributions of structure and solvent to protein thermodynamics62, the role of entropy remains obscure. The idea of using fast internal motion detected by solution NMR as a proxy for conformational entropy was introduced some time ago63,64, where motion evaluated assuming a specific motional model provided an indirect measure of the number of states visited. The detection of motion in proteins over a wide range of timescales is now well established, but it is the fast picosecond-to-nanosecond timescale that seems to be most important for the detection of conformational entropy65–67. The initial model-dependent interpretation of protein dynamics in terms of entropy was problematic65 but its use did reveal the probable presence of a large contribution from conformational entropy to the free energy of protein–ligand interactions68,69. To overcome many of the technical difficulties associated with using a dynamic proxy for conformational entropy, Wand and co-workers developed an empirical ‘entropy meter’ that appears able to quantitatively relate changes in fast internal protein motion measured by NMR relaxation to changes in conformational entropy54,70. This approach has been used to demonstrate the integral role of conformational entropy in molecular recognition by calmodulin70 and the catabolite activator protein (CAP)24. The calmodulin system provided illustrations of the various types of dynamical perturbation that can occur: purely local, long-range or directed dynamical transmission, which is perhaps the key to dynamically mediated allostery71. The CAP system has been particularly important in exploring the role of conformational entropy in allostery, as discussed below.
NMR analyses of allosteric proteins have revealed a cornucopia of ways that changes in dynamics are associated with allosteric transitions. Indeed, a continuum of dynamic allosteric phenomena has been observed, ranging from rigid body structural changes to intrinsically disordered proteins mediating the response (Fig. 2). The existence of this dynamic continuum suggests that although the two extremes of the spectrum represent radically different structural ‘pictures’ of allostery, they should nonetheless be amenable to a unified description.
Structured yet dynamic allosteric systems
The classic view of an allosteric protein is one of structure. The protein may have one or more domains or subunits whose structure can be determined, but which may show conformational dynamics when observed in NMR experiments. Many examples of structured allosteric proteins that show dynamic changes have been documented4,18,23–26,72. For reasons described below, these are ideal systems to probe ensemble properties. For now we simply note that the analyses of these types of systems have revealed a number of interesting paradoxes.
Since the initial prediction of dynamic allostery without conformational change by Cooper and Dryden50, two important experimental studies have gone on to show that allostery can indeed be manifested essentially without structural change. The first study focused on CAP, a homodimeric transcription factor consisting of a cyclic AMP (cAMP) binding domain coupled to a DNA binding domain (DBD) (Fig. 3a). The biological role of CAP is to bind cAMP, allowing CAP to activate transcription (Fig. 3a, right). Extensive studies have revealed that conformational entropy via backbone and side-chain dynamics is associated with the allosteric response of the system upon cAMP binding24–26. Whether the entire entropic penalty arises from quenching of dynamics upon ligand binding is an open question33, but certainly a significant contribution of the allosteric response is associated with the quenching of dynamics. Interestingly, high-resolution NMR spectroscopy has revealed that the population of binding-competent states for wild-type and mutants of CAP in solution is not proportional to the overall binding affinity. Instead, the entropic effects for a number of mutants were shown to correlate with the conformational entropy determined from NMR order-parameters24. In this case, the entropic penalty seems to be the basis of the negative cooperativity—binding cAMP increases dynamics in the adjacent domain that must be quenched to bind the second ligand. Interestingly, a recent study of CAP using elastic network models demonstrated that modulation of the normal modes can reproduce these results and can be used to rationally modulate the cooperativity of ligand binding73.
Figure 3. Allosteric systems from the dynamic continuum.

a, CAP homodimer with a cyclic nucleotide binding domain (CBD, blue domains with side chains) and a DNA binding domain (DBD, blue cylinders). Binding energetics of cAMP (purple ligand) quench dynamics in the bound-state ensemble and induce a 90° change in the conformation of the DBD, allowing it to bind DNA and turn on transcription24–26. b, Side-chain dynamics modulate the binding affinity of a canonical PDZ domain to its native ligand with and without α-helix 3 (Δα3, green arrows)23. c, AAC homodimer with each monomer represented as one purple domain. Binding of the allosteric effector acetyl-CoA (blue oval) is positively cooperative at low temperatures (green ‘+’), and negatively cooperative at high temperatures (red ‘−’)4. d, TetR homodimer27 depicted as a two-domain protein with a tetracycline binding domain (TBD, blue region, top) and a DBD (blue region, bottom). e, Doc/Phd toxin–antitoxin system equilibrium. Phd is depicted as a homodimer (top, blue and purple monomers) and Doc is depicted as the blue ligand28. f, Representation of variants of E1A30 shown from N to C terminus with binding sites for ligands in the two variants represented by blue rectangles (CBP) and green rectangles (pRb). g, Schematic representation of α-synuclein (AS) with its N-terminal membrane-binding domain (MBD) coupled to its C-terminal IDR. Upon oxidative stress (nitration) the affinity of the MBD decreases (bottom)29.
