

Abstract
Toxic liver injury causes necrosis and fibrosis, which may lead to cirrhosis and liver failure. Despite recent progress in understanding the mechanism of liver fibrosis, our knowledge of the molecular-level details of this disease is still incomplete. The elucidation of networks and pathways associated with liver fibrosis can provide insight into the underlying molecular mechanisms of the disease, as well as identify potential diagnostic or prognostic biomarkers. Towards this end, we analyzed rat gene expression data from a range of chemical exposures that produced observable periportal liver fibrosis as documented in DrugMatrix, a publicly available toxicogenomics database. We identified genes relevant to liver fibrosis using standard differential expression and co-expression analyses, and then used these genes in pathway enrichment and protein-protein interaction (PPI) network analyses. We identified a PPI network module associated with liver fibrosis that includes known liver fibrosis-relevant genes, such as tissue inhibitor of metalloproteinase-1, galectin-3, connective tissue growth factor, and lipocalin-2. We also identified several new genes, such as perilipin-3, legumain, and myocilin, which were associated with liver fibrosis. We further analyzed the expression pattern of the genes in the PPI network module across a wide range of 640 chemical exposure conditions in DrugMatrix and identified early indications of liver fibrosis for carbon tetrachloride and lipopolysaccharide exposures. Although it is well known that carbon tetrachloride and lipopolysaccharide can cause liver fibrosis, our network analysis was able to link these compounds to potential fibrotic damage before histopathological changes associated with liver fibrosis appeared. These results demonstrated that our approach is capable of identifying early-stage indicators of liver fibrosis and underscore its potential to aid in predictive toxicity, biomarker identification, and to generally identify disease-relevant pathways.
Introduction
Exposure to toxic chemicals can lead to liver injury through a variety of mechanisms, such as oxidative stress, the immune response, activation of apoptotic pathways, and necrosis [1]. Liver fibrosis is a common pathologic feature observed in a wide spectrum of liver injuries [2], [3] and is marked by inflammation and excessive accumulation of extracellular matrix (ECM) components [4]. Liver fibrosis results in scar formation and, if unresolved, leads to cirrhosis, portal hypertension, and liver failure [4]. Liver fibrosis typically starts with apoptosis or necrosis of hepatocytes, which causes reactive oxygen species generation. This process leads to the release of inflammatory mediators and ultimately results in activation of hepatic stellate cells [3], the main ECM-producing cells in the liver. This activation of hepatic stellate cells is the key pathogenic mechanism of liver fibrosis [3]–[6]. Activated hepatic stellate cells lead to further inflammation and ECM generation, which results in the replacement of liver parenchymal cells with ECM [5]. Despite recent progress, our understanding of the molecular mediators of liver fibrosis remains incomplete, and we are still in the process of identifying such mediators [7], [8].
Although fibrotic damage is reversible, there are no approved drugs or treatments for liver fibrosis. Key in understanding damage and control of fibrosis is accurate diagnosis or early indicators of damage. The gold standard for diagnosing fibrosis is currently via liver biopsy. This invasive method has many limitations, such as inter- and intra-observer variations and sampling variability [9]. Thus, there is a need to identify sensitive, specific, and non-invasive biomarkers of liver fibrosis. Identification of such biomarkers will improve diagnosis and allow better clinical management of the disease. In the military, this capability would aid in field assessment and potentially enable timely evacuation or guide return-to-duty decisions. Elucidation of the pathways and networks associated with liver fibrosis will provide insight into the molecular mechanisms of this disease and, importantly, help us to identify mechanism-based biomarkers of liver damage.
Computational systems biology approaches are now routinely used to analyze gene expression data and to gain insight into the molecular mechanisms of many diseases [10]–[15]. Pathway enrichment analysis provides a biological interpretation of gene lists obtained from microarray data using manually curated pathway databases, such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome [16], [17]. The BioSystems database [18], [19] provides an integrated resource of pathways from several major pathway databases, including KEGG and Reactome. Huang et al. [20] have summarized the various tools and statistical methods available for pathway enrichment analysis and their utility in elucidating the mechanisms of diseases [21]–[23]. In literature related to liver fibrosis, the work of Affo et al. [24] utilizes KEGG pathway analysis in identifying the role of focal adhesion and cytokine-cytokine receptor interaction pathways in alcoholic hepatitis. Although widely used, pathway analysis has some limitations [25]. Foremost, pathway analysis relies exclusively on experimentally confirmed, curated data. Only a small fraction of human genes map to curated pathway collections (e.g., KEGG [14]); thus, pathway analysis is inherently biased against the discovery of new molecular mediators. For example, we found that only 5,870 of the ∼21,000 human genes mapped to the 229 KEGG pathways [26]. Moreover, the gene networks comprising the pathway maps are not mutually exclusive, and the same gene can occur in many pathways [25]. Integrated analyses of gene expression data with protein-protein interaction (PPI) networks enable us to partly overcome the limitations associated with pathway analysis. The potential of this integrated approach has been shown in identifying biomarkers for breast cancer [27] and in understanding the molecular mechanisms of dilated cardiomyopathy [28], hepatitis C virus infection [29], and cancer and heart disease [30]–[34]. However, to the best of our knowledge, no such integrated analysis has been reported for liver fibrosis.
Thus, our goal in this study was to identify liver fibrosis-relevant pathways and networks based on an integrated gene expression and PPI network analysis. We analyzed the gene expression data from a range of chemical exposure conditions that produced periportal liver fibrosis in DrugMatrix, a publicly available toxicogenomics database [35]. We carried out differential expression and co-expression analysis using rank product and hierarchical clustering, respectively, to identify genes associated with liver fibrosis [36]. We then examined these genes in two separate analyses. In the first analysis, we identified the KEGG pathways associated with liver fibrosis. In the second analysis, we integrated the gene expression data with the high-confidence human PPI network to obtain liver fibrosis-relevant sub-networks [37]. We identified a PPI network module associated with liver fibrosis that includes known liver fibrosis-relevant genes like Timp1, Lgals3, Ctgf, and Lcn2, along with several new genes. We further analyzed the expression pattern of the genes in the PPI network across a wide range of 640 chemical exposure conditions in DrugMatrix and linked compounds such as carbon tetrachloride to potential fibrotic damage before histopathological evidence of liver fibrosis appeared. These results illustrate the potential of our approach to aid in toxicity prediction and biomarker discovery.
Materials and Methods
Figure 1 shows the overall workflows used in this study to identify liver fibrosis-relevant pathways and interaction networks. We used two separate but complementary approaches to map the overall gene transcriptional response to liver fibrosis via 1) enrichment analysis of knowledge-based pathways association, and 2) integration of gene expression data with PPI networks to identify interaction networks. These fibrosis-relevant interaction networks can be considered as de novo pathways.
Figure 1. Workflow used in this study to identify pathways and networks associated with liver fibrosis.

Data collection and processing
We used DrugMatrix, a publicly available toxicogenomics database [35], [38], for our analyses. DrugMatrix is a large collection of gene expression, hematology, histopathology, and clinical chemistry data obtained from Sprague-Dawley rats after exposure to a range of chemicals, including industrial chemicals, toxicants, and drugs with multiple time intervals, dose ranges, and tissues for each chemical [35]. A chemical exposure condition in DrugMatrix data refers to exposure to a particular chemical at a particular dose and time. We downloaded the DrugMatrix liver gene expression data generated using Affymetrix GeneChip Rat Genome 230 2.0 Arrays from the National Institute of Environmental Health Sciences (NIEHS) server (https://ntp.niehs.nih.gov/drugmatrix/index.html). Of the 2,218 microarray CEL files in this database, 1,941 were chemical exposures and 277 were controls. We performed background correction, quantile normalization, and summarization using the robust multi-array average method in R/BioConductor package affy [26], [39]–[41]. We then used the BioConductor package ArrayQualityMetrics to assess the quality of the microarray data and removed 155 outlier arrays [42]. We reprocessed the remaining arrays using the robust multi-array average method and used this final normalized data for all our analyses. With the BioConductor package genefilter, we carried out non-specific filtering of the genes [43]. We removed probe sets without Entrez ID or with low variance across chemical exposures based on inter-quartile range. We additionally filtered probe sets based on “Present” calls using the BioConductor package affy, retaining only the probe sets for which at least 25% of the chemical exposures had “Present” calls for all replicates within the chemical exposure condition. After calculating the average intensity between the replicates of a chemical exposure condition, we computed log-ratios for each gene between treatments and their corresponding controls. Our final log-ratio matrix contained 7,826 genes and 640 chemical exposure conditions.
