

Abstract
Neurodevelopmental disorders (NDDs) are caused by mutations in diverse genes involved in different cellular functions, although there can be crosstalk, or convergence, between molecular pathways affected by different NDDs. To assess molecular convergence, we generated human neural progenitor cell models of 9q34 deletion syndrome, caused by haploinsufficiency of EHMT1, and 18q21 deletion syndrome, caused by haploinsufficiency of TCF4. Using next-generation RNA sequencing, methylation sequencing, chromatin immunoprecipitation sequencing, and whole-genome miRNA analysis, we identified several levels of convergence. We found mRNA and miRNA expression patterns that were more characteristic of differentiating cells than of proliferating cells, and we identified CpG clusters that had similar methylation states in both models of reduced gene dosage. There was significant overlap of gene targets of TCF4 and EHMT1, whereby 8.3% of TCF4 gene targets and 4.2% of EHMT1 gene targets were identical. These data suggest that 18q21 and 9q34 deletion syndromes show significant molecular convergence but distinct expression and methylation profiles. Common intersection points might highlight the most salient features of disease and provide avenues for similar treatments for NDDs caused by different genetic mutations.
Introduction
Neurodevelopmental disorders (NDDs) include autism spectrum disorders, seizure disorders, and intellectual disability, among many others, although little is known about the precise molecular mechanisms that lead to disease. Currently, approximately 20%–45% of all NDDs are associated with variation in specific genes,1 most of which show variable expressivity and reduced penetrance, highlighting the phenotypic complexity of NDDs.2 Notably, NDDs are also characterized by locus heterogeneity, meaning that mutations in many different genes can lead to similar disease phenotypes.
Recent studies have suggested genotypic convergence across diagnostic categories of NDDs. For example, common variants in the same gene have been associated with two or more psychiatric disorders,3 and significant overlap between haploinsufficiency of a gene and more than one diagnostic category has also been shown.4 This leads to an important question in NDD genetics: do mutations in some or most genes associated with NDDs culminate on similar cellular functions, or do they affect distinct cell functions while having limited crosstalk between molecular pathways?
The purpose of the current study was to assess the degree that genes associated with similar phenotypes converge on the same cellular functions. To assess this question, we selected two genes that are unambiguously associated with NDDs, in which mutations cause disease by the same mechanism (haploinsufficiency), and for which the disease can be recapitulated in wild-type cells. Mutations in transcription factor 4 (TCF4 [MIM 602272]) cause 18q21 deletion syndrome (Pitt-Hopkins syndrome [MIM 610954]), characterized by moderate to severe intellectual disability, breathing difficulties, recurrent seizures, cupid-bow upper lip, distinct facial features, microcephaly, lack of speech, and psychiatric behavioral problems. Mutations in euchromatic histone-lysine N-methyltransferase 1 (EHMT1 [MIM 607001]) cause 9q34 deletion syndrome,5 characterized by severe intellectual disability, hypotonia, cupid-bow upper lip, microcephaly, lack of speech, distinct facial features, and psychiatric behavioral problems (MIM 610253). Both disorders have other symptoms, and not all subjects show all symptoms. We reasoned that modeling both disorders in the identical neural stem cell line derived from healthy human fetal brain might allow for an assessment of the degree of molecular convergence caused by reduced dosage of these two genes.
Material and Methods
All work was carried out with the approval of the research ethics board of the Douglas Hospital Research Institute.
Cell Culture
Fetal brain cells (FBCs) are ReNcells derived from the ventral mesencephalon of human fetal brain (Millipore SCC008). Cells were grown on 6-well plates coated with poly-L-ornithine/laminin (Sigma) and were maintained in 70% Dulbecco’s modified Eagle’s medium, 2% B27, 1% penicillin and streptomycin (Life Technologies), 30% Ham’s F12 (Mediatech Herndon), 20 ng/ml basic fibroblast growth factor (bFGF, R&D Systems), 20 ng/ml epidermal growth factor (EGF), 5 μg/ml heparin (Sigma), and 0.2 μg/ml puromycin (Sigma P8833). For studies involving differentiating FBCs, we triggered differentiation by removing both bFGF and EGF from cell media, leaving cells for 30 days, and changing media every 3 days.6
Generation of Stable Knockdown Human FBC Lines
All short hairpin RNA (shRNA) used in this study was designed, cloned into the pLKO.1 vector, and packaged into lentivirus at the Broad Institute. To create stable cell lines (i.e., cell lines where knockdown [KD] constructs are stably integrated into the cell genome), we transfected FBCs with lentivirus and then selected for cells where genomic integration occurred. For lentiviral transfection, FBCs were maintained at 30% confluency (∼400,000 cells/well) in a 6-well plate and then dosed with 20 μl viral media in 2 ml cell-culture media without penicillin and streptomycin. Puromycin (0.8 μl/ml, Sigma P8833), resistance to which is produced by the pLKO.1 vector, was added to cultures 48 hr after infection, and this followed an initial media change 24 hr after transfection. Stable cell lines were selected by continuous maintenance of low-dose puromycin in culture media (0.2 μl/ml). Cells that do not contain the KD construct also do not have the puromycin-resistance gene, so all cells that can survive in the media produce the KD construct. For controls, we used shRNAs targeting LacZ, GFP, RFP, and Luc mRNA. We refer to these controls as “nontarget” controls because they were generated in the same way as TCF4-KD and EHMT1-KD FBCs, but the stably integrated construct produces an shRNA that targets an mRNA not present in the human genome (i.e., LacZ, GFP, RFP, and Luc mRNA). After creation and selection of stable cell lines, FBCs were frozen down and regrown as required.
Immunocytochemistry
Cells were seeded in 6-well plates with glass coverslips and fixed with a 4% formaldehyde solution (Tousimis, 1008C) diluted in PBS when cells were 90% confluent. Fixed cells underwent a blocking and prepermeabilization step in 1% BSA and 0.1% Triton X solution for 1 hr at room temperature. Next, primary antibodies mouse monoclonal anti-EHMT1 (Abcam ab41969) and mouse monoclonal anti-TCF4 (Abcam ab60727) were diluted to 1/100 and 1/200, respectively, and incubated at room temperature for 2 hr. Cells were washed three times in 0.1% PBS and then incubated at room temperature for 1 hr with a goat anti-mouse secondary antibody coupled to Alexa Fluor 488 (Life Technologies A11001). Cells were washed three times in PBS, and coverslips were slide mounted with Vectashield with DAPI (VectorLabs H-1200).
Microscopy
Cells were imaged with an Olympus FluoView FV10i confocal microscope; high-magnification pictures (1,024 × 1,024 pixels) were taken with a 60× oil-immersion objective with a 2.4× numerical zoom with FluoView software (Olympus). In order to quantify TCF4 and EHMT1 signals, we acquired all pictures during a single session. Prior to image acquisition, we set laser intensities to control samples with the aim of eliminating saturating pixels equally and ensuring that the same laser intensities were applied equally to all samples. All images were exported as single black-and-white TIFFs for Alexa Fluor 488, DAPI, and phase-contrast channels and were imported into ImageJ (v.1.37c). For fluorescent intensities, we followed the step-by-step quantification procedures laid out in ImageJ. In brief, all quantification was done in gray scale with the corrected total-cell-fluorescence procedure after background correction. Raw values per cell were then exported to Excel (Microsoft), and a Student’s t test was calculated. Ten to fifteen cells were assessed per cell line.
Quantitative PCR
RNA from cells was extracted with the RNeasy MinElute Cleanup Kit (QIAGEN), and cDNA was synthetized with M-MLV Reverse Transcriptase (Invitrogen). Commercially available RNA from eight different tissues (Ambion Total RNA: liver, kidney, spinal cord, frontal lobe, fetal brain, lung, and testis) was used for analysis of EHMT1 (TaqMan Hs00226978_m1) and TCF4 (TaqMan Hs00162613_m1) expression, and TaqMan endogenous controls were used for targeting GAPDH (MIM 138400) or HPRT (MIM 308000). Primers for cell-characterization experiments were all TaqMan primers designed by Life Technologies. Real-time PCR reactions were run in triplicate with the ABI 7900HT Fast Real-Time PCR System, and data were collected with Sequence Detection System (SDS) software (Life Technologies).
miRNA was extracted with the QIAGEN miRNeasy Micro Kit, after which miRNA was reverse transcribed with gene-specific TaqMan RT-PCR miRNA assays according to the manufacturer’s (Life Technologies) instructions. Expression levels of MIR99A (MIM 614509; Applied Biosystems 000435) were calculated with the Absolute Quantitation standard-curve method with RNU6-2 (HGNC 34270; Applied Biosystems 001093) as the endogenous control. Real-time PCR reactions were run in quadruplicate with the ABI 7900HT Fast Real-Time PCR System, and data were collected with SDS software (Life Technologies). For each reaction, the quantitative PCR (qPCR) mix included 7 μl 2× No AmpErase UNG Master Mix (Applied Biosystems), 1 μl miRNA-specific primer and probe mix, 2 μl cDNA, and 20 μl H20.
RNA Sequencing
RNA sequencing (RNA-seq) libraries were prepared from high-quality RNA (RNA integrity number > 9; Agilent 2100 Bioanalyzer), and all libraries were prepared by expert technicians at the McGill University and Genome Quebec Innovation Center. Replicates for each shRNA construct were grown in different T75 flasks, and extraction of RNA was done independently for each flask. Prior to library preparation, we spiked in external RNA controls from the External RNA Control Consortium7 (Life Technologies) to assess sequencing depth and create standard curves to determine depth and quality of sequencing and library preparation. Three libraries were run per lane of an Illumina HiSeq 2000 flow cell (100 bp paired-end reads), which achieved an average of ∼65 million reads per library. For bioinformatic processing, we used FASTX-Toolkit, TopHat,8 Bowtie2,9 and Cufflinks210 with default parameters to preprocess, align, and assemble reads into transcripts, estimate abundance, and test differential expression. We used DAVID (Database for Annotation, Visualization and Integrated Discovery)11 set to default parameters for Gene Ontology (GO) analysis.
Whole-Genome miRNA Analysis
miRNAs were processed with the nCounter Human miRNA Expression Assay Kit (NanoString Technologies) at the NanoString facility at the Jewish General Hospital (Montreal), and all samples were run in duplicate. nCounter data were processed with NanoStringNorm in R, and all data were normalized to the geometric mean and miRNA spike-in controls according to the manufacturer’s instructions. All data were analyzed in R with DIANA miRPath.12
Reduced Representation Bisulfite Sequencing
We followed BisQC, the multiplexed bisulfite sequencing parameters that we developed.13,14 In brief, we used 5 μg of genomic DNA extracted from FBCs to carry out the MspI (New England Biolabs) digestion at 37°C for 7 hr (20 units of enzyme per microgram of DNA). We used the QIAGEN EpiTect Fast 96 Bisulfite Kit to carry out the bisulfite conversion of adaptor-ligated libraries and then sequenced four indexed samples per lane of an Illumina HiSeq 2000 by using 50 bp single-end reads. We used Trim Galore, a script to automate quality and adaptor trimming as well as quality control, with the added functionality of removing biased methylation positions for reduced representation bisulfite sequencing (RRBS) files. Afterwards, we used Bismark15 to map sequencing reads to the human genome (UCSC Genome Browser; we allowed two mismatches and used Bowtie2). For the postprocessing analysis, we developed an R script, including single-CpG and 500 bp tiling-window analyses. To assess differentially methylated regions (DMRs) within tiling windows, we needed at least two CpGs to be present in all KD samples and 75% of control samples. Only CpG sites with coverage greater than 5× were included, and we excluded the 0.1% of CpGs that showed the highest coverage for each sample. We determined significance by performing t tests of methylation frequency within identical windows between KD and control cells and then correcting these p values by using the Benjamini-Hochberg approach.
