

Abstract
Immunoglobulin G (IgG) immune complexes have been shown to modify immune responses driven by antigen presenting cells in either a pro- or anti-inflammatory direction depending upon the context of stimulation. However, the ability of immune complexes to modulate the inflammasome-dependent innate immune response is unknown. Here we show that IgG immune complexes suppress IL-1α and IL-1β secretion through inhibition of inflammasome activation. The mechanism by which this inhibition occurs is via immune complex ligation of activating Fcγ receptors (FcγR), resulting in prevention of both activation and assembly of the inflammasome complex in response to NLRP3, NLRC4, or AIM2 agonists. In vivo, administration of antigen in the form of an immune complex during priming of the immune response inhibited resultant adaptive immune responses in a NLRP3 dependent model of allergic airway disease. Our data reveal an unexpected mechanism regulating CD4+ T cell differentiation, whereby immune complexes suppress inflammasome activation and the generation of IL-1α and IL-1β from antigen presenting cells, which are critical for the antigen-driven differentiation of CD4+ T cells.
Introduction
IgG-antigen immune complexes have numerous effects on the host immune system driven by their signaling through Fcγ receptors (FcγR). Signals from FcγR can be either activating or inhibitory in nature. Signaling resulting in activation of the innate immune system leads to enhanced phagocytosis, the induction of antibody dependent cellular cytotoxicity, degranulation, as well as profound modifications in cytokine production by macrophages, dendritic cells and monocytes (1). Immune complexes have been shown to upregulate IL-10 production while concurrently suppressing IL-12 p40 generation (2–5). These changes induced in innate immune cytokine production have a major influence on the generation of an antigen specific adaptive response and have been shown to repress Th1 responses while enhancing Th2 responses (2, 6).
IL-1α and IL-1β are closely related cytokines that both signal through the IL-1R1. As potent pro-inflammatory cytokines the secretion of each is tightly regulated. IL-1β secretion is dependent upon the activation of caspase-1, which is activated as part of a multi-protein complex called the inflammasome (7, 8). In contrast, IL-1α processing and secretion can proceed in either an inflammasome-dependent or –independent manner depending on the particular agonist (9). Upon activation the nucleotide-binding domain leucine-rich repeat containing (NLR) family members (NLRP1, NLRP3, NLRC4) and the PYHIN family member (AIM2) can form an inflammasome complex, which typically contains an NLR (or AIM2), the adapter protein apoptosis-associated speck-like protein containing a CARD domain (ASC) and the cysteine protease caspase-1. Activation of the inflammasome requires a two step process; signal one, or priming, occurs in response to either microbial or endogenous danger signals (10, 11). Priming results in the generation of pro-IL-1α, pro-IL-1β and pro-IL-18 and also readies the inflammasome for activation through an unknown mechanism. Signal two can be provided via numerous stimuli resulting in the activation of the specific inflammasome complex and ultimately caspase-1 activation.
In this study we demonstrate that the ligation of FcγR by IgG immune complexes during priming inhibits the assembly and activation of the inflammasome complex. This in turn results in the markedly diminished production of IL-1α and IL-1β. We further show that in vivo alum-driven adaptive immune responses require the presence of both IL-1α and IL-1β and that IgG immune complex mediated suppression of IL-1α and IL-1β results in the inhibition of effector CD4+ T cell responses.
Materials and Methods
Mice
C57BL/6N and CD45.1 (B6Ly5.2Cr) mice were obtained from the NCI mouse repository (Frederick, MD). The generation of Nlrp3−/−, Casp1−/−, Il10−/−, Il1r1−/−, IL1a−/−, IL1b−/−, Fcer1g−/−, and Fcgr2b−/− mice have been described previously (12–19). OT-II (B6.Cg-Tg(TcraTcrb)425Cbn/J) transgenic mice (20) were purchased from Jackson Laboratories (Bar Harbor, ME). All protocols were approved by the Institutional Animal Use and Care Committees at the University of Iowa.
Immune complexes
IgG opsonized sheep erythrocytes were produced as described previously using a rabbit anti-sheep red blood cell antibody (Rockland, Gilbertsville, PA) (5). For IgG-Ova immune complexes, chicken egg ovalbumin (Ova) (Grade V) (Sigma, St. Louis, MO) and goat anti-Ova IgG (MP Cappel, Santa Ana, CA) were mixed at a 1:32 (μg Ova: μg IgG) ratio, and incubated for 30 min at room temperature. To produce IgG-opsonized C. albicans, log growth phase C. albicans (3–5 × 107 yeast/ml) were incubated with 0.5 mg/ml rabbit anti-C. albicans polyclonal IgG (Thermo Scientific) for 40 minutes at 4°C. The IgG-opsonized C. albicans was washed and resuspended in DPBS.
In vitro stimulation of BMDM
BMDM were generated as described previously (4). BMDM were either left unstimulated, primed with 50 ng/mL LPS (Invivogen, San Diego, CA), LPS and immune complexes, or LPS and particle control for 3–4 h. For studies using Ova or IgG-Ova, BMDM were treated with 1.6 μg/mL Ova equivalents. BMDM were then challenged with 5 mM ATP (Sigma), 50 μg/cm2 silica (Min-U-Sil-5; U.S. Silica), C. albicans FC20 strain at an MOI 10:1 for 6 h, P. aeruginosa PAK strain at an MOI of 1:1 for 6 h, or F. tularensis LVS strain at an MOI of 50:1 for 9 h. Supernatants were collected and assayed for IL-1α, IL-1β, IL-18, IL-10, and IL-12 p40. Antibody pairs for the IL-1β ELISAs were from R&D Systems. Antibody pairs for IL-1α, IL-10, and IL-12 p40 were from eBiosciences (San Diego, CA). IL-18 ELISA antibody pairs were from MBL (Woburn, MA).