Identifying genes relevant to liver fibrosis
The DrugMatrix database provides histopathology data associated with each chemical exposure condition. There were five chemical exposure conditions that produced liver periportal fibrosis with a histopathology score >1 (Figure S1). We carried out a quality check by clustering the replicates of these five chemical exposures, along with their respective controls. Ideally, the chemical exposures and controls would have clustered separately. But all replicates of 5-day exposures to Crotamiton-750 mg/kg clustered with controls rather than with other treatments (Figure S1). Hence, we excluded this condition from the liver fibrosis-producing condition set. Table 1 lists the four chemical exposure conditions that were used in this study as the liver fibrosis-producing condition set. All replicates of the four chemical exposure conditions had a histopathology score of 2. We used the rank product method to identify differentially expressed genes (DEG) [36]. Rank product is a non-parametric, permutation-based method that has been widely used in many studies [36], [44], [45]. With this method, the fold-change values were converted into rank, and then the significance of the obtained rank, including the false discovery rate (FDR) p-value, was calculated. We used the BioConductor package RankProd for this analysis [44]. This method produces separate lists of up-regulated and down-regulated genes. We considered all genes with an FDR <0.05 to be significantly differentially expressed. We carried out the rank product analysis separately for each of the four chemical exposure conditions; the genes that were significantly differentially expressed in at least two of the four chemical exposure conditions that produced periportal liver fibrosis were considered as fibrosis-relevant DEGs.
Table 1. Chemical exposure conditions that produced periportal liver fibrosis.
| Chemical | Dose (mg/kg) | Duration (days) | Histopathology (severity score) |
| 1-Naphthylisothiocyanate | 30 | 7 | 2 |
| 1-Naphthylisothiocyanate | 60 | 7 | 2 |
| 4,4′-Methylenedianiline | 81 | 5 | 2 |
| N-Nitrosodimethylamine | 10 | 5 | 2 |
We carried out hierarchical clustering using the R package hclust to identify co-expressed genes [21]. We clustered the genes in the log-ratio matrix using their log2 ratio values across 640 chemical exposure conditions. We used the Pearson correlation and the average linkage method to perform the clustering, and the R package dynamicTreeCut for automated extraction of clusters [46]. The dynamic tree cut algorithm implements an automated iterative process to identify and split sub-clusters from a dendrogram until the minimum cluster size threshold is reached [46]. We used the cutreeDynamic function in this package with minimum cluster size set to 16, method set to hybrid, and deepsplit set to True.
We calculated cluster activation scores to identify liver fibrosis-relevant clusters in order to identify clusters whose constituent genes show altered gene expression (either up- or down-regulation) in chemical exposure conditions that produce periportal liver fibrosis. To calculate a cluster activation score, we first normalized the log-ratio values of each gene across 640 chemical exposure conditions by converting them into Z-scores. The Z-score of gene i under chemical exposure condition j is given by
| (1) |
where 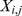 is the log-ratio value for gene i under chemical exposure condition j,
is the log-ratio value for gene i under chemical exposure condition j, 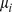 is the average log ratio for gene i across all 640 chemical exposure conditions, and
is the average log ratio for gene i across all 640 chemical exposure conditions, and 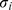 is the standard deviation of the log ratio for gene i across all 640 chemical exposure conditions. Next, we obtained the cluster activation scores for liver fibrosis by averaging the Z-scores of all genes within a cluster and across all chemical exposure conditions that produced periportal liver fibrosis. The activation score
is the standard deviation of the log ratio for gene i across all 640 chemical exposure conditions. Next, we obtained the cluster activation scores for liver fibrosis by averaging the Z-scores of all genes within a cluster and across all chemical exposure conditions that produced periportal liver fibrosis. The activation score 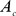 of cluster c in liver fibrosis is given by
of cluster c in liver fibrosis is given by
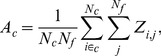 |
(2) |
where 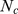 is the number of genes associated with cluster c,
is the number of genes associated with cluster c, 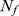 is the number of chemical exposure conditions that produce periportal liver fibrosis, and
is the number of chemical exposure conditions that produce periportal liver fibrosis, and 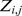 is the Z-score of gene i under chemical exposure conditions that produce periportal liver fibrosis j. We used an absolute cluster activation score cutoff of 2, corresponding to the 95th percentile of the probability density distribution, and selected genes in clusters above this threshold as liver fibrosis-relevant co-expressed genes.
is the Z-score of gene i under chemical exposure conditions that produce periportal liver fibrosis j. We used an absolute cluster activation score cutoff of 2, corresponding to the 95th percentile of the probability density distribution, and selected genes in clusters above this threshold as liver fibrosis-relevant co-expressed genes.
In general, to find disease-relevant genes, either differential expression or co-expression analysis is commonly used. Utilization of both approaches together will allow us to overcome the limitation associated with each approach. Hence, we combined the gene list from both approaches, i.e., differential expression and co-expression, to form the liver fibrosis-relevant gene set.
Pathway enrichment analysis
We used the Database for Annotation, Visualization, and Integrated Discovery (DAVID) tool to perform KEGG pathway enrichment analysis [47]. The pathways below a Benjamini-Hochberg FDR <0.05 were considered to be significantly enriched. We used the liver fibrosis-relevant gene set, i.e., the combined list of genes determined by differential expression and co-expression analysis to be relevant to liver fibrosis, for the pathway enrichment analysis. Then we separately used the up-regulated genes and down-regulated genes in this set and carried out pathway enrichment analysis.
PPI network analysis
Yu et al. [37] used the interaction detection based on shuffling (IDBOS) approach to generate a high-confidence PPI network and showed that the resulting high-confidence PPIs reduce the noise inherent in aggregated PPIs. In their work, they created experimentally derived high-confidence PPI networks for both rats and humans [37]. The rat high-confidence PPI network contained 1,001 nodes, whereas the human high-confidence PPI network consisted of 14,230 nodes. We chose to use the human high-confidence PPI network due to its much larger coverage. The rat probe-set identifiers were mapped to their corresponding human gene identifiers using orthology mapping tools [26], [48], [49]. This approach followed the work by Zhang et al. [50] for mapping other species' gene expression data to a human PPI network. We utilized the KeyPathwayMiner program in Cytoscape 2.8 to obtain the liver fibrosis-relevant sub-network [51]–[53]. KeyPathwayMiner attempts to find maximally connected sub-networks for the input query genes with gene expression data using the ant-colony optimization algorithm [51]. We used KeyPathwayMiner with ant-colony optimization algorithm, node exceptions (K) set to 100, and case exceptions (L) set to 0. The node exception (K) value provided the allowance for genes that were not present in the input gene set and the case exception (L) defined the number of conditions in which the input gene may not be active. We separately ran KeyPathwayMiner using the up-regulated and down-regulated genes in liver fibrosis-relevant DEGs and co-expressed genes and extracted the sub-networks. We then combined the four sub-networks into one final liver fibrosis-relevant sub-network. This sub-network was created by the union of the four sub-networks. We did not use the intersection of the four sub-networks, as it contained only two nodes. Finally, we clustered this liver fibrosis-relevant sub-network using the topology-based network clustering program in Cytoscape, Clusterviz. We used the EAGLE algorithm in Clusterviz with default parameters [54]. As implemented, Clusterviz generated 11 network modules.