Chromatin Immunoprecipitation Sequencing
For chromatin immunoprecipitation sequencing (ChIP-seq), 107 cells were dissociated by trypsin, crosslinked with 1% formaldehyde (Tousimis NC9611804) for 10 min, pelleted, and resuspended in lysis buffer with protease inhibitors. Samples were sonicated with the Labsonic M Ultrasonic Homogenizer (Labsonic Sartorius Stedim) for seven 30 s cycles at 30% power. Chromatin was sheared at 15 1 s pulses and subsequently rested for 2 min intervals at 50% power with the same ultrasonic homogenizer (200–1,000 bp). One hundred micrograms per microliter of DNA was incubated with antibodies (5 μg/μl) overnight at 4°C in dilution buffer and anti-EHMT1 (Abcam ab41969), anti-TCF4 (Abcam ab60727), or IgG (Millipore 1710109 kit) as a negative control. We tested all antibodies by immunoblot to ensure that a band of the correct size could be detected. Immunoprecipitation (IP) washes, elution, and crosslink reversal were performed with the Magna ChIP A – Chromatin Immunoprecipitation Kit (Millipore) according to the manufacturer’s instructions. IP and input DNA were purified by phenol-chloroform extraction, precipitated in ethanol, and resuspended in sterile water. We used the Illumina ChIP-Seq DNA Sample Prep Kit (IP-102-1001) for all experiments and followed instructions from the manufacturer. Libraries were sequenced with Illumina technology and single-end sequencing. We used Bowtie2 for alignment and MACS for peak calling with default parameters except for the background function, which we set to 10−6 instead of 10−5. To associate ChIP peaks with genes, we considered any regions 5 kb upstream of a gene and any region 2 kb downstream of gene, as we did to associate genomic regions for methylation analysis.
Case Samples
From Signature Genomics (SG), we analyzed a total of 36,938 probands referred for clinical oligonucleotide-based whole-genome array comparative genomic hybridization (aCGH) testing. Subject DNA was processed with arrays custom designed by SG—either a 105K-feature platform (SignatureChipOS v.1 or v.4, Agilent Technologies) or a 135K-feature platform (SignatureChipOS v.2 or v.3, Roche NimbleGen). The ethnic distribution in the samples was estimated from a sampling cross-section: 75% were white individuals, 7% were African American individuals, and 18% were individuals of other ethnicity. The sex distribution was 59% male and 41% female.
Control Samples
A total of 29,957 control samples were used in the copy-number variant (CNV) calling studies. All CNVs for control samples were performed with SNP arrays. Control samples were derived from the Database of Genomic Variants (DGV) from the following studies (n): Wellcome Trust Case Control Consortium (WTCCC)16,17 (7,878), Cooper et al.18 non-WTCCC (6,113), Lionel et al.19 (1,234), PopGen20 (1,123), Altshuler et al.21 (1,056), Pinto et al.22 (1,287), Abecasis et al.23 (1,092), Shaikh et al.24 (2,026), and Xu et al.25 (8,148).
Results
Expression and Generation of Reduced TCF4 and EHMT1 Dosage in FBCs
To assess the molecular convergence of two neurodevelopmental disorders, we first needed a reasonable cell line with which to model haploinsufficiency. We selected the ReNCell VM neural progenitor cell line, referred to here as FBCs, for several reasons: (1) they expressed appropriate neural markers in the proliferating and differentiating cell states (Figures 1A and 1B), (2) they expressed TCF4 and EHMT1 mRNA at levels comparable to those observed in fetal human brain (Figures 1C and 1D), although with very high expression of EHMT1, and (3) TCF4 and EHMT1 could be detected in all cells in a pattern consistent with expected function; that is, both proteins were in, or appeared to be clustered around, the nucleus, as expected for proteins that associate with DNA (Figures 1E–1G).
Figure 1.
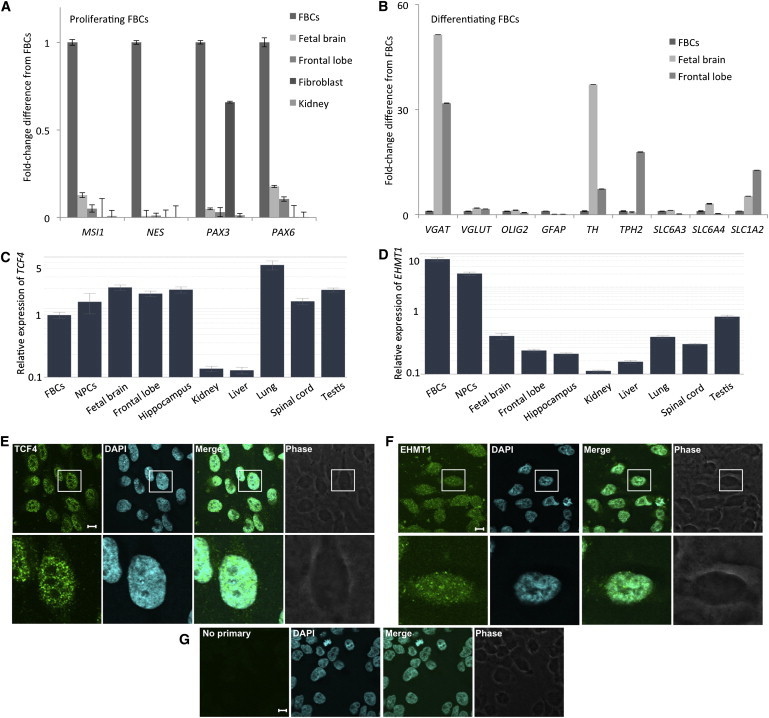
Characterization of Neural Progenitor Cells Derived from Human FBCs
(A) Expression of MSI1, NES, PAX3, and PAX6 (encoding neural progenitor cell markers) in proliferating FBCs and other human tissues.
(B) Characterization of genes encoding mature neuronal markers in differentiating FBCs and other human brain-derived tissues.
(C) qPCR expression of TCF4 in multiple human tissue types and cell lines. NPCs stands for neural progenitor cells derived from human skin.
(D) qPCR expression of EHMT1 in multiple human tissue types and cell lines.
(E–G) Immunocytochemical analysis of TCF4 (E) and EHMT1 (F) in FBCs and the no primary control (G). Note the punctate appearance of TCF4 and EHMT1 near and within DNA stained with DAPI. All scale bars represent 10 μm.
18q21 deletion syndrome is caused by haploinsufficiency of TCF4, and 9q34 deletion syndrome is caused by haploinsufficiency of EHMT1, so we modeled reduced dosage of each gene in FBCs in an attempt to recapitulate disease. We created stable cell lines (with KD constructs stably integrated in the cell genome, along with the puromycin-resistance gene, a selectable marker) for four shRNA KD constructs per gene and four nontarget control constructs. We identified two TCF4-KD lines that showed suitable reduced dosage (Figure 2A) and four constructs for EHMT1 KD (Figure 2B). To confirm reduced dosage at the protein level, we assessed TCF4-KD and EHMT1-KD cell lines by immunocytochemistry. We selected two KD cell lines per gene and one nontarget control and performed quantification. For the two TCF4-KD cell lines (Figure 2C), we found a significant decrease in TCF4 of 37% (p = 0.022) for construct 15036 and 40% (p = 0.009) for construct 15037. For two EHMT1-KD cell lines (Figure 2D), we found a significant decrease in EHMT1 of 38% (p = 0.002) for construct 229325 and 41% (p = 0.002) for construct 229326.
Figure 2.
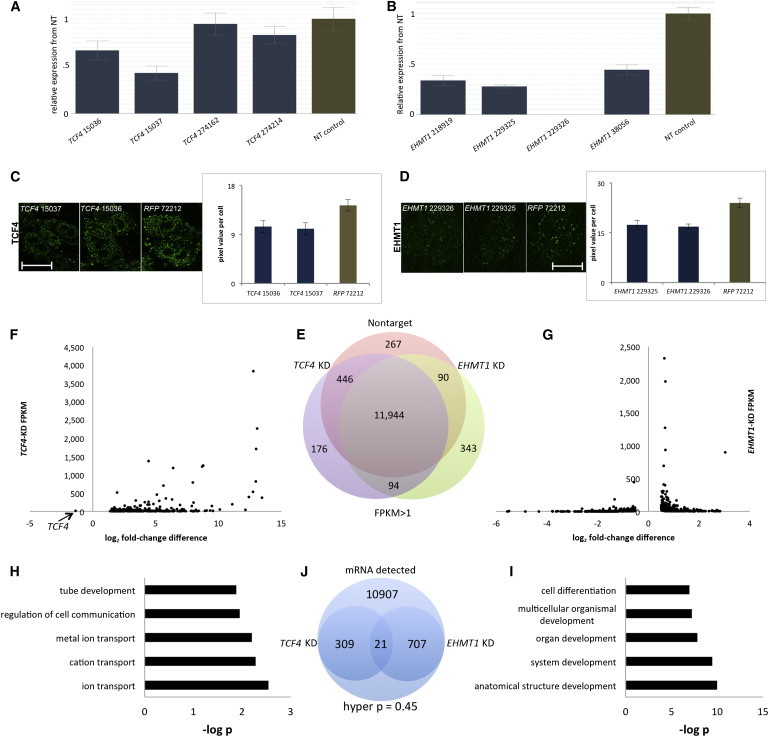
Creation, Validation, and RNA-Seq of Reduced Dosage of TCF4 and EHMT1 in FBCs
(A) Creation of four stable FBC lines with reduced dosage of TCF4. The graph compares qPCR results of the TCF4 expression level in these FBCs to that in four nontarget (NT) control cell lines (controls received a virus that produces a shRNA not known to bind to any known human mRNA). Expression in nontarget controls is represented by a single brown bar and includes expression from four independent cell lines in replicate.
(B) Creation of four stable FBC lines with reduced dosage of EHMT1. Color coding and sample numbers are identical to those in the TCF4 graph.
(C) Quantitative immunocytochemical analysis of TCF4 in one nontarget control (RFP) and two TCF4-KD cell lines. All scale bars represent 10 μm.
(D) Quantitative immunocytochemical analysis of EHMT1 in one nontarget control (RFP) and two EHMT1-KD cell lines.
(E) Venn diagram showing all detected RNA in nontarget control FBCs, FBCs with reduced TCF4 dosage (KD), and FBCs with reduced EHMT1 dosage with FPKMs > 1.
(F) A dot plot of significant genes from RNA-seq compares FBCs with reduced TCF4 dosage to nontarget control cell lines. All genome-wide-significant differentially expressed genes, except TCF4, showed increased expression. As a point of reference, the TCF4-KD FPKM value is 13.4 (34.4 in nontarget controls).