In a second important study, removal of an α-helix (Δα3) from a canonical PDZ domain resulted in an increase in side-chain dynamics of the distal binding site23 (Fig. 3b). The helix in question was presumed to be a molecular scaffold and not expected to affect binding, because it was not present in most other PDZ domains and was physically distal from the binding site23. Unexpectedly, deletion of α3 (depicted blue in Fig. 3b, right) resulted in a 25-fold reduction in binding affinity that was almost entirely entropic in nature, as determined by isothermal titration calorimetry. NMR studies revealed no global structural perturbations, and relaxation experiments showed no significant changes in the backbone dynamics. The only detectable differences were observed in the side-chain dynamics (Fig. 3b, top right) and the calorimetrically determined entropy change (in other words, the dynamics in the unliganded state). Both the CAP and the PDZ examples were pioneering studies that not only revealed the predicted importance of dynamics and the role of conformational entropy in allosteric mechanism, but also highlighted the limitations of deducing mechanism from static structure alone.
Local unfolding and intrinsic disorder
Moving down the dynamic continuum (bottom of Fig. 2), one of the more recent developments is the realization that local unfolding and intrinsic disorder61 can have a significant role in allosterically coupling domains to one another. As shown below, the hallmark feature of these systems is the apparently complex ‘tunable’ coupling between sites. An example in which local unfolding has a role in allostery is in the enzyme aminoglycoside N-(6′)-acetyltransferase II (AAC)—a homodimeric enzyme from Enterococcus faecium responsible for conferring bacterial resistance to amino-glycoside antibiotics4. An allosteric effector of AAC, acetyl-CoA, binds with positive cooperativity at low temperatures and switches to negative cooperativity at higher temperatures as determined by isothermal titration calorimetry (Fig. 3c). NMR reveals that certain backbone amides undergo dynamic broadening as a function of temperature, suggesting that local unfolding is responsible for this switching behaviour. This observation agrees with the change in enthalpy of binding as temperature is increased; the nonlinear temperature dependence at higher temperatures being a signature of local unfolding74.
Another example in which local unfolding has a role in allosteric coupling is the tetracycline repressor (TetR), a homodimeric transcription factor consisting of a Tet binding domain (TBD) and a DBD (Fig. 3d). TetR binds DNA in the absence of the antibiotic (Tet) and is released when Tet binds the TBD, inducing a reduction in binding affinity of the DBD for DNA. A paradoxical observation from structural data was that the inactive Tet-bound (holo) form shows the DBD α-helices, which are involved in binding DNA, to be in a more binding-competent conformation than when they are in the active apoform. Chaotropic denaturation and in vitro operator binding assays performed on TetR, as well as biologically relevant TetR mutants, revealed that local unfolding of the DBD in the native state ensemble was responsible for this paradox27. The protein functions because the DBD is locally unfolded in the absence of Tet, allowing enough conformational flexibility to bind DNA, and hence an increased binding affinity in the absence of Tet (Fig. 3d, left equilibrium). Upon binding Tet, the α-helices responsible for DNA binding are redistributed into an ensemble of conformations that are not completely binding competent. Interestingly, naturally occurring mutants exist that are Tet insensitive and/or have inverted responses, manifesting complexity and tunability like that seen in allostery of locally unfolded or intrinsically disordered sequences27.
Perhaps one of the fastest evolving fields in biophysics is protein intrinsic disorder59,60. IDPs stand as a challenge to the structure–function paradigm, as IDPs have no stable tertiary structure under physiological conditions yet are able to function58–60. Seminal bioinformatics and experimental work has revealed that intrinsically disordered regions (IDRs) and IDPs are hyper-abundant in transcription factors and cell signalling pathways. Aside from the fact that they lack a stable structure, IDPs nonetheless possess features that would otherwise make them suitable allosteric proteins. These include (1) high specificity for multiple targets resulting from their structural plasticity30; (2) high specificity and low-affinity binding18; (3) fast association and dissociation rates for proper turnover58; and (4) enrichment of post-translational modification sites75 and splicing isoforms76. To no surprise, several IDPs have been shown to exhibit allostery as part of their role in signalling. Interestingly, just as with proteins that exhibit local unfolding, allosteric IDPs exhibit complex coupling behaviour. Three examples highlight this complexity.
The first case involves the Phd/Doc toxin–antitoxin system (Fig. 3e) that inhibits the ribosome A site. As part of its regulatory cycle, Phd controls its own transcription by binding DNA at its own operon. Notably, increasing the ratio of Doc/Phd initially inhibits transcription, and as the ratio exceeds a certain biological threshold, transcription is activated again, exhibiting ‘conditional cooperativity’28. Conditional cooperativity results from the delicate balance of the ensemble of states present in solution. Phd has a locally unfolded carboxy terminus, which undergoes a disorder-to-order transition that exposes high-affinity (H) and low-affinity (L) binding sites (equilibrium 1, Fig. 3e). Increasing the ratio of Doc/Phd leads to partial saturation of the H and L sites by Doc, which in turn stabilizes the DBD of Phd, allowing DNA binding and transcription repression (equilibrium 2, Fig. 3e). Finally, increasing the Doc/Phd ratio further leads to saturation of the H and L binding sites, which leads to dissociation from DNA and activates transcription again (equilibrium 3, Fig. 3e).
Another recently reported example30 involves the IDP adenovirus early region 1A protein (E1A) (Fig. 3f). E1A can bind two different host proteins, CREB binding protein (CBP) and retinoblastoma protein (pRB), in its capacity to disrupt host cellular signalling. Single-molecule fluorescence resonance energy transfer (smFRET) was used to dissect the cooperative nature of the coupled folding and binding involving CBP and pRB. By measuring the cooperativity of binding using different truncated E1A constructs, these authors elegantly demonstrated cooperativity switching between the CBP and pRB sites, wherein positive coupling (green ‘+’ in Fig. 3f) becomes negative (red ‘−’ in Fig. 3f).
