

Abstract
An enantiospecific synthesis of a Nuphar alkaloid was achieved in 9 steps from N-Boc-(L)-proline. The alkaloid is a minor component of castoreum, the dried scent glands of the beaver. During the course of our study, the stereochemistry of three synthetic intermediates was verified by X-ray analysis, which contributes to resolving existing discrepancies among the literature reports regarding the synthesis of this particular compound. Based on our synthesis, we propose the structure of the natural product. Also, intrigued by castoreum’s therapeutic effect, which was used in ancient Greece and Rome for gynecological and other purposes, biological screening was conducted. We found that the alkaloid has affinity for the oxytocin receptor.
Keywords: nuphar, alkaloid, indolizidine, oxytocin, castoreum
Introduction
Asian traditional medicinal plants, especially from Japan and China, have been a rich source of plant-derived alkaloids. In this respect, Nuphar alkaloids are no exception. In 1931, an alkaloid was found in the rhizome of an aquatic plant, Nuphar japonicum DC, which has been traditionally used for medicinal purposes in Japan.1 Later, several similar alkaloids were isolated from other plants.2 Typically, those alkaloids possess a 3-furan group attached to quinolizidines, indolizidines, or piperidines. They have been the focus of chemical and biomedical research due to interesting biological activities such as apoptosis induction, metastatic arrest, and immunosuppression.3
Plants are not the only source of Nuphar alkaloids. They were also discovered in the extract of the dried scent glands of beavers, known as castoreum. Castoreum is a strong-smelling secretion produced in the castor sacs of beavers.4 It is believed that beavers use their secretion to mark territory.
Although it is unclear at what point people started using castoreum for any purpose, its history can be traced back to ancient Greece (Hippocrates,5 Herodotus6) and Rome (Lucretius,7 Pliny,8 Julius Caesar9). In those times, castoreum was used as a remedy for various medical conditions such as heart palpitation, vertigo, tremor, coma, and others. Among the many therapeutic effects of castoreum, however, it was best known as an emmenagogue in gynecology that promotes placental expulsion and menstruation.9,10 More recently, castoreum has been extensively used as an ingredient for perfume making and has been categorized by the FDA as “generally recognized as safe” (GRAS).11
In 1976, Maurer and Ohloff reported the isolation of fourteen nitrogen-containing compounds from castoreum, eight of which were (–)-castoramin and closely related analogs (Figure 1).12 Compound A (5-(3-furyl)-8-methyl-octahydroindolizin) is a minor component (<0.0002%) in castoreum (Figure 2). Due to insufficient quantities from natural sources, its structure was assigned based on mass fragmentation analysis. The relative and absolute stereochemistry remained ambiguous at the time and no biological activity of the compound has been reported to date.
Figure 1.

Structures of the major alkaloids in castoreum.
Figure 2.

Structures of minor alkaloid A in castoreum and proposed synthetic intermediate B.
During the last several years, our group has developed novel chemistry for the syntheses of monocyclic and bicyclic enaminones (2,3-dihydropyridin-4(1H)-ones).13 We and Comins have demonstrated their utility for the syntheses of natural products.13,14 In this regard, we envisioned that alkaloid A could be derived from enaminone B (Figure 2). Herein, we report our synthetic efforts and propose the structure of the natural product along with investigations into its biological activities.
Three isomers of alkaloid A have been synthesized in racemic form (Figure 3).15 In pursuit of structural determination of the natural product, four groups have analyzed their final products by GC/MS. Lalonde and Tufariello independently analyzed rac-1 and observed an identical fragmentation spectrum to that of the natural product. Ban noted in his paper that the GC/MS spectra of his compounds (rac-2 and rac-3) were almost identical to that of the natural product. Kurihara also observed in his synthesis of rac-3 that the mass spectrum of his final compound was identical to that of the natural product. This led us to hypothesize that the stereochemical difference of these compounds might not be reflected in their GC/MS fragmentation pattern, which is understandable considering the structural similarity of the alkaloids (rac-1, rac-2, and rac-3).
Figure 3.