Module characterization
We mapped the proteins in the PPI network modules to the rat gene expression Z-score matrix. We calculated the activation scores ( Equation 2 ) for the 11 network modules under conditions that caused periportal liver fibrosis. The method is the same as described above, except that here we used the genes in the module instead of cluster. We collected 28 known liver fibrosis-relevant genes from literature (Set 1) [3], [5], [55], [56]. Of these 28 genes, 26 mapped to the high-confidence human PPI network. We also collected genes that are known to be associated with liver cirrhosis from the Comparative Toxicogenomics Database (CTD) (Set 2) [57]. Of the 126 genes with direct evidence of an association with liver cirrhosis, 95 mapped to the high-confidence human PPI network. We used the Fisher exact test to calculate the enrichment of these genes (Set 1 and Set 2) in each PPI network module. The module genes were also characterized by gene ontology (GO) biological process-term enrichment using the DAVID tool. We used the Revigo tool to visualize the GO enrichment results [58] and analyzed the network modules in terms of activation in liver fibrosis, enrichment with known liver fibrosis-relevant genes, and enrichment of liver fibrosis-relevant GO terms. Based on this analysis, we prioritized one PPI network module (M5) as a liver fibrosis-relevant network module.
We used two statistical significance tests to analyze whether the network module M5 was obtained by random chance. Our null hypothesis was that the observed number of nodes (M5nodes) and edges (M5edges) in module M5 were obtained by random chance. In the first analysis, we randomly selected 92 proteins from the human PPI network and counted the number of nodes (Rnodes) and edges (Redges) of the largest connected component. This process was repeated 1,000 times. We computed the number of times Rnodes≥M5nodes, denoted as Nrandnode. Similarly, we computed the number of times Redges≥M5edges, denoted as Nrandedge. Then we computed the probability of obtaining a similar number of nodes by random chance using P = Nrandnode/1,000, and the probability of obtaining a similar number of edges by random chance using P = Nrandedge/1,000. In the second analysis, we shuffled the human PPI network and then mapped the proteins in the M5 network to this randomized network. We preserved the average node degree during network shuffling. Similar to the first analysis, we extracted the largest connected component, counted the number of nodes and edges, and calculated the probability of obtaining M5 parameters by random chance. We analyzed the overall robustness of the M5 module by comparing the modules generated from a reduced number of samples to those generated from the full dataset. We left out one quarter of the samples from the differential gene expression dataset and analyzed the remaining samples, repeating this procedure four times and leaving out each quarter of the data once. We then compared the overlap of the final module proteins from these four analyses with the module M5 proteins and found an average overlap of 72%. This showed that our method identified roughly the same genes even when samples were missing. We analyzed the expression of genes in module M5 in chemical exposures that produced periportal liver fibrosis across different time periods of exposures. Among the four chemical exposure conditions that produced periportal liver fibrosis, earlier time points were not available for exposure with N-nitroso dimethylamine at 10 mg/kg. Data were available for exposures to 1-naphthyl isothiocyanate at 30 mg/kg and 60 mg/kg at all time points, i.e., 0.25 day, 1 day, and 3 days, and for exposures to 4,4'-methylene dianiline at 1 day and 3 days. We mapped the expression profile of genes in module M5 across different time periods using the average log2 ratio of the available chemical exposure data at that time point. Finally, the genes in the prioritized network module M5 were used to cluster the 640 chemical exposure conditions in the DrugMatrix database. We used the clustering software cluster3 for this purpose [59]. We evaluated the specificity of network module M5 using the average Z-score across the genes in M5 for each of the 640 chemical exposure conditions.
External validation
In order to further demonstrate the relevance of network module M5 in liver fibrosis, we evaluated the M5 genes in two external datasets (GSE13747 and GSE6929) from the Gene Expression Omnibus (GEO). Both datasets used Affymetrix GeneChip Rat Genome 230 2.0 Arrays. In GSE13747, liver fibrosis was produced by bile duct ligation [60]. Six replicates of liver fibrosis samples and six controls were available. In GSE6929, liver cirrhosis was induced by inhalation of carbon tetrachloride [13]. Four replicates of liver cirrhosis controls and four replicates of sunitinib (SU11248)-treated samples were available. We used the same steps described above for the DrugMatrix database to preprocess these two external datasets and calculated the log2 ratio between the treatment and controls. Finally, we matched the genes in module M5 and calculated the correlation between the average log2 ratios in the four chemical exposure conditions that produced periportal liver fibrosis from the DrugMatrix database with the log2 ratio from external datasets.
Results and Discussion
Identification of liver fibrosis-relevant genes
We used differential gene expression and co-expression analysis to identify liver fibrosis-relevant genes. As outlined in the Materials and Methods section, we analyzed the chemical exposures in the DrugMatrix data that produced periportal liver fibrosis with a histopathological score >1. We used the rank product approach and identified 400 liver fibrosis-relevant DEGs, of which 192 genes were significantly up-regulated, and 208 genes were significantly down-regulated. Here, we used an FDR <0.05 as the cutoff value to select DEGs. This cutoff-based approach knowingly excludes DEGs to minimize false positives and may not capture the complete picture of the disease or processes being studied [61]–[63]. For example, many genes that do not meet the cutoff criteria can be involved in the same pathway as the DEGs and provide insights into the altered disease process [63]. Identifying co-expressed genes by means of gene clustering is an alternative approach that does not use cutoffs or thresholds at the individual gene level. Instead, genes are clustered based on their expression profiles across a wide range of exposures in order to identify gene sets that are expected to have similar functions, i.e., participate in related pathways [64]. We used hierarchical clustering to cluster 7,826 genes based on their log-ratio values across 640 chemical exposure conditions, which yielded 210 gene clusters containing an average of 37 genes each. Unlike differential expression analysis, these co-expressed genes were not linked or associated with liver fibrosis or any other particular disease. We used the cluster activation scores defined in Equation 2 to establish the connection between the gene clusters and liver fibrosis. We found 565 genes in the nine clusters with activation scores >2 and 42 genes in the two clusters with activation scores <−2. The genes in these clusters were used as liver fibrosis-relevant co-expressed genes. Finally, we combined the differentially expressed and co-expressed gene lists to create a liver fibrosis-relevant gene set with 897 genes. There were 110 genes in common between the differentially expressed and co-expressed genes, and we show the overlap between these two sets as a Venn diagram ( Figure 2 ). Table S1 provides the list of 897 genes, along with log-ratio values, in the four chemical exposure conditions that produced periportal liver fibrosis.
Figure 2. Number of fibrosis-relevant genes from differential and co-expression analysis.
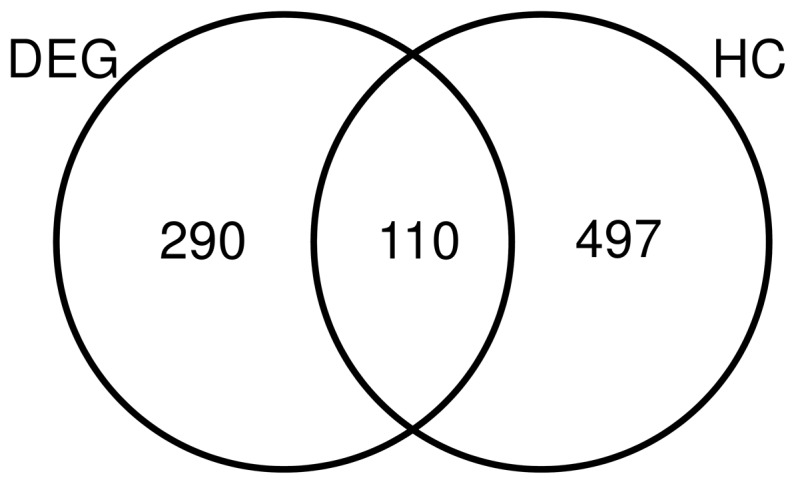
Number of genes in the liver fibrosis-relevant differentially expressed gene list and liver fibrosis-relevant co-expressed gene list and the overlap between them.
