

Abstract
Opioid drugs remain the gold standard for the treatment of severe pain, both acute/post-surgical and chronic. However, the utility of opioid drugs for the treatment of chronic pain is compromised by the development of analgesic tolerance which, in turn, leads to dose-escalation and increased likelihood of dangerous side effects, including dependence. Consequently, there remains resistance among clinicians and the general population to using opiates for pain management because of risk of “addiction.” These fears are not unwarranted. More than 2.5 million people begin abusing opioid painkillers each year, and prescription opioid abuse is now the second most common type of illegal drug use after marijuana. Some abusers become dependent due to recreational use of prescription painkillers. However, many abusers are among the 40 million people suffering from chronic pain, and developed dependence while using the drugs for legitimate purposes. Both of these trends highlight the need to develop opioid therapeutics with a reduced liability to cause tolerance, dependence and addiction. Identifying the ideal properties of opioid drugs that would retain analgesia but reduce these side-effects has been a goal of my laboratory for more than a decade. During this time, we have proposed the novel hypothesis that opioid drugs that promote desensitization, endocytosis and recycling of the mu-opioid-receptor (MOR) will retain analgesic efficacy, but will have a reduced liability to cause tolerance, dependence and addiction. We have generated substantial data, both pharmacological and genetic to suggest that our hypothesis is a valid one. These data are summarized in this review.
Keywords: opioid, G protein coupled receptor, analgesia, morphine, tolerance, dependence, addiction, trafficking, endocytosis, desensitization
1.0 Regulation of opioid receptor signaling by trafficking
Following activation by either small molecule alkaloid or peptide agonists, opioid receptors are regulated by multiple mechanisms, many of which have been implicated in receptor desensitization (Freedman and Lefkowitz, 1996). Signaling from many GPCRs is regulated by a well-characterized and highly conserved mechanism involving receptor phosphorylation by G protein-coupled receptor kinase (GRK) and subsequent arrestin recruitment (reviewed in (Ferguson et al., 1998)). These processes can contribute directly to GPCR desensitization by facilitating the uncoupling of receptor from G protein. Furthermore, following desensitization, receptors are endocytosed into an intracellular compartment from which they can be recycled to the membrane (resensitization) or targeted for degradation leading to receptor downregulation (Lefkowitz, 1998). Hence, these processes can contribute directly to tolerance by decreasing the number of functional cell surface receptors. Consequently, one view is that opioid receptor desensitization and endocytosis contribute directly to analgesic tolerance by reducing the number of functional receptors present in target neurons. However, morphine-activated MORs show substantially reduced arrestin binding and desensitization (Whistler and von Zastrow, 1998; Zhang et al., 1998) compared to MORs activated by endogenous ligand and some other opioids in all cases where this question has been examined. Specifically, morphine promotes little endocytosis of the wild-type (WT) MOR in cultured cells (Arden et al., 1995; Keith et al., 1996) and native neurons (Keith et al., 1998; Sternini et al., 1996), whereas endogenous peptide ligands such as endorphins and several opiate drugs such as methadone (Garrido and Troconiz, 1999) readily drive receptor endocytosis (Keith et al., 1998; Sternini et al., 1996). Even in systems where morphine has been seen to promote some endocytosis, it produces dramatically less endocytosis than that induced by other opioids (Haberstock-Debic et al., 2005; Haberstock-Debic et al., 2003).
Importantly, individual opiate drugs not only have varying propensities to promote endocytosis, they also have varying propensities to promote tolerance and dependence. Specifically, while heroin, morphine and many of its analogs (such as oxycontin) have a high liability for producing these side effects, the endogenous and modified enkephalins (such as the hydrolysis resistant enkephalin, [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO)) and the small molecule opioid methadone and etorphine, that do promote endocytosis, show reduced liability to produce tolerance (Figure 1A–C) and dependence (Figure 1D) when given at equi-analgesic doses as morphine in preclinical models (see also for example (Garrido and Troconiz, 1999; Kim et al., 2008) and even (Raehal and Bohn, 2011) which shows reduced tolerance to methadone compared to morphine even though that is not what is highlighted in this report). This is the case not only when drugs are administered systemically (Figure 1A), but also when the drugs are administered intracerebroventricularly (i.c.v, Figure 1B)) or intrathecally (i.t., Figure 1C), where peptide ligands and morphine can be directly compared, and where differences in the pharmacokinetics of the individual drugs are minimized. Of course, each time a new drug is used there are many parameters that have changed. Hence, it has been difficult to conclude based on these data that endocytosis “protects” against tolerance and dependence. Nevertheless, it is intriguing to speculate that drugs that promote endocytosis will have a reduced liability to produce tolerance and dependence, because they most closely resemble the endogenous ligand (Finn and Whistler, 2001; Whistler et al., 1999). Specifically, we propose that the regulation of opioid receptors by endocytosis serves a protective role in reducing the development of tolerance and dependence to opiate drugs specifically because this mechanism regulates signaling in a rapid and reversible way, as follows. First, as a consequence of endocytosis, cells are rapidly desensitized to agonist. Second, following endocytosis, receptors can be recycled to the cell surface in a fully active state, thereby resensitizing cells to agonist. This dynamic cycle of receptor regulation may be designed to mediate the actions of native opioid peptides, which are typically released in a phasic or pulsatile manner. Opiate drugs, in contrast, persist in the extracellular milieu for a prolonged period of time because of their slow clearance and, hence, can activate opioid receptors in an abnormally prolonged manner. Accordingly, opiate drugs that can induce the rapid desensitization and endocytosis of receptors followed by resensitization to agonist may more closely mimic the phasic actions and physiological adaptations observed with native peptide ligands. Clinically, tolerance to the effects of chronic opioids appear to develop at different rates for different tissues. For example analgesic tolerance appears to develop more quickly than tolerance to the constipatory effects of opioids (for review see (Collett, 1998)). It remains to be seen whether these difference in rates of tolerance development are related to differences in trafficking of the MOR, or whether they are a consequence of other difference in gut versus CNS responses to opioid drug.
Figure 1. Opioids have variable propensities to promote tolerance in mice and rats when given at equi-antinociceptive doses.
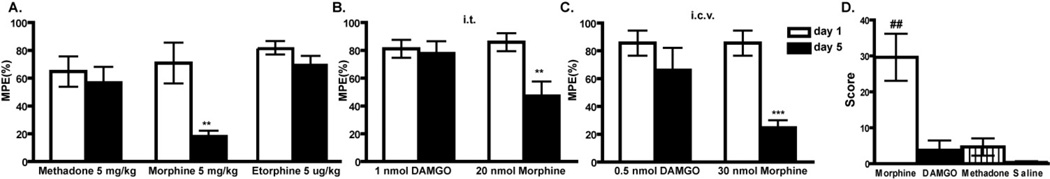
A. Mice were administered opioid drug s.c. at the dose stated twice per day for 5 days. Nociception was measured with the tail flick test on Day 1 and Day 5. Maximal possible effect (MPE 100% x [(drug response time – basal response time)/(cutoff time – basal response time)]) is shown. Mice developed tolerance to morphine but not methadone or etorphine, both of which promote MOR endocytosis (Whistler and von Zastrow, 1999). **p<0.01, ***p<0.001 compared to day 1. B,C. Rats were administered opioid drug at the dose stated, i.t.(B) or i.c.v. (C) twice per day for 5 days. Nociception was measured with the tail flick test on Day 1 and Day 5. Rats developed tolerance to morphine but not DAMGO both i.c.v. and i.t.. *p<0.05, ***p<0.001 compared to day 1. D. Dependence to opioids with different trafficking properties. Animals were administered drug for seven days (MS 30 nmol, DG 0.5 nmole, methadone 50 nmole). On day 7, withdrawal was precipitated with a subcutaneous (s.c.) injection of naloxone HCl (2 mg/kg). Four withdrawal signs (jumping, head shakes, teeth chattering, weight loss) were counted. A global withdrawal score was calculated for each animal giving equal weight to each individual sign. ##p<0.01 compared to other groups.
The ability of an agonist to promote endocytosis of the MOR does not always vary in a linear fashion with agonist activity (Whistler et al., 1999), indicating that MOR endocytosis is an independent functional property and providing a clear example of functional selectivity (for review see (Urban et al., 2007)). Since agonist activity and receptor endocytosis have opposing effects on receptor-mediated signaling, the net amount of signal transmitted to the cell is a function of both processes, a relationship we term “RAVE” for relative activity versus endocytosis. Thus, morphine would have a particularly high RAVE value as a consequence of its inability to promote receptor desensitization and endocytosis. Endorphins and opiate drugs that acutely signal with similar efficacy yet induce receptor desensitization and endocytosis would have lower RAVE values than morphine. These observations have led us to propose that drugs with high RAVE values, such as morphine, have an enhanced propensity to produce the adverse effects associated with prolonged drug exposure, precisely because they signal through the receptor for aberrantly long periods of time and qualitatively with a pattern quite different from than which occurs with an agonist that promotes endocytosis and recycling.
Figure 1 is simply meant to be illustrative of a body of published work (see for example Finn and Whistler, 2001; Whistler et al., 1999). Moreover, we do not mean to imply that drugs like methadone and etorphine cannot cause tolerance and dependence. Indeed, any drug given at a high enough dose for a long enough time will cause these side effects. The goal, therefore, is to identify the properties of an opioid drug that reduce the liability. Importantly, because each individual opiate drug differs in several properties, including selectivity, affinity, potency, efficacy and bioavailability/half life, it has been difficult to draw strong conclusions regarding liability for tolerance and dependence and a specific characteristic of an opioid drug—such as its efficacy, potency or its ability to promote endocytosis. In short, each time a different drug is used, for example morphine versus methadone, numerous aspects of signaling are modified—including but not limited to endocytosis. Thus, while different opioids have different “RA”s and different “VE”s, there are no two opioid ligands that differ only in their ability to endocytose the receptor, making it difficult to conclusively determine the contribution of endocytosis to tolerance and dependence.
2.0 Generation of RMOR mice
To eliminate as a factor the varying pharmacological properties amongst the opioid drugs and selectively focus on the role of trafficking per se on morphine tolerance and dependence, we chose to alter the RAVE of morphine by changing the receptor rather than using opioids with potentially different RAVEs (see Finn and Whistler, 2001; Whistler et al., 1999). Specifically, we created a mutant MOR, RMOR, in which the pharmacology of the receptor to endogenous ligands is unchanged, as is the affinity potency and efficacy of morphine, but which undergoes endocytosis in response to activation by morphine. We first generated substantial in vitro data using the RMOR (for recycling MOR; Finn and Whistler, 2001). We then created an RMOR knock-in mouse (Kim et al., 2008). The RMOR mice were viable, had no gross phenotypic abnormalities and showed normal baseline pain responses (hot-plate latency, 56°C: wild-type, 4.88 ± 0.33 seconds; mutant, 4.55 ± 0.32 seconds; see Figure 3 from Kim et al., 2008 saline treatments).
