

Abstract
Reinfections with respiratory viruses are common and cause significant clinical illness, yet precise mechanisms governing this susceptibility are ill defined. Lung Ag-specific CD8+ T cells (TCD8) are impaired during acute viral lower respiratory infection by the inhibitory receptor PD-1. To determine if PD-1 contributes to recurrent infection, we first established a model of reinfection by challenging B cell-deficient mice with human metapneumovirus (HMPV) several weeks after primary infection, and found that HMPV replicated to high titers in the lungs. A robust secondary effector lung TCD8 response was generated during reinfection, but these cells were more impaired and more highly expressed the inhibitory receptors PD-1, LAG-3, and 2B4 than primary TCD8. In vitro blockade demonstrated that PD-1 was the most dominant inhibitory receptor early after reinfection. In vivo therapeutic PD-1 blockade during HMPV reinfection restored lung TCD8 effector functions (i.e. degranulation and cytokine production) and enhanced viral clearance. PD-1 also limited the protective efficacy of HMPV epitope-specific peptide vaccination and impaired lung TCD8 during heterotypic influenza virus challenge infection. Our results indicate that PD-1 signaling may contribute to respiratory virus reinfection and evasion of vaccine-elicited immune responses. These results have important implications for the design of effective vaccines against respiratory viruses.
INTRODUCTION
The respiratory viruses human metapneumovirus (HMPV) and respiratory syncytial virus (RSV) are important causes of acute lower respiratory infection (LRI), which results in significant morbidity and mortality, especially in infants, the elderly, and the immunocompromised (1–8). Despite the need to protect these populations against serious LRI, no licensed vaccines or therapeutics exist for these viruses. The majority of LRI beyond infancy are actually reinfections, as nearly all individuals experience primary infection during early childhood (9). HMPV reinfection in children causes illness at a rate that equals primary infection (10) and can occur with both genetically heterologous and homologous viruses (11). Despite a high frequency of infection and minimal antigenic drift for both HMPV and RSV, protective immunity is poorly established, as individuals can be repeatedly reinfected throughout life (12–14). High anti-HMPV antibody titer in serum is insufficient to prevent reinfection in adults (15). Respiratory virus reinfections cause important clinical disease, but the mechanisms governing susceptibility to recurrent viral LRI are poorly understood. While much attention has been placed on humoral immunity, the above evidence argues that antibodies are not always associated with protection. Indeed, in animal models, both arms of the adaptive immune system contribute (16–18).
The anti-HMPV TCD8 response (19), like that against RSV (20) and influenza virus (19, 21), is functionally impaired in the respiratory tract. Virus-specific lung TCD8 do not optimally respond to stimulation by releasing lytic granules or producing anti-viral cytokines such as IFNγ. We recently demonstrated that during primary HMPV and influenza virus infections the inhibitory receptor programmed death-1 (PD-1) significantly contributes to this impairment by repressing TCD8 effector functions (19). Blockade of PD-1 signaling restored lung TCD8 functions and enhanced viral control. Prior to this, PD-1 had mainly been associated with T cell exhaustion during chronic infection and cancer, where prolonged T cell receptor (TCR) stimulation by persistent viral or tumor antigens maintains PD-1 expression (22). PD-1 ligand (PD-L) binding to PD-1 antagonizes TCR signaling by blocking PI3K/Akt activation, leading to decreased protein synthesis, cytokine production, proliferation and survival of effector T cells (23). PD-1 pathway blockade has recently proven effective at treating refractory malignancies (24, 25). Thus therapeutic targeting of this pathway has significant clinical potential.
A poorly explored aspect of PD-1 biology is its contribution to reinfection. We recently demonstrated that secondary effector TCD8 in wild type mice rapidly re-expressed PD-1 and were highly impaired (19). Secondary TCD8 also become exhausted during chronic infection in mice (26). In addition to PD-1, several other inhibitory receptors have been identified that contribute to functional TCD8 impairment or exhaustion in a variety of settings and therefore may also contribute to impairment during reinfection (27). Exhausted TCD8 express numerous inhibitory receptors, including TIM-3 (28), LAG-3 (29), 2B4 (26), and others (30). TIM-3 (31) and LAG-3 (32) also negatively regulate the TCD8 response to acute viral LRI. The role these receptors play in causing lung TCD8 functional impairment during respiratory virus reinfection has not been explored.
We hypothesized that inhibitory receptor signaling contributes to respiratory virus reinfections by causing lung TCD8 impairment during memory immune responses. We sought to elucidate PD-1’s contribution to HMPV reinfection and to determine whether PD-1 limits the effectiveness of potential vaccination strategies directed at the cellular immune response. To study secondary effector TCD8 impairment, we first established a model of HMPV reinfection using B cell-deficient mice, since rodents are normally protected against respiratory virus reinfection by neutralizing antibodies (17, 19, 34) and viral Ag directly modulates PD-1 expression through TCR signaling (19, 27, 33). B cell-deficient mice were susceptible to HMPV reinfection, but were unable to better control early viral replication as compared to primary infected mice. Secondary effector lung TCD8 expressed the inhibitory receptors PD-1, LAG-3, 2B4, and TIM-3, with the former three expressed more highly than during primary infection. However, in vitro experiments demonstrated that PD-1 was the dominant functional inhibitory receptor early during reinfection. Therapeutic blockade of PD-1 signaling during HMPV reinfection restored secondary lung TCD8 effector functions (i.e. degranulation and cytokine production) and reduced viral burdens in the lung. Furthermore, PD-1 limited the effectiveness of a peptide vaccination strategy against HMPV and also impaired secondary lung TCD8 during heterotypic influenza virus challenge. These results indicate the PD-1 contributes to lung TCD8 impairment during reinfection and prevents memory TCD8 from controlling viral replication. Given the severity and frequency of reinfections by respiratory viruses and the lack of effective vaccines, these studies have important implications for future vaccines and therapeutic interventions.