Induction and evaluation of airway inflammation
Mice were sensitized on day 0 by intraperitoneal injection with either 2 mg alum (Thermo Scientific) and 20 μg Ova or 2 mg alum and IgG-Ova (20 μg Ova). On days 15, 16, and 17 mice were intranasally challenged with 20 μg Ova in 50 μl PBS. Lymph nodes, lungs, blood, and BAL fluid were harvested on day 19. BAL was performed by delivering 1 ml cold PBS into the airway via a tracheal cannula and gently aspirating the fluid. The lavage was repeated three times. The cells were stained with trypan blue to determine viability and total nucleated cell counts were obtained using a hemocytometer. Cytospin slides were prepared and percentage of neutrophils, eosinophils, lymphocytes and DC/Macs was determined after HEMA3 staining (Fisher Scientific). Ova-specific IgG1 and IgG2c and total IgE in serum was determined by ELISA at day 19 as previously described (21, 22). Lungs were fixed, embedded in paraffin and 5 μM sections were stained with H&E.
T cell restimulation and proliferation
Cells from the draining LNs were cultured with 10 μM Ova for 72 h and supernatants were collected and analyzed by ELISA. Antibody pairs for IL-17A, IL-13 and IL-4 were from eBiosciences. For flow cytometric analysis of intracellular cytokines, Ova-restimulated LN cells were incubated for 4 h in the presence of 3 μg/ml brefeldin A and 2μM monensin (eBiosciences). Cells were fixed and permeabilized using Fixation/Permeabilization buffer (eBiosciences) and stained with anti-CD3, -CD4, -IL-13 (eBiosciences), and -IL-17A (BD Biosciences). Flow cytometric analysis was performed on a Becton Dickinson LSR II and data analyzed with FlowJo software (Tree Star Inc., Ashland, OR).
For proliferation analysis splenic CD4+ T cells from OT-II transgenic mice were prepared by positive selection using CD4 Miltenyi beads per the manufacturer’s instructions (Miltenyi Biotec, San Diego, CA). CD4+ T cells were labeled with 2.5 μM CFSE (Invitrogen) for 5–7 min at 37°C; 3 × 106 labeled CD4+ T cells were transferred intravenously into CD45.1 congenic mice. Mice were immunized with alum/Ova or alum/IgG-Ova as described above and T cell proliferation assessed at day 3 post-immunization by flow cytometry on an Accuri C6 flow cytometer (BD Biosciences).
Immunoblotting
Electrophoresis of lysates was performed using the NuPAGE system (Invitrogen) according to the manufacturers protocol. To detect caspase-1, IL-1β, ASC, NLRP3 and GAPDH, rabbit polyclonal anti-mouse caspase-1 p10 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-mouse IL-1β antibodies (Millipore, Billerica, MA), rabbit polyclonal anti-mouse ASC antibodies (Enzo Life Sciences, Farmingdale, NY), mouse monoclonal anti-mouse NLRP3 antibodies (Enzo Life Sciences) and mouse monoclonal anti-mouse GAPDH (Calbiochem) were used, respectively.
ASC speck assay
Immunofluorescence studies were carried out as previously described (23). Briefly, macrophages were seeded on to glass coverslips and challenged as described above. The cells were washed and fixed in 4% paraformaldehyde in PBS. The cells were then blocked and permeabilized in 5% FBS in PBS with 0.5% saponin. Cells were immunostained with rabbit anti-ASC (Enzo Life sciences) as the primary antibody and FITC conjugated donkey anti-rabbit IgG (BioLegend), actin was stained with rhodamine phalloidin (Molecular Probes). Coverslips were mounted on slides using VectaSheild with DAPI (Vector Laboratories, Burlingame, CA) and imaged using a confocal microscope (Zeiss 710, Carl Zeiss, Inc.).
Statistical analysis
Statistical data analysis was done using Prism 5.0a (GraphPad Software, La Jolla, CA). Unless otherwise noted, statistical significance for single comparisons was determined by Student’s t-test; ANOVA with a Bonferonni post-test was used for multiple comparisons.
Results
Immune complexes inhibit the secretion of IL-1α, IL-1β, and IL-18 in vitro
Cytokines secreted by antigen presenting cells are required to instruct the differentiation of CD4+ T cells; to assess if these cytokines were affected by immune complex uptake bone marrow-derived macrophages (BMDM) were primed with LPS in the presence of either IgG-opsonized sheep red blood cells (SRBC; eIgG) or unopsonized SRBC (e), the cells were then challenged with the NLRP3 inflammasome agonist silica. As expected stimulation of LPS-primed BMDM with silica resulted in the secretion of IL-1β and IL-18; however, the presence of eIgG immune complexes during LPS priming significantly inhibited both IL-1β and IL-18 secretion (Figure 1A). Consistent with previous studies immune complexes suppressed LPS-induced IL-12 p40 production, concurrently elevating IL-10 production (Figure 1A). This inhibition was not specific to the type of immune complex or the NLRP3 agonist as soluble IgG-Ova immune complexes were also capable of inhibiting IL-1β secretion induced by the NLRP3 agonists ATP and alum (Figure 1B and C). Treatment with IgG immune complexes did not appreciably alter IL-6 or TNF-α release (Supplemental Figure 1A and B) (4). Additionally, this inhibition required that IgG be complexed to an antigen as the addition of free IgG failed to inhibit IL-1β secretion (Supplemental Figure 1C).
Figure 1.

IgG immune complexes suppress Nlrp3 inflammasome activation. (A) BMDM were LPS primed in the presence of unopsonized (e) or IgG-opsonized erythrocytes (eIgG) for 4 h and then challenged with silica for 6 h. Cytokine secretion into culture supernatants was measured by ELISA. ***p ≤ 0.001 by Student’s t-test. (B, C) BMDM were LPS primed in the presence or absence of Ova or IgG-Ova and then challenged with either (B) ATP or (C) alum for 6 h. Culture supernatants were collected and analyzed for IL-1β secretion by ELISA. **p ≤ 0.01 by Student’s t-test. (D) BMDM were LPS primed in the presence or absence of eIgG; macrophages were then challenged with either silica or ATP. 6 h later cells lysates were analyzed for caspase-1 activation by immunoblot. (E, F) BMDM were LPS primed with or without IgG-ova, and then were challenged with ATP for 1 h; cells were fixed, permeabilized and stained for ASC. Representative confocal images are shown (E). Number of speck positive cells was quantified (F). ***p ≤ 0.001 by 1-way ANOVA with Bonferroni post-test. (G, H) BMDM were LPS primed with or without Ova or IgG-Ova; cells were then challenged with ATP (G) or alum (H). Culture supernatants were harvested and analyzed for IL-1α by ELISA. **p ≤ 0.01 by Student’s t-test. Determinations were performed in triplicate and are expressed as the mean±SD. Results shown are representative of at least three independent experiments.