Figure 3. Antinociception in WT MOR and RMOR knock-in mice.

A. Enhanced and prolonged morphine-induced antinociception in RMOR knock-in mice. Antinociceptive responses were measured with the hot-plate response latency test (56°C) after morphine treatment (10 mg/kg, sc). A response endpoint was defined as latency to either lick the fore- or hindpaws or flick the hindpaws. To avoid tissue damage, mice were exposed to the hot-plate for a maximum of 20 seconds. Data are reported as the mean ± SEM of percent maximum possible effect (MPE) using the following formula: 100% x [(drug response time – basal response time)/(20 s – basal response time)]. A two-way analysis of variance revealed that the MPE curve for RMOR mice (n=17) mice was significantly greater and prolonged relative to the MOR mice (n=17) as indicated by a significant genotype [p<.001, F(1,7)=28.05] and genotype X time interaction effect [p<.001, F(1,7)=4.97]. B. Dose-dependent morphine-antinociception. Antinociceptive responses were determined with the hot-plate test and data are reported as mean ± SEM of MPE (see A). Separate groups of mice for both genotypes (n=7–9) were injected with the doses of morphine indicated and assessed for antinociception 30 min later. To test whether the antinociceptive responses were mediated by opioid receptors, a final grouping was injected with morphine (10 mg/kg) followed by naloxone (2 mg/kg). RMOR knock-in mice showed enhanced antinociception at 3 and 10 mg/kg doses (RMOR vs. WT MOR scores for MPE at respective morphine doses, student’s t-test, *p<.03) with the latter dose inducing the maximum possible response (100%) in the mutant mice. At the highest dose tested (50 mg/kg) both genotypes exhibited the maximum possible response (100%). For both genotypes, antinociception induced by 10 mg/kg of morphine was reversed by treatment with 2 mg/kg of the opioid antagonist naloxone (morphine 10 mg/kg with and without naloxone 2 mg/kg treatment for each genotype respectively, student’s t-test +++p<.001). C. MOR desensitization in the brainstem following acute morphine treatment. Agonist-mediated [35S]GTPγS binding was measured in brainstem membranes of WT MOR and RMOR knock-in mice with increasing concentrations of DAMGO. Left Panel. Binding in WT MOR mice was significantly reduced (p<0.01) following acute morphine-treatment (10 mg/kg s.c. 30 min; EC50 = 428 ± 141 µM; open squares ) compared to vehicle-treated mice (EC50 = 2.35 ± 0.9 µM; closed squares). Right Panel. Binding in RMOR knock-in mice was not significantly changed (p>0.05) following acute morphine-treatment (EC50 = 3.29 ± 1.4 µM; open circles) compared to vehicle-treated mice (EC50 = 1.16 ± 0.6 µM; closed circles). Data were analyzed by nonlinear regression using GraphPad Prism software and are presented as means ± SEM of at least three experiments performed in triplicate with an investigator blind to genotype. D. Enhanced antinociception in RMOR knock-in mice is morphine-specific. Separate groups of mice for both genotypes (n=8–10) were injected with the doses of methadone indicated (1–10 mg/kg,) and assessed for antinociception. Methadone induced a dose-dependent increase in antinociceptive response with no genotypic differences. For both genotypes, antinociception induced by 4 mg/kg of methadone was reversed by treatment with 2 mg/kg of the opioid antagonist naloxone (methadone 4 mg/kg with and without naloxone 2 mg/kg treatment for each genotype respectively, student’s t-test +++p<.001). Thus, enhanced opioid-induced antinociception observed in the RMOR knock-in mice is agonist-specific, and naloxone-reversible. Reproduced with permission from Kim, J.A., Bartlett, S., He, L., Nielsen, C.K., Chang, A.M., Kharazia, V., Waldhoer, M., Ou, C.J., Taylor, S., Ferwerda, M., Cado, D., Whistler, J.L., 2008. Morphine-induced receptor endocytosis in a novel knockin mouse reduces tolerance and dependence. Curr. Biol. 18, 129–135.
Consistent with their equivalent baseline pain responses, there were no genotypic differences in receptor distribution, ligand affinity, potency, or efficacy for peptide ligands and morphine as assessed by G protein coupling in the WT MOR versus the RMOR mice (Kim et al., 2008), in line with our in vitro findings (Finn and Whistler, 2001). Furthermore, there are no differences in the potency or efficacy of either DAMGO or morphine for either a presynaptic or a postsynaptic response to opioid drug in brain slices of the ventral tegmental area and the locus coeruleus (Figure 2, from Madhavan et al., 2010 and data not shown). Specifically, the inhibitory effect of DAMGO on GABA IPSCs was recorded from dopamine neurons in WT and RMOR mice. Peak inhibition (black bar - baseline in Figure 2A inset) of GABA currents was plotted against the logarithm of agonist concentration (Figure 2A). The dose-response curves for inhibition by DAMGO (WT EC50 89.22 ± 0.28 nM, Emax 90.57 ± 7.6; RMOR EC50 112.6 ± 0.32 nM, Emax 86.96 ± 10.6; p = 0.9982; n = 33) was not significantly different between WT and RMOR mice. Thus, DAMGO is equally effective at inhibiting GABA IPSCs in VTA dopamine neurons in WT and RMOR mice. Additionally, there were no genotype differences in the presynaptic effects of DAMGO, which significantly decreased mIPSC frequency (Figure 2B), but not amplitude (Figure 2C), equivalently in both WT (Control: 13.34 ± 3.2 Hz, DAMGO: 7.76 ± 3 Hz; n=9; p<0.0001) and RMORs (Control: 13.92 ± 2.8 Hz, DAMGO: 7.0 ± 2.2 Hz; n=9; p=0.0058).
Figure 2. MOR and RMOR in the VTA show equivalent acute responses to opioid agonist.

A. DAMGO (DG) and morphine (MS for morphine sulfate) dose–response curves of peak inhibition (black bars, 3 min window, inset) of GABA IPSCs plotted against log10 (agonist in nanomolar). Sigmoidal dose–response curves fitted for WT and RMOR were not significantly different for DAMGO (WT EC50 of 89.22 ± 0.28 nm, E max of 90.57 ± 7.6; RMOR EC50 of 112.6 ± 0.32 nm, E max of 86.96 ± 10.6; n = 17 each) or morphine (WT EC50 of 106.2 ± 0.78 nm, E max of 52.82 ± 9.8; RMOR EC50 of 125.7 ± 0.51 nm, E max of 48.66 ± 6.52; n = 8 each). B. Cumulative inter-event interval distribution showing equivalent presynaptic inhibition of GABA frequency by 300 nm DAMGO (DG) in WT and RMOR mice. B. Inset, DAMGO significantly inhibits GABA mIPSC frequency in WT (n = 9) and RMOR (n = 9). C. Cumulative amplitude distribution of GABA mIPSCs shows no effect of DAMGO in either WT or RMOR. C. Inset, DAMGO does not inhibit GABA mIPSC amplitude in WT (n = 9) or RMOR (n = 9). The distributions of frequencies were not significantly different comparing WT and RMOR in either the absence (n = 9) or presence of DAMGO (n = 9). Reproduced from (Madhavan et al., 2009).
Thus, to date, we have found no difference between the MOR and RMOR receptor with regard to “classic” pharmacology. However, in RMOR mice, not only the endogenous peptide ligands but also morphine promotes receptor endocytosis, whereas in their WT mice morphine does not. Furthermore, RMOR receptors desensitize rapidly to morphine while WT receptors do not (Madhavan et al., 2010). In short, we have created a mutant mouse in which the pharmacology of the receptor to endogenous ligands is unchanged, as is the affinity potency and efficacy of morphine, but which, in response to activation by morphine, undergoes endocytosis and arrestin recruitment thereby presumably gaining, as well, any signaling biased for this pathway (for review see Rajagopal et al.). Endocytosis of the MOR likely serves at least two important functions in regulating signal transduction. First, desensitization and endocytosis act as an “OFF” switch by uncoupling receptors from their cognate G protein. Second, endocytosis and receptor recycling serve as an “ON” switch, resensitizing receptors by recycling them to the plasma membrane where agonist can regain access to the receptors. Thus, due to poor endocytosis, both the OFF and ON function of the WT MOR are altered in response to morphine compared to endogenous ligands, while in RMOR mice both the ON and OFF functions more closely resemble the endogenous ligand.
Importantly, RMOR mice show outstanding analgesic responses to morphine (Figure 3, from Kim et al., 2008) but develop neither tolerance or dependence to the drug in a paradigm where WT mice do (Figure 4, from Kim et al., 2008).
Figure 4. Opioid tolerance and dependence in WT and RMOR knock-in mice.

A. Mice were treated twice daily with morphine (10 mg/kg, sc) for 5 days and antinociception was assessed following the first injection of morphine each day. Mean ± SEM of MPE (Maximal possible effects) is presented. A two-way ANOVA revealed that mice given morphine (n=17) behaved differently as indicated by a significant group effect [F(2,42)=27.95, p<.001] and group X treatment days effect [F(4,84)=12.09, p<.001]. RMOR knock-in mice showed reduced tolerance compared to WT MOR mice treated with the same dose of morphine (10 mg/kg). [RMOR 10 vs. MOR 10 **p<.01]. RMOR mice chronically treated with an equi-antinociceptive dose of morphine (3 mg/kg) also showed reduced tolerance than WT MOR mice chronically treated with a higher dose of morphine (10 mg/kg) (RMOR 3 vs. MOR 10, ++p<.01). Only WT mice chronically treated with morphine showed a significant decrease in antinociception from Day 1 to Day 5 (MOR 10 Day 1 vs. Day 5, ###p<.001) indicative of tolerance. Reproduced from (Kim et al., 2008). B. Mice were treated with a cumulative dose response of morphine on day 1, then given morphine twice per day for 5 days. On the second dosing of day 5, mice were again treated with the cumulative dose response to measure the shift in potency of morphine. C. Naloxone precipitated withdrawal. Mice were treated with 10 mg/kg of morphine (black bars, n=10–12), 4 mg/kg of methadone (grey bars, n=9) or saline (white bars, n=6) as in A. Mice were challenged with naloxone (2 mg/kg, sc) 30 min following the final treatment. Standard withdrawal behaviors including jumping, wet-dog shakes, paw licks and paw tremors were scored by an observer blind to genotype. Total withdrawal scores (sum of all individual withdrawal behaviors) ± SEM are presented and group differences were analyzed with the LSD test. WT mice displayed significant withdrawal when chronically treated with morphine (MOR Morphine vs. MOR saline, ***p<.001; MOR Morphine vs. MOR methadone, ++p<.01). RMOR mice showed no significant withdrawal. Reproduced from (Kim et al., 2008). D. Magnitude of tolerance correlates with magnitude of withdrawal. Shown is a scatter plot of global withdrawal versus the magnitude of tolerance (100-MPE) for morphine-treated WT and RMOR mice. Correlation of tolerance and withdrawal in morphine-treated WT and RMOR mice shows that greater tolerance predicts increased somatic signs of withdrawal (solid black line, n=30, r=0.84, p<0.0001). This was also true for a correlation between tolerance and dependence in WT alone (dotted line, n=15, r=0.81, p=0.0003). Reproduced from Madhavan, A., He, L., Stuber, G.D., Bonci, A., Whistler, J., 2009. Morphine-induced mu opioid Receptor Endocytosis Prevents Adaptations in GABA Release During Withdrawal. International Narcotics Research Conference, Portland, Oregon, USA.