Pathway enrichment analysis
Table 2 lists the significantly enriched KEGG pathways derived from the liver fibrosis-relevant gene set. These pathways include leukocyte transendothelial migration, focal adhesion, chemokine signaling, regulation of the actin cytoskeleton pathway, and ECM-receptor interaction, and they mainly represent liver fibrosis-related processes. These processes are consistent with previous reports for liver fibrosis. Injured liver cells and activated hepatic stellate cells release chemokines that recruit leukocytes to the site of injury [65]. An elevated expression of chemokines and chemokine receptors has been reported in both animal models and clinical cases of liver fibrosis [66]. Hepatic stellate cell migration is an essential process in fibrosis [67]. Hepatic stellate cells are also known to express adhesion molecules and α-smooth muscle actin (SMA) [4]. Activated hepatic stellate cells are known to secrete prion protein [68], [69]. Innate immunity and adaptive immunity play a major role in hepatic stellate cell activation and propagation of liver fibrosis [70]. Anticoagulant drugs and peroxisome proliferator-activated receptor (PPAR)-γ agonists were reported to have an anti-fibrotic effect in experimental liver fibrosis, which is consistent with the enriched pathways, such as complement and coagulation cascades, and the PPAR signaling pathway [70].
Table 2. KEGGa pathway enrichment for all genes relevant to liver fibrosis.
| Pathway | Count | BHb |
| Prion diseases | 11 | 0.003 |
| Leukocyte transendothelial migration | 19 | 0.008 |
| Focal adhesion | 26 | 0.007 |
| Fc gamma R-mediated phagocytosis | 16 | 0.006 |
| Pyruvate metabolism | 10 | 0.010 |
| Viral myocarditis | 15 | 0.011 |
| Antigen processing and presentation | 14 | 0.035 |
| Systemic lupus erythematosus | 14 | 0.037 |
| Chemokine signaling pathway | 21 | 0.035 |
| Complement and coagulation cascades | 12 | 0.033 |
| Regulation of actin cytoskeleton | 24 | 0.031 |
| ECMc-receptor interaction | 13 | 0.030 |
| PPARd signaling pathway | 12 | 0.029 |
| Arginine and proline metabolism | 10 | 0.035 |
| Glycerolipid metabolism | 9 | 0.035 |
Kyoto Encyclopedia of Genes and Genomes
Benjamini-Hochberg false discovery rate
Extracellular matrix
Peroxisome proliferator-activated receptor
We analyzed genes involved in the enriched pathways to ascertain whether we could provide new, testable hypotheses. Genes were mapped to the enriched KEGG pathways ( Figure 3 ) and Table S2 provides the complete list of these genes with pathway information, and their average log2 ratio in chemical exposures that produced periportal liver fibrosis. The log2 ratio is commonly used in microarray data analysis, and a value of 0.6 corresponds to a ∼1.5 fold-change (up-regulation) in gene expression. Col1a1, Col1a2, Col4a1, Col4a2, Col5a2, Itgb1, Plat, Plau, Pdgfa, Ezr, and Msn were up-regulated and have an average log2 ratio >0.6. These genes are known to be altered in liver fibrosis [4], [67], [71]. We also analyzed the gene list (Table S2) for potential new candidates and found genes such as Lgmn and Limk2, which were up-regulated in all four chemical exposure conditions that produced periportal liver fibrosis. These genes are potential candidates for further exploration. Next, we carried out pathway enrichment analyses using the up- and down-regulated genes separately (Tables S3 and S4). The significantly up-regulated pathways were related to liver fibrosis-relevant processes, whereas the significantly down-regulated pathways were related to metabolism. The down-regulation of metabolism-related pathways could either be related to external factors, such as altered food intake, or could also be an indication of reduced liver function.
Figure 3. Genes that mapped to the enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.

The average log2 fold-change ratio across chemical exposures that produced periportal liver fibrosis was used as the gene expression value. Genes with average log2 fold-change ratios >0.6 are colored in red. Genes with average log2 fold-change ratios <−0.6 are colored in green. Genes whose average log2 fold-change ratios are between 0.6 and −0.6 are colored in grey.
Although pathway enrichment analyses are useful and provide an overview of biological processes associated with our gene list, the method has some well-known limitations. Pathway analysis is based on curated data and is limited to the information present in the underlying knowledge database. Moreover, the pathway enrichment based on an over-representation analysis approach treats pathways as simple gene lists without accounting for network connectivity [72]. As such, pathway analysis has limited utility in identifying new molecular mediators or new pathways. Using the connectivity information or pathway topology may provide us with alternative approaches to capture the most relevant pathways associated with a disease.
PPI network analysis
Based on the premise that proteins that are closely connected to each other in a network are more likely to be involved in similar processes, an integration of gene expression with PPI networks has been used to identify disease-specific networks [73]. Such networks have been proposed as de novo pathways and partly remedy the limitations of KEGG pathway analysis [25]. Consequently, we mapped the liver fibrosis-relevant genes to a high-confidence human PPI network. Out of 897 fibrosis-relevant genes, 606 mapped to the human PPI network. We extracted a liver fibrosis-relevant sub-network with 902 nodes (proteins) and 2,527 edges using KeyPathwayMiner. Out of the 606 fibrosis-relevant genes, 573 were present in this sub-network. Finally, we clustered the 902 proteins in the liver fibrosis-relevant sub-network into 11 PPI network modules. Data S1 – S3 provides all the input and Cytoscape session files associated with PPI network analysis. Table S5 provides the mapping of rat probe IDs to human gene IDs, and Table S6 provides the protein membership in the PPI network modules, along with their gene expression data.
Module characterization and potential application
We further analyzed the 11 network modules in terms of activation in chemical exposure conditions that produced periportal liver fibrosis, enrichment with known liver fibrosis-relevant genes, and enrichment of GO terms.
Table 3
shows the module activation scores 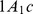 calculated using
Equation 2
for chemical exposure conditions that produced periportal liver fibrosis. Module M5 with 92 genes coding for proteins was the highest activated module with an activation score of 2.12. Next, we analyzed the enrichment of known fibrosis-relevant genes collected from the literature (Table S7) [3], [5], [55], [56].
Table 3
shows the p-values for enrichment of known fibrosis-relevant genes in the network modules. Modules M2, M3, and M5 were enriched with liver fibrosis-relevant genes with p-values <0.05. Module M5 had the lowest enrichment p-value with six known fibrosis-associated genes coding for proteins mapped to this module: TIMP1, APOA1, CTGF, LGALS3, TGFB1, and MMP-2. CTD provides curated information on genes associated with a disease [57]. Liver fibrosis was not curated in CTD, but genes associated with liver cirrhosis, the final stage of liver fibrosis, were available. We further analyzed the enrichment of genes associated with liver cirrhosis collected from CTD (Table S8) and found module M5 to be the module most enriched with liver cirrhosis-related genes (
Table 3
). We also characterized the 11 modules using the GO biological process-term enrichment analysis. We found that module M5 was enriched with liver fibrosis-relevant GO terms such as ECM organization and Wound healing. Figure S2 shows the enriched GO biological process-terms for module M5, and Table S9 provides the entire list of enriched GO terms for all modules. Based on activation in liver fibrosis-producing conditions, enrichment of known liver fibrosis-relevant genes, and liver fibrosis-relevant GO terms, we selected module M5 as the top liver fibrosis-relevant network module identified from this analysis.
calculated using
Equation 2
for chemical exposure conditions that produced periportal liver fibrosis. Module M5 with 92 genes coding for proteins was the highest activated module with an activation score of 2.12. Next, we analyzed the enrichment of known fibrosis-relevant genes collected from the literature (Table S7) [3], [5], [55], [56].
Table 3
shows the p-values for enrichment of known fibrosis-relevant genes in the network modules. Modules M2, M3, and M5 were enriched with liver fibrosis-relevant genes with p-values <0.05. Module M5 had the lowest enrichment p-value with six known fibrosis-associated genes coding for proteins mapped to this module: TIMP1, APOA1, CTGF, LGALS3, TGFB1, and MMP-2. CTD provides curated information on genes associated with a disease [57]. Liver fibrosis was not curated in CTD, but genes associated with liver cirrhosis, the final stage of liver fibrosis, were available. We further analyzed the enrichment of genes associated with liver cirrhosis collected from CTD (Table S8) and found module M5 to be the module most enriched with liver cirrhosis-related genes (
Table 3
). We also characterized the 11 modules using the GO biological process-term enrichment analysis. We found that module M5 was enriched with liver fibrosis-relevant GO terms such as ECM organization and Wound healing. Figure S2 shows the enriched GO biological process-terms for module M5, and Table S9 provides the entire list of enriched GO terms for all modules. Based on activation in liver fibrosis-producing conditions, enrichment of known liver fibrosis-relevant genes, and liver fibrosis-relevant GO terms, we selected module M5 as the top liver fibrosis-relevant network module identified from this analysis.