MATERIALS AND METHODS
Mice and Infections
C57BL/6 (B6) were purchased from the Jackson Laboratory. μMT mice were a kindly provided by Dr. Mark Boothby (Vanderbilt University, Nashville, TN). PD-1−/− mice were obtained with permission from Dr. Tasuku Honjo (Kyoto University, Kyoto, Japan). All animals were bred and maintained in specific pathogen-free conditions in accordance with the Vanderbilt Institutional Animal Care and Use Committee. Age- and gender-matched animals were used in all experiments. HMPV (pathogenic clinical strain TN/94-49, genotype A2) was grown and titered in LLC-MK2 cells as previously described (34). Influenza virus strains A/34/PR/8 (PR8; H1N1; ATCC) and HK/x31 (x31; H3N2; kindly provided by Dr. Jon McCullers and Paul Thomas, St. Jude Children’s Hospital, Memphis, TN) were grown in MDCK cells and titered on LLC-MK2 cells. Mice were anesthetized with ketamine-xylazine and infected intranasally (i.n.) with 1×106 plaque forming units (PFU) of HMPV. Viral titers in infected mouse lungs and nasal turbinates were measured by plaque titration as previously described (34). For influenza virus challenge experiments, mice were primed i.p. with 2×105 PFU of PR8 and challenged i.n. with 5×102 PFU of x31 at least 15 weeks later.
Flow Cytometry Staining
Lung lymphocytes were tetramer-stained or restimulated for intracellular cytokine staining (ICS) in parallel as previously described (19). Lung cells were stained for the inhibitory receptors PD-1 (clone RMP1-30), TIM-3 (clone RMT3-23), LAG-3 (clone C9B7W) and 2B4 (clone m2B4 (B6) 458.1) or with appropriate isotype control antibodies (all from Biolegend). Flow cytometric data were collected using an LSRII or Fortessa (BD Biosciences) and analyzed with FlowJo software (Tree Star). The Boolean gating function in FlowJo was used to assess inhibitory receptor co-expression and patterns were visualized using the SPICE program (NIAID).
IFNγ ELISPOT
ELISPOT assays were performed as previously described (35) with slight modifications. 5×104 lung cells were added to triplicate wells. Peptides were then added (10μM final concentration) followed by inhibitory receptor blocking antibodies (10μg/mL final concentration). The following blocking antibodies were used: isotype control (clone LTF-2, Bio X-cell), anti-PD-L1 (clone 10F.9G2, Bio X-cell), anti-PD-L2 (clone TY-25, Bio X-cell), anti-PD-1 (clone J43, Bio X-cell), anti-TIM-3 (clone RMT3-23, Bio X-cell), anti-LAG-3 (clone C9B7W, Bio X-cell), anti-2B4 (clone m2B4 (B6) 458.1, Biolegend), and anti-CD48 (clone HM48-1, Bio X-cell). Plates were incubated at 37C for 42–48 hours, developed, and then counted using an ImmunoSpot Micro Analyzer (Cellular Technology Limited). The average number of spots in wells stimulated with an irrelevant peptide was subtracted from each experimental value, which was then expressed as spot forming cells (SFC) per 106 lymphocytes.
In vivo Antibody Blockade
Mice were injected i.p. on days −1, 1, 3 and 5 p.i. with 200μg of rat isotype control antibody (clone LTF-2, Bio X-cell) or 200μg of both rat anti-mouse PD-L1 (clone 10F.9G2, Bio X-cell) plus rat anti-mouse PD-L2 (clone TY-25, Bio X-cell) to block PD-1 signaling.
Peptide Vaccination (TriVax)
Mice were injected intravenously with a mixture of 200μg Db/F528 peptide, 50μg anti-CD40 antibody (clone FGK4.5, Bio X-cell) and 50μg poly (I:C) (Invivogen). Peripheral blood was obtained to check immune responses in all vaccinated animals.
Statistical Analysis
Data analysis was performed using Prism v6.0 (GraphPad Software). Comparisons between tetramer staining and ICS within the same animals were performed using a paired, two-tailed t test. Comparisons between two groups were performed using an unpaired, two-tailed Student’s t test. Multiple group comparisons were performed using a one-way ANOVA with a Bonferroni post-test. Error bars on each graph represent standard error of the mean unless otherwise noted.
RESULTS
μMT mice are susceptible to reinfection with HMPV
Rodents are normally protected against reinfection with respiratory viruses due to the presence of neutralizing antibodies and the fact that they are semi-permissive hosts (34, 36, 37). We therefore utilized the B cell-deficient mouse strain μMT, which has been used to model reinfection with influenza virus and RSV (36, 38, 39), to test whether this strain was susceptible to reinfection with HMPV. HMPV demonstrated similar replication kinetics in the lungs and nasal turbinates during primary infection of μMT and WT mice (Fig. 1A), though there was a slight decrease in lung virus titers at day 7 in WT compared to μMT mice. Virus was cleared from the lungs by day 10 in both strains. Upon reinfection, WT mice were completely protected against viral replication, as expected (Fig. 1B). However, HMPV replicated to similarly high titers in reinfected μMT mice as in primary infection. Indeed, lung titers were indistinguishable between primary and secondary infection at day 5, the usual peak of viral replication (19). On day 7, lung titers were diminished in many μMT mice, with some undetectable, and by day 10, most had cleared the infection. μMT mice were also able to clear virus from the nasal epithelium during secondary infection, as mice had lower titers at days 5 and 7 post-challenge compared to primary infection. Thus, μMT mice are susceptible to HMPV reinfection, although they clear virus more rapidly than primary infection.
Figure 1. μMT mice are susceptible to HMPV reinfection.

(A) WT (open triangles) and μMT mice (closed squares) were infected with HMPV and viral titers were quantified for the lung (left panels) and nasal turbinates (right panels) in plaque forming units (pfu) per gram of tissue at the indicated days post-infection (p.i.). (B) WT and μMT mice were infected with HMPV and then challenged 8 weeks later and viral titers were quantified. Each symbol represents an individual mouse and horizontal lines denote the group mean. The dotted line indicates the limit of detection for the plaque assay. Data are from 2 separate experiments with 3–5 mice per time point.*p<0.05, student’s t-test.