2.1 Analgesia in RMOR mice
Morphine-induced antinociception was first evaluated by measuring response latencies in the hot-plate test. We tested a dose of morphine (10 mg/kg) known to induce robust antinociception in mice. The acute antinociceptive effect of this dose of morphine was significantly enhanced and prolonged in knock-in mice relative to their wild-type littermates (Figure 3A). A dose of 3 mg/kg in the mutant mouse was equi-antinociceptive to 10 mg/kg in the wild-type mouse (Figure 3B). Both genotypes reached a ceiling effect at the highest dose tested, 50 mg/kg. The opioid antagonist naloxone completely reversed the antinociceptive effects of morphine in both WT and RMOR mice (Figure 3B).
We propose that the enhanced antinociception in the mutant mice reflects the restoration of the ON function provided by receptor endocytosis and recycling. Specifically, we propose that morphine-occupied MORs become partially desensitized in WT mice and fail to resensitize due to poor endocytosis; whereas in the RMOR knock-in mice, receptors are also desensitized but are rapidly resensitized by endocytosis and recycling. Consistent with this hypothesis, MORs in WT mice given a single 10 mg/kg dose of morphine showed significant receptor-G protein uncoupling or “desensitization” (Figure 3C, left panel). Clearly, not all MORs in these mice were desensitized, since morphine is still an excellent acute antinociceptive agent in WT mice. Nevertheless, MORs in the brainstem of WT mice treated with a single dose of morphine showed a 200-fold shift in the EC50 of DAMGO (Figure 3C, left panel) compared to WT mice treated with vehicle. In contrast, receptors in RMOR mice given the same dose of morphine, showed no desensitization (Figure 3C, right panel). These data suggest that the reduced morphine antinociception in the WT compared to the RMOR mice reflects partial desensitization of MORs that is not reversed by endocytosis and recycling.
If this were the case, we would expect mice of both genotypes to show equivalent antinociception to an agonist that promotes endocytosis of the receptor in both genotypes. Indeed, there were no significant genotypic differences in antinociception induced by methadone (1–10 mg/kg; Figure 3D), a MOR agonist that promotes rapid internalization of both the WT MOR and mutant RMOR. Thus, the enhanced opioid antinociception observed in the RMOR knock-in mice is specific to morphine. Together with our immunohistochemical and pharmacological data from these mice (Kim et al., 2008), these data suggest that the enhanced morphine antinociception in the RMOR knock-in mice cannot be accounted for by differences in MOR distribution, ligand affinity, receptor number or receptor G-protein coupling. Rather, these data suggest that facilitating MOR endocytosis enhances morphine antinociception by enhancing arrestin recruitment, or reversing rapid desensitization.
2.2 Analgesic Tolerance in WT and RMOR mice
We next evaluated the development of analgesic tolerance following twice daily administrations of morphine (10 mg/kg) over 5 days (Kim et al., 2008). WT mice in this paradigm developed antinociceptive tolerance (Figure 4A, squares). In contrast, their RMOR littermates, treated with the same dose of morphine (10 mg/kg) at the same intervals, showed no evidence of tolerance, exhibiting as much antinociception on the last day of drug treatment as they did on the first day (Figure 4B, circles). To rule out the possibility that the lack of tolerance in the mutant mice was an artifact of enhanced morphine antinociception (Figure 3A,B), a separate group of knock-in mice were treated chronically with an equi-antinociceptive dose of morphine (3 mg/kg, see Figure 3B) given at the same intervals. These RMOR knock-in mice still showed reduced tolerance, maintaining similar levels of antinociception over the course of treatment (Fig 4B, triangles). Thus, reduced morphine tolerance in the RMOR relative to WT mice cannot be attributed to enhanced morphine antinociception. The enhanced tolerance in WT versus RMOR mice was also demonstrated as a shift in the dose response to morphine on day 5 versus day 1 in WT but not RMOR mice (Figure 4B).
These results suggest that endocytosis of the receptor reduces the development of antinociceptive tolerance. If this were the case, one would expect that opiate agonists, such as methadone, that promote endocytosis of the WT MOR would have reduced liability for promoting tolerance in WT mice (see Fig 1). In addition, WT and RMOR mice should show equivalent responsiveness to chronic methadone, which they do (see Kim et al., 2008). Thus, reduced chronic opioid tolerance in RMOR mice relative to WT mice is specific to morphine.
2.3 Assessing physical dependence in WT and RMOR mice
We next examined whether facilitating endocytosis in the RMOR mice affected the development of morphine dependence. Following chronic treatment with morphine, mice were challenged with the opioid antagonist, naloxone (2 mg/kg), 30 min following the final morphine injection. Global withdrawal responses were scored by an observer who was blind to genotype (Fig 4C). WT mice expressed robust withdrawal responses compared to RMOR mice, which were chronically treated with the same amount of morphine (10 mg/kg) but at a functionally higher dose (see Figure 4A, B). Consistent with the hypothesis that enhanced receptor endocytosis decreases withdrawal, chronic methadone treatment (4 mg/kg given at the same intervals as morphine), promoted substantially less withdrawal than did morphine in wild-type mice (Figure 4C). In fact, the moderate level of methadone withdrawal in wild-type mice was equivalent to that produced by either morphine or methadone in the RMOR mice (Figure 4C).
Individual mice show different magnitudes of tolerance and withdrawal, even with identical morphine treatment. In order to determine the relationship between tolerance and behavioral measures of withdrawal in animals that were treated with morphine, tolerance was plotted against the behavioral withdrawal score from individual mice. The magnitude of morphine tolerance in WT and RMORs correlated with withdrawal score, regardless of genotype (Figure 4D solid line; n=30 pairs; r=0.84; p<0.0001) with all RMOR mice showing less tolerance and dependence than any WT mouse. We also found that the magnitude of morphine tolerance in individual WT mice alone was a faithful predictor of the degree of dependence (Figure 4D dotted line; n=15 pairs; r=0.81; p=0.0003).
2.4 CNS Biomarkers of Tolerance/Dependence in WT and RMOR mice
CNS changes as diverse as cAMP super-activation, alterations in N-methyl-D-aspartate receptor (NMDAR) levels and glucocorticoid receptor (GR) levels have been implicated in the development of morphine tolerance and/or dependence (Inoue et al., 2003; Lim et al., 2005a; Lim et al., 2005b; Marinelli et al., 1998; Pasternak, 2007; Trujillo and Akil, 1991; Yang et al., 2004). Thus, although they are distinct phenomena, each is considered a biochemical hallmark, or “biomarker” of morphine tolerance and dependence, because manipulations of each of these systems influence these behaviors. However, whether these adaptations are related to one another, and how activity at a single receptor, the MOR, can dramatically affect so many diverse systems remains unknown. We assessed the consequences of promoting morphine-induced endocytosis on these biochemical changes utilizing the RMOR knock-in mouse model (Figure 5–Figure 7 from He et al., 2009). As expected, chronic morphine treatment of WT mice promoted superactivation of adenylyl cyclase (Figure 5A). This superactivation was brain region-specific, as it was detectable in striatum but not in other brain regions examined, including the thalamus, periacquductal grey (PAG) and brain stem (Figure 5A). Importantly, superactivation did not occur in any brain region in the RMOR mice (Figure 5B).
Figure 5. Adenylyl cyclase activity in WT and RMOR knock-in mice after chronic morphine treatment.

A. Adenylyl cyclase activity was assessed in membranes prepared from the striatum, thalamus, PAG and brain stem of WT mice after 5 days of morphine (10 mg/kg, s.c., twice daily) or saline treatment. B. Adenylyl cyclase activity was assessed in membranes prepared from the striatum of both WT and RMOR knock-in mice after 5 days of morphine or saline treatment as in (a). Data are the mean ± SEM from 4 independent experiments with each performed in triplicate. * p<0.05 versus saline. Reproduced with permission from He, L., Whistler, J.L., 2005. An opiate cocktail that reduces morphine tolerance and dependence. Curr. Biol. 15, 1028–1033.
Figure 7. Effects of chronic morphine treatment on GR levels.

A. Chronic morphine induced a significant increase in GR immunoreactivity in the thalamus and PAG of WT mice. B, C. Chronic morphine caused no change in RMOR mice in either thalamus (B) or PAG (C). In all cases, data are expressed as mean ± SEM of at least three separate experiments. NAc: nucleus accumbens; Str: striatum; Thal: thalamus; Amyg: amygdala; PAG: periaqueductal gray. * p< 0.05, ** p < 0.01, compared with saline group. Reproduced with permission from He, L., Kim, J.A., Whistler, J.L., 2009. Biomarkers of morphine tolerance and dependence are prevented by morphine-induced endocytosis of a mutant {micro}-opioid receptor. Faseb J. 23, 4327–4334.
We next assessed whether this morphine treatment regimen would promote changes in NMDAR or GR protein levels. Several brain regions were examined, including the striatum, nucleus accumbens (NAc), thalamus, amygdala and PAG, all regions that are thought to be important for morphine-induced antinociception and/or physical dependence. In WT mice, the most predominant changes in NMDAR number were observed in PAG with significant reductions in NR1, NR2A and NR2B levels after morphine treatment (Figure 6). We also observed a significant change in the thalamus where the NR2B subunit was up-regulated after morphine treatment (Figure 6). In WT mice, morphine treatment also caused a significant increase in GR protein levels in both the thalamus and PAG (Figure 7A). Importantly, none of these changes in NMDARs (Figure 6) or GRs (Figure 7) occurred in the RMOR mice. Together, these data demonstrate that morphine tolerance and dependence are mediated by multiple biochemical mechanisms and suggest that MOR endocytosis plays a critical role in each of these mechanisms. They also validate the RMOR mice as a model for evaluating biomarkers in the CNS associated with morphine dependence.