Importantly, IDPs are enriched in post-translational modifications75, telling of separate mechanisms that nature may use to regulate the allosteric response of the ensemble. An example of this occurs in α-synuclein, implicated in Parkinson’s disease29. α-Synuclein is composed of an amino-terminal membrane-binding domain and a C-terminal IDR (Fig. 3g) and exhibits reduced membrane-binding affinity under oxidative stress29. Using smFRET and NMR, it was shown that nitration of the C terminus shifts its conformational ensemble to a more globally extended conformation, which is accompanied by a reduction in binding affinity (Fig. 3g, bottom). Because the C-terminal IDR is known to interact with a variety of ligands, these results suggest that α-synuclein may use this allosteric coupling to regulate the biological response. Modulation of the conformational ensemble in IDPs and IDRs via these post-translational modifications may be a general mechanism for rapid, reversible control of biological response. How this may occur is discussed in more detail in the following section.
As the examples in Fig. 3 reveal, the hallmark feature of allostery in locally unfolded and intrinsically disordered proteins is a general mechanism whereby cooperativity between two sites can not only be tuned (high or low) by environmental conditions, but the actual sign of the coupling can change, transforming an activator into a repressor, or vice versa. A single static picture of the protein does not reveal how the same ligand, binding to the same site on the same protein, can transmit structural information from one site to another, causing a more favourable conformation to bind ligand under one circumstance and a less-favourable conformation under another. Restated more bluntly, it is not clear how the same ligand binding to the same site can cause either one biological effect or its ‘opposite’.
Here we show that by formulating the phenomenon of allostery in terms of thermodynamic ensembles, many of the seemingly complex and confounding properties of allosteric proteins, including the results of adding different allosteric ligands, emerge as a straightforward consequence of the heterogeneity in the system. From these allosteric ensembles, ground rules emerge that can be used to guide future experimental inquiry.
Allosteric ligands remodel the energy landscape
Much in same way that the ability of a protein to fold can be understood in terms of the conformational ‘energy landscape’ of the protein77–79, so too can allosteric coupling. According to this framework, all possible conformations of a protein are sampled (or populated) according to their energies; lower energy conformations are sampled more often than those of higher energy. Because the binding of any ligand to a state increases how often the state is sampled15,80, allosteric ligands effectively remodel the energy landscape of allosteric proteins18. This key point has been discussed, at least in qualitative terms, in a number of recent reviews5,15,18. The take-home message is that the energy landscape can be smooth with many accessible states in its conformational ensemble, discrete with few states, or something in between, and each of these scenarios can provide its own biological advantage.
However, stabilities and coupling energies in proteins are generally very modest, suggesting that no single state, or sets of allosteric states, will necessarily dominate the ensemble. In other words, allosteric mechanisms may be more statistical, and less deterministic, than classical models suggest. Indeed, a more quantitative look at the landscape, using ensemble models that divide the protein into interacting component parts61,81,82, illuminates the statistical nature of the signalling process, and reveals a wealth of regulatory opportunities directly resulting from the heterogeneity in the ensemble (Box 2).
BOX 2. The ensemble view as a framework to explain allostery.
a–c, Allosteric proteins can be viewed, to a first approximation, as a functional domain coupled to one or a number of effector-binding domains that interact with each other. To illustrate how an ensemble can switch the outcome of binding a particular effector molecule, we consider a protein with two effector-binding domains, which can interact with each other, as well as the functional site (a). Each domain can be inactive or active, together with (or separately from) the other domain, producing an ensemble of eight possible states (b, first column). In each state, the effector 1 binding site is in the active conformation (b, third column) either with or without the functional site in the active conformation (b, second column). Thus, the ensemble consists of states in which effector 1 site is positively coupled to the functional site (states 1 and 3; b, fifth column) and negatively coupled (states 4 and 5; b, sixth column)—that is, the effector 1 and functional sites are both active, or the effector 1 site is active while the functional site is inactive, respectively. Which set of states will govern the overall observed allosteric transition is determined primarily by the magnitude and sign of the interaction energies, Δgint, between each domain (a)61. To see how this affects function, we consider the energy of all the states in the ensemble under a particular set of conditions (c)8. In the absence of effector 1, protein conformations in which the functional site is active have high energy, and so the probability of the functional site being active is low. Effector 1 binding lowers the energies (dotted lines in c) of all states in which the effector 1 binding site is active, including conformations that contain the active form of the functional site. This increases the probability that the functional site will be active. The sizes of the depicted structures indicate the probability of their formation; larger structures are more likely to form. Addition of effector 2, in the absence of effector 1, redistributes the ensemble to a new starting position (d, left) so that the two lowest energy conformations now have active functional sites. Addition of effector 1 binds to and lowers the energy of the same states as described above (dotted lines in d). In this instance, however, only one of the two lowest energy states has the functional site in the active conformation, lowering the probability that the functional site will be active. As a result, effector 1 switches from being an activator to a repressor84.
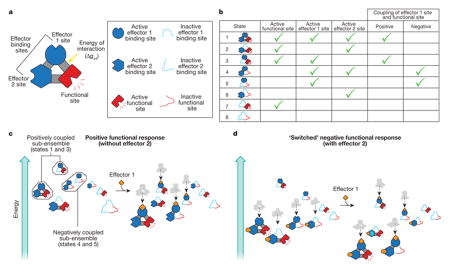
Of course, treating allosteric proteins as ensembles is not a new idea. Early work on the lambda phage repressor, for example, demonstrated that consideration of all the accessible ligated states was necessary to understand its biological function83. However, when conformational heterogeneity at each binding site is explicitly considered in addition to the degeneracy of ligated species, important ground rules for allostery emerge8.