Table 3. Activation of network modules and enrichment of known genes relevant to liver fibrosis.
| Module | Activation score in Liver fibrosis | No. of genes in module | No. of fibrosis genes (set 1)a | p-value (set 1)a | No. of cirrhosis genes (set 2)b | p-value (set 2)b |
| M1 | 1.47 | 150 | 1 | 0.24 | 1 | 1 |
| M2 | 0.95 | 144 | 3 | 0.002 | 5 | 0.003 |
| M3 | 1.67 | 127 | 2 | 0.02 | 0 | 1 |
| M4 | 1.92 | 110 | 1 | 0.18 | 1 | 0.52 |
| M5 | 2.12 | 92 | 6 | 1.28E-08 | 13 | 3.25E-14 |
| M6 | 0.77 | 81 | 1 | 0.14 | 2 | 0.10 |
| M7 | 1.09 | 58 | - | 1 | 1 | 0.32 |
| M8 | 1.38 | 49 | - | 1 | 2 | 0.04 |
| M9 | 0.36 | 46 | - | 1 | 1 | 0.27 |
| M10 | 0.87 | 40 | - | 1 | 1 | 0.24 |
| M11 | 1.38 | 34 | - | 1 | 1 | 0.20 |
First, we ascertained whether the network module M5 could be observed by random chance. As outlined in the Materials and Methods section, we carried out two statistical significance tests based on random sampling and permutation of the network. The average node degree of the network was 12.76 and it was preserved during network shuffling. Our null hypothesis was that the number of nodes and edges in M5 could be obtained by random chance. In both tests, M5 was significantly different from random occurrence, and the probability of finding this module by random chance was essentially zero ( Figure 4 ).
Figure 4. Statistical significance analysis of network module M5.

A) The comparison of number of nodes in M5 to that from random sampling analysis. B) The comparison of number of edges in M5 to that from random sampling analysis. C) The comparison of number of nodes in M5 to the number present in shuffled protein-protein interaction (PPI) networks. D) The comparison of number of edges in M5 to the number present in shuffled PPI networks.
Second, we plotted the PPI network for the protein products encoded by the genes in module M5 ( Figure 5 ). Many of the molecular interactions captured were already reported to be associated with liver fibrosis, validating the computational approach. For example, the network map shows connectivity between the genes coding for matrix metalloproteinases and the up-regulated genes encoding TIMP1, COLs and FBN1 ( Figure 5 ) [4]. This network map supports a published disease mechanism where increased expression of the gene coding for TIMP1, the negative regulator of matrix metalloproteinases, leads to an increased accumulation of ECM proteins, e.g., collagens (COLs). The network module M5 included other genes encoding the proteins implicated in the pathogenesis of fibrosis (e.g., LUM, CTGF, LGALS3, LCN2, PDGFR, PLAT, and LOX) [4], [74]–[76]. Genes coding for the integrin receptors (ITG) in this network interact with ECM proteins that support their known role as mediators of pro-fibrogenic signaling of ECM proteins [77]. In addition to retrieving genes coding for proteins that are already known to be associated with liver fibrosis, this network module also retrieved some potential new candidate protein products. One such candidate is LGMN (average log2 ratio of 1.24), a cysteine protease that functions in ECM remodeling, but has no known associations with liver fibrosis [78]. Another candidate is the gene encoding PLIN3 (mannose-6-phosphate receptor binding protein), with an average log2 ratio of 1.29; it interacts with IGF2R in the M5 network. PLIN3 is known to play a role in the pathogenesis of steatosis, and is also reported to play a role in PGE2 production [79], [80].
Figure 5. Liver fibrosis-relevant network module M5.

Proteins encoded by genes with average log2 fold-change ratios>0.6 are colored in red. Proteins encoded by genes with average log2 fold-change ratios <−0.6 are colored in green. Proteins encoded by genes with average log2fold-change ratios between 0.6 and −0.6<−0.6 are colored in grey. Proteins without corresponding gene expression data are shown as white circles.
Third, we used the network analysis to identify proteins encoded by genes that do not change in expression, but may form PPI networks. Among the 92 proteins in M5, 30 are K-node exceptions obtained using KeyPathwayMiner, based on their connection to fibrosis-relevant proteins. In protein network analysis, the concept of guilt by association is well known [14]. If a protein is known to be associated with many proteins involved in a biological process, it can be hypothesized to play a role or to be related to the biological process associated with these proteins. For example, the genes encoding osteopontin (SPP1) and vitronectin (VTN) are known to be associated with liver fibrosis [4], [81]. These genes did not reach the fold-change threshold in our preprocessed expression dataset, but the network interactions with other fibrosis-relevant protein products that did change in expression predicted them to be fibrosis-relevant ( Figure 5 ). In addition to retrieving known proteins, the network analysis also identified MYOC, a protein with no reported association with liver fibrosis. MYOC is a secreted glycoprotein involved in the pathogenesis of glaucoma [82]. MYOC interacts with many up-regulated genes in the high-confidence human PPI network, including genes that encode TIMP1, LGALS3, FBN1, COL1A2, and COL3A1 ( Figure 6 ). Based on its connection with many liver fibrosis-relevant proteins, MYOC is a new testable candidate in fibrosis diagnosis and/or pathogenesis. We also analyzed the expression profile of genes in module M5 in chemical exposures that produced periportal liver fibrosis across different time periods of exposures ( Figure 7 ). Only a few genes were activated (i.e., log2 ratio >0.6) after 0.25 day and 1 day, but at ≥3 days, most of the genes exhibited increased expression ( Figure 7 ).
Figure 6. Myocilin interaction network.

First neighbors of myocilin (MYOC) in the entire high-confidence human protein-protein interaction (PPI) network.
Figure 7. Activation of genes encoding proteins in liver fibrosis-relevant network module M5 at different time points.

Genes encoding proteins with average log2 fold-change ratios>0.6 are colored in red. Genes encoding proteins with average log2 fold-change ratios <−0.6 are colored in green. Genes encoding proteins with average log2fold-change ratios between 0.6 and −0.6<−0.6 are colored in grey. A) Activation at 0.25-day exposure. The mapped expression profile is the average log2 ratio in 1-naphthyl isothiocyanate 30 mg/kg and 60 mg/kg, at 0.25-day exposure. B) Activation at 1 day of exposure. The mapped expression profile is the average log2 ratio in 1-naphthyl isothiocyanate 30 mg/kg and 60 mg/kg, and 4,4'-Methylenedianiline 81 mg/kg, at 1 day of exposure. C) Activation at 3 days of exposure. The mapped expression profile is the average log2 ratio in 1-naphthyl isothiocyanate 30 mg/kg and 60 mg/kg, and 4,4'-Methylenedianiline 81 mg/kg, at 3 days of exposure. D) Activation at >3 days of exposure. The mapped expression profile is the average log2 ratio across chemical exposures that produced liver fibrosis.