Caspase-1 activation involves autocatalytic processing of the 45 kDa pro-caspase-1 to generate two subunits, p20 and p10. Caspase-1 activation in silica or ATP stimulated LPS-primed BMDM was detected in immunoblots by the appearance of the p10 cleavage product (Figure 1D). However, if BMDM were LPS primed in the presence of eIgG immune complexes caspase-1 activation was not observed in response to silica or ATP challenge (Figure 1D). To elucidate the mechanism by which immune complexes block inflammasome activation we examined the oligomerization of inflammasomes upon receiving an activating signal. Upon activation of the inflammasome the adapter protein ASC forms cytosolic aggregates, or specks, that co-localize with caspase-1, and these foci can be visualized using confocal microscopy (23, 24). We tested if IgG immune complexes were able to block the formation of ASC specks. BMDM were challenged with ATP following LPS priming in the presence or absence of immune complexes. We observed the formation of ASC specks upon ATP challenge in BMDM that had been LPS primed in the absence of immune complexes (Figure 1E and F). However, ATP challenged BMDM that had been primed in the presence of immune complexes had a significant reduction in ASC specks (Figure 1E and F). These data indicate that immune complexes block caspase-1 activation and subsequent processing and secretion of IL-1β by preventing NLRP3 inflammasome assembly.
Unlike IL-1β, cleavage and secretion of IL-1α can be either inflammasome-dependent or -independent depending on the NLR agonist (9). In general, crystalline activators of the NLRP3 inflammasome like alum induce caspase-1-independent IL-1α secretion, whereas soluble activators like ATP induce caspase-1-dependent IL-1α secretion. Consistent with inhibition of caspase-1 activation by IgG immune complexes, we observed a suppression of ATP-induced, but not alum-induced, IL-1α secretion (Figure 1G and H). Taken together these data suggest that immune complex mediated suppression of IL-1β secretion and ATP-induced IL-1α secretion is through inhibition of caspase-1 activation.
Immune complexes do not prevent the synthesis of pro-IL-1β, NLRP3, ASC or caspase-1
We next asked at what step immune complexes acted to interfere with inflammasome activation. Inhibition of IL-1β secretion occurred if immune complexes were added up to 3 h following the addition of LPS, but not if immune complexes were added concurrently with the NLRP3 agonist, suggesting that immune complexes interfered with the priming signal required for inflammasome activation (Figure 2A). Additionally, we found that immune complex mediated inhibition of IL-1β secretion was dependent upon the number of immune complexes used as well as the concentration of antibody used to opsonize the target (Figure 2B and C).
Figure 2.
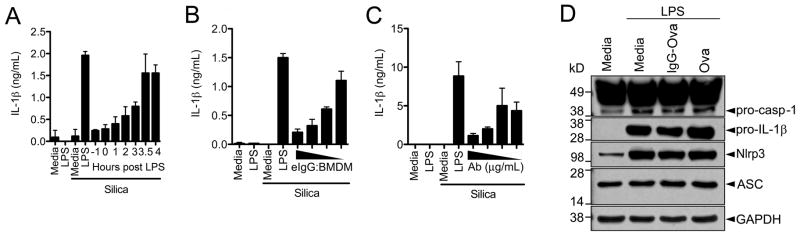
IgG immune complexes do not effect the synthesis of inflammasome components. (A) BMDM were LPS primed; eIgG were added at the indicated time points after LPS treatment. BMDM were then challenged with silica for 6 h. Supernatants were harvested and IL-1β measured by ELISA. (B) BMDM were LPS primed in the presence of increasing numbers of eIgG (eIgG:BMDM ratio of 10:1, 5:1, 2.5:1, and 1:1). BMDM were challenged with silica for 6 h and culture supernatants collected and analyzed for IL-1β by ELISA. (C) BMDM were primed with LPS in the presence of erythrocytes that were IgG opsonized with decreasing concentrations of anti-SRBC IgG (400, 200, 100, and 50 μg/ml). BMDMs were then challenged with silica for 6 h and culture supernatants analyzed for IL-1β release by ELISA. (D) BMDM were LPS primed with or without Ova or IgG-Ova. After 4 h lysates were collected and analyzed by immunoblot for pro-caspase-1, pro-IL-1β, NLRP3, ASC, and GAPDH. Determinations were performed in triplicates and are expressed as the mean±SD. Results shown are representative of at least three independent experiments.
The precise mechanism by which priming readies the inflammasome for activation is unknown, however it has been suggested that upregulation of NLRP3 expression is one factor in the priming process. To determine if immune complexes inhibited the expression of NLRP3, or other inflammasome components, BMDM were LPS-primed in the presence or absence of IgG immune complexes for 4 h and cell lysates subjected to immunoblot. Similar amounts of pro-caspase-1, pro-IL-1β, NLRP3 and ASC were detected in the absence or presence of immune complexes (Figure 2D) suggesting that inhibition of inflammasome complex component expression is not responsible for immune complex-mediated caspase-1 inhibition.