(G) Dot plot of significant differentially expressed genes in cells with reduced EHMT1 dosage.
(H and I) GO analyses of significant differentially expressed genes from cells with reduced dosage of TCF4 (H) and EHMT1 (I).
(J) Statistical analysis of mRNA overlaps between TCF4-KD and EHMT1-KD cell lines. Hyper p refers to the cumulative hypergeometric p value and was calculated with the numbers shown.
Whole-Genome Gene-Expression Patterns in FBCs with Reduced Dosage of EHMT1 and TCF4
We asked how reduced expression of either TCF4 or EHMT1 might affect the transcriptome of FBCs. We performed an RNA-seq experiment by using two KD lines per gene as well as replicates grown in independent flasks (two shRNAs per gene for a total of four samples per gene) and four nontarget controls with replicates for each (n = 4 shRNA controls, n = 8 samples) in proliferating FBCs. We assessed the number of annotated genes detectable across all cell lines and found 11,944 genes with FPKM (fragments per kilobase of transcript per million fragments mapped) > 1, and very few were expressed in a cell-line-specific manner (Figure 2E). We identified 330 genome-wide-significant differentially expressed annotated genes in TCF4-KD cells, which we show pictorially in Figure 2F. Immediately apparent is that 329/330 genes all showed increased expression in TCF4-KD cells, suggesting that TCF4 is a negative regulator of gene expression. Only a single gene showed significantly reduced expression, and this was TCF4. For EHMT1-KD cells, there were 728 differentially expressed genes that did not show a strong directional bias; compared to nontarget controls, these cells showed increased expression of 409 genes and decreased expression of 318 genes (Figure 2G), despite the fact that EHMT1 action (H3K9 methylation) was associated with gene repression.26 To understand what pathways might be affected in each deletion syndrome, we used DAVID to perform GO analysis of significantly differentially expressed genes from each analysis. Notably, the three highest GO clusters representing independent categories for TCF4-KD cells were ion transport (e.g., GABA, glutamate, and calcium receptor subunits), regulation of cell communication (more specifically, TGF-β and MAPK signaling), and neural-tube development (Figure 2H). Genes from GO terms included noggin (NOG [MIM 602991]), TGFB-induced factor homeobox 1 (TGIF1 [MIM 602630]), and WNT5A (MIM 164975). Similarly, an EHMT1-KD GO analysis for those genes that were significantly differentially expressed (Figure 2I) suggested involvement of pathways important in development, and differentially expressed gens included BMP7 (MIM 112267), WNT7A (MIM 601570), CTNNB1 (MIM 116806), and TGFB2 (MIM 190220). From 11,944 expressed genes, 21 were differentially expressed in both EHMT1-KD cells (728 genes with q values < 0.05) and TCF4-KD cells (330 genes with q values < 0.05). To find the probability that this overlap occurred by chance, we calculated the hypergeometric distribution27 and found a nonsignificant value of 0.45, suggesting that any overlap at the gene level between TCF4-KD cells and EHMT1-KD cells might be random (Figure 2J). Still, there were ten intersecting genes that overlapped and showed the same directional change (increased expression) in both EHMT1-KD and TCF4-KD cells (Table 1).
Table 1.
FPKM Values from Genes Significantly More Expressed in TCF4-KD and EHMT1-KD Cells Than in Nontarget Controls
| Gene | EHMT1 KD | Nontarget Control | Log2Fold Change | q Value | TCF4 KD | Nontarget Control | Log2Fold Change | q Value |
|---|---|---|---|---|---|---|---|---|
| CDH6 | 11.93 | 1.81 | −2.72 | 0.00 | 92.71 | 1.22 | −6.25 | 0.002385 |
| CHD3 | 2.63 | 1.19 | −1.15 | 0.02 | 16.86 | 0.76 | −4.47 | 0.002385 |
| CHRNB1 | 3.91 | 1.95 | −1.01 | 0.01 | 5.27 | 1.32 | −2.00 | 0.0133388 |
| COL5A3 | 1.29 | 0.50 | −1.37 | 0.01 | 9.16 | 0.34 | −4.76 | 0.002385 |
| FAM181A | 10.54 | 3.36 | −1.65 | 0.00 | 19.61 | 2.27 | −3.11 | 0.00823788 |
| JPH2 | 1.25 | 0.20 | −2.61 | 0.00 | 6.63 | 0.14 | −5.59 | 0.002385 |
| LPAR1 | 5.71 | 1.66 | −1.78 | 0.00 | 44.93 | 1.12 | −5.33 | 0.002385 |
| SLC38A3 | 6.52 | 2.92 | −1.16 | 0.01 | 7.70 | 1.92 | −2.00 | 0.0133388 |
| TFAP2C | 3.22 | 1.17 | −1.46 | 0.00 | 4.60 | 0.79 | −2.54 | 0.002385 |
| TOX1 | 8.92 | 5.95 | −0.59 | 0.04 | 22.47 | 4.04 | −2.47 | 0.002385 |
The q value is the genome-wide corrected p value and was calculated by the Benjamini-Hochberg method.
Assessment of Duplications of Genes with Significantly Increased Expression in FBCs with Reduced Dosage of EHMT1 and TCF4
We identified ten different genes that showed increased expression in both EHMT1-KD and TCF4-KD FBCs, which might suggest that some of these genes are important to the NDD phenotype for both 18q21 and 9q34 deletion syndromes. We therefore reasoned that given that decreased expression of either TCF4 or EHMT1 might lead to increased expression of any of these genes, duplication in affected subjects might show a similar clinical phenotype to 18q21 or 9q34 deletion syndromes, even though gene duplications are not always associated with dosage changes and gene duplications can be complicated by duplication breakpoint location and even lead to decreased gene expression. Notwithstanding these caveats, we screened a large cohort of affected subjects referred for genetic testing with duplications in any of these genes to see whether the phenotype overlapped that of 18q21 or 9q34 deletion syndromes (Table 2). We observed only a small number of subjects for each genomic region when selecting duplication CNVs that were >100 kb and <3 Mb and encompassed the gene of interest. Genes for which there were at least two subjects with similar phenotypes related to 18q21 and 9q34 deletion syndromes included COL5A3 (MIM 120216), SLC38A3 (MIM 604437), CHRNB1 (MIM 100710), and TFAP2C (MIM 601602), suggesting that duplications of these genes might contribute to abnormal phenotypes and warrant further investigation. Eight of 14 CNV duplications were considered pathogenic in a Clinical Laboratory Improvement Amendments-certified laboratory and were reported as such to referring physicians, whereas the six remaining CNVs were variants of unknown significance but were considered valid by American Board of Medical Genetics-certified geneticists. Nine of ten genes assessed had complete gene duplications; only CDH6 (MIM 603007) could not be unambiguously described as having a complete gene duplication as a result of probe density over this region on the array. To ensure these were not CNVs that occurred in the general population, we utilized the DGV to determine whether complete duplications in these same genes could be identified. Because these control data are derived exclusively from SNP array data (unlike case data, which were derived from aCGH arrays), we restricted this analysis to CNVs > 100 kb, meaning that these genes in the general population might contain duplication CNVs that are below the detection resolution of the technology. One CNV duplication in each of TFAP2C (DGV nsv525425) and SLC38A3 (DGV nsv876768) in the control data set met these criteria, suggesting that duplications in these two genes are unlikely to be pathogenic. Still, we identified so few affected subjects for the other genes possibly related to the clinical phenotype that no significance level could be ascertained. Sample sizes orders of magnitude larger will be required for assessing any potential role for these genes in disease.
Table 2.
Clinical Information for Those Subjects with Duplications < 3 Mb in Genes Upregulated in Both EHMT1-KD and TCF4-KD Cells
| ID | Sex | Phenotype | Inheritance | Array | Coordinates | Age | Other Potentially Significant CNVs | Validation |
|---|---|---|---|---|---|---|---|---|
| CDH6 | ||||||||
| 38742 | F | congenital diaphragmatic hernia | unknown | SignatureChipOS v.2.0 12-plex | chr5: 31,247,785–31,508,304a | prenatal | none | – |
| 68773 | M | velocardiofacial syndrome | unknown | SignatureChipOS v.3.1 12-plex | chr5: 31,247,785–31,538,938a | 16 years | arr[hg18] 4p16.3(33,860–2,349,973) ×1 | – |
| CHD3 | ||||||||
| GC45076 | M | congenital heart disease | unknown | SignatureChipOS v.2.0 12-plex | chr17: 7,696,576–8,579,933 | 0 months | none | RP11-769H22×3 |
| GC70690 | F | developmental delay, seizure disorder | de novo | SignatureChipOS v.2.0 12-plex | chr17: 6,716,420–8,264,897 | 6 years | none | RP11-298H4×3 |
| CHRNB1 | ||||||||
| GC39423 | F | encephalopathy | unknown | SignatureChipOS v.1.1 Rev. B 2-plex | chr17: 6,904,478–7,628,740 | 6 years | none | CTD-3054O5×2b |
| GC70690 | F | developmental delay, seizure disorder | de novo | SignatureChipOS v.2.0 12-plex | chr17: 6,716,420–8,264,897 | 6 years | none | – |
| COL5A3 | ||||||||
| GC24840 | M | dysmorphic features, seizure disorder | unknown | SignatureChipOS v.1.0 2-plex | chr19: 9,147,542–10,279,708 | 14 years | none | RP11-365L4×2b |
| GC25160 | M | developmental delay, dysmorphic features | maternal | SignatureChipOS v.1.0 2-plex | chr19: 9,147,542–11,061,034 | 6 years | none | RP11-365L4×3 |
| GC93811 | M | brain abnormality (posterior fossa), partial agenesis of cerebellar vermis | unknown | SignatureChipOS v.4.0 4-plex | chr19: 7,668,882–10,208,159 | prenatal | none | CTD-2102F19×3 |
| SLC38A3 | ||||||||
| 40532 | F | glycogenosis, other specific developmental learning difficulties, unspecified lack of normal physiological development, disorders of mitochondrial metabolism | unknown | SignatureChipOS v.2.0 12-plex | chr3: 50,162,650–50,286,670 | 12 years | arr[hg18] 1p36.31p36.23(6,151,213–8,039,703)×1 | – |
| 43079 | M | unspecified disturbance of conduct | unknown | SignatureChipOS v.2.0 12-plex | chr3: 50,162,650–50,286,670 | 5 years | none | – |
| TFAP2C | ||||||||
| 37732 | M | developmental delay | unknown | SignatureChipOS v.2.0 12-plex | chr20: 54,033,017–54,914,766 | 4 years | arr[hg18] 2q31.3q32.3(180,784,421–192,194,263) ×1 | – |
| GC66182 | M | developmental delay, dysmorphic features | unknown | SignatureChipOS v.2.0 12-plex | chr20: 54,558,953–54,744,904 | 18 months | none | RP11-361G16×2b |
| MIRLET7E | ||||||||
| GC66320 | F | obesity | unknown | SignatureChipOS v.2.0 12-plex | chr19: 56795726-57581342 | 8 years | none | – |
Phenotypes are those listed by referring physicians; thus, subjects might have more than what is listed. Coordinates listed are according to the hg18 assembly of the UCSC Human Genome Browser. American Board of Medical Genetics-certified geneticists reassessed all CNVs to ensure nonartifactual calls. For subjects with IDs starting with “GC,” these findings were reported as abnormal and therefore adhered to all quality-assurance and quality-control requirements for a Clinical Laboratory Improvement Amendment-certified laboratory. Missing genes were FAM181A (smallest duplication was at least 7.5 Mb, although arrays did not have any direct coverage of this gene, so small, whole-gene duplications might remain undetected), JPH2 (smallest duplication was at least 18.5 Mb), LPAR1 (smallest duplications were part of more complex 9q rearrangements involving at least ∼15 Mb), and TOX1 (smallest duplication was at least 27 Mb). Abbreviations are as follows: F, female; and M, male.