Kinetics and magnitude of CD8+ T cell responses are similar in WT and μMT mice
Since the μMT mouse represents a new model of HMPV reinfection, we next tested whether the kinetics and functionality of TCD8 during primary and secondary infection were similar to WT B6 mice. WT and μMT mice were either primarily infected with HMPV, or for secondary infection, mice were infected with HMPV, allowed to clear the infection, and then challenged with HMPV 16 weeks later. Lung lymphocytes were collected on days 7, 10, and 14 after primary or secondary infection and enumerated by tetramer staining and intracellular cytokine staining (ICS) for the two dominant HMPV epitopes Db/F528–536 (F528) and Kb/N11–19 (N11) (19) (Figure 2). During primary infection, the percentage of F528-specific TCD8 was similar between μMT and WT mice; in both groups, a minority of the TCD8 degranulated or produced IFNg, with the % functional declining over time as previously described (19) (Fig. 2 A, B, top panels). Interestingly, the percentage of N11-specific TCD8 increased from day 7 to day 14 in both groups, though functionality also declined over time (Fig. 2 A, B, bottom panels). The results were similar for both populations of epitope-specific TCD8 during secondary infection of μMT and WT mice (Fig. 2 C, D). While the magnitude of the N11-specific TCD8 response was higher in both groups during secondary infection as judged by tetramer staining, the percentage that were functional was even lower than during primary infection. Thus, the μMT mouse serves as an appropriate model of TCD8 responses compared to WT B6 mice.
Figure 2. Kinetics and magnitude of primary and secondary CD8+ T cell responses are similar in μMT mice.

μMT or WT B6 mice either underwent primary infection (A, B) or were infected with HMPV and then reinfected 16 weeks later (C, D). The lung F528- (top) and N11-specific (bottom) TCD8 responses were quantified via tetramer staining (black bars), CD107a mobilization (gray bars), and intracellular IFNγ (white bars) at days 7, 10, and 14 p.i. (A) μMT mice, primary infection. (B) WT mice, primary infection. (C) μMT mice, secondary infection. (D) WT mice, secondary infection. Data are combined from two independent experiments with 3–5 mice per group per experiment.
Secondary effector lung TCD8 express multiple inhibitory receptors
The ability of μMT mice to clear HMPV during secondary infection was presumably due to cellular immunity (i.e., CD4+ and CD8+ T cells). However, we were surprised to see that at days 5 and 7 after reinfection lung viral titers were equivalent to peak titers during primary infection. We therefore quantified inhibitory receptor expression during both primary and secondary HMPV infection of μMT mice to search for an explanation for the large degree of TCD8 impairment observed during reinfection. Reinfected μMT mice generated a three-fold greater F528-specific response compared to primary infected mice (Fig. 3A). Lung F528-specific TCD8 expressed the inhibitory receptors PD-1, LAG-3, TIM-3, and 2B4 (Fig. 3B); however, PD-1, LAG-3 and 2B4 were more highly expressed by secondary TCD8 as compared to primary (Fig. 3B, C).
Figure 3. Secondary effector lung TCD8 express multiple inhibitory receptors.

Lung lymphocytes were isolated at day 7 after primary HMPV infection of μMT mice (1o) or reinfection of μMT mice infected with HMPV 10 weeks earlier (2o). (A) The total number of Db/F528-specific lung TCD8 was calculated based on tetramer staining. (B) Cell surface expression of the inhibitory receptors PD-1, TIM-3, LAG-3 and 2B4 was quantified on 1o (blue squares) or 2o (red triangles) F528 tetramer+ lung TCD8. (C) Histograms showing representative inhibitory receptor expression. (D) Co-expression of inhibitory receptors is displayed as the total # expressed by 1o or 2o F528 tetramer+ lung TCD8. (E) Patterns of inhibitory receptor expression are shown. Data in (A–E) are combined from two independent experiments with 4–5 mice per group per experiment. * p<0.05, student’s t-test.
Co-expression of inhibitory receptors is important in TCD8 exhaustion during chronic infection, as the number of receptors expressed on the cell surface corresponds to the degree of functional impairment (29). During HMPV LRI, most F528-specific lung TCD8 expressed ≥2 inhibitory receptors and a large fraction expressed ≥3 (Fig. 3D). A greater proportion of lung TCD8 expressed 3 or 4 inhibitory receptors during secondary infection compared to primary infection (Fig. 3D). While the expression pattern of these receptors was similar during primary and secondary infection, the percentage of F528-specific TCD8 expressing all four receptors or PD-1, TIM-3 and LAG-3 was increased during reinfection (Fig. 3E). Additionally, the fraction of cells that expressed only PD-1 or no inhibitory receptors was decreased during secondary infection (Fig. 3E), confirming a skewing towards augmented inhibitory receptor expression during reinfection. These results indicate that secondary TCD8 express numerous inhibitory receptors, with several of these receptors more frequently expressed than primary TCD8 at the same time post-infection.
PD-1 is the dominant inhibitory receptor early during reinfection
Given the co-expression of numerous inhibitory receptors during reinfection, we next sought to determine their potential contribution to lung TCD8 impairment. To do so, we isolated lung cells from reinfected μMT mice and added them to an IFNγ-detecting ELISPOT assay. Cells were restimulated with F528-peptide and incubated with blocking monoclonal antibodies against each inhibitory receptors or its ligand(s). F528 peptide restimulation of lung cells plus PD-L1 blockade resulted in significantly more IFNγ-secreting cells than isotype-treated cells (i.e. more spots) (Fig. 4A), confirming our previous in vivo results during primary infection (19). Blockade of the PD-1 receptor itself also resulted in more spot-forming cells (Fig. 4B). Blocking PD-L2, TIM-3, LAG-3, 2B4, or CD48 (the ligand for 2B4) alone did not result in any significant changes. There was a trend toward decreased responsiveness during 2B4 blockade. Combined blockade of TIM-3, LAG-3, or 2B4 with PD-L1 did not result in greater responses than PD-L1 or PD-1 blockade alone. Anti-PD-L1 and anti-PD-1 treatment also resulted in larger spots (Fig. 4C), indicating a greater amount of IFNγ secreted by each cell compared to isotype treatment. Again, blockade of other inhibitory receptors or their ligands failed to increase spot size. These results indicate that despite co-expression of numerous inhibitory receptors, PD-1 is the dominant mediator of lung TCD8 impairment early during respiratory virus reinfection.