Inflammasome inhibition by IgG immune complexes requires signaling through the FcRγ chain
FcγRs can be either activating (FcγRI, FcγRIII and FcγRIV) or inhibitory (FcγRIIb). The activating receptors require the ITAM containing FcRγ chain (encoded by the Fcer1g gene) for signal propagation (25), whereas the inhibitory FcγRIIb signals through a cytosolic ITIM motif (26). To determine the contribution of activating and inhibitory FcγR in immune complex-mediated inflammasome inhibition we utilized BMDM from FcRγ chain-deficient (Fcer1g−/−) or FcγRIIb-deficient (Fcgr2b−/−) mice. Immune complexes failed to suppress the secretion of IL-1β in response to silica or ATP in BMDM from Fcer1g−/− mice (Figure 3A). In contrast immune complexes mediated suppression of IL-1β secretion remained intact in BMDM from Fcgr2b−/− mice (Figure 3B). Together these data suggest that inhibition of inflammasome activation by immune complexes is not mediated by signaling through the inhibitory receptor FcγRIIb and instead that signaling through the FcRγ chain is required, implicating an activating FcγR in this process.
Figure 3.
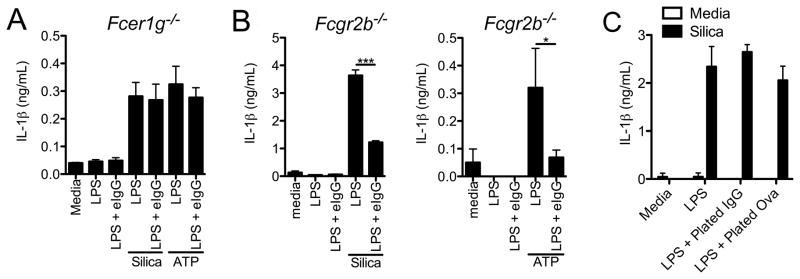
Signaling through the FcRγ chain but not FcγRIIb is required for inflammasome suppression. BMDM from Fcer1g−/− (A) or Fcgr2b−/− (B) were LPS primed with or without eIgG and challenged with silica or ATP for 6 h. Culture supernatants were harvested and analyzed for IL-1β by ELISA. *p ≤ 0.05, ***p ≤ 0.001 by Student’s t-test. (C) BMDM were seeded on tissue culture plates that had been coated with 2 μg/mL IgG or Ova. The cells were then LPS primed for 4 h and subsequently challenged with silica for 6 h. Culture supernatants were collected and IL-1β secretion was assessed by ELISA. Determinations were performed in triplicate and are expressed as the mean±SD. Results shown are representative of at least three independent experiments.
The initiation of phagocytosis is an important function of signaling through the FcRγ chain. To determine if uptake of IgG immune complexes was required to block IL-1β secretion we plated macrophages on tissue culture plates that had been coated with IgG or Ova. Exposure of macrophages to plate bound IgG did not inhibit IL-1β secretion in response to silica (Figure 3C) suggesting that FcγR ligation alone is not sufficient to inhibit inflammasome activation. In contrast, exposure of macrophages to plate bound IgG did inhibit IL-12p40 production and enhance IL-10 secretion in response to LPS stimulation (data not shown) (4).
Immune complexes inhibit activation of NLRP3, NLRC4 and AIM2 inflammasomes
To determine if inflammasome inhibition by immune complexes is specific to the NLRP3 inflammasome or affects other inflammasomes as well we utilized three pathogens, Candida albicans, Pseudomonas aeruginosa, and Francisella tularensis LVS that activate the NLRP3, NLRC4 and AIM2 inflammasomes respectively. Consistent with our observations with other NLRP3 agonists we observed a marked inhibition of IL-1β secretion in response to C. albicans in BMDM that had been LPS-primed in the presence of immune complexes (Figure 4A). Direct IgG opsonization of C. albicans also resulted in significantly diminished IL-1β secretion from bone marrow-derived dendritic cells (BMDC) as compared to challenge with unopsonized C. albicans (Figure 4B). Systemic infection of mice with C. albicans in the presence of IgG-Ova immune complexes also resulted in diminished serum IL-1β levels compared to mice challenged with C. albicans in the presence Ova alone (Figure 4C).
Figure 4.

IgG immune complexes suppress NLRP3, NLRC4 and AIM2 inflammasome activation. (A) BMDM were LPS primed with or with e or eIgG and then challenged with C. albicans (MOI 10:1) for 6 h. Culture supernatants were collected and IL-1β was measured by ELISA. (B) BMDC were challenged with either unopsonized or IgG-opsonized C. albicans (MOI 10:1) for 6 h. Culture supernatants were collected and analyzed for IL-1β secretion by ELISA. **p ≤ 0.01 by Student’s t-test. (C) WT mice were infected i.v. with 2 × 106 C. albicans yeast, resuspended in the presence of Ova or IgG-Ova. After 6 h serum IL-1β levels were determined by ELISA. *p ≤ 0.05 by Mann-Whitney test (n=14 mice per group). (D, E) BMDM were LPS primed with or without e or eIgG and then challenged with either P. aeruginosa PAK strain (MOI 1:1) (D) or F. tularensis LVS (MOI 50:1) (E) for 6 and 9 h respectively. IL-1β in supernatants was measured by ELISA. (F) BMDM were LPS primed with or without IgG-Ova and then challenged with P. aeruginosa PAK strain (MOI 1:1) for 6 h. Cells were washed, fixed, permeabilized and stained for ASC. The number of ASC specks was quantified by microscopy. ***p ≤ 0.001 by 1-way ANOVA with Bonferroni post-test. Determinations were performed in triplicate and are expressed as the mean±SD (A, B, D–F) or mean±SEM (C). Results shown are representative of at least three independent experiments (A, B, D–F) or are pooled from four independent experiments (C).
IgG immune complexes were also capable of effectively suppressing IL-1β secretion from BMDM challenged with P. aeruginosa or F. tularensis LVS, which activate the NLRC4 and AIM2 inflammasomes respectively (Figure 4D and E). Similar to our findings with NLRP3 agonists we also observed that P. aeruginosa challenged BMDM that had been primed in the presence of immune complexes had a significant reduction in ASC specks (Figure 4F). These results demonstrate that immune complexes inhibit caspase-1 activation induced by multiple different inflammasomes suggesting that inhibition occurs at a point discrete from the individual NLR or AIM2 receptors.