Figure 6. NMDAR subunit levels are altered in WT but not RMOR knock-in mice after chronic morphine treatment.

Protein levels of the NMDAR subunits NR1 (A), NR2A (B) and NR2B (C), were assessed in several mouse brain regions by immunoblot in both WT and RMOR knock-in mice treated with morphine or saline. Data are expressed as mean± SEM. Tissue samples were analyzed from at least three separate experiments. Thal: thalamus; PAG: periaqueductal gray. * p< 0.05, ** p < 0.01 and *** p< 0.001, compared with saline group. Reproduced with permission from He, L., Kim, J.A., Whistler, J.L., 2009. Biomarkers of morphine tolerance and dependence are prevented by morphine-induced endocytosis of a mutant {micro}-opioid receptor. Faseb J. 23, 4327–4334.
3.0 Morphine-induced changes in physiology are affected by MOR trafficking
We also examined synaptic plasticity in the ventral tegmental area (VTA) that occurs in a paradigm that produces morphine tolerance and dependence in WT but not RMOR mice. Acute opioid activation of the MOR modulates dopamine neuron activity by inhibiting GABA release onto dopamine neurons (Johnson and North, 1992a, b). And, as mentioned above, chronic morphine exposure promotes a compensatory up-regulation of adenylyl cyclase activity and a rebound increase in cAMP levels (also called “cAMP superactivation”; (Nestler, 2004a, b; Williams et al., 2001). Interestingly, withdrawal from chronic morphine promotes an increased probability of GABA release onto dopamine neurons in the VTA, which is dependent on cAMP superactivation (Bonci and Williams, 1997). We hypothesized that the RMOR mutation, that reduces cAMP superactivation in vitro (Finn and Whistler, 2001) and in vivo (He et al., 2009), would reduce cAMP activation in the VTA and prevent these changes in plasticity. To test this, we examined the effects of morphine withdrawal on GABA signaling in WT and RMOR mice. These results are published (Madhavan et al., 2010), and select data is shown here.
WT and RMOR mice were treated twice a day for 5 days with saline or 10 mg/kg morphine as for all experiments above. The frequency and amplitude of GABA spontaneous inhibitory post-synaptic currents (sIPSCs) were then recorded from VTA dopamine neurons from saline or morphine treated mice (Figure 8A). There was a higher frequency of GABA sIPSCs following morphine withdrawal in tyrosine hydroxylase positive (TH+) dopamine neurons from WT but not RMOR mice (WT control: 13.75 ± 1.67 Hz, n = 32; WT Chronic morphine: 26.11 ± 1.68 Hz, n = 28; p<0.0001; RMOR Control: 12.52 ± 1.82 Hz, n = 15; RMOR Chronic morphine: 12.74 ± 1.82 Hz, n = 31; Figure 8B, D). The frequency of mIPSCs was also enhanced in morphine-withdrawn WTs but not RMORs (data not shown, see Madhavan et al., 2010). The paired pulse ratio (PPR=IPSC2/IPSC1) of GABA IPSCs was also significantly enhanced exclusively in dopamine neurons from WT and not RMOR mice following morphine withdrawal (WT control: 0.75 ± 0.04, n = 30; WT Chronic morphine: 0.60 ± 0.04, n = 34; RMOR Control: 0.72 ± 0.04, n = 29; RMOR Chronic morphine: 0.73 ± 0.02, n = 37; p=0.015; Figure 8F). These differences were not due to genotype-specific differences in the ability of the MOR and RMOR to transmit an opioid signal since dose response curves for GABA IPSC inhibition by DAMGO (WT EC50 of 89.22 ± 0.28 nM, Emax of 90.57 ± 7.6, n = 17; RMOR EC50 of 112.6 ± 0.32 nM, Emax of 86.96 ± 10.6, n = 17; t(11) = 0.06; p = 0.958) and morphine (WT EC50 of 106.2 ± 0.78 nM, Emax of 52.82 ± 9.8, n = 8; RMOR EC50 of 125.7 ± 0.51 nM, Emax of 48.66 ± 6.52, n = 8; t(8) = 0.06; p = 0.954) were not significantly different between WT and RMOR mice (see also Figure 2 and Madhavan et al., 2010). Additionally, there were no genotype differences in the presynaptic effects of opioids, which significantly decreased mIPSC frequency, but not amplitude, equivalently in both WT (control, 13.34 ± 3.2 Hz; DAMGO, 7.76 ± 3 Hz; n = 9 neurons, n = 5 mice; t(8) = 6.9; p < 0.0001) and RMOR (control, 13.92 ± 2.8 Hz; DAMGO, 7.0 ± 2.2 Hz; n = 9 neurons, n = 5 mice; t(8) = 3.7; p = 0.0058) (and see Figure 2 and (Madhavan et al., 2010)). These data suggest that increased probability of GABA release during morphine withdrawal that occurs in WT but not RMOR mice cannot be explained by differences in potency of opioid inhibition of GABA release. They also suggest that, at least in the VTA, there are no differences in the ability of the MOR versus the RMOR to signal in response to either morphine or DAMGO.
Figure 8. NLX-precipitated withdrawal from chronic morphine treatment enhances frequency of GABA sIPSCs in VTA dopamine neurons from WT but not RMOR mice.

A. GABA sIPSCs recorded in WT, WT treated with 10 mg/kg morphine for 5 days (WT Chronic Morphine), or RMOR knock-in (RMOR), and RMOR treated with 10 mg/kg morphine for 5 days (RMOR Chronic Morphine). Calibration: 13.3 pA, 153.6 ms. B. Cumulative inter-event distribution of sIPSCs shows that events in morphine-withdrawing WTs are more frequent than in other conditions. C. Cumulative amplitude distribution of sIPSCs shows that larger-amplitude events are more frequent in WT morphine-withdrawing slices. D. Mean frequency of GABA sIPSCs is significantly higher after NLX-precipitated withdrawal in WT mice (WT Chronic Morphine, n = 32) compared with WT controls (WT, n = 28). Mean frequency of GABA sIPSCs is not significantly different in RMOR controls (RMOR, n = 15) and morphine-treated RMOR (RMOR Chronic Morphine, n = 31) or compared with WT controls (WT). E. Mean amplitude of GABA sIPSCs is higher during NLX-precipitated withdrawal in morphine-treated WT (WT Chronic Morphine) than in the other three conditions. F. Paired pulse ratio of GABA IPSCs is significantly reduced during NLX-precipitated withdrawal in morphine-treated WT mice (WT Chronic Morphine; n = 30) than in control conditions (WT, n = 29; RMOR, n = 34; RMOR Chronic Morphine, n = 37). *p < 0.05; **p < 0.01. Reproduced from (Madhavan et al., 2009).
4. Measuring reward in WT and RMOR mice
Importantly, more recently we have utilized the RMOR mice to show that facilitating MOR trafficking not only enhances morphine reward (Figure 9 from Berger and Whistler, 2011), but, despite this, also reduces the development of addiction-like behaviors (Figure 10, from Berger and Whistler, 2011). To demonstrate this, we developed a novel model of the transition from controlled to compulsive drug use that recapitulates many features of human addiction, including persistent drug seeking despite adverse consequences and a decreased preference for alternative rewards. These behaviors emerged spontaneously in WT but not RMOR mice, and their intensity predicted the reinstatement of morphine seeking after extended abstinence, while prior morphine intake did not (Figure 9, from Berger and Whistler, 2011).
Figure 9. Opioid reward in opioid-naïve WT and RMOR mice.

A. Mice were treated with morphine (MS) in a chamber with distinctive environmental cues. Their preference for the MS-paired context was later assessed during a drug-free state. Lower doses of MS produced a positive place preference in RMOR (gray bars) than in WT mice (black bars). Data are presented as the mean + SEM. *p<0.05 WT v. RMOR. #p<0.05, ##p<0.01, ###p<0.001 v. baseline preference. B. CPP induced by methadone (MD). No genotype differences were detected. #p<0.05, ##p<0.01, ###p<0.001 v. baseline preference. Data are presented as the mean + SEM. Reproduced with permission from Berger, A.C., Whistler, J.L., 2011. Morphine-induced mu opioid receptor trafficking enhances reward yet prevents compulsive drug use. EMBO Mol. Med. 3, 385–397.
Figure 10. Longitudinal measures of motivation and compulsivity and relapse in WT and RMOR mice.

A. Lever pressing during the VI period. WT (black) and RMOR mice (gray) expended similar amounts of effort to obtain MS at the start of drinking; however, over time, only WT mice exhibited a progressive increase in responding on the active lever (solid line) (Genotype effect:F[1,82]=16.1,p<0.001 and Genotype x Time interaction: F[2,82]=11.2, p<0.0001). There was no change in responding on the inactive lever (dashed line) in either genotype. B. Latency to earn MS. The latency of WT and RMOR mice to earn morphine was similar at the start of drinking; however, over time, RMOR but not WT mice exhibited a progressive increase in latency (Genotype effect: F[1,82]=13.6, p<0.001 and Genotype x Time interaction: F[2,82]=6.2, p<0.01). C. MS earned during the operant session. The amount of MS earned by WT mice did not change over time despite increased lever pressing. In contrast, RMOR mice showed a progressive decline in MS consumption during the operant session (Genotype effect F[1,82]=57.1, p<0.0001 and Genotype x Time interaction: F[2,82]=8.2, p<0.001). D. Lever pressing during the time-out. WT and RMOR mice initially showed similar amounts of unreinforced lever pressing; however, over time, only WT mice exhibited an increase (Genotype effect: F[1,41]=12.4, p<0.01 and Genotype x Time interaction: F[1,41]=10.6, p<0.01). This effect was restricted to the active lever. E. Suppression of MS seeking by a shock-predictive conditioned stimulus (CS). A CS that had previously been paired with a footshock suppressed MS retrieval to a similar extent in WT and RMOR mice at the start of drinking; however, over time, WT mice became less inhibited compared to RMOR mice (Genotype x Time interaction: F[1,37]=4.3, p<0.05). F. Preference for MS+saccharin versus saccharin alone. RMOR mice initially showed a higher MS preference than WT mice; however, over time, MS preference rose in WT but not RMOR mice (Genotype x Time interaction: F[1,41]=12.5, p<0.01). G. Reinstatement. Reinstatement was induced by the non-contingent delivery of a single MS reinforcer and the contingent presentation of a MS-paired CS. RMOR mice showed significantly less reinstatement than WT mice (p<0.01). (A–F) Data are presented as the mean + SEM. *p<0.05, **p<0.01, ***p<0.001 WT v. RMOR; #p<0.05, ##p<0.01, ###p<0.001 v. earliest time point. Reproduced with permission from Berger, A.C., Whistler, J.L., 2011. Morphine-induced mu opioid receptor trafficking enhances reward yet prevents compulsive drug use. EMBO Mol. Med. 3, 385–397.