Figure 4. PD-1 is the dominant inhibitory receptor early during reinfection.
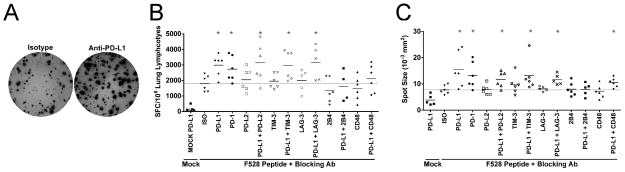
Lung suspension cells were isolated at day 6 post-reinfection of μMT mice, added to anti-IFNγ-coated ELISPOT plates and restimulated with F528 peptide in the presence of the indicated inhibitory receptor blocking antibodies or isotype control antibody (ISO). (A) Representative wells are shown. Each spot indicates a single IFNγ-secreting cell. (B) Combined data are expressed as the number of spot forming cells (SFC) per 106 lung lymphocytes. (C) The mean spot size is displayed for treatment with each receptor or ligand-blocking antibody. (B–C) Each symbol represents the mean from one of 5 or 6 total independent experiments with 2–3 mice per experiment. Mock indicates wells stimulated with an irrelevant peptide. Dotted line indicates the mean number of spots from F528-stimulated, isotype control antibody treated cells as a reference. *p<0.05, student’s t-test compared to isotype treatment.
Therapeutic PD-1 blockade restores function to impaired secondary lung TCD8 and enhances virus clearance
To directly test whether PD-1 signaling contributes to lung TCD8 impairment during HMPV reinfection, we therapeutically blocked this pathway by injecting μMT mice undergoing secondary infection with blocking antibodies directed against PD-1 ligands or with isotype control antibody. Naïve μMT mice were also infected for comparison of TCD8 responses and viral titers. Both the F528-specific and N11-specific TCD8 responses were quantified. Primary infected μMT mice possessed F528-specific TCD8 that were mostly functional (Figure 5A,B), as previously reported for early times p.i. (19). Secondary lung TCD8 in isotype-treated, reinfected mice were more impaired than primary TCD8 (Figure 5A,B), indicating that elevated inhibitory receptor co-expression during reinfection (Figure 3) is correlated with decreased functionality. Anti-PD-L treatment resulted in enhanced degranulation ability (i.e., CD107a mobilization) and greater IFNγ production for N11-specific TCD8 compared to isotype-treated mice (Figure 5A). Treatment also resulted in a greater percentage of functional F528- and N11-specific TCD8 (Figure 5B). TNF production was significantly enhanced for F528-specific TCD8 and trended higher for N11-specific cells. The absolute numbers of TCD8 were similar between isotype- and anti-PD-L treated mice, though the number of functional N11-specific TCD8 was higher in anti-PD-L treated mice (Figure 5C). PD-1 was more highly expressed during secondary infection and PD-L blockade further increased PD-1 expression (Figure 5D), as previously described for primary infection (19). Surprisingly, at day 6 p.i., isotype-treated, reinfected mice still exhibited lung viral titers indistinguishable from primary infected μMT mice (Figure 5E). Anti-PD-L treatment reduced lung viral titers ~3-fold in reinfected mice, suggesting that PD-1 signaling impairs the ability of secondary effector lung TCD8 to effectively reduce viral replication. Despite the increased numbers of functional TCD8, anti–PD-L was not associated with exacerbated lung histopathology (data not shown). These results indicate that PD-1 signaling impairs the T cell response during reinfection and may therefore contribute to respiratory virus reinfection.
Figure 5. Therapeutic PD-1 blockade restores function to impaired secondary lung TCD8 and enhances virus clearance.

μMT mice either underwent primary infection (1o Only) or were infected with HMPV and then reinfected 16 weeks later. Of the reinfected mice, one group received 200μg of anti-PD-L1 and anti-PD-L2 antibodies i.p. (2o + Anti-PD-L) while another group received the same amount of isotype control antibody (2o + ISO). (A) The lung Db/F528-(left) and Kb/N11-specific (right) TCD8 responses were quantified via tetramer staining (black bars), CD107a mobilization (gray bars), and intracellular IFNγ (white bars) or TNF production (white dotted bars) at day 6 p.i. BLD = below limit of detection. (B) The percentage of functional HMPV-specific TCD8 was calculated by dividing the percentage that degranulate or make IFNγ by the percentage that stained tetramer+. (C) Absolute number of F528-specific (left) and N11-specific (right) TCD8 is shown. (D) PD-1 MFI for F528-specific TCD8 is shown. (E) Lung viral titers were quantified by plaque assay. Data are combined from three independent experiments with 3–5 mice per group per experiment. A–D, * p<0.05, student’s t-test for TCD8 data. E,* p<0.05, ANOVA for viral titers.
PD-1 limits the effectiveness of vaccine-elicited anti-viral TCD8
Vaccine strategies that only elicit humoral immune responses against the related virus RSV have thus far proven unsuccessful and potentially hazardous (40), highlighting the need for better understanding of the contribution of T cells to protective immunity against RSV and HMPV. Recently, a peptide vaccination strategy against RSV proved highly effective in mice when given close to the time of challenge infection; however, the efficacy waned when mice were challenged several weeks later (41). We therefore tested whether PD-1 was responsible for the decreased effectiveness of TCD8-directed peptide vaccination. WT and PD-1−/− mice were immunized i.v. with F528 TriVax (F528 peptide + Anti-CD40 Ab + Poly[I:C]) and the TCD8 response was monitored in peripheral blood. PD-1−/− mice were employed to ensure PD-1 signaling did not occur throughout the entire experiment, from vaccine prime to the end of viral challenge. We found that WT mice generated more F528-specific TCD8 than PD-1−/− mice, which was true in three independent experiments (Fig. 6A). Five days post-HMPV challenge, we detected a greater overall F528-response in WT mice compared to PD-1−/− mice as determined by tetramer staining (Fig. 6B), which corresponded to the greater magnitude of the initial immunization. There were similar percentages of degranulating or IFNγ-producing F528-specific TCD8 in both groups. However, calculation of the percentage of functional lung TCD8 (which takes into account the different magnitude of tetramer response) revealed that F528-specific TCD8 were more functional in PD-1−/− mice (Fig. 6C). The absolute numbers of TCD8 were similar between WT and PD-1−/− mice (Figure 6D).