Antigen-IgG immune complexes suppress the development of alum-driven Th2 and Th17 immune responses in vivo
To examine the effect of immune complexes on the generation of adaptive immune responses we utilized a murine model of allergic airway disease. Mice were immunized intraperitoneally with either ovalbumin (Ova) or IgG-Ova immune complexes along with the adjuvant alum. 15 days later mice were challenged intranasally with Ova for three consecutive days; 48 h later the extent of airway disease was assessed by histology as well as determining the inflammatory cell composition of the bronchoalveolar lavage (BAL) fluid (Figure 5A). As expected, mice immunized with alum/Ova had an eosinophilic response to intranasal challenge with Ova (Figure 5B and C). In contrast, mice immunized with alum/IgG-Ova had markedly diminished eosinophilic influx into the lungs following intranasal challenge with unopsonized Ova (Figure 5B and C).
Figure 5.

Immunization with IgG immune complexes suppresses adaptive immune responses. (A) Schematic illustration of the allergic airway disease model. (B–F) WT mice were injected intraperitoneally with either alum/Ova or alum/IgG-Ova on day 0; mice were then intranasally challenged with Ova on days 15, 16 and 17. 48 h after the final intranasal challenge hematoxylin and eosin lung histology sections (B) and differential cell counts in bronchoalveolar lavage fluid were analyzed (C). **p ≤ 0.01 by 2-way ANOVA with a Bonferroni post-test. Representative sections from 4 mice per group are shown; upper panel bar = 50 μm; lower panel bar = 20 μm. (D) Mediastinal lymph nodes were collected and restimulated in vitro with Ova protein. 72 h later supernatants were collected and analyzed for IL-17A, IL-13 and IL-4 by ELISA. *p ≤ 0.05, ***p ≤ 0.001 by Student’s t-test. (E) Intracellular cytokine analysis of LN cells stimulated in vitro with Ova for 72 h. (F) Total IgE and Ova specific IgG1 and IgG2c in serum was measured by ELISA. **p ≤ 0.01, ***P ≤ 0.001 by Mann-Whitney test. (G) Congenically mismatched CFSE labeled OTII T cells were transferred i.v. into WT mice; 24 h later mice were immunized with either alum/Ova or alum/IgG-Ova. 72 h later draining lymph nodes were collected and CFSE dilution was analyzed by flow-cytometry. Determinations were performed in triplicate and are expressed as the mean±SD. Data shown are representative of two (G) or three (C–F) independent experiments each with a minimum of 3 mice per group.
To determine if subsequent adaptive immune responses to the antigen were affected by presentation of antigen within an immune complex we restimulated mediastinal lymph node (LN) cells with Ova ex vivo. LN cells from mice immunized with alum/Ova produced IL-13, IL-4, and IL-17A upon Ova restimulation, consistent with an alum-driven Th2 and Th17 response (Figure 5D and E). Surprisingly LN cells from mice immunized with alum/IgG-Ova secreted significantly less IL-13, IL-4, and IL-17A upon Ova restimulation (Figure 5D and E). No difference in IFN-γ production was observed following Ova restimulation of LN cells from mice immunized with alum/Ova or alum/IgG-Ova (data not shown). Ova-specific IgG1 and total IgE antibody induction was also significantly diminished in mice immunized with alum/IgG-Ova compared to alum/Ova (Figure 5F). Ova-specific IgG2c levels were not different between alum/Ova and alum/IgG-Ova immunized mice (Figure 5F).
To determine whether the impaired adaptive response was due to a failure of antigen presenting cells to process or present the IgG-Ova appropriately we adoptively transferred CFSE labeled T-cell receptor (TCR) transgenic OT-II CD4+ T cells into mice immunized with either alum/Ova or alum/IgG-Ova. OT-II CD4+ T cells proliferated normally in both alum/Ova and alum/IgG-Ova immunized mice suggesting that the processing and presentation of antigen remained intact (Figure 5G). We also found that IgG-Ova immune complexes could inhibit Th2 and Th17 immune responses against keyhole limpet hemocyanin (KLH), an antigen that was not part of the IgG-Ova immune complex (Supplemental Figure 2). Taken together these data demonstrate that antigen-IgG immune complexes suppress the development of both Th2 and Th17 immune responses.
Enhanced IL-10 production induced by immune complexes does not suppress Th2 and Th17 responses
IL-10 is a potent anti-inflammatory mediator that affects a number of cell types and is important in limiting inflammation (27). To determine if the enhanced IL-10 production by BMDM following challenge with immune complexes was required for the inhibition of Th2 and Th17 responses in vivo we utilized IL-10-deficient mice. LN cells from Il10−/− mice immunized with alum/IgG-Ova suppressed IL-17A and IL-13 production upon Ova restimulation (Figure 6A). These data suggest that the enhanced IL-10 production elicited by immune complexes is dispensable for the inhibition of alum-driven Th2 and Th17 responses. We next asked whether the IL-10 release associated with immune complexes was responsible for the inhibition of IL-1β secretion in vitro. Similar to the findings for the Th2 and Th17 responses impaired IL-1β secretion remained intact in cells from Il10−/− mice, suggesting immune complex–mediated IL-1β suppression does not require IL-10 (Figure 6B).
Figure 6.

Alum-induced Th2 and Th17 responses require the NLRP3 inflammasome and signaling through IL-1R1. (A) Il10−/− mice were injected intraperitoneally with either alum/Ova or alum/IgG-Ova on day 0; mice were then intranasally challenged with Ova on days 15, 16 and 17. 48 h after the final intranasal challenge mediastinal lymph nodes were restimulated in vitro with Ova protein. 72 h later supernatants were collected and analyzed by ELISA for IL-17A and IL-13. *p ≤ 0.05, **p ≤ 0.01 by Student’s t-test. (B) Il10−/− BMDM were LPS primed with or without eIgG and then challenged for 6 h with silica or ATP; supernatants were collected and IL-1β measured by ELISA. ***p ≤ 0.001 by Student’s t-test. (C–F) WT, Nlrp3−/−, Il1r1−/−, Casp1−/−, Il1a−/−, and Il1b−/− mice were immunized with alum/Ova as described in (A). Mediastinal lymph nodes were restimulated in vitro with Ova protein. 72 h later supernatants were collected and IL-17A and IL-13 was measured by ELISA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by (C) 1-way ANOVA with Bonferroni post-test or (D–F) Student’s t-test. Determinations were performed in triplicate and are expressed as the mean±SD. Data shown are representative of three independent experiments each with a minimum of 3 mice per group.