Morphine reward was measured using a conditioned place preference (CPP) paradigm. In brief, mice were treated with various doses of morphine in one chamber and with saline in an adjoining chamber with distinct contextual cues. Only one drug and one saline conditioning session were conducted in order to minimize any influence of tolerance and dependence. Following conditioning, mice were allowed to freely explore both chambers in a drug-free state. A preference for the morphine-paired side indicated a positive reinforcing effect of the drug. RMOR mice exhibited greater sensitivity to the rewarding effects of morphine, indicated by a significant Genotype x Dose interaction (F[3,90]=2.8, p<0.05). Thus, a low dose (0.3 mg/kg) that was insufficient to produce CPP in WT mice (Figure 9A, black, p=0.34) produced robust CPP in RMOR mice (Figure 9A, gray, p<0.05). An intermediate dose (3 mg/kg) produced greater CPP in RMOR than in WT mice (p<0.05; Figure 9A). In contrast, the CPP produced by methadone was equivalent in the two genotypes (Figure 9B). This demonstrates that the mutant RMOR receptor functions equivalently to the WT receptor in response to ligands that promote MOR trafficking and indicates that the potentiation of morphine reward (as well as analgesia (Figure 3; Kim et al., 2008), in RMOR mice is due to a selective enhancement of morphine-induced trafficking and not some other non-specific gain-of-function of the RMOR.
5. Measuring “addiction” in WT and RMOR mice
To monitor the transition from impulsive to compulsive drug seeking, we combined elements of operant self-administration and the two bottle choice test to track development of like behaviors (Figure 10 from Berger and Whistler, 2011). Repeated measures of drug seeking behavior were taken for each subject to determine whether that animal's motivation to consume morphine increased over time. We followed each animal for approximately four months, a time frame that is much longer than is possible with intravenous self-administration. Mice were allowed to self-administer morphine in an operant session on the first day of each week, and they were given unlimited access to both morphine and water in their home cages on days 2–5. On the last two days of the week, only water was provided in the home cage. Thus, operant sessions were always conducted in an otherwise drug-free state. The length of operant sessions was also restricted to 30 minutes in order to minimize any effect of morphine on performance. Each operant session consisted of three components to measure five different aspects of compulsivity:
High motivation to obtain drug. We measured the rate of lever pressing to earn a morphine reward. Lever presses were reinforced on a variable interval (VI) (25 second) schedule, meaning that morphine was delivered for the first response after an unpredictable amount of time, averaging 25 seconds, had passed. In a VI schedule, the total amount of drug available is held constant, and there is no direct relationship between the number of responses emitted and the number of reinforcers received. VI schedules thus produce steady rates of lever pressing that reflect how hard a subject is willing to work for a given amount of drug.
Futile drug seeking. We measured lever pressing during a time-out period when it was signaled to the animals that morphine was not available.
Persistent drug seeking in the face of adverse consequences. We measured conditioned suppression of drug seeking (Vanderschuren and Everitt, 2004). Mice learned that they could retrieve a morphine reinforcer if they entered the reward port during the presentation of a light discriminative stimulus (DS). During separate fear conditioning sessions, they also learned that an auditory cue predicted the delivery of a footshock. This auditory cue was played during presentation of the DS to determine whether it suppressed morphine retrieval.
Preference for drug over alternative rewards (measured during home cage drinking sessions). Mice were normally presented with the choice between sweetened morphine and water; however, they were occasionally given the choice between morphine plus saccharin and saccharin alone (0.2%).
Reinstatement of drug seeking following extinction and 15 days of abstinence. Morphine seeking was provoked by the presentation of a drug-associated conditioned stimulus and the delivery of a single, non-contingent morphine reinforcer.
5.1 High motivation to obtain drug and futile drug seeking
The rate of lever pressing was measured in both a VI and a time-out task, in which it was signaled to the mice that morphine was not available. Statistical analysis revealed a significant Genotype x Time interaction and a main effect of Genotype on lever pressing in both tasks (Figure 10A, VI task: F[2,82]=11.2, p<0.0001 and F[1,82]=16.1, p<0.001; Figure 10D, Time-out task: F[1,41]=10.6, p<0.01 and F[1,41]=12.4, p<0.01), the latency to earn morphine (Figure 10B F[2,82]=6.2, p<0.01 and F[1,82]=13.6, p<0.001), and the amount of morphine earned (Figure 10C, F[2,82]=8.2, p<0.001 and F[1,82]=57.1, p<0.0001). After only two weeks of drinking experience (a length of time that would be possible with “classic” addiction models using i.v. self administration), RMOR and WT mice did not differ in their drug seeking behavior. However, over time, WT mice progressively increased their responding on the active lever during both drug and no drug periods (Figure 10A, p<0.001; Figure 10D p<0.05); whereas, RMOR mice decrease their responding. These changes were specific to the active lever, indicating that they were related to drug seeking and not due to non-specific changes in motor activity (Lever x Time interaction in VI task, WT: F[2,96]=10.1, p<0.001 and RMOR: F[2,68]=3.7, p<0.05). Importantly, the increase in lever pressing in WT mice was not associated with any increase in the amount of morphine delivered, suggesting that WT mice were willing to work harder to obtain the same amount of drug (Figure 10C). This was not due to a ceiling effect, as WT mice were only earning about 50% of possible reinforcers.
5.2 Persistent drug seeking in the face of adverse consequences
We next assessed whether morphine seeking could be suppressed by an auditory cue that had previously been paired with a footshock. All fear conditioning sessions were carried out in a drug-free state. RMOR and WT mice showed similar conditioned suppression of drug seeking after only two weeks of morphine drinking experience. However, after prolonged experience, drug seeking became more resistant to suppression in the WT mice than in the RMOR mice (Figure 10E, p<0.05). Statistical analysis revealed a significant Genotype x Time interaction (F[1,37]=4.3, p<0.05). This cannot be explained by a difference in learning, as the two genotypes showed equivalent increases in cue-induced freezing following fear conditioning (data not shown).
5.3 Preference for drug over alternative rewards
We measured shifts in the preference for sweetened morphine versus a highly palatable saccharin solution. Again, there was a significant Genotype x Time interaction (Figure 10F, F[1,41]=12.5, p<0.01). RMOR mice initially showed a significantly higher preference for morphine than WT mice (p<0.01), and a substantial fraction, 40%, preferred morphine outright compared to only 12% of WTs. After extended drinking experience, however, WT mice showed a significantly higher preference for morphine than RMOR mice (p<0.05), and 44% preferred morphine outright compared to only 22% of RMORs.
5.4 Propensity to relapse
We measured the reinstatement of morphine seeking following extinction and 15 days of abstinence. Morphine seeking was provoked by the presentation of a drug-associated conditioned stimulus and the delivery of a single, non-contingent morphine reinforcer. The rate of lever pressing under these conditions is thought to be indicative of the propensity to relapse (Zernig et al., 2007). The level of reinstatement was significantly greater in WT than in RMOR mice (Figure 10G, p<0.01).
5.5 Correlation of relapse to other behaviors
This variability in the WT population provided an opportunity to examine whether the longitudinal measures above (Figure 10) were valid behavioral markers for relapse probability. In order to determine whether individual differences in all these addiction-like behaviors were predictive of the propensity to relapse across all WT mice, we quantified the relationship between reinstatement and a compulsivity score that incorporated all four measures (Figure 11, from Berger and Whistler, 2011). There was a significant positive correlation between the compulsivity score and reinstatement (Figure 11A, p<0.01, r[20]=0.62). All in all, these data suggest that changes in the four longitudinal markers do, in fact, model the progression from controlled drug use to addiction and that, over the course of the study, addiction developed in WT but not RMOR mice. Given the wide disparity in morphine consumption between WT and RMOR mice, we next examined whether differences in addiction-like behaviors or reinstatement could be explained by differences in consumption. In sharp contrast to the relationship between the compulsivity score and reinstatement, there was no significant relationship between cumulative morphine intake and either reinstatement (p=0.27) or the compulsivity score in WT mice (Figure 11B, p=0.51).
Figure 11. The relationship of compulsivity to reinstatement and consumption in WT and RMOR mice.

A. Compulsivity v. reinstatement in WT mice. A compulsivity score was calculated for each WT mouse that quantified how much its behavior deviated from the mean for all WT mice in the four longitudinal measures in Figure 4. Note that since the average WT mouse showed an increase in compulsivity, a negative score does not necessarily indicate a decline in compulsivity. B. Compulsivity v. consumption in WT mice. Reproduced with permission from Berger, A.C., Whistler, J.L., 2011. Morphine-induced mu opioid receptor trafficking enhances reward yet prevents compulsive drug use. EMBO Mol. Med. 3, 385–397.
6. Other Correlates of Abuse Liability
Clearly the longitudinal tests above (Figure 10) cannot be used to determine the abuse liability for every new chemical entity, since compound synthesis alone for such a study would be prohibitively expensive. In addition, it is clear from these studies that neither preference as measured by CPP (Figure 9) nor consumption (Figure 11) are at all predictive of the liability to progress to compulsive drug seeking. Consequently, we examined two other behavioral aspects of repeated drug use “affective withdrawal” and “reward sensitization” in WT and RMOR mice to determine whether these correlated with “addiction” (Figure 12, from Berger and Whistler, 2011).
Figure 12. Morphine reward in WT and RMOR mice following chronic morphine treatment.

A. Mice treated chronically with MS (solid bars) or saline (open bars) were injected with naloxone (NX) in a chamber with distinctive environmental cues. Their preference or avoidance of the NX-paired side was measured one week later. A low dose of NX (0.1 mg/kg) blocked MS CPP in WT but not RMOR mice. Similarly, a high dose (1 mg/kg) produced a strong aversion, indicative of affective dependence, only in WT mice. Data are presented as the mean + SEM. *p<0.05 WT v. RMOR. #p<0.05 v. baseline preference. B. Sensitization of MS reward during protracted abstinence. Mice were treated chronically with MS or saline. CPP was measured as in Figure 3 two weeks after spontaneous withdrawal. Data are presented as the mean + SEM. *p<0.05 WT v. RMOR. #p<0.05 v. baseline preference. Reproduced with permission from Berger, A.C., Whistler, J.L., 2011. Morphine-induced mu opioid receptor trafficking enhances reward yet prevents compulsive drug use. EMBO Mol. Med. 3, 385–397.
6.1 “Affective withdrawal.”