First, all ensembles have tunable sensitivity, wherein the same perturbation can produce a range of results, simply based on where a given equilibrium is poised before perturbation. This reality not only affects function but also the interpretation of conformational entropy changes in allosteric transitions, which, in principle, can change from positive to negative, even for the same transition (Box 1).
Second, ensembles contain a plurality of potential allosteric mechanisms, a point that can be demonstrated by noting that allosteric proteins often segregate different functions (for example, effector-binding and activity) into different domains. This allows for a straightforward enumeration of the ensemble that accounts for each functional domain to be in the active or inactive conformation8,61,84, both with and without other domains in their functional states. Binding of the effector molecule stabilizes the active form of the effector-binding domain, which in turn can stabilize (or destabilize) the functional domain (Box 2)61. This is so because, according to landscape theory77–79, all of the possible states in the ensemble have some finite probability based on their energies. Thus, if landscape theory holds, within any ensemble, there exist sub-ensembles in which two different binding sites are either positively coupled (both domains active) or negatively coupled (one domain active and the other inactive) (Box 2). In other words, the potential for different allosteric mechanisms is already pre-encoded in the ensemble, a property that is uniquely provided by an ensemble allosteric model8.
The question remains as to what determines positive or negative coupling between two binding sites. In the context of the ensemble, the answer is the relative stability of the different states, which is determined by the equilibrium between the active and inactive state of each domain and the interaction energy between domains. In fact, the signs of the interaction energies are the key parameters in determining whether allosteric proteins can switch between activation and repression61,84. The critical insight is that the overall allosteric behaviour is an ensemble-weighted contribution of all the states and thus cannot be reconciled in the context of a single structure85. Instead the effect is statistical. Under one set of conditions, the ensemble could be poised such that two domains would be positively coupled, because the properties of the positively coupled sub-ensemble dominate the average. However, any perturbation of the ensemble that remodels the energy landscape may change which sub-ensemble dominates, and thus, transform activation into repression, or vice versa (Box 2).
Importantly, perturbations can arise from any number of sources, including post-translational modification (for example, phosphorylation, methylation, sumoylation, nitration29), alternative splicing or translation start sites76, truncation30, temperature4 and the binding of a second allosteric effector to a different effector-binding domain (Box 2). In short, redistributing the ensemble can modulate the mechanism. As shown in Box 2, remodelling of the ensemble with effector 2 corresponds to dialing-in (in a rheostat-like fashion) the negative coupling contribution to the overall allosteric coupling.
Discussion
The importance of an ensemble representation of allostery is that it highlights the statistical nature of the allosteric coupling process. The classic notion of an allosteric pathway, through a static picture of the protein, often conveys a deterministic (or at least homogeneous) picture of the signal propagation process, wherein each molecule at any instant is in the same conformation or at least can be equally well represented by the same average conformation. The ensemble model states that, in the most general case, activity is an ensemble-weighted contribution of all species present in solution, regardless of how structurally dissimilar they may be. As such, the important questions shift from identification of the single active conformation, or the allosteric pathway through the protein, to questions that focus on identifying the fraction of molecules that have each domain in the active conformation, and/or the fraction of time a particular set of interactions exist when the functional site (and/or the effector-binding domains) is in its active conformation. An important point critical to characterizing an allosteric ensemble is that, although a given effector molecule may bind to only a small fraction of the states in an ensemble, thus directly stabilizing only those states, stabilization will redistribute the entire ensemble, changing the probability of every state (Box 2). Consequently, every state in the ensemble can be regarded as a potential allosteric state, regardless of whether it binds one or a number of effectors or is functionally silent. Such silent states, simply as a consequence of being populated, attenuate the impact of effector binding on the activity (Box 2). Indeed, the numerous examples that show complex, stability-dependent and tunable coupling28,30,86 suggest that the principles outlined here are at play in real allosteric systems.
An important question is how to practically leverage the ensemble view of allostery. The most immediate impact could be refocusing inquiry into allosteric mechanism, particularly as it pertains to IDPs, in which structural methods have limited applicability. The ensemble allosteric model described here and elsewhere8,61,84 suggests that experimental determination of stabilities and coupling energies represents a viable strategy for determining the relevant states in both disordered and structured ensembles and the relative stabilities of each. Of course, this should not be misconstrued as minimizing the importance of structural characterization of IDPs. Significant progress has been made in modelling disordered ensembles at atomic resolution using various experimental constraints87–91. The future challenge will be for these computed ensembles to reproduce the observed highly variable and complex switching behaviour seen in many IDPs.
With regard to structured proteins, the ensemble model of allostery provides insight into important lines of inquiry. Specifically, because the sensitivity of an allosteric protein is maximal at the threshold of a transition (Box 1), many important states are likely to be minutely, albeit measurably, populated under functional conditions (that is, between 1 and 5%). As such, experimental and computational strategies to identify low-probability states are critical. In addition to NMR55,92–95, fundamental progress has been made in detecting such low-populated states combining other structural biology techniques51,96, with at least one X-ray crystallography study corroborating the states detected by NMR51. The ability to observe alternative conformations masked by the cryogenic temperatures typically used in structural analysis51,97, and to resolve, using ensemble refinement, multiple states from crystal data collected at room temperature98, reveal a much richer dynamic picture. Similarly, new data analysis methods applied to electron microscopy images quite expectedly reveal multiple conformational states of macromolecules99.