We performed hierarchical clustering of the 640 chemical exposure conditions present in the DrugMatrix database using the expression data for the genes in module M5. We identified a single cluster that included all four chemical exposure conditions that were initially identified with grade 2 periportal liver fibrosis ( Figure 8a ). Most of the 17 conditions in the cluster were associated with compounds that cause fibrosis. Vinblastine, carmustine, and allyl alcohol all had conditions within the drug matrix dataset that caused periportal fibrosis, but they did not meet the histopathology grade 2 threshold used for our initial analysis. Carbon tetrachloride and lipopolysaccharide exposures were present in this cluster ( Figure 8a ). Analysis of the literature shows that both carbon tetrachloride and lipopolysaccharide are well known agents that cause liver fibrosis [66]. In the DrugMatrix database, a 28-day exposure study with carbon tetrachloride showed histopathological evidence of liver fibrosis; however, gene expression data were not available for this time point. Furthermore, all but one (crotamiton-750 mg/kg exposure) of the chemicals in the DrugMatrix database that caused periportal fibrosis and had Affymetrix arrays for the liver were identified in this cluster. The data associated with the crotamiton-750 mg/kg exposure most likely represents an outlier in the underlying dataset. Thus, our analysis has the potential to generate gene sets that could predict the early onset of liver fibrosis, consistent with earlier reports that gene expression analysis of liver biopsy samples could complement histopathological data [83].
Figure 8. Analysis of genes in liver fibrosis-relevant network module M5.

A) Hierarchical clustering of 640 chemical exposures using genes in liver fibrosis-relevant network module M5. The conditions that clustered with four liver fibrosis-producing conditions are highlighted and listed. Genes with Z-scores>2 are colored in red. Genes with Z-scores <−2 are colored in green. Genes with Z-scores between 2 and -2 are colored in yellow. NA in the table represents that histopathological data was not available for that chemical exposure condition. B) Average Z-scores across the genes in module M5 for each of the 640 chemical exposure conditions.
To evaluate whether network module M5 activation was specific for liver fibrosis, we plotted the average Z-scores of M5 genes in each of the 640 chemical exposure conditions ( Figure 8b ). The M5 genes were mostly activated in liver fibrosis-related chemical exposure conditions and showed very low activation in other chemical exposure conditions ( Figure 8b ). Exposure to rosiglitazone, a PPAR-γ agonist, produced an opposing effect on expression of the genes encoding the protein products in M5 (i.e., down-regulation as measured by log2 ratio values; Figure S3). This result was consistent with the evidence that PPAR-γ agonists prevented liver fibrosis in experimental models [70].
Finally, we used two external gene expression datasets (GEO datasets GSE13747 and GSE6929) to evaluate the relevance of module M5 in liver fibrosis. The overlap of genes between the M5 module and the processed GSE13747 and GSE6929 datasets was 66 and 65, respectively. In the study associated with the GSE13747 dataset, liver fibrosis was induced using bile duct ligation. We observed the predicted positive correlation of gene expression data between the DEGs in this dataset and the genes in module M5 (r = 0.78, Figure 9a ). In the GSE6929 dataset, sunitinib, a multi-kinase inhibitor, was used to treat experimental liver cirrhosis. As expected, we observed a negative correlation (r = −0.46) between log2 ratio gene expression data from GSE6929 and M5 ( Figure 9b ). Thus, the external dataset further supports the validity of M5 genes in liver fibrosis.
Figure 9. Validation with external datasets.

M5 gene expression compared with external datasets. A) GSE13747 represents liver fibrosis produced by bile duct ligation. B) GSE6929 represents sunitinib (SU11248) treatment in liver cirrhosis.
In this work, we showed the utility of integrating gene expression and PPI network analyses to understand the molecular-level details of liver fibrosis and to identify biomarker candidates. The presented computational approach has general applicability for characterizing adverse health effects. For example, our approach can be used to computationally predict toxicity pathways associated with specific diseases. Recent work has suggested that the body's response to toxic exposures and the resulting disease progression proceed through a finite set of toxicity pathways [84]. As stated earlier, the available knowledge-based pathway databases are limited in coverage and susceptible to annotation biases towards well-studied diseases. The computational approach presented in this work enabled us to partly overcome these limitations and develop de novo pathways linked to a specific disease. In particular, we believe that our approach could be used to further understand the molecular basis of adverse health effects such as acute kidney injury, liver cholestasis, liver steatosis, and myocardial infarction. More recently, the concept of adverse outcome pathways (AOPs) has been proposed as a novel tool in mechanism-based predictive toxicology [85]. The AOPs provide a flow-chart-like mechanistic representation of adverse health effects and consist of a molecular initiating event and a series of intermediate key events that lead to the final adverse outcome [85]. Our computational approach will be applicable to the development of novel AOPs; we believe that network modules, such as the liver fibrosis module (M5) identified in this work, could be used either as a key molecular event or used to quantify the reversibility or point of departure of key events in AOPs.
Conclusion
We have carried out systems-level analyses of gene expression data for periportal liver fibrosis. We found that both pathway and network analyses provided insights into the molecular mechanisms of liver fibrosis. Network analyses allowed us to generate de novo pathways and overcome the limitations of analyses based on KEGG pathways. We identified a liver fibrosis-relevant network module that was enriched with known liver fibrosis-relevant genes predictive of liver fibrosis and validated it with external data. The systems approach used in this study allowed us to generate liver fibrosis-relevant pathways and have the potential to predict mechanism-based biomarker candidates.
Supporting Information
Dendrogram from the clustering of chemical exposure conditions that produced periportal liver fibrosis with histopathological scores >1 and their controls.
(DOCX)
Treemap view of gene ontology (GO) biological process-term enrichment for genes in the network module M5.
(DOCX)
Activation of proteins in liver fibrosis-relevant network module M5 in rosiglitazone-1,800 mg/kg, at 5 days of exposure.
(DOCX)
The list of fibrosis-relevant genes from differential expression and co-expression analysis.
(XLSX)
The list of genes that mapped to enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and their gene expression data in chemical exposures that produced liver fibrosis.
(XLSX)
The list of enriched up-regulated pathways for the liver fibrosis-relevant gene set.
(XLSX)
The list of enriched down-regulated pathways for the liver fibrosis-relevant gene set.
(XLSX)
Mapping of rat probe IDs to human gene IDs.
(XLSX)
Protein membership in the protein-protein interaction (PPI) network modules and their gene expression data.
(XLSX)
List of 28 fibrosis-relevant genes collected from literature.
(XLSX)
List of 126 experimental liver cirrhosis-relevant genes collected from the Comparative Toxicogenomics Database (CTD).
(XLSX)
List of enriched gene ontology (GO) terms for the protein-protein interaction (PPI) network modules, M1-M11.
(XLSX)
Cytoscape session file associated with network module generation. The Cytoscape session file includes 1) high-confidence human PPI network data, 2) the liver fibrosis-relevant parent sub-network, and 3) 11 modules obtained by clustering the liver fibrosis-relevant parent sub-network.
(ZIP)
KeyPathwayMiner input gene list.
(XLSX)
Cytoscape session file associated with generation of the liver fibrosis-relevant parent sub-network. The Cytoscape session file includes 1) four sub-networks obtained from KeyPathwayMiner, 2) merged nodes of four sub-networks, 3) high-confidence human PPI network data, and 4) the final liver fibrosis-relevant parent sub-network.