Figure 6. PD-1 limits the effectiveness of vaccine-elicited anti-viral TCD8.

WT or PD-1 −/− mice were immunized i.v. with F528 TriVax (see Materials and Methods). (A) The F528-specific response to immunization was measured in the peripheral blood via tetramer staining in WT (open squares) and PD-1−/− mice (closed triangles). (B) The lung F528-specific TCD8 response was quantified via tetramer staining (black bars), CD107a mobilization (gray bars) and intracellular IFNγ production (white bars) at day 5 p.i. (C) The percentage of functional F528-specific TCD8 was calculated. (D) Absolute number of F528-specific TCD8 is shown. (E) Lung viral titers were quantified by plaque assay. UnVax indicates mice that were not immunized with TriVax. NP366 is the immunodominant epitope for influenza virus in C57BL/6 mice and serves as a control for the other components of TriVax. Data are combined from three independent experiments with 5 mice per group per experiment.
To determine whether PD-1 affects the ability of the vaccine-elicited TCD8 to control viral replication, we measured viral titers in vaccinated WT and PD-1−/− mice, as well as in unvaccinated or control vaccinated mice (Fig. 6E). We found that in all the groups examined, only PD-1−/− mice receiving F528 TriVax had decreased viral titers at day 5 post-challenge. Despite the increased percentage of functional TCD8, lung histopathology was comparable in the WT and PD-1−/− mice (data not shown). These results suggest that PD-1 limits the effectiveness of anti-viral TCD8 elicited by peptide vaccination. Interestingly, F528 was not protective in WT mice, unlike an immunodominant RSV epitope tested using TriVax in BALB/c mice (41). (42). PD-1 may temper some of the strong stimulatory signals received during priming that potentially cause activation-induced cell death (43).
PD-1 limits the response to secondary influenza infection
To determine whether PD-1 impairs secondary effector TCD8 responses during reinfection with other respiratory viruses, we primed mice with PR8 influenza (H1N1) virus and challenged with x31 (H3N2) influenza virus. The percent and absolute number of H2-Db/NP366–374 tetramer-specific TCD8 was similar between WT and PD-1−/− mice (Fig. 7 A, B). However, the percent and number of functional TCD8 was higher in PD-1−/− mice (Fig. 7A–D).
Figure 7. PD-1 limits the response to secondary influenza infection.

PD-1−/− and control WT mice were primed i.p. with influenza virus strain PR8 (H1N1) and then challenged i.n. with strain x31 (H3N2). The percentage (A) and absolute number (B) of the lung NP366-specific TCD8 response was quantified via tetramer staining (black bars), CD107a mobilization (gray bars) and intracellular IFNγ production (white bars) at day 7 post-challenge. The percentage (C) and absolute number (D) of CD107a+ or IFNγ+ NP366-specific TCD8 was calculated. Data are representative of one out of two independent experiments with 5 mice per experiment.* p<0.05, unpaired t-test.
DISCUSSION
Reinfections with respiratory viruses are extremely common among humans of all ages. For influenza virus, progressive evolution through RNA genome mutations leads to antigenic drift and immune escape (44). However, paramyxoviruses, including HMPV and RSV, do not exhibit the same degree of genetic evolution and remain antigenically stable over decades (11, 45, 46). Healthy adults who have experienced natural RSV infection have been experimentally challenged with the same RSV strain and productively reinfected repeatedly over months, even within two months of the first infection (14). Thus, there is limited protection against reinfection with antigenically identical viruses. Many theories have been proposed to explain this phenomenon, including waning humoral antibody, insufficient IgA production at mucosal surfaces, and viral evasion of innate immunity (40). HMPV reinfection in children causes illness at an equal rate to primary infection (10), highlighting the importance of better understanding the mechanisms that contribute to this proclivity. Reinfection with HMPV occurs in healthy and immunocompromised humans, despite the presence of serum antibody (8, 15, 48). This may occur due to limited cross-protective immunity between different strains of HMPV, or may indicate that antibody-mediated protection is not sufficient to prevent HMPV infection.
T cell immunity is important for respiratory virus clearance and resolution of infection. HMPV infections are more severe, and even fatal, in HIV-infected or immunocompromised patients (2, 7, 49, 50). The contribution of T cells to protection against HMPV in humans remains poorly defined. However, we previously discovered that TCD8 are significantly impaired in the respiratory tract during HMPV infection of mice and this impairment was mediated by the inhibitory receptor PD-1 (19), suggesting that optimal T cell immunity may be compromised by inhibitory pathways present in the immune system. Furthermore, the PD-1 pathway is activated in humans with severe viral LRI (19), indicating a potential role for PD-1 signaling during human respiratory virus infections. A previous report utilizing a mouse model of vaccinia virus infection found that primary and memory TCD8 were increased in number and function in PD-1−/− mice (51). However, that study used primary i.n. vaccinia virus infection but secondary i.p. infection, and thus did not directly address reinfection of the respiratory tract. Thus, a key unanswered question is whether inhibitory receptor-mediated T cell impairment contributes to respiratory virus reinfection.
We found that lung TCD8 highly express numerous inhibitory receptors, including PD-1, LAG-3, TIM-3, and 2B4. PD-1, LAG-3, and 2B4 were more highly expressed by secondary lung TCD8. TIM-3 (31) and LAG-3 (32) have been shown to limit the magnitude of the TCD8 response in models of LRI caused by influenza virus and Sendai virus, respectively. The NK cell receptor 2B4 can deliver inhibitory or co-stimulatory signals to TCD8 depending on the isoform expressed (51), but the role of NK-expressed 2B4 during acute LRI has not been explored. All of these receptors are co-expressed with PD-1 and contribute to T cell exhaustion during chronic infection (26, 28, 29). The mechanisms regulating expression and signaling via these receptors and the role of PD-1 signaling in CD4+ T cells during respiratory viral infection is not known.