Interestingly, the ensemble model also provides a potential framework for interpreting long-time scale molecular dynamics simulations of allosteric proteins100. If such simulations can provide the amount of time that specific interactions are made (and the co-occurrence with other interactions), molecular dynamics could be used to map the energy landscape of the allosteric process, possibly even illuminating the multitude of mechanisms pre-encoded in the ensemble. Indeed, a recent long-time molecular dynamics simulation101 reveals the statistical nature of the interactions associated with allosteric drugs of the muscarinic acetylcholine receptor (M2 receptor), indicating that interactions are not obligatorily associated with ligand-bound or -unbound states. Other computational approaches, such as coarse-grained modelling of the normal modes of proteins, yield predictive cooperative energies even in the absence of structural change73. Ongoing and future efforts to integrate computation, structure and even different models of allostery and function102 are likely to be critical to understanding signalling within large macromolecular complexes103.
Finally, the ensemble representation of allostery is particularly appealing when considering protein design and evolution. The genetic record clearly shows that nature has evolved and adopted a finite number of functional and binding domains, and has combined them in different ways to make new proteins with different functions and regulatory properties104,105. The ensemble representation suggests that by simply manipulating either or both the active/inactive equilibrium for different domains and the coupling between domains, evolution or design of allosteric proteins may be much more straightforward than a detailed inspection of the atomic structure of the protein suggests. Future research directions should include whether allosteric proteins can be designed or evolved without detailed consideration of the structure, and whether positively coupled domains can be transformed to negatively coupled ones, simply by identifying the thermodynamic ground rules for switching84 and rationally changing the relevant parameters106–108. Although the answers to such questions are uncertain, what is certain is that as more dynamic and disordered systems have become amenable to experimental inquiry, the ensemble nature of allostery has become clear.
Acknowledgments
This work was supported by National Science Foundation grant MCB1330211 and by National Institutes of Health grants GM63747 and T32-GM008403.
Footnotes
Author Contributions V.J.H. conceived the manuscript; H.N.M., J.O.W., J.L. and V.J.H. wrote and edited the manuscript.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Monod J, Jacob F. Teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harb Symp Quant Biol. 1961;26:389–401. doi: 10.1101/sqb.1961.026.01.048. [DOI] [PubMed] [Google Scholar]
- 2.Changeux JP. The feedback control mechanisms of biosynthetic L-threonine deaminase by L-isoleucine. Cold Spring Harb Symp Quant Biol. 1961;26:313–318. doi: 10.1101/sqb.1961.026.01.037. [DOI] [PubMed] [Google Scholar]
- 3.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 4.Freiburger LA, et al. Competing allosteric mechanisms modulate substrate binding in a dimeric enzyme. Nature Struct Mol Biol. 2011;18:288–294. doi: 10.1038/nsmb.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nussinov R, Tsai CJ, Ma B. The (still) underappreciated role of allostery in the cellular network. Annu Rev Biophys. 2013;42:169–189. doi: 10.1146/annurev-biophys-083012-130257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monod J. Chance and Necessity: Essay on the Natural Philosophy of Modern Biology. Penguin Books; 1977. [Google Scholar]
- 7.Fenton AW. Allostery: an illustrated definition for the “second secret of life”. Trends Biochem Sci. 2008;33:420–425. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hilser VJ, Wrabl JO, Motlagh HN. Structural and energetic basis of allostery. Ann Rev Biophys. 2012;41:585–609. doi: 10.1146/annurev-biophys-050511-102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perutz MF, et al. Structure of haemoglobin: a three-dimensional Fourier synthesis at 5.5-Å resolution, obtained by X-ray analysis. Nature. 1960;185:416–422. doi: 10.1038/185416a0. [DOI] [PubMed] [Google Scholar]
- 10.Perutz MF. Stereochemistry of cooperative effects in haemoglobin. Nature. 1970;228:726–734. doi: 10.1038/228726a0. [DOI] [PubMed] [Google Scholar]
- 11.Dickerson RE. X-ray studies of protein mechanisms. Annu Rev Biophys Chem. 1972;41:815–842. doi: 10.1146/annurev.bi.41.070172.004123. [DOI] [PubMed] [Google Scholar]
- 12.Laskowski RA, Gerick F, Thornton JM. The structural basis of allosteric regulation in proteins. FEBS Lett. 2009;583:1692–1698. doi: 10.1016/j.febslet.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 13.Perutz MF, Wilkinson AJ, Paoli M, Dodson GG. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu Rev Biophys Biomol Struct. 1998;27:1–34. doi: 10.1146/annurev.biophys.27.1.1. [DOI] [PubMed] [Google Scholar]
- 14.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 15.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins Struct Funct Bioinf. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 16.Tzeng SR, Kalodimos CG. Protein dynamics and allostery: an NMR view. Curr Opin Struct Biol. 2011;21:62–67. doi: 10.1016/j.sbi.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 17.Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Smock RG, Gierasch LM. Sending signals dynamically. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai CJ, del Sol A, Nussinov R. Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms. Mol Biosyst. 2009;5:207–216. doi: 10.1039/b819720b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daily MD, Gray JJ. Allosteric communication occurs via networks of tertiary and quaternary motions in proteins. PLOS Comput Biol. 2009;5:e1000293. doi: 10.1371/journal.pcbi.1000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swain JF, et al. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuiderweg ER, et al. Allostery in the Hsp70 chaperone proteins. Top Curr Chem. 2013;328:99–153. doi: 10.1007/128_2012_323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, Lee AL. Hidden dynamic allostery in a PDZ domain. Proc Natl Acad Sci USA. 2009;106:18249–18254. doi: 10.1073/pnas.0904492106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tzeng SR, Kalodimos CG. Protein activity regulation by conformational entropy. Nature. 2012;488:236–240. doi: 10.1038/nature11271. [DOI] [PubMed] [Google Scholar]