Alum-induced Th2 and Th17 responses require the NLRP3 inflammasome and signaling through IL-1R1
The NLRP3 inflammasome has been implicated in the generation of alum-driven Th2 responses in vivo (28, 29). Furthermore, signaling through the IL-1R1 on CD4+ T cells enhances their expansion and differentiation into Th2 and Th17 effector cells (30, 31). However the role of NLRP3 inflammasome activation in alum-driven Th17 responses is unclear. To evaluate if the NLRP3 inflammasome was required for Th17 responses in the alum/Ova allergic airway model we evaluated mice deficient in NLRP3, caspase-1, or IL-1R1. Consistent with the role for IL-1R1 in Th17 differentiation, LN cells from Il1r1−/− mice immunized with alum/Ova had significantly diminished production of IL-17A upon Ova restimulation (Figure 6C). LN from mice deficient in either NLRP3 or caspase-1 also had significantly diminished IL-17A production upon Ova restimulation ex vivo (Figure 6C and D). As expected, mice deficient in NLRP3, caspase-1, or IL-1R1 also displayed defective IL-13 responses (Figure 6C and D). While these data suggest NLRP3-dependent IL-1 production plays an important role in alum-driven Th2 and Th17 responses, the further diminution of IL-17A production in Il1r1−/− mice indicates inflammasome-independent IL-1 production is also likely to contribute to alum-driven Th17 responses in vivo.
Both IL-1α and IL-1β signal through the IL-1R1, however their individual relevance in driving alum-induced Th2 and Th17 responses is unclear. To assess the separate contribution of each of these cytokines, mice deficient in either IL-1α or IL-1β were immunized with alum/Ova and subsequently challenged intranasally with Ova. Diminished IL-17A and IL-13 production was observed in LN cells restimulated ex vivo with Ova from both Il1a−/− and Il1b−/− mice compared to WT mice (Figure 6E and F).
Suppression of CD4+ T cell responses by immune complexes is rescued by exogenous IL-1α or IL-1β
To ascertain if the suppression of macrophage IL-1α and IL-1β production by immune complexes observed in vitro contributed to the ability of immune complexes to inhibit the generation of Th2 and Th17 responses in vivo, mice were immunized with alum/IgG-Ova in the presence or absence of recombinant IL-1α or IL-1β. The presence of exogenous IL-1α or IL-1β during the sensitization phase with alum/IgG-Ova was able to overcome the effects of immune complexes and restore the alum-driven Th17 response (Figure 7A and B). However, exogenous IL-1α, but not IL-1β, was able to overcome the effects of immune complexes and restore the alum-driven Th2 response (Figure 7A and B). Taken together these data suggest that both IL-1α and IL-1β play roles in the generation of alum-induced Th2 and Th17 responses and that immune complex mediated suppression of IL-1α and IL-1β is capable of dampening these adaptive immune responses. In addition, these data suggest differential functions of IL-1α versus IL-1β in the generation of the Th2 response.
Figure 7.

Immune complex mediated downregulation of adaptive immune responses is rescued by exogenous IL-1. (A, B) WT mice were immunized as in Figure 6 with an additional group receiving rIL-1α (A) or rIL-1β (B). Mediastinal lymph nodes were collected and restimulated in vitro with Ova protein. 72 h later supernatants were collected, and IL-17A and IL-13 measured by ELISA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by 1-way ANOVA with Bonferroni post-test. Determinations were performed in triplicate and are expressed as the mean±SD. Data shown are representative of three independent experiments each with a minimum of 3 mice per group. (C) Predicted model depicting the suppression of Th2 and Th17 responses by immune complexes in the context of the adjuvant alum.
Discussion
Swift and robust innate immune responses are required for the control of microbial pathogens but a continued or disproportionate innate response can cause collateral tissue damage and lead to autoimmunity. Considering the strong pro-inflammatory activity of IL-1α and IL-1β and their critical role in the initiation of adaptive immune responses their processing and secretion must be tightly regulated. In this study we demonstrate a novel pathway by which inflammasome activation and assembly in macrophages can be modulated: through the internalization of immune complexes. While previous studies have shown immune complexes can modify innate and adaptive immune responses through regulating the release of IL-10 and IL-12, their impact on inflammasome activation had not been determined. Here we show a novel function for immune complexes wherein the presence of immune complexes inhibits activation of inflammasomes and thus the release of inflammasome-dependent IL-1α and IL-1β (Figure 7C). These findings suggest the precise effect induced by immune complexes is specific and unique to the innate-activating signal it modifies. These findings of immunologic suppression induced by immune complexes are unexpected as earlier studies have suggested immune complexes enhance rather than suppress immunogenicity (32–34).
Previous studies suggest that the modulation of IL-10 and IL-12 p40 by IgG-immune complexes occurs at the level of transcription (4). Interestingly, immune complexes do not appear to suppress inflammasome activation in a similar manner, rather immune complexes appear to act at a post-translational level by preventing inflammasome assembly and subsequent caspase-1 activation. Additionally, the inhibition of inflammasome activation by immune complexes is not specific to the NLRP3 inflammasome but also applies to both the NLRC4 and AIM2 inflammasomes, as the presence of immune complexes inhibited IL-1β release induced by both P. aeruginosa and F. tularensis. Thus, this novel inhibition of early inflammasome activation by immune complexes appears to apply globally to multiple types of inflammasomes. Importantly, we show that the initial steps of priming occur in cells treated with immune complexes, as determined by the upregulation of inflammasome components, is intact in our immune complex treated cells. However the degree of inflammasome inhibition is dependent upon the timing of the addition of immune complexes, wherein immune complexes inhibit inflammasome activation only if added prior to the addition of the inflammasome agonist. Hence inhibition of inflammasome activation by immune complexes occurs following initial priming, but prior to assembly of the inflammasome complex as well as the subsequent cleavage of caspase-1.