These duplications might or might not include the entire gene; the last nonduplicated probe (chr5: 31,221,930–31,221,990) is outside the gene, and the first duplicated probe (chr5: 31,247,782–31,247,845) is within the gene.
Below resolution of detection by fluorescence in situ hybridization.
Expression Differences that Define Proliferating FBCs with Reduced Dosage of TCF4 and EHMT1 Are More Characteristic of Differentiating than of Proliferating Cells
GO analysis for FBCs with reduced dosage of TCF4 and EHMT1 suggested that genes important in development, cell differentiation, and ion transport might be affected in these cell models of human deletion syndromes. Given this observation, we wondered whether reduced dosage of TCF4 or EHMT1 in FBCs might affect the balance of genes involved in cell proliferation and cell differentiation, collectively referred to here as cell state. To test this idea, we generated RNA-seq data from nontarget controls in both a proliferating state (n = 4 shRNA control constructs, n = 8 samples in duplicate) and a differentiating state (n = 4 shRNA control constructs, n = 8 samples in duplicate). We defined differentiating FBCs as cells that are maintained in culture for 30 days in the absence of bFGF or EGF and proliferating cells as those maintained in culture with bFGF and EGF (Figure 3A), a technique that has been accepted for many years.6,28 We identified 12,378 transcripts with FPKM > 1 (Figure 3B) in common between both proliferating and differentiating FBCs. We performed differential-expression analysis on these two cell states and found that 7,683 genes were significantly differentially expressed and that there was a slight overrepresentation of genes showing decreased expression in differentiating FBCs (n = 4,094 cells with decreased expression in differentiating FBCs; Figure 3C). Performing GO analysis with genes downregulated in differentiating FBCs (Figure 3D) or upregulated in differentiating FBCs (Figure 3E) gave predictable results: genes involved in the cell cycle had lower expression in differentiating FBCs, whereas genes involved in neurodevelopment had higher expression. If reduced dosage of TCF4 leads FBCs to an altered proliferation or differentiation state, we might expect an overlap of gene-expression changes in the TCF4-KD and cell-state experiments. We intersected the differentially expressed genes from these experiments (Figure 3F) and found a significant overlap beyond what would be expected by chance (n = 236 genes). GO terms associated with these overlapping genes were involved in cell-projection assembly and ion transport (Figure 3G). We plotted the log2 fold-change differences for the common differentially expressed genes in the TCF4-KD and the cell-state experiments and observed a significant positive correlation between gene-expression patterns (Pearson p = 0.0001; Figure 3H)—the increased expression of genes in the TCF4-KD experiment were correlated with increased gene expression in differentiating cells, suggesting that genes whose expression is altered by reduced dosage of TCF4 are similarly altered in normal cells as they differentiate. We performed identical analyses for EHMT1 by using the same rationale and found a significant overlap of significantly differentially expressed genes between the EHMT1-KD experiment and the cell-state experiment (n = 674 genes; Figure 3I), and GO terms for these overlapping genes were all related to neurodevelopment (Figure 3J). As for the TCF4 results, we observed a significant positive Pearson correlation for log2 fold-change differences between EHMT1 KD and differentiating FBCs (p = 1.4 × 10−11; Figure 3K). These data suggest that genes that show differential expression in FBCs with reduced dosage of EHMT1 or TCF4 are more characteristic of genes that define a differentiating cell state than of genes that define a proliferating cell state, although the genes that make up these cell states are different for cells with reduced dosage of TCF4 and EHMT1. Of note, we did not observe any cell-proliferation phenotype of TCF4-KD or EHMT1-KD cells in culture, meaning that the expression changes observed here do not suffice to drive cells out of a proliferating state in the presence of growth factors.
Figure 3.
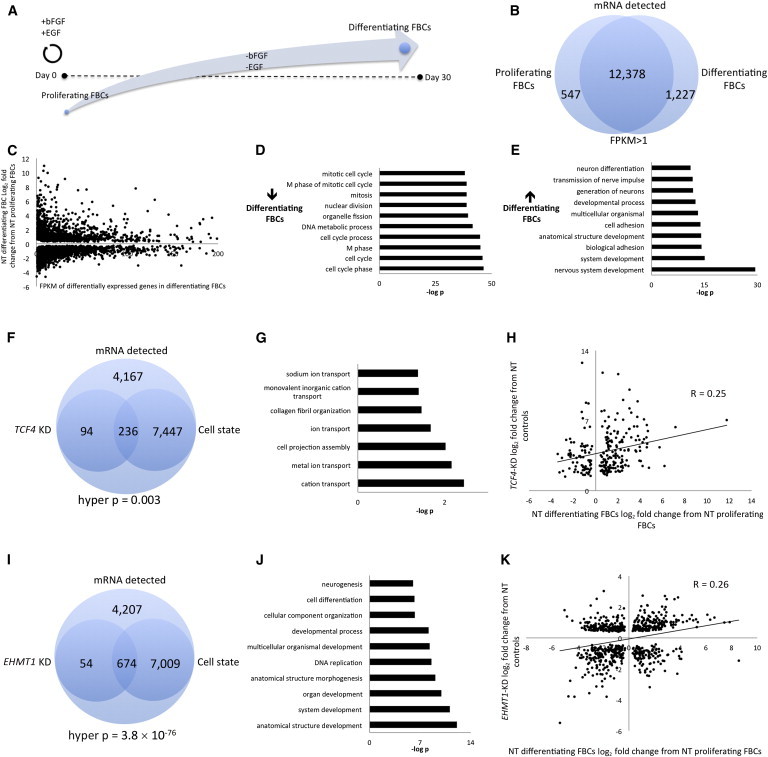
Gene-Expression Patterns in FBCs with Reduced Dosage of TCF4 or EHMT1 Are More Characteristic of Differentiating Cells Than of Proliferating FBCs
(A) Diagram outlining the experimental procedures to define proliferating and differentiating FBCs.
(B) RNA-seq analysis of four nontarget control (GFP, LacZ, RFP, Luc) FBC lines in a proliferating state or a differentiating state. The Venn diagram shows the number of detected transcripts that were common to both cells states with FPKMs > 1.
(C) Dot plot plotting the log2 fold-change differences of FPKMs in proliferating and differentiating nontarget (NT) FBCs against the FPKM value of differentiating FBCs. The graph was truncated at FPKM = 200.
(D and E) GO analysis for significantly differentially expressed genes that showed lower (D) or higher (E) expression in differentiating FBCs than in proliferating FBCs.
(F) This Venn diagram demonstrates the overlap of all significantly differentially expressed genes identified in the TCF4-KD and cell-state experiments. We used 11,944 as the total mRNAs detectable.
(G) GO analysis of the 236 overlapping genes from (F).
(H) Dot plot demonstrating that those genes common to both the TCF4-KD analysis and the cell-state analysis (n = 236 genes) have a significant positive correlation. Plotted are the log2 fold-change differences for each analysis.
(I) This Venn diagram demonstrates the overlap of all significantly differentially expressed genes identified in the EHMT1-KD and cell-state experiments.
(J) GO analysis of the 674 genes from (I).
(K) Dot plot demonstrating that those genes common to both the EHMT1-KD analysis and the cell-state analysis (n = 674 genes) have a significant positive correlation. Plotted are the log2 fold-change differences for each analysis.
miRNA Convergence in FBCs with Reduced Dosage of TCF4 and EHMT1
To further understand the impact of reduced dosage of both EHMT1 and TCF4, we performed another analysis on genome-wide expression in proliferating FBCs, this time of miRNA, by using the same design as for the RNA experiment (two KD shRNAs for each of EHMT1-KD and TCF4-KD FBCs in replicate and four nontarget shRNA controls in replicate). Of the 800 miRNAs present on NanoString arrays, only 254 were detectable in at least 75% of samples in the nontarget control group or in 100% of KD cell lines. For both TCF4-KD and EHMT1-KD FBCs, compared to nontarget FBCs, only a single miRNA, MIRLET7E (MIM 611250), passed Benjamini-Hochberg false-discovery-rate correction, suggesting that reduced dosage of both EHMT1 and TCF4 increases expression of this gene. A total of six genes were identical between TCF4-KD and EHMT1-KD FBCs when we selected miRNAs with p values < 0.10, and we calculated that the probability that this overlap occurred by chance to be 0.027 (Figure 4A). When we focused more closely on these six miRNAs common to both TCF4-KD and EHMT1-KD FBCs (Figure 4B), we found that the expression pattern was equivalent across all genes; for example, when a miRNA was increased in TCF4-KD cells, it was also increased in EHMT1-KD cells. Two of these miRNAs (MIRLET7E and MIR99A) are involved in cell differentiation,29–31 and two of the remaining four (MIR302D [MIM 614599] and MIR378E [MIM 611957]) have known roles in cell proliferation.32–34 We confirmed the validity of the NanoString data by performing targeted qPCR on one miRNA, MIR99A (ranked the second and third most significant miRNA for both TCF4-KD and EHMT1-KD cells, respectively), which had a unique probe available and whose specificity we could confirm. The directional changes we observed in this qPCR experiment were identical to those in the NanoString arrays (TCF4 KD, p = 0.01; EHMT1 KD, p = 0.09).
Figure 4.
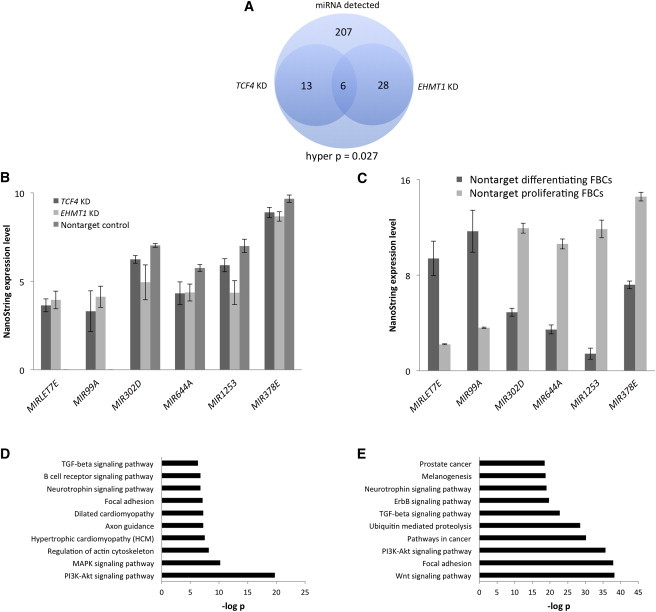
miRNA Analysis in FBCs with Reduced Dosage of TCF4 and EHMT1
(A) Venn diagram showing overlap of differentially expressed miRNAs common to FBCs with reduced dosage of TCF4 and EHMT1.