Interesting, PD-1 is poised for rapid re-expression at the gene expression level due to demethylation of its promoter during primary infection (33). Such epigenetic alterations could explain the higher and more rapid expression of these receptors by secondary effector TCD8. In addition, inhibitory receptor expression increases over time during primary infection (J.E. unpublished observations), suggesting that the increased expression by secondary effector TCD8 could be due to more rapid differentiation from memory precursors compared to naïve TCD8 during primary infection.
Despite co-expression of multiple inhibitory receptors, we found that only PD-1 impairs TCD8 early during reinfection. Blockade of these receptors at later time points does result in increased IFNγ production by lung TCD8 (data not shown), indicating that they are indeed capable of delivering inhibitory signals. One possible explanation for why these receptors showed no functional role could be that their ligands are not expressed at a high enough level in the lung to mediate receptor interaction. We previously showed that PD-L1 is upregulated by infection and peaks at day 7 (19). The ligands for LAG-3, TIM-3, and 2B4 are MHC-II, galectin-9, and CD48, respectively. It may take a longer amount of time for each of these ligands to be sufficiently upregulated to cause inhibitory signaling than the PD-1:PD-L1 interaction. Alternatively, the downstream mediators of the signaling pathways utilized by TIM-3, LAG-3, and 2B4 may take additional time to be expressed and trafficked to the correct subcellular location. It is interesting to note that while each of these receptors can contribute to T cell dysfunction, PD-1 is capable of limiting all of the cellular functions associated with T cell exhaustion single-handedly. A recent study showed that increasing the level of surface-expressed PD-1 could directly reduce the ability of T cells to produce cytokines, proliferate and be cytotoxic (52). Therefore, PD-1 may solely contribute to early impairment, but additional inhibitory receptors may be activated to maintain this impairment at later time points. This theory requires further investigation.
Importantly, we show that blockade of PD-1 signaling significantly improves TCD8 functionality and viral clearance in a mouse model of HMPV reinfection. This suggests that PD-1-mediated TCD8 impairment directly contributes to respiratory virus reinfection. Use of the μMT mouse strain overcame the major barrier to studying HMPV reinfection in rodents: antibody-mediated virus neutralization. A limitation to this model is the complete lack of a B cell compartment, which could have unintended effects on the immune system. For instance, the microbiota of these mice are significantly different compared to WT mice (53), which could affect numerous other immune cell types. Care must be taken in extrapolating these results to humans who are not B cell deficient. B cells can also provide co-stimulation to T cells, which could alter the functionality and magnitude of the T cell response. However, by directly comparing primary infected and reinfected μMT mice, we demonstrated that despite a quantitatively greater secondary TCD8 response, virus titers at day 5 and 6 post-reinfection were indistinguishable from primary infection. Blocking PD-1 overcame this, allowing for better viral control. Furthermore, PD-1−/− mice reinfected with heterosubtypic influenza virus exhibited greater TCD8 functionality compared to WT mice.
Additionally, PD-1 limited the effectiveness of a peptide vaccination formula. TriVax was previously shown to completely protect against RSV infection when mice were challenged soon after the time of vaccination (41). The TCD8 response in that report was high in magnitude and mostly functional, suggesting that a large anti-viral TCD8 response that prevents initial viral infection also prevents TCD8 impairment. This may be because of low levels of viral antigen due to viral clearance. Indeed, rapid clearance of LCMV prevents functional impairment as well (54, 55). However, when mice were challenged with RSV several weeks after TriVax administration, the efficacy was greatly reduced. In contrast, we found that F528 TriVax was not protective in WT mice. However, when PD-1 signaling was removed, we found that HMPV-specific TCD8 were more functional and viral replication was reduced. This suggests that the effect on viral titers might be even more pronounced if the PD-1−/− mice generated a similar immune response to F528 TriVax. It is unclear why the WT mice responded with a higher number of epitope-specific cells. TriVax is a potent vaccination strategy that elicits a robust TCD8 response (42). PD-1 may temper some of the strong stimulatory signals received during priming that potentially cause activation-induced cell death (43). We demonstrate that this may be due to impairment of secondary TCD8, as viral control was only improved in the absence of PD-1 signaling.
The exact mechanisms allowing respiratory viruses to repeatedly reinfect individuals throughout life have been unclear. Taken together, our findings indicate that PD-1 inhibits secondary effector lung TCD8 during respiratory virus reinfection. We provide data to suggest that rapid re-expression of multiple inhibitory receptors contributes to the functional impairment of secondary effector TCD8 in the respiratory tract. In vitro studies only uncovered a role for PD-1 in impairing these cells; thus it remains to be determined if TIM-3, LAG-3, and 2B4 play a role in vivo. We focused on the dominant inhibitory receptor PD-1, and found that PD-1 potently inhibits TCD8 effector functions during reinfection. These results highlight the importance of better understanding the role of PD-1 and other inhibitory receptors in modulating lung TCD8 effector functions in order to design more effective vaccines and therapeutics against respiratory viruses. Further work is warranted to explore whether PD-1-mediated TCD8 impairment contributes to respiratory virus reinfection in humans.
Acknowledgments
Funding: Supported by AI085062 (JVW) and GM007347 for the Vanderbilt Medical Scientist Training Program (JJE, MCR). The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404).
We thank Drs. T. Honjo and M. Boothby for providing mice used in these experiments. We also thank Dr. Jennifer Schuster for assistance with manuscript preparation.
Footnotes
Conflict of interest: JVW serves on the Scientific Advisory Board of Quidel. All other authors have declared that no conflict of interest exists.