FcγRs can be either activating (FcγRI, FcγRIII and FcγRIV) or inhibitory (FcγRIIb). Activating FcγRs all signal through the common ITAM-containing FcRγ chain. We show the mechanism by which immune complexes abrogate inflammasome activation depends upon the presence of activating Fc receptors, as macrophages from FcRγ chain-deficient mice are incapable of immune complex-mediated downregulation of inflammasome responses. Interestingly, Nlrp3 inflammasome activation by Schistosoma mansoni and C. albicans requires the c-type lectin receptor (CLR) dectin-2 (35–37), which couples with the FcRγ chain for signal propagation (38). Hence signal propagation through the FcRγ chain can either be activating or inhibitory for the NLRP3 inflammasome depending on the upstream receptor that initiates the signaling.
Engagement of the IL-1R1 causes a marked expansion of T cells in response to their cognate antigen. Th17 differentiation requires signaling through the IL-1R1 as cells deficient in the receptor fail to develop into Th17 effectors in an EAE model (30). Similarly, IL-1R1 signaling has been shown to be required for development of Th2 responses as lymph node cells from IL-1α- or IL-1β-deficient mice failed to differentiate in vitro into Th2 cells in the presence of IL-4 alone but required the addition of exogenous IL-1α or IL-1β (39). Previous studies have shown a requirement for the NLRP3 inflammasome in the development of alum-antigen dependent Th2 responses (28, 29). Here we expand on this association and show the NLRP3 inflammasome and IL-1R1 signaling are similarly required for alum-dependent Th17 responses. Further, the development of specific CD4+ T cell responses is diminished in the absence of either IL-1α or IL-1β, suggesting both are required for full CD4+ T cell effector function. IL-1α is often thought to be interchangeable with IL-1β in terms of function, likely due to the fact that both molecules signal through the IL-1R1 on T cells (40). However, many important differences between IL-1α and IL-1β exist and suggest that their impact on T helper cell commitment is more complex than previously appreciated. Engagement of the IL-1R1 on T cells by either IL-1α or IL-1β typically results in downstream signaling through MyD88 to activate NF-κB (41), yet alternative signaling pathways involving the recruitment of phosphoinositide 3-kinase and activation of the protein tyrosine kinase Akt have been described following IL-1R1 ligation (42). Interestingly, the addition of exogenous IL-1α during initial activation in vivo is sufficient to restore both Th2 and Th17 effector cytokine production while addition of exogenous IL-1β is sufficient to restore only Th17 responses. These data concur with studies that show a critical role for IL-1R1 and IL-1α in Th2 sensitization in a house dust mite allergic airway disease model (43). In addition, our studies show that IgG-immune complexes were not able to suppress alum-induced IL-1α production in vitro, consistent with previous findings that alum-induced IL-1α production occurred in an inflammasome-independent manner (9). However, the mechanism by which alum activates the NLRP3 inflammasome in vivo is unclear and may involve additional indirect mechanisms including the release of extracellular ATP (44).
Our data support a mechanism whereby immune complexes block inflammasome activation and in doing so prevent the generation of effective adaptive immune responses (Figure 7C). The presence of immune complexes is a marker for the successful development of an adaptive immune response, thus the ability of immune complexes to shut off inflammasomes represents a negative feedback loop. Another example of adaptive immune downregulation of innate responses is seen by the ablation by CD4+ T effector and memory T cells of inflammasome activation in a CD40/CD40L dependent manner (45). Together our data suggest that the presence of an effective adaptive immune response through antigen-antibody immune complexes acts as a negative feedback loop, shutting off inflammasome activation and hence providing the signal to terminate the inflammatory response.
Supplementary Material
Acknowledgments
NIH grants R01 AI087630 (F.S.S.), R01 AI104706 (S.L.C.), T32 AI007485 (J.R.J.), T32 AI007511 (S.H.), an Edward Mallinckrodt, Jr. Foundation scholarship (F.S.S.), and an Asthma and Allergy Foundation of America fellowship (S.L.C.) supported this work.
We thank David Mosser and Gail Bishop for helpful discussion and critical review of this manuscript; Richard Flavell, David Chaplin, Dmitry Shayakhmetov, and Millennium Pharmaceuticals for providing knockout mice; and Vickie Knepper-Adrian for technical assistance.