(B) Expression values of each of the six miRNAs common to FBCs with reduced dosage of TCF4 and EHMT1 at p < 0.10; the direction of change is identical for all six miRNAs.
(C) NanoString results for the miRNA experiment performed in nontarget control cell lines in a proliferating and differentiating state.
(D and E) GO analysis for TCF4-KD (D) and EHMT1-KD (E) differentially expressed miRNAs (p < 0.10).
RNA-seq data as well as the direction of expression changes of four of the six miRNAs (Figure 4B) suggested that miRNAs important in cell proliferation or cell differentiation might be affected when TCF4 or EHMT1 expression is reduced in FBCs. Two of the miRNAs in this list of six genes (MIR1253 [HGNC 35318] and MIR644A [HGNC 32900]) are of unknown function, but their expression patterns in the KD FBCs might suggest that they are important in cell proliferation, given the expression differences of the four known miRNAs. We performed another NanoString expression analysis by using four nontarget control FBCs in the proliferating state and the same nontarget control FBCs in a differentiating state (n = 4 shRNAs per group, in duplicate) in an attempt to identify miRNAs important in each cell state. With these data, we would then be able to better determine where the two miRNAs of unknown function (MIR1253 and MIR644A) have a role. We found 394 miRNAs that could be detected in 75% of samples in either the differentiating state or the proliferating state, and 210 miRNAs had a p value < 0.01. With these newly generated miRNA expression maps of differentiating and proliferating FBCs, we graphed the values for the six miRNAs that we previously identified as common to both TCF4-KD and EHMT1-KD FBCs. We found the directional patterns of these six miRNAs to be identical to the results from the gene-KD experiments, although the magnitude was much more drastic in the cell-state experiment (Figure 4C). These results support the idea that miRNA patterns in cells with reduced dosage of TCF4 or EHMT1 are more characteristic of a differentiating cell state than of a proliferating cell state. Finally, we performed a GO analysis for differentially expressed miRNA for both gene-KD conditions and implicated developmental and signaling pathways (Figures 4D and 4E).
Methylation Differences Caused by Reduced Dosage of TCF4 or EHMT1 in FBCs
Chromatin modifiers and methyl binding proteins are implicated in NDDs,35,36 suggesting that DNA-methylation patterns might reflect cell state. Methylation of DNA is not a function of TCF4, but it might be a function of EHMT1,37 and reduced dosage of either gene might have downstream effects that culminate in DNA-methylation changes. We therefore hypothesized that reduced dosage of EHMT1 or TCF4 would lead to methylation changes in identical genomic regions. These DMRs might reflect regulatory regions for genes important in proliferation or differentiation, for example.
To do this analysis, we performed RRBS by using our established pipeline13 with two shRNAs per KD cell line and four nontarget controls. For the analysis, we segregated the genome into 500 bp windows and assessed those windows with at least two CpGs, and that showed a significant difference between methylation frequencies in KD and nontarget controls (mean methylation differences were >2%). A total of 15,433 windows were detected and used for statistical testing, and Figures 5A and 5B show the Manhattan plots of the chromosomal distribution of significant windows for TCF4 KD and EHMT1 KD. For TCF4 KDs, 89 DMRs were genome-wide significant (q values < 0.05), whereas for EHMT1 KDs, 96 DMRs were genome-wide significant, and these DMRs appeared more frequently in the 5′ UTR and upstream regions than in the total group of all windows (Figures 5C–5F). Twenty DMRs were identical between TCF4-KD and EHMT1-KD cells, which has a hypergeometric probability of occurring by chance of 8.3 × 10−26 (Figure 5G). We observed more hypomethylation in the KD groups; TCF4 KD had 55/89 hypomethylated DMRs (Figure 5H), whereas EHMT1 KD had 60/93 hypomethylated DMRs (Figure 5I). Importantly, 18/20 DMRs were directionally identical (Pearson = 0.65, p = 0.001; Figure 5J), and three DMRs showed hypermethylation. To ensure that a single CpG of strong effect did not influence these DMRs, we plotted all CpGs for each of these 20 clusters for which we observed cluster-specific methylation patterns, meaning that multiple CpGs appeared to contribute equally to the signal (Figure 5K). A GO analysis of overlapping genes in which these DMRs were identified strongly suggested a role for cell-differentiation processes (Figure 5L), and we selected two DMRs (one in NCAM2 [MIM 602040] and one in RUNX2 [MIM 600211]) for demonstration (Figure 5M).
Figure 5.
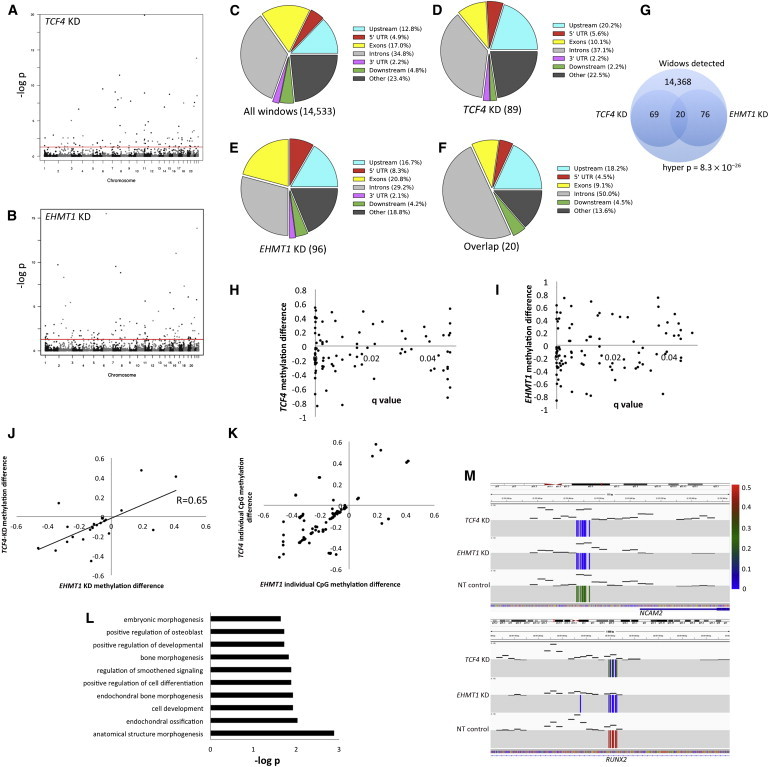
Methylation Analysis of FBCs with Reduced Dosage of TCF4 and EHMT1
(A and B) Manhattan plots showing genomic regions with genome-wide-significant differential methylation in reduced-dosage FBCs. This analysis was done with 500 bp windows and by assessment of the mean methylation per CpG in the interval. The red lines represent q value = 0.05 (i.e., genome-wide-corrected p values).
(C) Genomic distribution of 500 bp windows observed (i.e., at least two CpGs detected in all cell groups) in RRBS.
(D) Genomic distribution of DMRs from FBCs with reduced TCF4 dosage.
(E) Genomic distribution of DMRs from FBCs with reduced EHMT1 dosage.
(F) Genomic distribution of DMRs common to FBCs with reduced dosage of TCF4 and EHMT1.
(G) Venn diagram of statistical assessment of overlapping DMRs in TCF4 KD and EHMT1 KD.
(H and I) Dot plots showing the relationship between p value and direction of change (either hyper- or hypomethylated) for FBCs with reduced dosage of TCF4 or EHMT1.
(J) Correlation analysis of those DMRs common to FBCs with reduced dosage of EHMT1 and TCF4.
(K) Plot of all individual CpGs that contribute to each cluster plotted in (J); each dot represents a single CpG site.
(L) GO analysis for DMRs common to FBCs with reduced dosage of TCF4 and EHMT1.
(M) Integrative Genomics Viewer (IGV) image showing two examples of DMRs common to FBCs with reduced dosage of TCF4 and EHMT1. Compared to nontarget (NT) controls, both reduced-dosage cell lines were hypomethylated. Each individual colored line reflects a single CpG at this locus, and black dashes represent read coverage over the region (the scale is set from 0 to 100 reads). The colored scale bar refers to methylation frequency.
Data from the mRNA and miRNA experiments suggest that TCF4 KD and EHMT1 KD might affect genes important in cell proliferation or differentiation. To test this idea with methylation data, we performed an RRBS experiment by using nontarget control cells in proliferating (n = 4 nontarget controls) and differentiating (n = 4 nontarget controls) cell states and identified 134 DMRs. We wanted to assess whether those DMRs significantly differentially methylated between proliferating FBCs and differentiating FBCs were also significantly differentially methylated in the gene-KD experiments. For TCF4 KD, we found 11 DMRs that were common between the KD and cell-state experiments (Figure 6A); we graph the distribution of these differential methylation patterns in Figure 6B. Six of 11 DMRs were more similar to the differentiating nontarget FBCs; this ratio is not above what would be expected by chance, although it is in the direction expected if DNA-methylation patterns are more similar to nontarget differentiating FBCs than to nontarget proliferating FBCs. All of these DMRs occurred in the introns of genes, except for those in MIR34 (MIM 611172) and TMEM240 (HGNC 25186), which were within the promoter and less than 1 kb from the transcription start site (TSS). For EHMT1 KD, we found seven DMRs that were common between the reduced-dosage analysis and the cell-state analysis (Figure 6C); five of seven DMRs matched the pattern observed in differentiating FBCs (Figure 6D), supporting the notion that DNA-methylation patterns are more similar to a differentiating cell state than to a proliferating one, although this was not statistically assessed because of the small number of overlapping DMRs observed. However, analyzing significantly differentially methylated CpG dinucleotides did support the hypothesis that methylation states are more characteristic of differentiating cells than of proliferating cells (Figure S1, available online). Three DMRs (in ETV6 [MIM 600618], RUNX2 [MIM 600211], and GNAS [MIM 139320]) were common to both TCF4 KD and EHMT1 KD and were identified in the cell-state experiment (Figure 6E), and only one, in RUNX2, showed similar methylation patterns among EHMT1-KD, TCF4-KD, and differentiating nontarget FBCs. To provide perspective to these data, we also show the ratio of hypermethylation to hypomethylation in nontarget differentiating and proliferating FBCs (Figure 6F).
Figure 6.
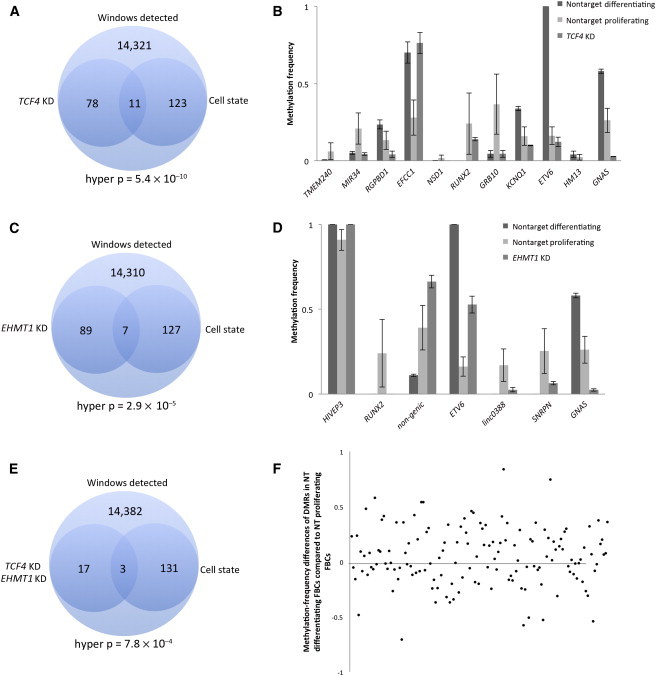
Comparison of Methylation Patterns in Differentiating and Proliferating Nontarget Control FBCs and FBCs with Reduced Dosage of EHMT1 and TCF4
(A) Venn diagram showing the overlap of DMRs identified in the TCF4-KD and the cell-state experiments. The analysis included a total of 14,533 windows.