Repeated morphine exposure can induce a state of dependence with separate somatic and affective components (Koob, 1992). The affective symptoms, in particular, are extremely sensitive to small changes in opioid tone. RMOR mice exhibit a less intense physical withdrawal syndrome than WT mice (Figure 4; Kim et al., 2008). To determine whether the affective symptoms of dependence are also reduced, we measured the place aversion induced by naloxone-precipitated withdrawal (Figure 12A). A low dose of naloxone (0.1 mg/kg) blocked morphine CPP in WT (p=0.15) but not RMOR mice (p<0.01), indicating that the WT mice were more sensitive to naloxone. A high dose of naloxone (1 mg/kg), not only blocked morphine reward in WT mice, but also revealed a strong aversive effect of withdrawal (p<0.05) (Figure 12A). In contrast, the same dose of naloxone in RMOR mice reversed morphine CPP but had no negative reinforcing effect (p=0.9). The highest dose of naloxone (1 mg/kg) had no motivational effects in opioid-naïve mice of either genotype (Figure 12A, open bars). These data suggest that chronic morphine induces affective dependence in WT but not RMOR mice, and that the aversive effects of withdrawal in WT mice provide a strong incentive to maintain morphine consumption.
6.2 “Reward Sensitization.”
The symptoms of opioid withdrawal peak within hours to days of the cessation of drug use; however, the risk of relapse remains high weeks to months and even years later (Hunt et al., 1971). The negative reinforcing effects of acute withdrawal may contribute to the intense motivation to obtain drug during active drug use, and conditioned withdrawal may contribute to craving in extended abstinence. Relapse may also result from sensitization to the positive reinforcing effects of drug (Aston-Jones and Harris, 2004); for example, repeated morphine treatment results in an increase in heroin self-administration at low doses that do not normally maintain responding well beyond the abatement of withdrawal (Gerak et al., 2009). To determine whether chronic morphine exposure induced a lasting change in morphine reward, we measured CPP during protracted abstinence (Figure 12B). RMOR and WT mice were treated with twice daily injections of morphine (10 mg/kg) or saline for five days. Two weeks later, morphine-induced place preference was measured as in Figure 9. In this paradigm, a low dose of morphine (0.3 mg/kg), that was insufficient to produce CPP in opioid-naive WT mice (p=0.34), produced robust CPP in morphine-treated WT mice that was equivalent to that generated by a 10-fold higher dose in naive mice (p<0.05; Figure 12B). In contrast, repeated morphine administration did not induce any shift in the threshold dose of morphine required to elicit CPP in RMOR mice. Thus, a relatively short course of chronic morphine treatment caused a long lasting sensitization to morphine's rewarding effects in WT but not RMOR mice.
In short, we have found that extensive self-administration experience is necessary for the emergence of compulsive morphine seeking. Importantly, while a long duration of drug use was critical, a high amount of drug consumption was neither necessary nor sufficient. WT mice with both high and low levels of reinstatement and addiction-like behaviors did not differ in either their cumulative morphine intake or their early pattern of escalation (Fig 11, and see Berger and Whistler, 2011), and there were heavy and light drinkers in both groups. In addition, “reward” measured by CPP was in no way predictive of “addiction” since RMOR mice showed greater reward than WT mice but did not progress to compulsive drug seeking. It will be interesting in future experiment to determine whether opioid agonists that promote MOR endocytosis and recycling in both MOR and RMOR mice, such as methadone, show comparable levels of reward sensitization and affective withdrawal in both genotypes, especially since they show equivalent analgesia (Kim et al., 2008) and reward (Berger and Whistler, 2011) in these two genotypes.
While we cannot completely rule out the possibility that there is an as yet unidentified difference in the signaling profile of the RMOR and WT MOR that accounts for the morphine-specific differences in tolerance, dependence, and transition to compulsive drug seeking, multiple lines of evidence suggest that it is changes in trafficking that are responsible. First, responses of MOR and RMOR mice/cells to drugs other than morphine, including DAMGO and methadone, are indistinguishable at the cellular (Finn and Whistler, 2001), synaptic (Madhavan et al., 2010) and behavioral (Kim et al., 2008) level. Second, several completely distinct manipulations that alter trafficking on the MOR selectively for morphine prevent cellular and/or behavioral manifestations of tolerance and dependence. For example, a naturally-occurring mutation in the human MOR, in a region of the receptor distinct from the cytoplasmic tail, produces a receptor that undergoes morphine-induced endocytosis and prevents morphine-induced cAMP superactivation in vitro (Ravindranathan et al., 2009). In addition, opioid cocktails containing morphine and a second opioid (either DAMGO (He et al., 2002) or methadone (He and Whistler, 2005), at doses that do not promote endocytosis on their own), promote endocytosis of the WT receptor and also produce reduced tolerance and dependence (see Figure 13–14 below). Third, in the setting of tissue injury, endorphins and enkephalins released by inflammatory cells have been shown to promote MOR trafficking in peripheral sensory neurons and to inhibit tolerance to locally applied morphine (Zollner et al., 2008). Fourth, while all opioid drugs can produce tolerance and dependence when given at a high enough dose for a long enough period of time, drugs such as methadone, when given at equi-antinociceptive doses, produce substantially reduced tolerance and dependence compared to morphine (Kim et al., 2008 and see Figure 1). What each of these manipulations has in common is that they alter trafficking of the MOR selectively in response to morphine. Thus, while it is possible that all these manipulations produce some other as yet to be identified change in signaling, we know for certain that they all effect the trafficking of the receptor selectively in response to morphine.
Figure 13. Trafficking of the MOR with morphine and the cocktail.

A. Biotin protection assay to quantify endocytosis. HEK293 cells stably expressing FLAG-tagged MORs were examined for endocytosis by biotin protection. Methadone enhanced morphine-induced endocytosis. Saturating concentrations of morphine alone (1 µM) or d-methadone (1 µM), as well as low concentrations of dl-methadone (100 nM, 10 nM) failed to promote endocytosis. A saturating concentration (1µM) of dl-methadone alone promotes endocytosis. B. Immunocytochemical staining. HEK293 cells stably expressing FLAG-tagged MORs receptors were fed antibody, exposed to 1µM morphine, 10nM methadone or both then fixed and stained for receptor. Methadone enhanced morphine-induced MOR endocytosis. C. cAMP superactivation. CRE-luciferase expression was assessed in HEK293 cells stably expressing both the MOR and a CRE-luciferase reporter gene. Cells were treated with drug or drug combinations for 14 hrs, washed then treated with 2 µM forskolin for 4 hrs. Morphine induced cAMP superactivation, while methadone decreased morphine-induced cAMP superactivation (***p<.001 NT vs. MS, ###p<.001 MS vs. MS+MD, 1xANOVA, Tukey post-test.) MD; dl-methadone, MS morphine sulfate, NT no treatment. D, E. Immunohistochemistry in rat brain. MOR distribution in PAG (D) and VTA (E) neurons was assessed 30 mins after acute i.c.v. administration of drug. Neither methadone (50 nmol) nor morphine (30 nmol) promoted endocytosis of the MOR, while co-administration of both induced MOR endocytosis. For quantification, slides were encoded and vesicles counted by a second party blind to treatment. (***p<.001: MS 30 nmol+ MD 50 nmol vs. MS 30 nmol or MD 50 nmol). Reproduced with permission from He, L., Whistler, J.L., 2005. An opiate cocktail that reduces morphine tolerance and dependence. Curr. Biol. 15, 1028–1033.
Figure 14. Tolerance and dependence to morphine and the cocktail.

A. Morphine tolerance development was measured with a tail-flick assay over a 5-day period. Rats treated with morphine (30 nmol) developed substantial tolerance by day 5, whereas rats receiving the same dose of morphine plus varying doses of dl-methadone showed no tolerance at all but the lowest dose of methadone (*** p< 0.001: MS 30 nmol alone vs. MS 30 nmol +MD 50 nmol or MS 30 nmol + MD 20 nmol. p< 0.001: MS 30 nmol on day 5 vs. day 1; MD 50 nmol alone; MS 30 nmol+ MD 5 nmol). B. Rats from A. were injected with naloxone (3 mg/kg) s.c. after the final tail-flick assay on day and withdrawal signs were monitored. Withdrawal was significantly attenuated with several doses of dl-methadone compared with morphine alone (*** p< 0.001 MD 50 nmol vs. MS 30 nmol; ** p< 0.01: MS 30 nmol MD 50 nmol vs. MS 30 nmol. NS, no significance vs. MS 30 nmol) NS: no significance vs. MS 30 nmol). MS: morphine sulfate; MD: dl-methadone. C. Rats were implanted with an i.c.v. cannula and morphine tolerance development was measured as above. Rats treated with morphine alone or morphine plus d-methadone (MS 30 nmol + d-MD 50 nmol) developed substantial tolerance (***p< 0.001: day 5 vs. day 1 MS 30 nmol + d-MD 50 nmol). d-methadone alone did not produce analgesia. D. Withdrawal was not significantly attenuated by d-methadone (*** p< 0.001 d-MD 50 nmol vs. MS 30 nmol; NS: no significance vs. MS 30 nmol). MS: morphine sulfate; dl-MD: dl-methadone d-MD: d-methadone. MPE = maximal possible effect. Reproduced with permission from He, L., Whistler, J.L., 2005. An opiate cocktail that reduces morphine tolerance and dependence. Curr. Biol. 15, 1028–1033.
7. Properties of existing opioid therapeutics
We predict that the ideal opioid would be highly selective for the MOR, have a potency and pharmacokinetic properties similar to morphine, but promote MOR desensitization and trafficking similar to endogenous opioids. Unfortunately, among the opioids used to treat pain, no such drug currently exists as outlined below:
Morphine and codeine – These two drugs are the primary active ingredients of raw opium. Codeine is a pro-drug that is converted in vivo into codeine-6-glucuronide and morphine, through which it exerts the majority of its analgesic effect. Compared to endogenous opioid peptides, both codeine and morphine induce significantly less MOR internalization in vitro and in vivo even when they are administered at higher doses for an extended period of time (see for example Keith et al., 1998).
Hydromorphone, hydrocodone, oxycodone, and oxymorphone – Along with morphine, these drugs are the most commonly prescribed to chronic pain patients. They are all semi-synthetic drugs that are derived from naturally occurring opiate alkaloids, such as morphine, and structurally, they are very similar to morphine. It is not surprising then that, like morphine, they are poor internalizers of the MOR (see for example Koch et al., 2005), and their use is associated with the development of both substantial tolerance and dependence.