- 25.Tzeng SR, Kalodimos CG. Dynamic activation of an allosteric regulatory protein. Nature. 2009;462:368–372. doi: 10.1038/nature08560. [DOI] [PubMed] [Google Scholar]
- 26.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nature Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. The first experimental demonstrations of dynamically mediated protein allostery in the CAP using relaxation dispersion NMR and NMR-detected hydrogen exchange. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reichheld SE, Yu Z, Davidson AR. The induction of folding cooperativity by ligand binding drives the allosteric response of tetracycline repressor. Proc Natl Acad Sci USA. 2009;106:22263–22268. doi: 10.1073/pnas.0911566106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Pino A, et al. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell. 2010;142:101–111. doi: 10.1016/j.cell.2010.05.039. This article demonstrates how increasing the relative concentration of one ligand can result in conditional cooperativity in an intrinsically disordered protein; in other words, the same protein can initially be an on-switch but then an off-switch at higher concentrations. [DOI] [PubMed] [Google Scholar]
- 29.Sevcsik E, Trexler AJ, Dunn JM, Rhoades E. Allostery in a disordered protein: oxidative modifications to α-synuclein act distally to regulate membrane binding. J Am Chem Soc. 2011;133:7152–7158. doi: 10.1021/ja2009554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreon ACM, Ferreon JC, Wright PE, Deniz AA. Modulation of allostery by protein intrinsic disorder. Nature. 2013;498:390–394. doi: 10.1038/nature12294. This article directly demonstrates cooperative ‘switching’ behaviour in an intrinsically disordered protein, via allosteric effects from truncation of the amino acid sequence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monod J, Changeux JP, Jacob F. Allosteric proteins and cellular control systems. J Mol Biol. 1963;6:306–329. doi: 10.1016/s0022-2836(63)80091-1. [DOI] [PubMed] [Google Scholar]
- 32.Koshland DE, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 33.Cui Q, Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. A thoughtful, comprehensive review that synthesizes the ‘old’ and ‘new’ views of allostery with experimental and computational case studies from the literature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koshland DE. Enzyme flexibility and enzyme action. J Cell Comp Physiol. 1959;54:245–258. doi: 10.1002/jcp.1030540420. [DOI] [PubMed] [Google Scholar]
- 35.Whitley MJ, Lee AL. Frameworks for understanding long-range intra-protein communication. Curr Protein Pept Sci. 2009;10:116–127. doi: 10.2174/138920309787847563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Changeux JP. Allostery and the Monod–Wyman–Changeux model after 50 years. Ann Rev Biophys. 2012;41:103–133. doi: 10.1146/annurev-biophys-050511-102222. [DOI] [PubMed] [Google Scholar]
- 37.Eaton WA, et al. Evolution of allosteric models for hemoglobin. IUBMB Life. 2007;59:586–599. doi: 10.1080/15216540701272380. [DOI] [PubMed] [Google Scholar]
- 38.Eaton WA, Henry ER, Hofrichter J, Mozzarelli A. Is cooperative oxygen binding by hemoglobin really understood? Nature Struct Biol. 1999;6:351–358. doi: 10.1038/7586. [DOI] [PubMed] [Google Scholar]
- 39.Erman B. A fast approximate method of identifying paths of allosteric communication in proteins. Proteins Struct Funct Bioinf. 2013;81:1097–1101. doi: 10.1002/prot.24284. [DOI] [PubMed] [Google Scholar]
- 40.Tang S, et al. Predicting allosteric communication in myosin via a pathway of conserved residues. J Mol Biol. 2007;373:1361–1373. doi: 10.1016/j.jmb.2007.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.England JL. Allostery in protein domains reflects a balance of steric and hydrophobic effects. Structure. 2011;19:967–975. doi: 10.1016/j.str.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 42.VanWart AT, Eargle J, Luthey-Schulten Z, Amaro RE. Exploring residue component contributions to dynamical network models of allostery. J Chem Theory Comput. 2012;8:2949–2961. doi: 10.1021/ct300377a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 44.Süel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nature Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 45.Colombo MF, Rau DC, Parsegian VA. Protein solvation in allosteric regulation: a water effect on hemoglobin. Science. 1992;256:655–659. doi: 10.1126/science.1585178. Groundbreaking experimental work demonstrating the large energetic effects of hydration on haemoglobin conformation and thus protein allostery. [DOI] [PubMed] [Google Scholar]
- 46.Elber R. Simulations of allosteric transitions. Curr Opin Struct Biol. 2011;21:167–172. doi: 10.1016/j.sbi.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 47.Weinkam P, Chen YC, Pons J, Sali A. Impact of mutations on the allosteric conformational equilibrium. J Mol Biol. 2013;425:647–661. doi: 10.1016/j.jmb.2012.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marcos E, Crehuet R, Bahar I. Changes in dynamics upon oligomerization regulate substrate binding and allostery in amino acid kinase family members. PLOS Comput Biol. 2011;7:e1002201. doi: 10.1371/journal.pcbi.1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silva MM, Rogers PH, Arnone A. A third quaternary structure of human hemoglobin A at 1.7-Å resolution. J Biol Chem. 1992;267:17248–17256. [PubMed] [Google Scholar]