Structures of isomers of alkaloid A and prior racemic (top) and enantiospecific syntheses (bottom).
As for the absolute stereochemistry of the alkaloid, to date three different groups have reported enantiospecific syntheses of alkaloids 1 and 2.16 Assuming that the biosynthetic pathways of the compounds in castoreum are similar to each other, Kunz suggested that either alkaloid 1 or 2 is the natural product, and embarked on their syntheses. With both alkaloids in hand, Kunz analyzed them using GC/MS and observed that the fragmentation of 2 is in agreement with that of the natural product, whereas the fragmentation of 1 had several missing peaks in comparison to the GC/MS reported for the natural product.12,17 This observation, however, does not agree with the published reports by LaLonde, Tufariello, Ban, and Kurihara, which demonstrated that all three isomers display essentially the same GC/MS fragmentation patterns.
Upon close inspection of the supporting information from prior publications, we found that the 1H NMR data for compound 1 reported by LaLonde or Tufariello are partly different from the ones reported by Barluenga.18 Also, the 1H NMR data of compound 1 in Davis’ report16b are different from the ones by Barluenga and Kunz.19 Interestingly the 13C NMR data for compound 1 reported by Kunz, Barluenga, and LaLonde matched well. We also noticed that the 1H NMR data of compound 2 reported by Ban differ from the ones reported by Kunz.15b, 16c Considering the relatively simple structure of the target compound, this inconsistency between the 1H NMR spectra is quite surprising. In order to resolve these existing contradictions, we recognized that our synthetic approach must have a way to determine these structures in an unambiguous manner.
Results and Discussion
Our synthesis started from N-Boc-(L)-proline, which was converted to the homologated Weinreb amide 4 in two steps using a reported procedure (Scheme 1).14e Weinreb amide 4 was reacted with MeLi to furnish methylketone 5. Next, this ketone underwent a boron-aldol reaction with 3-furancarboxaldehyde, providing β-hydroxyketone 6.20 The hydroxyl group of β-hydroxyketone 6 was oxidized to diketone 7 with IBX.21 This diketone was reacted with formic acid to deprotect the Boc group. After evaporation of the formic acid, the reaction mixture was treated with K2CO3 in MeOH, which led to the spontaneous cyclization of the intermediate amino ketone, affording the desired enaminone 8. The structure of enaminone 8 was confirmed by X-ray analysis.
Scheme 1.

Synthesis of enaminone 8.
The α-alkylation of enaminone 8 was found to be somewhat challenging (Scheme 2). However, after screening several electrophiles, an ester group was successfully installed on enaminone 8 using Mander’s reagent, affording β-ketoester 9. The structure of 9 was confirmed by X-ray analysis. Next, the α-tertiary center of 9 was alkylated using MeI. The resulting diastereomeric mixture of β-ketoesters was subjected to a Krapcho decarboxylation in a microwave reactor at 160 °C.22 The α-methylated enaminones 10a (73%) and 10b (21%) were separated by column chromatography. The coupling constants of the two geminal hydrogens clearly indicated the relative stereochemistry of the two stereocenters.
Scheme 2.

Synthesis of α-methylenaminones 10a and 10b.
The major diastereomer 10a was first reacted with Tf2O, and the resulting iminium enol triflate was reduced with LAH to provide the corresponding triflate (Scheme 3).23 This crude triflate was directly subjected to Pd-catalyzed reduction to afford the final product 1. The minor diastereomer 10b was subjected to similar conditions, furnishing the epimer 2. The 1H NMR and 13C NMR data of both alkaloids were identical to those reported by Kunz. The specific optical rotations of alkaloids 1 and 2 were −121 and −96 respectively (both in CHCl3). Although the magnitude of the rotations was slightly different, the negative signs of the rotations were consistent with the data from Kunz (1: −94.5; 2: −57.9, both in CHCl3), Barluenga (1: −99.0 in CH2Cl2), and Davis (1: −98.4 in CH2Cl2). Given the inconsistency of the 1H NMR data, however, we decided to further investigate our compounds.
Scheme 3.