References
- 1.Papenburg J, Hamelin ME, Ouhoummane N, Carbonneau J, Ouakki M, Raymond F, Robitaille L, Corbeil J, Caouette G, Frenette L, De Serres G, Boivin G. Comparison of Risk Factors for Human Metapneumovirus and Respiratory Syncytial Virus Disease Severity in Young Children. J Infect Dis. 2012 doi: 10.1093/infdis/jis333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shahda S, Carlos WG, Kiel PJ, Khan BA, Hage CA. The human metapneumovirus: a case series and review of the literature. Transpl Infect Dis. 2011;13:324–328. doi: 10.1111/j.1399-3062.2010.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sloots TP, Mackay IM, Bialasiewicz S, Jacob KC, McQueen E, Harnett GB, Siebert DJ, Masters BI, Young PR, Nissen MD. Human metapneumovirus, Australia, 2001–2004. Emerging infectious diseases. 2006;12:1263–1266. doi: 10.3201/eid1208.051239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh EE, Peterson DR, Falsey AR. Human metapneumovirus infections in adults: another piece of the puzzle. Arch Intern Med. 2008;168:2489–2496. doi: 10.1001/archinte.168.22.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Widmer K, Zhu Y, Williams JV, Griffin MR, Edwards KM, Talbot HK. Rates of Hospitalizations for Respiratory Syncytial Virus, Human Metapneumovirus, and Influenza Virus in Older Adults. J Infect Dis. 2012 doi: 10.1093/infdis/jis309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams JV, Harris PA, Tollefson SJ, Halburnt-Rush LL, Pingsterhaus JM, Edwards KM, Wright PF, Crowe JE., Jr Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N Engl J Med. 2004;350:443–450. doi: 10.1056/NEJMoa025472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams JV, Martino R, Rabella N, Otegui M, Parody R, Heck JM, Crowe JE., Jr A prospective study comparing human metapneumovirus with other respiratory viruses in adults with hematologic malignancies and respiratory tract infections. J Infect Dis. 2005;192:1061–1065. doi: 10.1086/432732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams JV, Wang CK, Yang CF, Tollefson SJ, House FS, Heck JM, Chu M, Brown JB, Lintao LD, Quinto JD, Chu D, Spaete RR, Edwards KM, Wright PF, Crowe JE., Jr The role of human metapneumovirus in upper respiratory tract infections in children: a 20-year experience. J Infect Dis. 2006;193:387–395. doi: 10.1086/499274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, Osterhaus AD. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med. 2001;7:719–724. doi: 10.1038/89098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pavlin JA, Hickey AC, Ulbrandt N, Chan YP, Endy TP, Boukhvalova MS, Chunsuttiwat S, Nisalak A, Libraty DH, Green S, Rothman AL, Ennis FA, Jarman R, Gibbons RV, Broder CC. Human metapneumovirus reinfection among children in Thailand determined by ELISA using purified soluble fusion protein. J Infect Dis. 2008;198:836–842. doi: 10.1086/591186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang CF, Wang CK, Tollefson SJ, Piyaratna R, Lintao LD, Chu M, Liem A, Mark M, Spaete RR, Crowe JE, Jr, Williams JV. Genetic diversity and evolution of human metapneumovirus fusion protein over twenty years. Virol J. 2009;6:138. doi: 10.1186/1743-422X-6-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kroll JL, Weinberg A. Human metapneumovirus. Semin Respir Crit Care Med. 2011;32:447–453. doi: 10.1055/s-0031-1283284. [DOI] [PubMed] [Google Scholar]
- 13.Johnson KM, Chanock RM, Rifkind D, Kravetz HM, Knight V. Respiratory syncytial virus. IV. Correlation of virus shedding, serologic response, and illness in adult volunteers. JAMA. 1961;176:663–667. [PubMed] [Google Scholar]
- 14.Hall CB, Walsh EE, Long CE, Schnabel KC. Immunity to and frequency of reinfection with respiratory syncytial virus. J Infect Dis. 1991;163:693–698. doi: 10.1093/infdis/163.4.693. [DOI] [PubMed] [Google Scholar]
- 15.Okamoto M, Sugawara K, Takashita E, Muraki Y, Hongo S, Nishimura H, Matsuzaki Y. Longitudinal course of human metapneumovirus antibody titers and reinfection in healthy adults. J Med Virol. 2010;82:2092–2096. doi: 10.1002/jmv.21920. [DOI] [PubMed] [Google Scholar]
- 16.Skiadopoulos MH, Biacchesi S, Buchholz UJ, Riggs JM, Surman SR, Amaro-Carambot E, McAuliffe JM, Elkins WR, St Claire M, Collins PL, Murphy BR. The two major human metapneumovirus genetic lineages are highly related antigenically, and the fusion (F) protein is a major contributor to this antigenic relatedness. J Virol. 2004;78:6927–6937. doi: 10.1128/JVI.78.13.6927-6937.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wyde PR, Chetty SN, Jewell AM, Schoonover SL, Piedra PA. Development of a cotton rat-human metapneumovirus (hMPV) model for identifying and evaluating potential hMPV antivirals and vaccines. Antiviral Res. 2005;66:57–66. doi: 10.1016/j.antiviral.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 18.Kolli D, Bataki EL, Spetch L, Guerrero-Plata A, Jewell AM, Piedra PA, Milligan GN, Garofalo RP, Casola A. T lymphocytes contribute to antiviral immunity and pathogenesis in experimental human metapneumovirus infection. J Virol. 2008;82:8560–8569. doi: 10.1128/JVI.00699-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erickson JJ, Gilchuk P, Hastings AK, Tollefson SJ, Johnson M, Downing MB, Boyd KL, Johnson JE, Kim AS, Joyce S, Williams JV. Viral acute lower respiratory infections impair CD8+ T cells through PD-1. J Clin Invest. 2012;122:2967–2982. doi: 10.1172/JCI62860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang J, Braciale TJ. Respiratory syncytial virus infection suppresses lung CD8+ T-cell effector activity and peripheral CD8+ T-cell memory in the respiratory tract. Nat Med. 2002;8:54–60. doi: 10.1038/nm0102-54. [DOI] [PubMed] [Google Scholar]
- 21.Vallbracht S, Unsold H, Ehl S. Functional impairment of cytotoxic T cells in the lung airways following respiratory virus infections. Eur J Immunol. 2006;36:1434–1442. doi: 10.1002/eji.200535642. [DOI] [PubMed] [Google Scholar]