- 50.Cooper A, Dryden DTF. Allostery without conformational change. Eur Biophys J. 1984;11:103–109. doi: 10.1007/BF00276625. The first explicit articulation of dynamic allostery, demonstrating the theoretical relevance of an entirely entropic energetic contribution to biological function. [DOI] [PubMed] [Google Scholar]
- 51.Fraser JS, et al. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462:669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lukin JA, et al. Quaternary structure of hemoglobin in solution. Proc Natl Acad Sci USA. 2003;100:517–520. doi: 10.1073/pnas.232715799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sekhar A, Kay LE. NMR paves the way for atomiclevel descriptions of sparsely populated, transiently formed biomolecular conformers. Proc Natl Acad Sci USA. 2013;110:12867–12874. doi: 10.1073/pnas.1305688110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wand AJ. The dark energy of proteins comes to light: conformational entropy and its role in protein function revealed by NMR relaxation. Curr Opin Struct Biol. 2013;23:75–81. doi: 10.1016/j.sbi.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manley G, Rivalta I, Loria JP. Solution NMR and computational methods for understanding protein allostery. J Phys Chem B. 2013;117:3063–3073. doi: 10.1021/jp312576v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu J, et al. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873–6888. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uversky VN. Intrinsically disordered proteins from A to Z. Int J Biochem Cell Biol. 2011;43:1090–1103. doi: 10.1016/j.biocel.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 58.Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18:343–384. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 59.Wright PE. Intrinsically unstructured proteins: re-assessing the structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 60.Tompa P. Unstructural biology coming of age. Curr Opin Struct Biol. 2011;21:419–425. doi: 10.1016/j.sbi.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 61.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci USA. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. The first paper to propose and demonstrate that intrinsic disorder can be used by proteins to mediate allosteric coupling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luque I, Freire E. Structural parameterization of the binding enthalpy of small ligands. Proteins. 2002;49:181–190. doi: 10.1002/prot.10208. [DOI] [PubMed] [Google Scholar]
- 63.Li Z, Raychaudhuri S, Wand AJ. Insights into the local residual entropy of proteins provided by NMR relaxation. Protein Sci. 1996;5:2647–2650. doi: 10.1002/pro.5560051228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang D, Kay LE. Contributions to conformational entropy arising from bond vector fluctuations measured from NMR-derived order parameters: application to protein folding. J Mol Biol. 1996;263:369–382. doi: 10.1006/jmbi.1996.0581. [DOI] [PubMed] [Google Scholar]
- 65.Igumenova TI, Frederick KK, Wand AJ. Characterization of the fast dynamics of protein amino acid side chains using NMR relaxation in solution. Chem Rev. 2006;106:1672–1699. doi: 10.1021/cr040422h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jarymowycz VA, Stone MJ. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem Rev. 2006;106:1624–1671. doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- 67.Frederick KK, Marlow MS, Valentine KG, Wand AJ. Conformational entropy in molecular recognition by proteins. Nature. 2007;448:325–329. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee AL, Kinnear SA, Wand AJ. Redistribution and loss of side chain entropy upon formation of a calmodulin–peptide complex. Nature Struct Biol. 2000;7:72–77. doi: 10.1038/71280. [DOI] [PubMed] [Google Scholar]
- 69.Palmer AG, Kroenke CD, Loria JP. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 70.Marlow MS, Dogan J, Frederick KK, Valentine KG, Wand AJ. The role of conformational entropy in molecular recognition by calmodulin. Nature Chem Biol. 2010;6:352–358. doi: 10.1038/nchembio.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Igumenova TI, Lee AL, Wand AJ. Backbone and side chain dynamics of mutant calmodulin–peptide complexes. Biochemistry. 2005;44:12627–12639. doi: 10.1021/bi050832f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Laine O, Streaker ED, Nabavi M, Fenselau CC, Beckett D. Allosteric signaling in the biotin repressor occurs via local folding coupled to global dampening of protein dynamics. J Mol Biol. 2008;381:89–101. doi: 10.1016/j.jmb.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rodgers TL, et al. Modulation of global low-frequency motions underlies allosteric regulation: demonstration in CRP/FNR family transcription factors. PLoS Biol. 2013;11:e1001651. doi: 10.1371/journal.pbio.1001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schrank TP, Bolen DW, Hilser VJ. Rational modulation of conformational fluctuations in adenylate kinase reveals a local unfolding mechanism for allostery and functional adaptation in proteins. Proc Natl Acad Sci USA. 2009;106:16984–16989. doi: 10.1073/pnas.0906510106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao J, Xu D. Correlation between posttranslational modification and intrinsic disorder in protein. Pac Symp Biocomput. 2012:94–103. [PMC free article] [PubMed] [Google Scholar]
- 76.Romero PR, et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci USA. 2006;103:8390–8395. doi: 10.1073/pnas.0507916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 78.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nature Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 79.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 80.Pan H, Lee JC, Hilser VJ. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc Natl Acad Sci USA. 2000;97:12020–12025. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bray D, Duke TA. Conformational spread: the propagation of allosteric states in large multiprotein complexes. Annu Rev Biophys Biomol Struct. 2004;33:53–73. doi: 10.1146/annurev.biophys.33.110502.132703. [DOI] [PubMed] [Google Scholar]