(B) Genomic region in which the 11 DMRs from (A) were located, as well as the mean methylation level for each.
(C) Venn diagram showing the overlap of DMRs identified in the EHMT1-KD and cell-state experiments.
(D) Genomic region in which the seven DMRs were located, as well as the mean methylation level for each analysis.
(E) Venn diagram showing the overlap of DMRs that intersected the TCF4-KD, EHMT1-KD, and cell-state experiments.
(F) Distribution of methylation-frequency differences of 134 DMRs in the cell-state experiment.
EHMT1 and TCF4 DNA-Binding Sites in Human FBCs
Both EHMT1 and TCF4 either interact with DNA through large protein complexes or directly bind DNA, so we next performed genome-wide ChIP-seq to identify binding sites in fetal brain. For TCF4, we identified 750 peaks, and 47% of targets were associated with a gene (Figure 7A). We first asked whether genes that are differentially expressed are also associated with DNA ChIP target regions in TCF4. We found that COL5A3, DGKZ (MIM 601441), GRK4 (MIM 137026), MPP7 (MIM 610973), PI4KA (MIM 600286), PLEKHA5 (MIM 607770), and QPCT (MIM 607065) were bound by TCF4 and showed differential expression (Table 3), suggesting that the explicit cause of increased expression of these particular genes might be reduced binding by TCF4, although the probability of observing seven genes common to both the RNA-seq data (n = 330 genes) and the ChIP-seq (n = 349 unique genes) by chance is only modestly significant (hypergeometric p = 0.022) when the 41,566 annotated genes in GRCh37 are used as the global pool of genes. We provide the significant de novo binding motifs as predicted by the HOMER (Hypergeometric Optimization of Motif Enrichment) algorithm for TCF4 in Figure S2. TCF4 peaks intersected with some genomic regions that are known to associate with transcription factors, at least according to ENCODE transcription factor maps generated with the H1 stem cell line (Figure 7C). For EHMT1 ChIP-seq, we identified 1,218 peaks, of which 56% were associated with a gene (Figure 7B); a significant proportion of peaks, at least compared to TCF4 peaks, were in close proximity to the TSS. Gene-expression analysis of EHMT1 KD showed 728 different genes with differential expression, whereas 683 unique genes were associated with ChiP-seq peaks; 34 of these genes were common between EHMT1-KD RNA-seq and ChIP-seq experiments, corresponding to a hypergeometric probability of p < 7.1 × 10−8 (Table 4). Sixteen of these 34 genes showed binding directly in the promoter or 5′ UTR (within 1 kb of the TSS), suggesting not only that EHMT1 (compared to TCF4) preferentially targets promoter regions but also that these EHMT1 target regions might also affect gene expression. Figure S3 shows the DNA motifs that are most commonly associated with EHMT1 sequencing reads. EHMT1 ChIP peaks showed more overlap with ENCODE-identified transcription factor binding regions (Figure 7C) than did TCF4 ChIP peaks. Finally, we assessed whether TCF4 and EHMT1 target the same genes, though not necessarily at the same genomic loci and irrespective of expression effects. We intersected genes that were associated with ChIP peaks for both TCF4 and EHMT1 and identified 29 overlapping genes between TCF4 and EHMT1 ChIP-seq experiments, corresponding to a hypergeometric probability of 5.3 × 10−7(Figure 7D and Table 5). GO terms associated with these 29 overlapping peaks were consistent with previous results in that neuron development was again a significant term (Figure 7E). In Figures 7F and 7G, we show two examples of TCF4 and EHMT1 binding regions in the same gene. For TCF4, we observed binding in overlapping inward- or outward-facing reads, suggesting that TCF4 binds in dimer at some distance from TSSs. For EHMT1, we observed a clustering of reads in 500–800 bp regions often close to TSSs. These data suggest that TCF4 and EHMT1 target some of the same genes in the human genome, which might reflect biological effects on these genes, although we did not observe expression differences for any overlapping target genes in both KD cell models.
Figure 7.
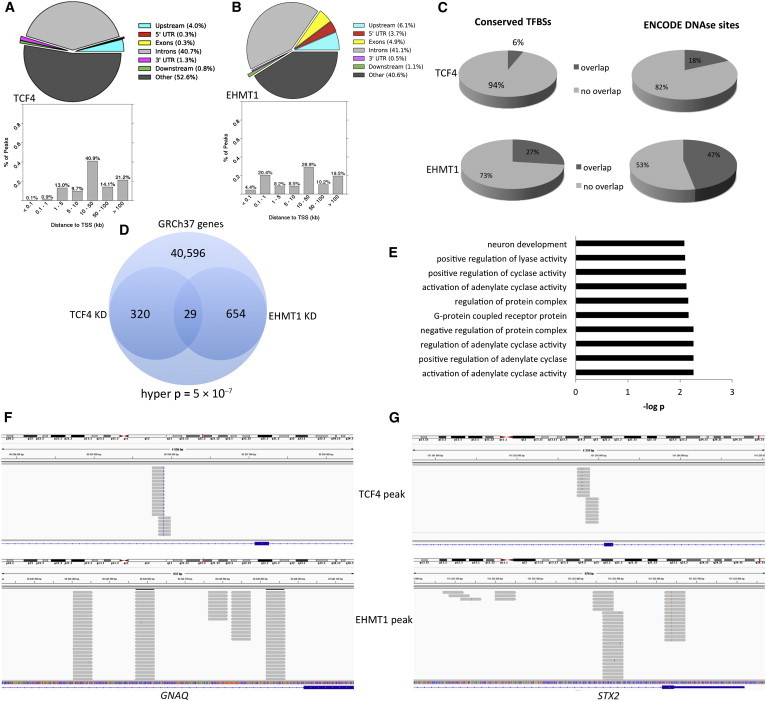
DNA-Binding Analysis of FBCs with Reduced Dosage of TCF4 and EHMT1
(A) Genomic distribution of peaks from TCF4 ChIP-seq (colored pie chart); the bar graph represents the distribution of TCF4 ChIP peaks from the nearest TSS and was generated with ChIP-Enrich.
(B) Genomic distribution of peaks from EHMT1 ChIP-seq (colored pie chart); the bar graph represents the distribution of EHMT1 ChIP peaks from the nearest TSS.
(C) Pie chart showing the degree of overlap among ChIP peaks, conserved transcription factor binding sites (TFBSs), and ENCODE DNase hypersensitivity sites from the H1 stem cell line.
(D) Venn diagram showing the degree of overlap between TCF4 ChIP peaks and EHMT1 ChIP peaks in the same genes. Hyper p refers to the cumulative hypergeometric p value.
(E) GO analysis of the 29 overlapping genes common to both TCF4 and EHMT1 ChIP peaks.
(F and G) IGV images showing ChIP peaks from TCF4 and EHMT1. Peaks are in different regions of the same gene and show different binding patterns. Grey bars represent sequencing reads, and blue represents the gene of interest.
Table 3.
TCF4 RNA-Seq and TCF4 ChIP-Seq Intersection
| Gene | RNA-Seq Log2Fold Change | TCF4 Peak (Distance from TSS in bp) | Peak Coordinates (hg19) |
|---|---|---|---|
| COL5A3 | 4.75 | 20,129 | chr19: 10,100,917–10,101,120 |
| DGKZ | 2.99 | −3,649 | chr11: 46,350,704–46,350,906 |
| GRK4 | 2.07 | 50,743 | chr4: 3,015,789–3,016,381 |
| MPP7 | 6.06 | 251,199 | chr10: 28,340,695–28,340,898 |
| PI4KA | 3.74 | 91,421 | chr22: 21,121,578–21,121,780 |
| PLEKHA5 | 4.16 | 242,739 | chr12: 19,525,263–19,525,465 |
| QPCT | 5.42 | 1,8791 | chr2: 37,590,397–37,590,689 |
Table 4.
EHMT1 RNA-Seq and EHMT1 ChIP-Seq Intersection
| Gene | RNA-Seq Log2Fold Change | EHMT1 Peak (Distance from TSS in bp) | Peak Coordinates (hg19) |
|---|---|---|---|
| ANKRD52 | −0.77 | −90 | chr12: 56,652,025–56,652,442 |
| ARIH1 | −0.67 | 638 | chr15: 72,767,080–72,767,528 |
| B3GALTL | 0.64 | 95 | chr13: 31,773,895–31,774,518 |
| BCL2L1 | 0.62 | 25,036 | chr20: 30,285,526–30,285,714 |
| CA2 | −0.65 | −193 | chr8: 86,375,713–86,376,162 |
| CCBE1 | 1.96 | 79 | chr18: 57,364,328–57,364,802 |
| CEP70 | −0.73 | 24,421 | chr3: 138,288,449–138,288,968 |
| DGKG | 0.67 | 121,476 | chr3: 185,958,454–185,958,641 |
| E2F2 | 0.7 | 374 | chr1: 23,857,001–23,857,675 |
| GREB1L | 1.2 | 262 | chr18: 18,822,248–18,822,680 |
| HNRNPM | 0.49 | 40,988 | chr19: 8,550,410–8,551,171 |
| ICAM5 | −2.05 | 40 | chr19: 10,400,338–10,401,051 |
| KDM5B | −0.88 | 48,817 | chr1: 202,728,529–202,728,936 |
| KIF11 | 0.48 | 46,478 | chr10: 94,399,209–94,399,396 |
| LARGE | −1.17 | 125,911 | chr22: 34,192,579–34,192,767 |
| LASP1 | 0.55 | 212 | chr17: 37,025,935–37,026,711 |
| MCM2 | 0.58 | −117 | chr3: 127,317,003–127,317,268 |
| MXI1 | −0.68 | 3,168 | chr10: 111,970,325–111,970,735 |
| NFIC | 0.54 | 16,756 | chr19: 3,376,188–3,376,444 |
| NUMB | −0.59 | 137 | chr14: 73,924,911–73,925,388 |
| NUP210 | 0.48 | 7,606 | chr3: 13,453,958–13,454,448 |
| PCCA | −1.09 | 171,972 | chr13: 100,913,147–100,913,334 |
| PDE4B | 0.61 | 572 | chr1: 66,258,549–66,258,979 |
| PDLIM5 | 0.97 | 111 | chr4: 95,372,954–95,373,343 |
| PPP1R14C | 1.15 | 46,422 | chr6: 150,510,516–150,510,703 |
| PPP4R1 | 0.66 | 361 | chr18: 9,614,127–9,614,352 |
| REV3L | 0.78 | 1,241 | chr6: 111,803,282–111,804,073 |
| SEZ6 | −0.89 | 329 | chr17: 27,332,931–27,333,327 |
| SHCBP1 | 0.65 | 14,486 | chr16: 46,640,732–46,640,919 |
| SLC35F1 | 0.73 | 314,827 | chr6: 118,543,422–118,543,609 |
| SLC45A3 | −1.06 | 41,003 | chr1: 205,608,456–205,608,799 |
| VGLL4 | −0.85 | 931 | chr3: 11,760,893–11,761,686 |
| VIPR1 | 2.13 | 13,499 | chr3: 42,543,989–42,544,590 |
| WDR76 | 0.76 | 19,668 | chr15: 44,138,686–44,138,873 |
Table 5.