References
- 1.Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nature Reviews Immunology. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 2.Anderson CF, Gerber JS, Mosser DM. Modulating macrophage function with IgG immune complexes. Journal of Endotoxin Research. 2002;8:477–481. doi: 10.1179/096805102125001118. [DOI] [PubMed] [Google Scholar]
- 3.Gerber JS, Mosser DM. Reversing lipopolysaccharide toxicity by ligating the macrophage Fcγ receptors. The Journal of Immunology. 2001;166:6861–6868. doi: 10.4049/jimmunol.166.11.6861. [DOI] [PubMed] [Google Scholar]
- 4.Sutterwala FS, Noel GJ, Clynes R, Mosser DM. Selective suppression of interleukin-12 induction after macrophage receptor ligation. J Exp Med. 1997;185:1977–1985. doi: 10.1084/jem.185.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutterwala FS, Noel GJ, Salgame P, Mosser DM. Reversal of proinflammatory responses by ligating the macrophage Fcg Receptor Type 1. Journal of Experimental Medicine. 1998;188:217–222. doi: 10.1084/jem.188.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bandukwala HS, Clay BS, Tong J, Mody PD, Cannon JL, Shilling RA, Verbeek JS, Weinstock JV, Soloway J, Sperling AI. Signaling through the FcγRIII is required for optimal T helper type (Th)2 responses and Th2-mediated airway inflammation. The Journal of Experimental Medicine. 2007;204:1875–1889. doi: 10.1084/jem.20061134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 8.Kuida K, Lippke JA, Ku G, HMW, Livingston DJ, Su MS-S, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 9.Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, Quadroni M, Drexler SK, Tschopp J. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 10.Franchi L, Eigenbrod T, Nunez G. Cutting Edge: TNF-α Mediates Sensitization to ATP and Silica via the NLRP3 Inflammasome in the Absence of Microbial Stimulation. The Journal of Immunology. 2009;183:792–796. doi: 10.4049/jimmunol.0900173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavelle RA, Leemans JC, Sutterwala FS. Necrotic cells trigger a sterile inflammatory response through the NLRP3 inflammasome. PNAS. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 13.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW, Flavell RA. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 14.Labow M, Shuster D, Zetterstrom M, Nunes P, Terry R, Cullinan EB, Bartfai T, Solorzano C, Moldawer LL, Chizzonite R, McIntyre KW. Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor-deficient mice. J Immunol. 1997;159:2452–2461. [PubMed] [Google Scholar]
- 15.Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, Takahashi M, Iwakura Y. Production of mice deficient in genes for interleukin (IL)-1α, IL-1β, IL-1α/β, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med. 1998;187:1463–1475. doi: 10.1084/jem.187.9.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shornick LP, De Togni P, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, Ferguson TA, Chaplin DD. Mice deficient in IL-1beta manifest impaired contact hypersensitivity to trinitrochlorobenzone. J Exp Med. 1996;183:1427–1436. doi: 10.1084/jem.183.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 18.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 19.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 20.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 21.Seymour BW, Gershwin LJ, Coffman RL. Aerosol-induced immunoglobulin (Ig)-E unresponsiveness to ovalbumin does not require CD8+ or T cell receptor (TCR)-gamma/delta+ T cells or interferon (IFN)-gamma in a murine model of allergen sensitization. J Exp Med. 1998;187:721–731. doi: 10.1084/jem.187.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herrick CA, MacLeod H, Glusac E, Tigelaar RE, Bottomly K. Th2 responses induced by epicutaneous or inhalational protein exposure are differentially dependent on IL-4. J Clin Invest. 2000;105:765–775. doi: 10.1172/JCI8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. The Journal of Experimental Medicine. 2010;207:1645–1655. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistrabution of the Apoptotic Speck-like Protein Containing a Capsase Recruitment Domain. The Journal of Immunology. 2009;182:3173–3182. doi: 10.4049/jimmunol.0802367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takai T, et al. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 26.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature. 1994;368:70–73. doi: 10.1038/368070a0. [DOI] [PubMed] [Google Scholar]
- 27.Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- 28.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A. 2009;106:7119–7124. doi: 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Regnault A, Lankar D, Lacabanne V, Rodriguez A, Thery C, Rescigno M, Saito T, Verbeek S, Bonnerot C, Ricciardi-Castagnoli P, Amigorena S. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. 1999;189:371–380. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuurhuis DH, van Montfoort N, Ioan-Facsinay A, Jiawan R, Camps M, Nouta J, Melief CJ, Verbeek JS, Ossendorp F. Immune complex-loaded dendritic cells are superior to soluble immune complexes as antitumor vaccine. J Immunol. 2006;176:4573–4580. doi: 10.4049/jimmunol.176.8.4573. [DOI] [PubMed] [Google Scholar]
- 34.Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV. Antitumor monoclonal antibodies enhance cross-presentation ofcCellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med. 2002;195:125–133. doi: 10.1084/jem.20011097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gross O, Peock H, Bscheider M, Dostert C, Hannesschlager N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V, Mocsai A, Tschopp J, Ruland J. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defense. Nature. 2009;459:433–436. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 36.Ritter M, Gross O, Kays S, Ruland J, Nimmerjahn F, Saijo S, Tschopp J, Layland LE, da Costa CP. Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune system. PNAS. 2010;107:20459–20464. doi: 10.1073/pnas.1010337107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson MJ, et al. Dectin-2 is a syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. The Journal of Experimental Medicine. 2009;206:2037–2051. doi: 10.1084/jem.20082818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato K, Yang X, Yudate T, Chung J, Wu J, Luby-Phelps K, Kimberly R, UD, Cruz PD, Jr, Ariizumi K. Dectin-2 Is a Pattern Recognition Receptor for Fungi that Couples with the Fc Receptor γ Chain to Induce Innate Immune Responses. The Journal of Biological Chemistry. 2006;281:38854–38866. doi: 10.1074/jbc.M606542200. [DOI] [PubMed] [Google Scholar]
- 39.Helmby H, Grencis RK. Interleukin 1 plays a major role in the development of Th2-mediated immunity. Eur J Immunol. 2004;34:3674–3681. doi: 10.1002/eji.200425452. [DOI] [PubMed] [Google Scholar]
- 40.Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77:1627–1652. [PubMed] [Google Scholar]
- 41.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Molecular cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 42.Davis CN, Mann E, Behrens MM, Gaidarova S, Rebek M, Rebek J, Jr, Bartfai T. MyD88-dependent and -independent signaling by IL-1 in neurons probed by bifunctional Toll/IL-1 receptor domain/BB-loop mimetics. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2953–2958. doi: 10.1073/pnas.0510802103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, Hammad H. Interleukin-1α controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med. 2012;209:1505–1517. doi: 10.1084/jem.20112691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riteau N, Baron L, Villeret B, Guillou N, Savigny F, Ryffel B, Rassendren F, Le Bert M, Gombault A, Couillin I. ATP release and purinergic signaling: a common pathway for particle-mediated inflammasome activation. Cell Death Dis. 2012;3:e403. doi: 10.1038/cddis.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guarda G, Dostert C, Staehli F, Cabalzar K, Castillo R, Tardivel A, Schneider P, Tschopp J. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature. 2009;460:269–273. doi: 10.1038/nature08100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.