- 82.Luque I, Leavitt SA, Freire E. The linkage between protein folding and functional cooperativity: two sides of the same coin? Annu Rev Biophys Biomol Struct. 2002;31:235–256. doi: 10.1146/annurev.biophys.31.082901.134215. [DOI] [PubMed] [Google Scholar]
- 83.Ackers GK, Johnson AD, Shea MA. Quantitative model for gene regulation by the lambda phage repressor. Proc Natl Acad Sci USA. 1982;79:1129–1133. doi: 10.1073/pnas.79.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Motlagh HN, Hilser VJ. Agonsim/antagonism switching in allosteric ensembles. Proc Natl Acad Sci USA. 2012;109:4134–4139. doi: 10.1073/pnas.1120519109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bai F, et al. Conformational spread as a mechanism for cooperativity in the bacterial flagellar switch. Science. 2010;327:685–689. doi: 10.1126/science.1182105. [DOI] [PubMed] [Google Scholar]
- 86.Gekko K, Obu N, Li J, Lee JC. A linear correlation between the energetics of allosteric communication and protein flexibility in the Escherichia coli cyclic AMP receptor protein revealed by mutation-induced changes in compressibility and amide hydrogen-deuterium exchange. Biochemistry. 2004;43:3844–3852. doi: 10.1021/bi036271e. [DOI] [PubMed] [Google Scholar]
- 87.Fisher CK, Stultz CM. Constructing ensembles for intrinsically disordered proteins. Curr Opin Struct Biol. 2011;21:426–431. doi: 10.1016/j.sbi.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Forman-Kay JD, Mittag T. From sequence and forces to structure, function, and evolution of intrinsically disordered proteins. Structure. 2013;21:1492–1499. doi: 10.1016/j.str.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mittag T, Forman-Kay JD. Atomic-level characterization of disordered protein ensembles. Curr Opin Struct Biol. 2007;17:3–14. doi: 10.1016/j.sbi.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 90.Bernadó P, et al. A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc Natl Acad Sci USA. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jensen MR, et al. Quantitative determination of the conformational properties of partially folded and intrinsically disordered proteins using NMR dipolar couplings. Structure. 2009;17:1169–1185. doi: 10.1016/j.str.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 92.Cavalli A, Salvatella X, Dobson CM, Vendruscolo M. Protein structure determination from NMR chemical shifts. Proc Natl Acad Sci USA. 2007;104:9615–9620. doi: 10.1073/pnas.0610313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clore GM. Visualizing lowly-populated regions of the free energy landscape of macromolecular complexes by paramagnetic relaxation enhancement. Mol Biosyst. 2008;4:1058–1069. doi: 10.1039/b810232e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lindorff-Larsen K, Best RB, Depristo MA, Dobson CM, Vendruscolo M. Simultaneous determination of protein structure and dynamics. Nature. 2005;433:128–132. doi: 10.1038/nature03199. [DOI] [PubMed] [Google Scholar]
- 95.Tang C, Louis JM, Aniana A, Suh JY, Clore GM. Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease. Nature. 2008;455:693–696. doi: 10.1038/nature07342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu B, et al. Structural and energetic mechanisms of cooperative autoinhibition and activation of Vav1. Cell. 2010;140:246–256. doi: 10.1016/j.cell.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fraser JS, et al. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci USA. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burnley TB, Afonine PV, Adams PD, Gros P. Modelling dynamics in protein crystal structures by ensemble refinement. eLife. 2012;1:e00311. doi: 10.7554/eLife.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Penczek PA, Kimmel M, Spahn CMT. Identifying conformational states of macromolecules by eigen-analysis of resampled cryo-EM images. Structure. 2011;19:1582–1590. doi: 10.1016/j.str.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dror RO, Dirks RM, Grossman JP, Xu H, Shaw DE. Biomolecular simulation: a computational microscope for molecular biology. Annu Rev Biophys. 2012;41:429–452. doi: 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- 101.Dror RO, et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- 102.Qian H. Cyclic conformational modification of an enzyme: serial engagement, energy relay, hysteretic enzyme, and Fischer’s hypothesis. J Phys Chem B. 2010;114:16105–16111. doi: 10.1021/jp102400u. [DOI] [PubMed] [Google Scholar]
- 103.Ward AB, Sali A, Wilson IA. Integrative structural biology. Science. 2013;339:913–915. doi: 10.1126/science.1228565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Andreeva A, et al. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36:D419–D425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cesareni G, Gimona M, Sudol M, Yaffe M. Modular Protein Domains. Wiley-VCH; 2005. [Google Scholar]
- 106.Loh SN, Ha JH. Protein conformational switches: from nature to design. Chemistry. 2012;18:7984–7999. doi: 10.1002/chem.201200348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Choi JH, San A, Ostermeier M. Non-allosteric enzyme switches possess larger effector-induced changes in thermodynamic stability than their non-switch analogs. Protein Sci. 2013;22:475–485. doi: 10.1002/pro.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zayner JP, Antoniou C, French AR, Hause RJ, Jr, Sosnick TR. Investigating models of protein function and allostery with a widespread mutational analysis of a light activated protein. Biophys J. 2013;105:1027–1036. doi: 10.1016/j.bpj.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]