Gene Intersection of TCF4 ChIP-Seq and EHMT1 ChIP-Seq Data
| Gene | TCF4 Peak (Distance to TSS in bp) | Peak Coordinates (hg19) | EHMT1 Peak (Distance to TSS in bp) | Peak Coordinates (hg19) |
|---|---|---|---|---|
| ADCY10 | 25,347 | chr1: 167,858,004–167,858,209 | 61,837 | chr1: 167,821,523–167,821,710 |
| CALN1 | 477,279 | chr7: 71,434,732–71,434,982 | 605,204 | chr7: 71,306,839–71,307,026 |
| CDH8 | 128,864 | chr16: 61,941,774–61,941,976 | 187,576 | chr16: 61,883,070–61,883,257 |
| CLPP | 8,311 | chr19: 63,69,672–63,69,874 | 129 | chr19: 6,361,242–6,361,940 |
| CRAMP1L | 28,848 | chr16: 1,693,387–1,693,590 | 2,441 | chr16: 1,666,987–1,667,175 |
| CYB5B | 1,753 | chr16: 69,460,128–69,460,372 | 3,977 | chr16: 69,462,350–69,462,598 |
| DGKG | 12,293 | chr3: 186,067,593–186,067,867 | 121,476 | chr3: 185,958,454–185,958,641 |
| DMXL2 | 18,399 | chr15: 51,896,467–51,896,669 | 153,878 | chr15: 517,60,989–51,761,190 |
| DONSON | 277,890 | chr21: 35,006,697–35,006,930 | 295,407 | chr21: 34,989,085–34,989,507 |
| EXOC4 | 94,228 | chr7: 133,031,940–133,032,160 | 357,929 | chr7: 133,295,574–133,295,929 |
| FRMD4B | 320,008 | chr3: 69,271,624–69,271,826 | 292,358 | chr3: 69,299,280–69,299,471 |
| GNAQ | 47,397 | chr9: 80,598,629–80,599,016 | 467 | chr9: 80,645,405–80,646,100 |
| GPR124 | 8,578 | chr8: 37,662,852–37,663,105 | −4,997 | chr8: 37,649,284–37,649,523 |
| ITCH | 51,309 | chr20: 33,002,269–33,002,471 | 6,587 | chr20: 32,957,508–32,957,789 |
| KIF11 | 31,106 | chr10: 94,383,829–94,384,032 | 46,478 | chr10: 94,399,209–94,399,396 |
| LOC100507412 | 2,999 | chrun_gl000220: 99,910–100,344 | 7,145 | chrun_gl000220: 104,089–104,457 |
| MAP4 | 12,792 | chr3: 48,117,876–48,118,079 | 158,853 | chr3: 47,971,803–47,972,030 |
| PCDH7 | 235,654 | chr4: 30,957,571–30,957,809 | 210,386 | chr4: 30,932,249–30,932,596 |
| PLEKHA5 | 242,739 | chr12: 19,525,263–19,525,465 | −9 | chr12: 19,282,326–19,282,906 |
| SEMA3D | 86,757 | chr7: 84,696,180–84,696,416 | 67,000 | chr7: 84,715,960–84,716,151 |
| SLC25A15 | 4,876 | chr13: 41,368,302–41,368,543 | 53 | chr13: 41,363,363–41,363,836 |
| SOBP | 109,434 | chr6: 107,920,576–107,920,925 | 47,647 | chr6: 107,858,849–107,859,078 |
| SORCS1 | 85,549 | chr10: 108,838,816–108,839,019 | −306 | chr10: 108,924,603–108,924,942 |
| SPPL2A | 21,599 | chr15: 51,036,210–51,036,412 | 20,242 | chr15: 51,037,574–51,037,762 |
| SPTBN4 | 38,508 | chr19: 41,010,453–41,010,857 | 47,050 | chr19: 41,018,905–41,019,490 |
| STX2 | 41,021 | chr12: 131,282,689–131,282,891 | 440 | chr12: 131,323,071–131,323,672 |
| UNC5D | 413,175 | chr8: 35,506,046–35,506,252 | 145 | chr8: 35,092,687–35,093,551 |
| WDFY3 | 3,010 | chr4: 85,884,433–85,884,635 | 5,271 | chr4: 85,882,147–85,882,400 |
| WDR34 | 8,261 | chr9: 131,410,767–131,410,969 | 238 | chr9: 131,418,757–131,419,025 |
Discussion
In this study, we assessed the molecular convergence of two neurodevelopmental disorders that show similar phenotypes in affected individuals: 18q21 and 9q34 deletion syndromes, which are caused by TCF4 and EHMT1 haploinsufficiency, respectively. To do this, we assessed the degree to which TCF4 KD and EHMT1 KD had similar effects on neural stem cells. In identifying the expression, methylation, or DNA-binding convergence points, we reasoned that we might better dissect the pathways that lead to neurodevelopmental disease caused by haploinsufficiency of TCF4 or EHMT1.
We modeled the two syndromes by knocking down TCF4 and EHMT1 in a human FBC line and using well-controlled comparison constructs. Using stringent RNA-seq parameters, we found that all differentially expressed genes in the 18q21 deletion syndrome model (with the exception of the target gene, TCF4) were upregulated, a strong indication that this disease causes release from gene repression. However, this was not observed in the cell model of 9q34 deletion syndrome, where we found approximately equal up- and downregulation of differentially expressed genes. We found ten different genes that intersected these two syndromes from the RNA-seq analysis and that showed increased expression; all have the potential to be dosage sensitive and cause a phenotype, at least on the basis of their published function. Still, this represented very few common targets between the two diseases, and statistical analysis of this overlap suggested that ten genes might arise by chance.
We found ten intersecting genes that code for mRNA and that showed increased expression in both EHMT1-KD and TCF4-KD FBCs, and we used a very large cohort of clinical case samples to attempt to support a role for these genes in disease. We cannot say, however, that duplications in any gene cause all or part of the clinical phenotype of 9q34 or 18q21 deletion syndromes. That said, the data support COL5A1 and CHRNB1, both of which are genes expressed in muscle; complete duplications of each gene were associated with a neurodevelopmental phenotype in at least two affected subjects and absent from control subjects. We cannot rule out that this NDD phenotype is due to dosage effects of neighboring genes.
GO terms and the changes in direction of expression of fundamental neurodevelopmental genes, such as NOG38 and WNT7A,39 in EHMT1-KD and TCF4-KD cells suggested to us that these two disease models might share a similar cell state, namely becoming more like differentiating cells. Both disease models were always assayed from a proliferating cell state, yet our gene-expression analysis suggested that KD FBCs share characteristics with differentiating nontarget control FBCs. We did not observe a cell-proliferation phenotype in culture in the KD FBCs, so we do not think that reducing the dosage of TCF4 or EHMT1 causes neural progenitor cells to spontaneously differentiate. Rather, only some genes important in cell state appear to be affected, and we hypothesize that this puts cells with reduced dosage of TCF4 and EHMT1 in a suboptimal or compromised position: the processes that regulate the timing of differentiation40 or the check points established to ensure that neural progenitor cells continue to divide might be impaired. This might cause neural progenitor cells to differentiate too early in response to outside signaling cues, for example. In human subjects with either deletion syndrome, one might speculate that some neural progenitor cells differentiate prematurely and thus lead to neurodevelopmental anomalies, such as improper integration into cellular networks. This might be a convergence point for cells with reduced dosage of TCF4 and EHMT1, but we note that the genes affected in each disorder differ.
miRNA-expression data provide convergence points for reduced dosage of TCF4 and EHMT1—the same miRNA, MIRLET7E, was genome-wide significant for both disease models and had a role in cell differentiation.31 Our analysis of the six miRNAs differentially expressed in both disease models further supports findings from the RNA-seq experiment, namely that differentially expressed miRNAs were important in cell state and seemed to match expression patterns of differentiating neural progenitor cells. One of these miRNAs, MIR302D, can drive somatic cells to pluripotency,41 and this gene was downregulated in both EHMT1-KD and TCF4-KD FBCs in comparison to nontarget control FBCs. We were able to provide supporting evidence for this idea by generating miRNA-expression maps from proliferating and differentiating nontarget control FBCs and could show that the six miRNAs differentially expressed and common to both deletion-syndrome models showed increased expression of genes characteristic of a differentiating state and lower expression of miRNAs characteristic of a proliferating cell state.
We suspected that DNA-methylation changes might be similar in 18q21 and 9q34 deletion syndromes. We found specific methylation clusters that converged between cells with reduced dosage of TCF4 and EHMT1 and observed highly significant correlations between methylation direction (hyper- or hypomethylation) and valence (extent of the change) of both disease models in comparison to nontarget control cells. This suggests that DNA-methylation patterns in both disease states might be similarly altered and that the diseases might converge at these genomic locations. We also performed a methylation sequencing experiment in nontarget controls in proliferating and differentiating cell states to assess whether methylation patterns in both disease models are more characteristic of the differentiating state, as was suggested by the RNA-seq and miRNA analyses. Too few DMRs were common between disease models and the cell-state experiment for us to determine whether the direction of methylation differences was more similar to that in proliferating or differentiating cells, although we did observe the expected pattern when we analyzed individual CpG sites. Several DMRs were identified within genes that were either lowly expressed in FBCs or not expressed at all. For example, in FBCs with reduced dosage of TCF4 and EHMT1, a DMR in RUNX2 matched the methylation pattern of differentiating FBCs and differed from the increased methylation observed in proliferating nontarget control FBCs. RUNX2 is a transcription factor important in osteogenesis and has been implicated in the bone morphology and craniofacial anomalies of 9q34 deletion syndrome.42 Interestingly, RUNX2 is also thought to be one target of SATB2 (SATB2 [MIM 608148] has been implicated in 2q33.1 deletion syndrome [MIM 612313], a disease not modeled here) and might be important in a bone morphology phenotype43 in subjects with 2q33.1 haploinsufficiency. Why there is a DMR in this gene in cells with reduced dosage of EHMT1 and TCF4 is not known, and FPKMs for this gene range from 3 to 6 in either proliferating or differentiating FBCs. It could be that the aberrant methylation pattern observed in FBCs is recapitulated in osteoblasts but that the expression of RUNX2 is so much higher in osteoblasts that this DMR affects expression only in those cells and not neural progenitor cells.