(ZIP)
Acknowledgments
We thank Drs. Xueping Yu and Nela Zavaljevski for valuable discussions and comments on the manuscript. Disclaimer: The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Army or of the U.S. Department of Defense. Citations of commercial organizations or trade names in this report do not constitute an official Department of the Army endorsement or approval of the products or services of these organizations. This paper has been approved for public release with unlimited distribution.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors were supported by the Military Operational Medicine Research Program and the U.S. Army's Network Science Initiative, U.S. Army Medical Research and Materiel Command (USAMRMC, http://mrmc.amedd.army.mil), Ft. Detrick, MD. This research was supported in part by an appointment to the Postgraduate Research Participation Program at the U.S. Army Center for Environmental Health Research (USACEHR, http://usacehr.amedd.army.mil) administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and USAMRMC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Malhi H, Gores GJ (2008) Cellular and molecular mechanisms of liver injury. Gastroenterology 134: 1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sebastiani G (2012) Serum biomarkers for the non-invasive diagnosis of liver fibrosis: the importance of being validated. Clin Chem Lab Med 50: 595–597. [DOI] [PubMed] [Google Scholar]
- 3. Brenner DA (2009) Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc 120: 361–368. [PMC free article] [PubMed] [Google Scholar]
- 4. Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Invest 115: 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baranova A, Lal P, Birerdinc A, Younossi ZM (2011) Non-invasive markers for hepatic fibrosis. BMC Gastroenterology 11: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kisseleva T, Brenner DA (2008) Mechanisms of fibrogenesis. Exp Biol Med (Maywood) 233: 109–122. [DOI] [PubMed] [Google Scholar]
- 7. Mukhopadhyay P, Rajesh M, Cao Z, Horváth B, Park O, et al. (2014) Poly (ADP-ribose) polymerase-1 is a key mediator of liver inflammation and fibrosis. Hepatology 59: 1998–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qiu L, Lin J, Ying M, Chen W, Yang J, et al. (2013) Aldose reductase is involved in the development of murine diet-induced nonalcoholic steatohepatitis. PLOS ONE 8: e73591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adams LA (2011) Biomarkers of liver fibrosis. J Gastroenterol Hepatol 26: 802–809. [DOI] [PubMed] [Google Scholar]
- 10. Miller JA, Oldham MC, Geschwind DH (2008) A systems level analysis of transcriptional changes in Alzheimer's disease and normal aging. J Neurosci 28: 1410–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Afshari CA, Hamadeh HK, Bushel PR (2011) The evolution of bioinformatics in toxicology: advancing toxicogenomics. Toxicol Sci 120 Suppl 1 S225–S237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shi Z, Derow CK, Zhang B (2010) Co-expression module analysis reveals biological processes, genomic gain, and regulatory mechanisms associated with breast cancer progression. BMC Syst Biol 4: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tugues S, Fernandez-Varo G, Munoz-Luque J, Ros J, Arroyo V, et al. (2007) Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology 46: 1919–1926. [DOI] [PubMed] [Google Scholar]
- 14. Feala JD, Abdulhameed MD, Yu C, Dutta B, Yu X, et al. (2013) Systems biology approaches for discovering biomarkers for traumatic brain injury. J Neurotrauma 30: 1101–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tawa GJ, AbdulHameed MD, Yu X, Kumar K, Ippolito DL, et al. (2014) Characterization of chemically induced liver injuries using gene co-expression modules. PLOS ONE 9: e107230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40: D109–D114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, et al. (2009) Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res 37: D619–D622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Geer LY, Marchler-Bauer A, Geer RC, Han L, He J, et al. (2010) The NCBI BioSystems database. Nucleic Acids Res 38: D492–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pan Y, Cheng T, Wang Y, Bryant SH (2014) Pathway analysis for drug repositioning based on public database mining. J Chem Inf Model 54: 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang da W, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim MJ, Pelloux V, Guyot E, Tordjman J, Bui LC, et al. (2012) Inflammatory pathway genes belong to major targets of persistent organic pollutants in adipose cells. Environ Health Perspect 120: 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, et al. (2008) Subtypes of breast cancer show preferential site of relapse. Cancer Res 68: 3108–3114. [DOI] [PubMed] [Google Scholar]
- 23. Huang L, Heinloth AN, Zeng ZB, Paules RS, Bushel PR (2008) Genes related to apoptosis predict necrosis of the liver as a phenotype observed in rats exposed to a compendium of hepatotoxicants. BMC Genomics 9: 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Affo S, Dominguez M, Lozano JJ, Sancho-Bru P, Rodrigo-Torres D, et al. (2013) Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut 62: 452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lehne B, Schlitt T (2012) Breaking free from the chains of pathway annotation: de novo pathway discovery for the analysis of disease processes. Pharmacogenomics 13: 1967–1978. [DOI] [PubMed] [Google Scholar]
- 26. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chuang HY, Lee E, Liu YT, Lee D, Ideker T (2007) Network-based classification of breast cancer metastasis. Mol Syst Biol 3: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu W, Yang L, Du Z (2009) Layered functional network analysis of gene expression in human heart failure. PLOS ONE 4: e6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reiss DJ, Avila-Campillo I, Thorsson V, Schwikowski B, Galitski T (2005) Tools enabling the elucidation of molecular pathways active in human disease: application to hepatitis C virus infection. BMC Bioinformatics 6: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Camargo A, Azuaje F (2007) Linking gene expression and functional network data in human heart failure. PLOS ONE 2: e1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Azuaje F, Devaux Y, Wagner DR (2010) Coordinated modular functionality and prognostic potential of a heart failure biomarker-driven interaction network. BMC Syst Biol 4: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Azuaje FJ, Dewey FE, Brutsaert DL, Devaux Y, Ashley EA, et al. (2012) Systems-based approaches to cardiovascular biomarker discovery. Circ Cardiovasc Genet 5: 360–367. [DOI] [PubMed] [Google Scholar]
- 33. Xiao Y, Xu C, Xu L, Guan J, Ping Y, et al. (2012) Systematic identification of common functional modules related to heart failure with different etiologies. Gene 499: 332–338. [DOI] [PubMed] [Google Scholar]
- 34. Huan J, Wang L, Xing L, Qin X, Feng L, et al. (2014) Insights into significant pathways and gene interaction networks underlying breast cancer cell line MCF-7 treated with 17beta-estradiol (E2). Gene 533: 346–355. [DOI] [PubMed] [Google Scholar]
- 35. Ganter B, Snyder RD, Halbert DN, Lee MD (2006) Toxicogenomics in drug discovery and development: mechanistic analysis of compound/class-dependent effects using the DrugMatrix database. Pharmacogenomics 7: 1025–1044. [DOI] [PubMed] [Google Scholar]
- 36. Breitling R, Armengaud P, Amtmann A, Herzyk P (2004) Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett 573: 83–92. [DOI] [PubMed] [Google Scholar]
- 37. Yu X, Wallqvist A, Reifman J (2012) Inferring high-confidence human protein-protein interactions. BMC Bioinformatics 13: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DrugMatrix (nd) National Institute of Environmental Health Sciences. Available: https://ntp.niehs.nih.gov/drugmatrix/index.html.
- 39. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, et al. (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) Affy – analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315. [DOI] [PubMed] [Google Scholar]
- 41.R Development Core Team (2011) R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. Available: http://www.R-project.org/.
- 42. Kauffmann A, Gentleman R, Huber W (2009) ArrayQualityMetrics – a bioconductor package for quality assessment of microarray data. Bioinformatics 25: 415–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gentleman R, Carey V, Huber W, Hahne F (2013) genefilter: methods for filtering genes from microarray experiments, R. package version 1.40.0. Seattle, WA: Bioconductor. Available: http://bioc.ism.ac.jp/2.11/bioc/html/genefilter.html.
- 44. Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, et al. (2006) RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 22: 2825–2827. [DOI] [PubMed] [Google Scholar]
- 45. Vinuela A, Snoek LB, Riksen JA, Kammenga JE (2010) Genome-wide gene expression analysis in response to organophosphorus pesticide chlorpyrifos and diazinon in C. elegans . PLOS ONE 5: e12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Langfelder P, Zhang B, Horvath S (2008) Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 24: 719–720. [DOI] [PubMed] [Google Scholar]
- 47. Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 48. Yu C, Desai V, Cheng L, Reifman J (2012) QuartetS-DB: a large-scale orthology database for prokaryotes and eukaryotes inferred by evolutionary evidence. BMC Bioinformatics 13: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE, et al. (2012) The Mouse Genome Database (MGD): comprehensive resource for genetics and genomics of the laboratory mouse. Nucleic Acids Res 40: D881–D886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang J, Yang Y, Wang Y, Zhang J, Wang Z, et al. (2011) Identification of hub genes related to the recovery phase of irradiation injury by microarray and integrated gene network analysis. PLOS ONE 6: e24680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alcaraz N, Friedrich T, Kotzing T, Krohmer A, Muller J, et al. (2012) Efficient key pathway mining: combining networks and OMICS data. Integr Biol (Camb) 4: 756–764. [DOI] [PubMed] [Google Scholar]
- 52.Baumbach J, Friedrich T, Kotzing T, Krohmer A, Muller J, et al. (2012) Efficient algorithms for extracting biological key pathways with global constraints. Proceedings of the 14th Annual Conference on Genetic and Evolutionary Computation. Philadelphia, Pennsylvania, USA: Association for Computing Machinery. pp. 169–176.