Fentanyl – Fentanyl is a fully synthetic agonist that is structurally unrelated to morphine. It is commonly used as an induction agent for surgical anesthesia and, in transdermal and buccal preparations, as a treatment for chronic pain, particularly cancer pain. Fentanyl is two orders of magnitude more potent than morphine, and, compared to morphine, it does induce a greater degree of MOR desensitization and endocytosis when applied at saturating concentrations in vitro (Keith et al., 1998; Koch et al., 2005), an observation that seems, at face value, to nullify the RAVE hypothesis. However, importantly, at clinically relevant doses, fentanyl does not promote significant MOR endocytosis, despite the fact it is both a high efficacy and high potency ligand. We propose that the failure of fentanyl to produce endocytosis at low doses reflects the oligomeric nature of the opioid receptors as follows: At clinically relevant doses of fentanyl, the majority of receptors in any oligomeric complex will be unoccupied by agonist and therefore in an “inactive” conformation. A few receptors will be occupied by fentanyl and thus be in an active conformation. However, having only one receptor in an active conformation, while sufficient for coupling to G protein, is not sufficient to recruit the endocytic machinery. For all opioid ligands tested to date, the EC50 for G protein coupling is left shifted 2–3 log units compared to the EC50 for endocytosis, supporting this hypothesis (see for example Alvarez et al., 2002). This shift in potency for endocytosis versus G protein coupling presents a particular issue for high potency/efficacy ligands where receptor occupancy is low. Hence, contrary to the prevailing hypothesis that high efficacy ligands will promote less tolerance (because they need fewer receptors to be analgesic so downregulation is less of a problem), we predict that high efficacy ligands will actually promote more tolerance and dependence, because they have high RAVE values due to a high “RA” and very little “VE” at clinically-relevant doses. Indeed, the high potency/efficacy ligand fentanyl appears to fit this hypothesis since its use is also associated with the rapid development of hyperalgesia, dependence and tolerance, frequently after even a single exposure to fentanyl (Angst et al., 2003; Laulin et al., 2002).
Methadone – Methadone has a potency similar to morphine but, in contrast, is quite effective at stimulating receptor endocytosis (Keith et al., 1998; Whistler et al., 1999). However, methadone has a very long half-life, and unfortunately, there is a great deal of both pharmacokinetic and pharmacodynamic variability in methadone metabolism and response across the human population (Eap et al., 2002). In particular, in many patients, the effective half-life of methadone for analgesia does not mirror the half-life for respiratory suppression and cardiac side effects, making methadone difficult to dose consistently and safely. Therefore, methadone is only rarely used as a first line therapy for chronic pain.
8. Using the oligomeric state of MOR to promote endocytosis
We have found that WT MOR forms heterodimers/oligomers with mutant MORs, including RMOR (Finn and Whistler, 2001; He et al., 2002). Importantly, in the presence of RMOR, co-expressed WT MOR also showed significant endocytosis in response to morphine (He et al., 2002). We called this phenomenon “dragging” because it appeared that the RMORs could drag the MORs into the cell in response to morphine, presumably because these receptors were making heterodimers. Based on these results, and the proposed existence of MOR as an oligomeric structure (He et al., 2002; Jordan and Devi, 1999), we designed a novel pharmacological cocktail that facilitates endocytosis of the MOR in response to morphine (Figure 13, from He and Whistler, 2005). This cocktail consists of morphine and a small, non-analgesic dose of methadone. Importantly, this cocktail, while retaining full analgesic potency, does not promote morphine tolerance or dependence in rats when drugs were administered i.c.v. (Figure 14; He and Whistler, 2005).
Methadone is a unique opioid drug in that it possesses not only MOR agonist activity but also antagonist activity at NMDA receptors (Ebert et al., 1995). This fact was relevant to the study in Figure 13–14, because there is much evidence that the NMDA receptor system is involved in the manifestation of morphine tolerance and dependence (for review see Raith and Hochhaus, 2004; Trujillo, 2000)). Methadone is a mixed enantiomer of d- and l-methadone. l-Methadone is the enantiomer primarily responsible for analgesia (Scott et al., 1948) and has a high affinity for the MOR (Kristensen et al., 1995). d-Methadone has a low affinity for the MOR (Kristensen et al., 1995), is a poor analgesic (Ingoglia and Dole, 1970), and does not promote MOR endocytosis (Figure 13A). However, previously, d-methadone has been shown to prevent morphine tolerance (Davis and Inturrisi, 1999), and this effect of d-methadone has been attributed specifically to the NMDA receptor antagonist properties of this enantiomer (kd ~5–7µM; Gorman et al., 1997). Although the doses of methadone used in Figure 14A,B were substantially lower than those reported to block tolerance, these observations raise the reasonable question as to whether NMDA antagonism by d-methadone contributed to the anti-tolerance and/or -dependence effects of the morphine-methadone cocktail, since the methadone utilized for our studies contains both the d- and l-enantiomers.
To examine this possibility, we utilized a morphine/d-methadone cocktail, which should retain any NMDAR antagonism properties of our morphine/dl-methadone cocktail but not enhance MOR endocytosis. As expected, d-methadone (50 nmol) showed no analgesia (Figure 14C, gray line, closed circle), nor did it enhance acute morphine analgesia on day one (Figure 13C, green line, open square). Furthermore, at the dose administered in this study, d-methadone did not prevent morphine tolerance (Figure 14C, green line, open square) or dependence (Figure 14D). This strongly suggests that significant NMDAR antagonism is not occurring at this methadone dose, and that NMDAR antagonism is not responsible for the ability of the mixed dl-methadone in our cocktail to prevent morphine tolerance and dependence. Thus, while NMDAR antagonists have been shown to attenuate morphine tolerance and dependence, taken together, our data suggest that it is the enhanced endocytosis of the MOR, not NMDA antagonism that is preventing morphine tolerance and dependence during administration of the cocktail.
9. Conclusions
Opioids are exceptionally effective analgesics and the mainstay for the treatment of severe pain in many settings both acute and chronic. Morphine and other clinically important opiate drugs, including oxycodone (OxyContin), hydrocodone (Vicodin), codeine and hydormorphone (Dilaudid), exert their effects primarily through the mu opioid receptor (MOR). However, the utility of these drugs for the treatment of chronic pain is compromised by the development of tolerance to the analgesic effects of drug and by rebound hyperalgesia and allodynia. Importantly, the dose escalation necessary to overcome tolerance and hyperalgesia in chronic pain conditions not only puts patients at a greater risk for severe side-effects, such as respiratory depression, but also increases the liability for physical dependence and/or addiction to the drug. Despite decades of research into the causes of tolerance and dependence to opioids, and billions of dollars spent on the development of new opioid drugs, little headway has been made in discovering opioid therapeutics with reduced liability to cause tolerance and dependence. For decades the field has hypothesized that tolerance to opioids was mediated by loss or “desensitization” of opioid receptors. Hence, the therapeutic focus has been on developing ligands with greater potency and/or efficacy that could overcome tolerance by exerting a full analgesic effect in the presence of fewer functional receptors, or by increasing the effective half life through improved formulation of existing opioids. In fact, morphine and its derivatives are unique agonists because they do not promote desensitization and endocytosis of their target receptor while, somewhat paradoxically, the endogenous ligands at the MOR, which are themselves excellent analgesics, do promote MOR endocytosis. In light of these observations, we have proposed a new and fundamentally different hypothesis for the cell biological mechanisms underlying the development of tolerance and dependence to opiate drugs that suggests a new approach to the pharmacotherapy of chronic pain. We propose that the regulation of opioid receptors by endocytosis serves a protective role in reducing the development of tolerance and dependence to opiate drugs. We have used both genetic and pharmacological approaches to assess this hypothesis as outlined above. Based on the strength of the data summarized in this review and presented at the BBC meeting in San Antonio on March 5th, 2011, we are hopeful that there will be renewed effort to identify novel opioid therapeutic leads with low “RAVE” values that could provide excellent antinociceptive relief with reduced liability to produce tolerance and dependence. Indeed, there are screening platforms generally available which could identify opioid ligands with selectivity and potency similar to morphine but with the ability to drive receptor trafficking (for review see Allen and Roth, 2011).
Acknowledgements
Role of Funding Source: This work was supported by the National Institute on Drug Abuse (NIDA) grants DA015232 and DA019958 and funds provided by the state of California for medical research on alcohol and substance abuse through the University of California, San Francisco (UCSF). The NIH and UCSF had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributors: This review represents work done over the past decade in my laboratory. It represents the work of many people, and was previously published as noted. The review was compiled from the published data by Jennifer L. Whistler.
Conflict of Interest: No conflict declared.
This paper was presented in a symposium at the Behavior, Biology, and Chemistry: Translational Research in Addiction meeting on March 5, 2011 in San Antonio, TX entitled "New concepts in mu-opioid pharmacology - implications for addiction and its management.”