- 22.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 23.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.West EE, Youngblood B, Tan WG, Jin HT, Araki K, Alexe G, Konieczny BT, Calpe S, Freeman GJ, Terhorst C, Haining WN, Ahmed R. Tight regulation of memory CD8(+) T cells limits their effectiveness during sustained high viral load. Immunity. 2011;35:285–298. doi: 10.1016/j.immuni.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. J Immunol. 2012;188:2957–2965. doi: 10.4049/jimmunol.1100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK, Ahmed R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010;107:14733–14738. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamoto N, Cho H, Shaked A, Olthoff K, Valiga ME, Kaminski M, Gostick E, Price DA, Freeman GJ, Wherry EJ, Chang KM. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog. 2009;5:e1000313. doi: 10.1371/journal.ppat.1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma S, Sundararajan A, Suryawanshi A, Kumar N, Veiga-Parga T, Kuchroo VK, Thomas PG, Sangster MY, Rouse BT. T cell immunoglobulin and mucin protein-3 (Tim-3)/Galectin-9 interaction regulates influenza A virus-specific humoral and CD8 T-cell responses. Proc Natl Acad Sci U S A. 2011;108:19001–19006. doi: 10.1073/pnas.1107087108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. 2004;172:5450–5455. doi: 10.4049/jimmunol.172.9.5450. [DOI] [PubMed] [Google Scholar]
- 33.Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, Wei Z, Lu P, Austin JW, Riley JL, Boss JM, Ahmed R. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. 2011;35:400–412. doi: 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams JV, Tollefson SJ, Johnson JE, Crowe JE., Jr The cotton rat (Sigmodon hispidus) is a permissive small animal model of human metapneumovirus infection, pathogenesis, and protective immunity. J Virol. 2005;79:10944–10951. doi: 10.1128/JVI.79.17.10944-10951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rock MT, Crowe JE., Jr Identification of a novel human leucocyte antigen-A*01-restricted cytotoxic T-lymphocyte epitope in the respiratory syncytial virus fusion protein. Immunology. 2003;108:474–480. doi: 10.1046/j.1365-2567.2003.01619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graham BS, Bunton LA, Wright PF, Karzon DT. Reinfection of mice with respiratory syncytial virus. J Med Virol. 1991;34:7–13. doi: 10.1002/jmv.1890340103. [DOI] [PubMed] [Google Scholar]
- 37.Prince GA, Prieels JP, Slaoui M, Porter DD. Pulmonary lesions in primary respiratory syncytial virus infection, reinfection, and vaccine-enhanced disease in the cotton rat (Sigmodon hispidus) Lab Invest. 1999;79:1385–1392. [PubMed] [Google Scholar]
- 38.Graham MB, Braciale TJ. Resistance to and recovery from lethal influenza virus infection in B lymphocyte-deficient mice. J Exp Med. 1997;186:2063–2068. doi: 10.1084/jem.186.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Epstein SL, Lo CY, Misplon JA, Bennink JR. Mechanism of protective immunity against influenza virus infection in mice without antibodies. J Immunol. 1998;160:322–327. [PubMed] [Google Scholar]
- 40.Collins PL, Melero JA. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res. 2011;162:80–99. doi: 10.1016/j.virusres.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee S, Stokes KL, Currier MG, Sakamoto K, Lukacs NW, Celis E, Moore ML. Vaccine-elicited CD8+ T cells protect against respiratory syncytial virus strain A2-line 19F-induced pathogenesis in BALB/c mice. J Virol. 2012;86:13016–13024. doi: 10.1128/JVI.01770-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho HI, Celis E. Optimized peptide vaccines eliciting extensive CD8 T-cell responses with therapeutic antitumor effects. Cancer Res. 2009;69:9012–9019. doi: 10.1158/0008-5472.CAN-09-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunol Rev. 2003;193:70–81. doi: 10.1034/j.1600-065x.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 44.Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305:371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- 45.Garcia O, Martin M, Dopazo J, Arbiza J, Frabasile S, Russi J, Hortal M, Perez-Brena P, Martinez I, Garcia-Barreno B, et al. Evolutionary pattern of human respiratory syncytial virus (subgroup A): cocirculating lineages and correlation of genetic and antigenic changes in the G glycoprotein. J Virol. 1994;68:5448–5459. doi: 10.1128/jvi.68.9.5448-5459.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang CF, Wang CK, Tollefson SJ, Lintao LD, Liem A, Chu M, Williams JV. Human metapneumovirus G protein is highly conserved within but not between genetic lineages. Arch Virol. 2013;158:1245–1252. doi: 10.1007/s00705-013-1622-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hall CB, Walsh EE, Long CE, Schnabel KC. Immunity to and frequency of reinfection with respiratory syncytial virus. J Infect Dis. 1991;163:693–698. doi: 10.1093/infdis/163.4.693. [DOI] [PubMed] [Google Scholar]
- 48.Falsey AR, Hennessey PA, Formica MA, Criddle MM, Biear JM, Walsh EE. Humoral immunity to human metapneumovirus infection in adults. Vaccine. 2010;28:1477–1480. doi: 10.1016/j.vaccine.2009.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Englund JA, Boeckh M, Kuypers J, Nichols WG, Hackman RC, Morrow RA, Fredricks DN, Corey L. Brief communication: fatal human metapneumovirus infection in stem-cell transplant recipients. Ann Intern Med. 2006;144:344–349. doi: 10.7326/0003-4819-144-5-200603070-00010. [DOI] [PubMed] [Google Scholar]
- 50.Madhi SA, Ludewick H, Kuwanda L, van Niekerk N, Cutland C, Klugman KP. Seasonality, incidence, and repeat human metapneumovirus lower respiratory tract infections in an area with a high prevalence of human immunodeficiency virus type-1 infection. Pediatr Infect Dis J. 2007;26:693–699. doi: 10.1097/INF.0b013e3180621192. [DOI] [PubMed] [Google Scholar]
- 51.Chlewicki LK, Velikovsky CA, Balakrishnan V, Mariuzza RA, Kumar V. Molecular basis of the dual functions of 2B4 (CD244) J Immunol. 2008;180:8159–8167. doi: 10.4049/jimmunol.180.12.8159. [DOI] [PubMed] [Google Scholar]
- 52.Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, Freeman GJ, Krogsgaard M, Riley JL. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1305394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shulzhenko N, Morgun A, Hsiao W, Battle M, Yao M, Gavrilova O, Orandle M, Mayer L, Macpherson AJ, McCoy KD, Fraser-Liggett C, Matzinger P. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med. 2011;17:1585–1593. doi: 10.1038/nm.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 55.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]