- 53. Saito R, Smoot ME, Ono K, Ruscheinski J, Wang PL, et al. (2012) A travel guide to Cytoscape plugins. Nat Methods 9: 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shen H (2009) Detect overlapping and hierarchical community structure in networks. Physica A 388: 1706–1712. [Google Scholar]
- 55. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, et al. (2006) Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci U S A 103: 5060–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Page S, Birerdinc A, Estep M, Stepanova M, Afendy A, et al. (2013) Knowledge-based identification of soluble biomarkers: hepatic fibrosis in NAFLD as an example. PLOS ONE 8: e56009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Davis AP, Murphy CG, Johnson R, Lay JM, Lennon-Hopkins K, et al. (2013) The Comparative Toxicogenomics Database: update 2013. Nucleic Acids Res 41: D1104–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Supek F, Bosnjak M, Skunca N, Smuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLOS ONE 6: e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. de Hoon MJ, Imoto S, Nolan J, Miyano S (2004) Open source clustering software. Bioinformatics 20: 1453–1454. [DOI] [PubMed] [Google Scholar]
- 60. Moreno M, Chaves JF, Sancho-Bru P, Ramalho F, Ramalho LN, et al. (2010) Ghrelin attenuates hepatocellular injury and liver fibrogenesis in rodents and influences fibrosis progression in humans. Hepatology 51: 974–985. [DOI] [PubMed] [Google Scholar]
- 61. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pan KH, Lih CJ, Cohen SN (2005) Effects of threshold choice on biological conclusions reached during analysis of gene expression by DNA microarrays. Proc Natl Acad Sci U S A 102: 8961–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nam D, Kim SY (2008) Gene-set approach for expression pattern analysis. Brief Bioinform 9: 189–197. [DOI] [PubMed] [Google Scholar]
- 64. Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P (2004) Coexpression analysis of human genes across many microarray data sets. Genome Res 14: 1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Friedman SL (2000) Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 275: 2247–2250. [DOI] [PubMed] [Google Scholar]
- 66. Liedtke C, Luedde T, Sauerbruch T, Scholten D, Streetz K, et al. (2013) Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair 6: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li L, Wang JY, Yang CQ, Jiang W (2012) Effect of RhoA on transforming growth factor beta1-induced rat hepatic stellate cell migration. Liver Int 32: 1093–1102. [DOI] [PubMed] [Google Scholar]
- 68. Ikeda K, Kawada N, Wang YQ, Kadoya H, Nakatani K, et al. (1998) Expression of cellular prion protein in activated hepatic stellate cells. Am J Pathol 153: 1695–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kitada T, Seki S, Ikeda K, Nakatani K, Sakaguchi H, et al. (2000) Clinicopathological characterization of prion: a novel marker of activated human hepatic stellate cells. J Hepatol 33: 751–757. [DOI] [PubMed] [Google Scholar]
- 70. Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Okayama T, Kikuchi S, Ochiai T, Ikoma H, Kubota T, et al. (2008) Attenuated response to liver injury in moesin-deficient mice: impaired stellate cell migration and decreased fibrosis. Biochim Biophys Acta 1782: 542–548. [DOI] [PubMed] [Google Scholar]
- 72. Khatri P, Sirota M, Butte AJ (2012) Ten years of pathway analysis: current approaches and outstanding challenges. PLOS Comput Biol 8: e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ulitsky I, Krishnamurthy A, Karp RM, Shamir R (2010) DEGAS: de novo discovery of dysregulated pathways in human diseases. PLOS ONE 5: e13367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krishnan A, Li X, Kao WY, Viker K, Butters K, et al. (2012) Lumican, an extracellular matrix proteoglycan, is a novel requisite for hepatic fibrosis. Lab Invest 92: 1712–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schuppan D, Kim YO (2013) Evolving therapies for liver fibrosis. J Clin Invest 123: 1887–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang LP, Takahara T, Yata Y, Furui K, Jin B, et al. (1999) Increased expression of plasminogen activator and plasminogen activator inhibitor during liver fibrogenesis of rats: role of stellate cells. J Hepatol 31: 703–711. [DOI] [PubMed] [Google Scholar]
- 77. Mallat A, Lotersztajn S (2013) Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am J Physiol Cell Physiol 305: C789–C799. [DOI] [PubMed] [Google Scholar]
- 78. Morita Y, Araki H, Sugimoto T, Takeuchi K, Yamane T, et al. (2007) Legumain/asparaginyl endopeptidase controls extracellular matrix remodeling through the degradation of fibronectin in mouse renal proximal tubular cells. FEBS Lett 581: 1417–1424. [DOI] [PubMed] [Google Scholar]
- 79. Okumura T (2011) Role of lipid droplet proteins in liver steatosis. J Physiol Biochem 67: 629–636. [DOI] [PubMed] [Google Scholar]
- 80. Nose F, Yamaguchi T, Kato R, Aiuchi T, Obama T, et al. (2013) Crucial role of perilipin-3 (TIP47) in formation of lipid droplets and PGE2 production in HL-60-derived neutrophils. PLOS ONE 8: e71542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Koukoulis GK, Shen J, Virtanen I, Gould VE (2001) Vitronectin in the cirrhotic liver: an immunomarker of mature fibrosis. Hum Pathol 32: 1356–1362. [DOI] [PubMed] [Google Scholar]
- 82. Tamm ER (2002) Myocilin and glaucoma: facts and ideas. Prog Retin Eye Res 21: 395–428. [DOI] [PubMed] [Google Scholar]
- 83. Heinloth AN, Boorman GA, Foley JF, Flagler ND, Paules RS (2007) Gene expression analysis offers unique advantages to histopathology in liver biopsy evaluations. Toxicol Pathol 35: 276–283. [DOI] [PubMed] [Google Scholar]
- 84. Hartung T, McBride M (2011) Food for Thought… on mapping the human toxome. ALTEX 28: 83–93. [DOI] [PubMed] [Google Scholar]
- 85. Vinken M (2013) The adverse outcome pathway concept: a pragmatic tool in toxicology. Toxicology 312: 158–165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dendrogram from the clustering of chemical exposure conditions that produced periportal liver fibrosis with histopathological scores >1 and their controls.
(DOCX)
Treemap view of gene ontology (GO) biological process-term enrichment for genes in the network module M5.
(DOCX)
Activation of proteins in liver fibrosis-relevant network module M5 in rosiglitazone-1,800 mg/kg, at 5 days of exposure.
(DOCX)
The list of fibrosis-relevant genes from differential expression and co-expression analysis.
(XLSX)
The list of genes that mapped to enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and their gene expression data in chemical exposures that produced liver fibrosis.
(XLSX)
The list of enriched up-regulated pathways for the liver fibrosis-relevant gene set.
(XLSX)
The list of enriched down-regulated pathways for the liver fibrosis-relevant gene set.
(XLSX)
Mapping of rat probe IDs to human gene IDs.
(XLSX)
Protein membership in the protein-protein interaction (PPI) network modules and their gene expression data.
(XLSX)
List of 28 fibrosis-relevant genes collected from literature.
(XLSX)
List of 126 experimental liver cirrhosis-relevant genes collected from the Comparative Toxicogenomics Database (CTD).
(XLSX)
List of enriched gene ontology (GO) terms for the protein-protein interaction (PPI) network modules, M1-M11.
(XLSX)
Cytoscape session file associated with network module generation. The Cytoscape session file includes 1) high-confidence human PPI network data, 2) the liver fibrosis-relevant parent sub-network, and 3) 11 modules obtained by clustering the liver fibrosis-relevant parent sub-network.
(ZIP)
KeyPathwayMiner input gene list.
(XLSX)
Cytoscape session file associated with generation of the liver fibrosis-relevant parent sub-network. The Cytoscape session file includes 1) four sub-networks obtained from KeyPathwayMiner, 2) merged nodes of four sub-networks, 3) high-confidence human PPI network data, and 4) the final liver fibrosis-relevant parent sub-network.
(ZIP)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.