Our final analysis to assess convergence between TCF4 and EHMT1 function was to determine whether DNA-binding targets of TCF4 and EHMT1 overlap in normal FBCs (i.e., those without viral transfections). We observed a strongly significant overlap between TCF4 and EHMT1 target genes, although this only represented 29 different target genes. EHMT1 appeared to bind near the TSS and formed large (500–800 bp) clusters; TCF4 was more likely to associate with introns and seemed to bind in dimer given the pattern of sequenced reads. When we calculated the overlap of genes whose expression differed from that of those targeted by either TCF4 or EHMT1, we only found significant effects for EHMT1, meaning that TCF4 might exert its effects by binding directly to target genes. This is noteworthy because of the remarkable repressive effect that TCF4 clearly has on gene expression—all differentially expressed genes from the TCF4-KD RNA-seq experiment had increased expression. How TCF4 accomplishes this can thus not be explained by its DNA-binding patterns; however, TCF4 rarely bound promoter regions, so it could be that there is a direct association between TCF4 DNA binding and expression but that TCF4 exerts its repressive effects from some distance.
These data suggest that 18q21 and 9q34 deletion syndromes have significant molecular convergence with respect to DNA methylation, miRNA, and DNA-binding targets but that each maintains a distinct expression, epigenetic, and DNA-binding profile. Our studies comparing analyses of disease-model cells and control proliferating and differentiating cells suggest that disease-model cells share some characteristics with differentiating cells. Collectively, these data lead us to suggest that neural progenitor cells with reduced dosage of TCF4 and EHMT1 might share characteristics with differentiating cells that might make them vulnerable to aberrant timing of cell differentiation when they respond to external signaling cues during human brain development. This hypothesis should be addressed in future studies of reduced dosage of TCF4 and EHMT1, as well as of other genetically defined NDDs.
Acknowledgments
This work was funded by a grant from the Scottish Rite Charitable Foundation and the Banting Foundation to C.E., who is supported by the Canada Research Chairs program. C.O.G. is supported by a Conselho Nacional de Desenvolvimento Científico e Tecnológico scholarship, E.S.C. is supported by a fellowship from Fundação de Amparo à Pesquisa do Estado de São Paulo, K.V. and J.P.L. are supported by the Canadian Institute of Health Research, and L.C. is supported by the Fonds de Recherche de Quebec Santé. We are grateful to Maria Antonietta Davoli and Naguib Mechawar for help and advice on performing fluorescent microscopy and microscopy analysis.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Calculation of the hypergeometric distribution, http://nemates.org/MA/progs/overlap_stats.html
Chip-Enrich, http://chip-enrich.med.umich.edu/chipMain.jsp
Database of Genomic Variants, http://dgv.tcag.ca/dgv/app/home
HOMER, http://homer.salk.edu/
ImageJ, http://imagej.nih.gov/ij/
Integrative Genomics Viewer, http://www.broadinstitute.org/igv/home
The RNAi Consortium, http://www.broadinstitute.org/rnai/trc
Trim Galore, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
UCSC Human Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Krumm N., O’Roak B.J., Shendure J., Eichler E.E. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37:95–105. doi: 10.1016/j.tins.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pinto D., Delaby E., Merico D., Barbosa M., Merikangas A., Klei L., Thiruvahindrapuram B., Xu X., Ziman R., Wang Z. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014;94:677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams H.J., Craddock N., Russo G., Hamshere M.L., Moskvina V., Dwyer S., Smith R.L., Green E., Grozeva D., Holmans P. Most genome-wide significant susceptibility loci for schizophrenia and bipolar disorder reported to date cross-traditional diagnostic boundaries. Hum. Mol. Genet. 2011;20:387–391. doi: 10.1093/hmg/ddq471. [DOI] [PubMed] [Google Scholar]
- 4.Talkowski M.E., Rosenfeld J.A., Blumenthal I., Pillalamarri V., Chiang C., Heilbut A., Ernst C., Hanscom C., Rossin E., Lindgren A.M. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleefstra T., Kramer J.M., Neveling K., Willemsen M.H., Koemans T.S., Vissers L.E., Wissink-Lindhout W., Fenckova M., van den Akker W.M., Kasri N.N. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am. J. Hum. Genet. 2012;91:73–82. doi: 10.1016/j.ajhg.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reynolds B.A., Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev. Biol. 1996;175:1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- 7.Baker S.C., Bauer S.R., Beyer R.P., Brenton J.D., Bromley B., Burrill J., Causton H., Conley M.P., Elespuru R., Fero M., External RNA Controls Consortium The External RNA Controls Consortium: a progress report. Nat. Methods. 2005;2:731–734. doi: 10.1038/nmeth1005-731. [DOI] [PubMed] [Google Scholar]
- 8.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vlachos I.S., Kostoulas N., Vergoulis T., Georgakilas G., Reczko M., Maragkakis M., Paraskevopoulou M.D., Prionidis K., Dalamagas T., Hatzigeorgiou A.G. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012;40(Web Server issue):W498–W504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen G.G., Diallo A.B., Poujol R., Nagy C., Staffa A., Vaillancourt K., Lutz P.E., Ota V.K., Mash D.C., Turecki G., Ernst C. BisQC: an operational pipeline for multiplexed bisulfite sequencing. BMC Genomics. 2014;15:290. doi: 10.1186/1471-2164-15-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ernst C., McGowan P.O., Deleva V., Meaney M.J., Szyf M., Turecki G. The effects of pH on DNA methylation state: In vitro and post-mortem brain studies. J. Neurosci. Methods. 2008;174:123–125. doi: 10.1016/j.jneumeth.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 15.Krueger F., Andrews S.R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Craddock N., Hurles M.E., Cardin N., Pearson R.D., Plagnol V., Robson S., Vukcevic D., Barnes C., Conrad D.F., Giannoulatou E., Wellcome Trust Case Control Consortium Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lionel A.C., Crosbie J., Barbosa N., Goodale T., Thiruvahindrapuram B., Rickaby J., Gazzellone M., Carson A.R., Howe J.L., Wang Z. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci. Transl. Med. 2011;3:95ra75. doi: 10.1126/scitranslmed.3002464. [DOI] [PubMed] [Google Scholar]
- 20.Krawczak M., Nikolaus S., von Eberstein H., Croucher P.J., El Mokhtari N.E., Schreiber S. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006;9:55–61. doi: 10.1159/000090694. [DOI] [PubMed] [Google Scholar]
- 21.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Peltonen L., International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaikh T.H., Gai X., Perin J.C., Glessner J.T., Xie H., Murphy K., O’Hara R., Casalunovo T., Conlin L.K., D’Arcy M. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu H., Poh W.T., Sim X., Ong R.T., Suo C., Tay W.T., Khor C.C., Seielstad M., Liu J., Aung T. SgD-CNV, a database for common and rare copy number variants in three Asian populations. Hum. Mutat. 2011;32:1341–1349. doi: 10.1002/humu.21601. [DOI] [PubMed] [Google Scholar]
- 26.Ogawa H., Ishiguro K., Gaubatz S., Livingston D.M., Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- 27.Roy P.J., Stuart J.M., Lund J., Kim S.K. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature. 2002;418:975–979. doi: 10.1038/nature01012. [DOI] [PubMed] [Google Scholar]
- 28.Pollard S.M., Conti L., Sun Y., Goffredo D., Smith A. Adherent neural stem (NS) cells from fetal and adult forebrain. Cereb. Cortex. 2006;16(Suppl 1):i112–i120. doi: 10.1093/cercor/bhj167. [DOI] [PubMed] [Google Scholar]
- 29.Baek S.T., Kerjan G., Bielas S.L., Lee J.E., Fenstermaker A.G., Novarino G., Gleeson J.G. Off-target effect of doublecortin family shRNA on neuronal migration associated with endogenous microRNA dysregulation. Neuron. 2014;82:1255–1262. doi: 10.1016/j.neuron.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turcatel G., Rubin N., El-Hashash A., Warburton D. MIR-99a and MIR-99b modulate TGF-β induced epithelial to mesenchymal plasticity in normal murine mammary gland cells. PLoS ONE. 2012;7:e31032. doi: 10.1371/journal.pone.0031032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viñas J.L., Ventayol M., Brüne B., Jung M., Sola A., Pi F., Mastora C., Hotter G. miRNA let-7e modulates the Wnt pathway and early nephrogenic markers in mouse embryonic stem cell differentiation. PLoS ONE. 2013;8:e60937. doi: 10.1371/journal.pone.0060937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng Z., Du W.W., Fang L., Shan S.W., Qian J., Lin J., Qian W., Ma J., Rutnam Z.J., Yang B.B. The intermediate filament vimentin mediates microRNA miR-378 function in cellular self-renewal by regulating the expression of the Sox2 transcription factor. J. Biol. Chem. 2013;288:319–331. doi: 10.1074/jbc.M112.418830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anokye-Danso F., Trivedi C.M., Juhr D., Gupta M., Cui Z., Tian Y., Zhang Y., Yang W., Gruber P.J., Epstein J.A., Morrisey E.E. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376–388. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lipchina I., Studer L., Betel D. The expanding role of miR-302-367 in pluripotency and reprogramming. Cell Cycle. 2012;11:1517–1523. doi: 10.4161/cc.19846. [DOI] [PubMed] [Google Scholar]
- 35.Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 36.Talkowski M.E., Mullegama S.V., Rosenfeld J.A., van Bon B.W., Shen Y., Repnikova E.A., Gastier-Foster J., Thrush D.L., Kathiresan S., Ruderfer D.M. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 2011;89:551–563. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Myant K., Termanis A., Sundaram A.Y., Boe T., Li C., Merusi C., Burrage J., de Las Heras J.I., Stancheva I. LSH and G9a/GLP complex are required for developmentally programmed DNA methylation. Genome Res. 2011;21:83–94. doi: 10.1101/gr.108498.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sasai Y., De Robertis E.M. Ectodermal patterning in vertebrate embryos. Dev. Biol. 1997;182:5–20. doi: 10.1006/dbio.1996.8445. [DOI] [PubMed] [Google Scholar]
- 39.Qu Q., Sun G., Murai K., Ye P., Li W., Asuelime G., Cheung Y.T., Shi Y. Wnt7a regulates multiple steps of neurogenesis. Mol. Cell. Biol. 2013;33:2551–2559. doi: 10.1128/MCB.00325-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korzh V.P. Genetic control of early neuronal development in vertebrates. Curr. Opin. Neurobiol. 1994;4:21–28. doi: 10.1016/0959-4388(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 41.Suh M.R., Lee Y., Kim J.Y., Kim S.K., Moon S.H., Lee J.Y., Cha K.Y., Chung H.M., Yoon H.S., Moon S.Y. Human embryonic stem cells express a unique set of microRNAs. Dev. Biol. 2004;270:488–498. doi: 10.1016/j.ydbio.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 42.Balemans M.C., Ansar M., Oudakker A.R., van Caam A.P., Bakker B., Vitters E.L., van der Kraan P.M., de Bruijn D.R., Janssen S.M., Kuipers A.J. Reduced Euchromatin histone methyltransferase 1 causes developmental delay, hypotonia, and cranial abnormalities associated with increased bone gene expression in Kleefstra syndrome mice. Dev. Biol. 2014;386:395–407. doi: 10.1016/j.ydbio.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 43.Dobreva G., Chahrour M., Dautzenberg M., Chirivella L., Kanzler B., Fariñas I., Karsenty G., Grosschedl R. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell. 2006;125:971–986. doi: 10.1016/j.cell.2006.05.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.