References
- Allen JA, Roth BL. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2011;51:117–144. doi: 10.1146/annurev-pharmtox-010510-100553. [DOI] [PubMed] [Google Scholar]
- Alvarez VA, Arttamangkul S, Dang V, Salem A, Whistler JL, Von Zastrow M, Grandy DK, Williams JT. mu-Opioid receptors: ligand-dependent activation of potassium conductance, desensitization, and internalization. J. Neurosci. 2002;22:5769–5776. doi: 10.1523/JNEUROSCI.22-13-05769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angst MS, Koppert W, Pahl I, Clark DJ, Schmelz M. Short-term infusion of the mu-opioid agonist remifentanil in humans causes hyperalgesia during withdrawal. Pain. 2003;106:49–57. doi: 10.1016/s0304-3959(03)00276-8. [DOI] [PubMed] [Google Scholar]
- Arden JR, Segredo V, Wang Z, Lameh J, Sadee W. Phosphorylation and agonist-specific intracellular trafficking of an epitope-tagged mu-opioid receptor expressed in HEK 293 cells. J. Neurochem. 1995;65:1636–1645. doi: 10.1046/j.1471-4159.1995.65041636.x. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Harris GC. Brain substrates for increased drug seeking during protracted withdrawal. Neuropharmacology. 2004;47(Suppl. 1):167–179. doi: 10.1016/j.neuropharm.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Berger AC, Whistler JL. Morphine-induced mu opioid receptor trafficking enhances reward yet prevents compulsive drug use. EMBO Mol. Med. 2011;3:385–397. doi: 10.1002/emmm.201100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J. Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collett BJ. Opioid tolerance: the clinical perspective. Br. J. Anaesth. 1998;81:58–68. doi: 10.1093/bja/81.1.58. [DOI] [PubMed] [Google Scholar]
- Davis AM, Inturrisi CE. d-Methadone blocks morphine tolerance and N-methyl-D-aspartate-induced hyperalgesia. J. Pharmacol. Exp. Ther. 1999;289:1048–1053. [PubMed] [Google Scholar]
- Eap CB, Buclin T, Baumann P. Interindividual variability of the clinical pharmacokinetics of methadone: implications for the treatment of opioid dependence. Clin. Pharmacokinetics. 2002;41:1153–1193. doi: 10.2165/00003088-200241140-00003. [DOI] [PubMed] [Google Scholar]
- Ebert B, Andersen S, Krogsgaard-Larsen P. Ketobemidone, methadone and pethidine are non-competitive N-methyl-D-aspartate (NMDA) antagonists in the rat cortex and spinal cord. Neurosci. Lett. 1995;187:165–168. doi: 10.1016/0304-3940(95)11364-3. [DOI] [PubMed] [Google Scholar]
- Ferguson SS, Zhang J, Barak LS, Caron MG. Molecular mechanisms of G protein-coupled receptor desensitization and resensitization. Life Sci. 1998;62:1561–1565. doi: 10.1016/s0024-3205(98)00107-6. [DOI] [PubMed] [Google Scholar]
- Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32:829–839. doi: 10.1016/s0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- Freedman NJ, Lefkowitz RJ. Desensitization of G protein-coupled receptors. Recent Prog. Horm. Res. 1996;51:319–351. discussion 352-313. [PubMed] [Google Scholar]
- Garrido MJ, Troconiz IF. Methadone: a review of its pharmacokinetic/pharmacodynamic properties. J. Pharmacol. Toxicol. Methods. 1999;42:61–66. doi: 10.1016/s1056-8719(00)00043-5. [DOI] [PubMed] [Google Scholar]
- Gerak LR, Galici R, France CP. Self administration of heroin and cocaine in morphine-dependent and morphine-withdrawn rhesus monkeys. Psychopharmacology (Berl.) 2009;204:403–411. doi: 10.1007/s00213-009-1471-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman AL, Elliott KJ, Inturrisi CE. The d- and l-isomers of methadone bind to the non-competitive site on the N-methyl-D-aspartate (NMDA) receptor in rat forebrain and spinal cord. Neurosci. Lett. 1997;223:5–8. doi: 10.1016/s0304-3940(97)13391-2. [DOI] [PubMed] [Google Scholar]
- Haberstock-Debic H, Kim KA, Yu YJ, von Zastrow M. Morphine promotes rapid, arrestin-dependent endocytosis of mu-opioid receptors in striatal neurons. J. Neurosci. 2005;25:7847–7857. doi: 10.1523/JNEUROSCI.5045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberstock-Debic H, Wein M, Barrot M, Colago EE, Rahman Z, Neve RL, Pickel VM, Nestler EJ, Von Zastrow M, Svingos AL. Morphine acutely regulates opioid receptor trafficking selectively in dendrites of nucleus accumbens neurons. J. Neurosci. 2003;23:4324–4332. doi: 10.1523/JNEUROSCI.23-10-04324.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Fong J, Von Zastrow M, Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. 2002;108:271–282. doi: 10.1016/s0092-8674(02)00613-x. [DOI] [PubMed] [Google Scholar]
- He L, Kim JA, Whistler JL. Biomarkers of morphine tolerance and dependence are prevented by morphine-induced endocytosis of a mutant {micro}-opioid receptor. Faseb J. 2009;23:4327–4334. doi: 10.1096/fj.09-133223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Whistler JL. An opiate cocktail that reduces morphine tolerance and dependence. Curr. Biol. 2005;15:1028–1033. doi: 10.1016/j.cub.2005.04.052. [DOI] [PubMed] [Google Scholar]
- Hunt WA, Barnett LW, Branch LG. Relapse rates in addiction programs. J. Clin. Psychol. 1971;27:455–456. doi: 10.1002/1097-4679(197110)27:4<455::aid-jclp2270270412>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Ingoglia NA, Dole VP. Localization of d- and l-methadone after intraventricular injection into rat brains. J. Pharmacol. Exp. Ther. 1970;175:84–87. [PubMed] [Google Scholar]
- Inoue M, Mishina M, Ueda H. Locus-specific rescue of GluRepsilon1 NMDA receptors in mutant mice identifies the brain regions important for morphine tolerance and dependence. J. Neurosci. 2003;23:6529–6536. doi: 10.1523/JNEUROSCI.23-16-06529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci. 1992a;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J. Physiol. 1992b;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DE, Anton B, Murray SR, Zaki PA, Chu PC, Lissin DV, Monteillet-Agius G, Stewart PL, Evans CJ, von Zastrow M. mu-Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol. Pharmacol. 1998;53:377–384. [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von ZM. Morphine activates opioid receptors without causing their rapid internalization. J. Biol. Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Kim JA, Bartlett S, He L, Nielsen CK, Chang AM, Kharazia V, Waldhoer M, Ou CJ, Taylor S, Ferwerda M, Cado D, Whistler JL. Morphine-induced receptor endocytosis in a novel knockin mouse reduces tolerance and dependence. Curr. Biol. 2008;18:129–135. doi: 10.1016/j.cub.2007.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N, Beyer A, Grecksch G, Hollt V. Receptor endocytosis counteracts the development of opioid tolerance. Mol. Pharmacol. 2005;67:280–287. doi: 10.1124/mol.104.004994. [DOI] [PubMed] [Google Scholar]
- Koob GF. Neural mechanisms of drug reinforcement. Ann. N.Y. Acad. Sci. 1992;654:171–191. doi: 10.1111/j.1749-6632.1992.tb25966.x. [DOI] [PubMed] [Google Scholar]
- Kristensen K, Christensen CB, Christrup LL. The mu1, mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. 1995;56:PL45–PL50. doi: 10.1016/0024-3205(94)00426-s. [DOI] [PubMed] [Google Scholar]
- Laulin JP, Maurette P, Corcuff JB, Rivat C, Chauvin M, Simonnet G. The role of ketamine in preventing fentanyl-induced hyperalgesia and subsequent acute morphine tolerance. Anesth. Analg. 2002;94:1263–1269. doi: 10.1097/00000539-200205000-00040. table of contents. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J. Biol. Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- Lim G, Wang S, Zeng Q, Sung B, Mao J. Spinal glucocorticoid receptors contribute to the development of morphine tolerance in rats. Anesthesiology. 2005a;102:832–837. doi: 10.1097/00000542-200504000-00020. [DOI] [PubMed] [Google Scholar]
- Lim G, Wang S, Zeng Q, Sung B, Yang L, Mao J. Expression of spinal NMDA receptor and PKCgamma after chronic morphine is regulated by spinal glucocorticoid receptor. J. Neurosci. 2005b;25:11145–11154. doi: 10.1523/JNEUROSCI.3768-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan A, He L, Stuber GD, Bonci A, Whistler J. Morphine-induced mu opioid Receptor Endocytosis Prevents Adaptations in GABA Release During Withdrawal. Portland, Oregon, USA. International Narcotics Research Conference.2009. [Google Scholar]
- Madhavan A, He L, Stuber GD, Bonci A, Whistler JL. micro-Opioid receptor endocytosis prevents adaptations in ventral tegmental area GABA transmission induced during naloxone-precipitated morphine withdrawal. J. Neurosci. 2010;30:3276–3286. doi: 10.1523/JNEUROSCI.4634-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli M, Aouizerate B, Barrot M, Le Moal M, Piazza PV. Dopamine-dependent responses to morphine depend on glucocorticoid receptors. Proc. Nat. Acad. Sci. USA. 1998;95:7742–7747. doi: 10.1073/pnas.95.13.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Historical review: molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol. Sci. 2004a;25:210–218. doi: 10.1016/j.tips.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular mechanisms of drug addiction. Neuropharmacol. 2004b;47(Suppl. 1):24–32. doi: 10.1016/j.neuropharm.2004.06.031. [DOI] [PubMed] [Google Scholar]
- Pasternak GW. When it comes to opiates, just say NO. J. Clin. Invest. 2007;117:3185–3187. doi: 10.1172/JCI34035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacol. 2011;60:58–65. doi: 10.1016/j.neuropharm.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raith K, Hochhaus G. Drugs used in the treatment of opioid tolerance and physical dependence: a review. Int. J. Clin. Pharmacol. Ther. 2004;42:191–203. doi: 10.5414/cpp42191. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nature Rev. 9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranathan A, Joslyn G, Robertson M, Schuckit MA, Whistler JL, White RL. Functional characterization of human variants of the mu-opioid receptor gene. Proc. Nat. Acad. Sci. USA. 2009;106:10811–10816. doi: 10.1073/pnas.0904509106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CC, Robbins EB, Chen KK. Pharmacologic comparison of the optical isomers of methadone. J. Pharmacol. Exp. Ther. 1948;93:282–286. [PubMed] [Google Scholar]
- Sternini C, Spann M, Anton B, Keith DJ, Bunnett NW, von ZM, Evans C, Brecha NC. Agonist-selective endocytosis of mu opioid receptor by neurons in vivo. Proc. Nat. Acad. Sci. USA. 1996;93:9241–9246. doi: 10.1073/pnas.93.17.9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo KA. Are NMDA receptors involved in opiate-induced neural and behavioral plasticity? A review of preclinical studies. Psychopharmacology (Berl.) 2000;151:121–141. doi: 10.1007/s002130000416. [DOI] [PubMed] [Google Scholar]
- Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, Von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJ, Everitt BJ. Drug seeking becomes compulsive after prolonged cocaine self-administration. Science. 2004;305:1017–1019. doi: 10.1126/science.1098975. [DOI] [PubMed] [Google Scholar]
- Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- Whistler JL, von Zastrow M. Morphine-activated opioid receptors elude desensitization by beta-arrestin. Proc. Nat. Acad. Sci. USA. 1998;95:9914–9919. doi: 10.1073/pnas.95.17.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, von Zastrow M. Dissociation of functional roles of dynamin in receptor-mediated endocytosis and mitogenic signal transduction. J. Biol. Chem. 1999;274:24575–24578. doi: 10.1074/jbc.274.35.24575. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol. Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zheng X, Wang Y, Cao J, Dong Z, Cai J, Sui N, Xu L. Stress enables synaptic depression in CA1 synapses by acute and chronic morphine: possible mechanisms for corticosterone on opiate addiction. J. Neurosci. 2004;24:2412–2420. doi: 10.1523/JNEUROSCI.5544-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zernig G, Ahmed SH, Cardinal RN, Morgan D, Acquas E, Foltin RW, Vezina P, Negus SS, Crespo JA, Stockl P, Grubinger P, Madlung E, Haring C, Kurz M, Saria A. Explaining the escalation of drug use in substance dependence: models and appropriate animal laboratory tests. Pharmacology. 2007;80:65–119. doi: 10.1159/000103923. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc. Nat. Acad. Sci. USA. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollner C, Mousa SA, Fischer O, Rittner HL, Shaqura M, Brack A, Shakibaei M, Binder W, Urban F, Stein C, Schafer M. Chronic morphine use does not induce peripheral tolerance in a rat model of inflammatory pain. J. Clin. Invest. 2008;118:1065–1073. doi: 10.1172/JCI25911. [DOI] [PMC free article] [PubMed] [Google Scholar]