Synthesis of alkaloids 1 and 2.
To establish the stereochemistry of the methyl and furan substituents unambiguously, the major diastereomer 10a was again reacted with Tf2O to furnish the corresponding crude triflate (Scheme 4). Instead of reducing the triflate functionality with hydrogen, this triflate was hydrolyzed to ketone 11, which had already been reported by Davis.16b Although we observed a significant difference in the 1H NMR data to those reported by Davis, the structure of our compound was confirmed by X-ray analysis, thus providing definitive proof that our synthesis indeed produced the desired alkaloids.
Scheme 4.
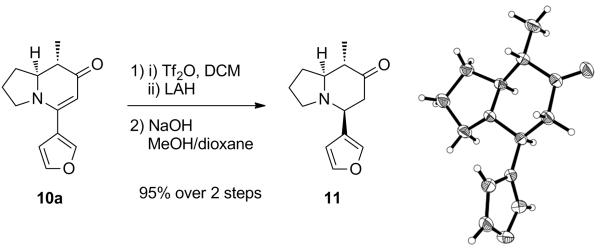
Synthesis of known ketone 11.
Given the inconsistent GC/MS information among the prior reports, we analyzed our compounds 1 and 2 using EI-GC/MS (70 eV). Unlike what was reported by Kunz, however, the fragmentation of both compounds was essentially identical to that of the natural product,19 which leaves the relative configuration of the methyl-bearing carbon in the natural product undetermined. Although we have little information to speculate why the fragmentation of Kunz’s compound 1 produced a spectrum different from that of the natural product,17 our results are supported by considering LaLonde, Tufariello, and Ban’s earlier observations, who found that their compounds rac-1, rac-2 and rac-3 showed fragmentations similar to the natural product.
Since the GC/MS did not provide information about the structure of the natural product, we realized that the only way to identify the structure was to isolate the natural product and analyze by 1H NMR. Although we have attempted the isolation from locally- purchased castor sacs several times, we were not successful in isolating the natural product. Thus, we looked for clues elsewhere. Firstly, in our synthesis, we found that alkaloid 2 is relatively unstable and volatile. Compound 1 formed white crystals at 0 °C whereas compound 2 was a clear oil. Secondly, the other alkaloid components in castoreum were taken into consideration. All three major alkaloids (castoramin and its epimers) share a configuration in which the methyl group is in a trans relationship to the furan group. Although biosynthetic pathways of these compounds have not been reported, it is a reasonable conjecture that the minor alkaloid A underwent a similar biosynthetic route as the other castoramins and therefore shares a similar structure. These two factors – 1) stability of the compound, and 2) the structure of castoramin – led us to the conclusion that alkaloid 1 is most likely the natural product.
The final compound 1, synthetic intermediates thereof, and a few derivatives were tested in an MCF-7 cancer cell line at 10 μM for their cytotoxic activity. As expected from the industrial application of castoreum, no cytotoxicity was observed for any of the compounds.
Our final compound 1 was also submitted to the NIMH-Psychoactive Drug Screening Program at the University of North Carolina and was tested in CNS receptor assays.24 Although compound 1 was inactive in most of the assays, binding to three receptors was observed: oxytocin receptor (Ki = 0.6 μM), sigma 1 receptor (0.23 μM), and sigma 2 receptor (1.8 μM). Our compound was found to be selective for the oxytocin receptor over vasopressin receptors (Ki > 10 μM), which have structural similarity with oxytocin receptors. It is quite interesting that our small non-peptidic compound (molecular weight = 205), possesses a certain level of selectivity between those two receptors.25
The oxytocin receptor is a member of GPCR and expressed in brain, uterus and others.26 Its natural ligand, oxytocin, is a nonapeptide hormone and involved in parturition. It can stimulate milk ejection and uterine contractions, and also can modulate mood and promote human bondings.27 Although we can only conjecture as to how castoreum was obtained and applied to humans in ancient Greece and Rome, the affinity of compound 1 to the oxytocin receptor is quite intriguing, and could potentially explain the therapeutic effects of castoreum in gynecological applications, considering the structural similarities of the components in castoreum.
With regards to the sigma receptors, a number of synthetic ligands are known to bind to this receptor, most of which have basic amine functionalities, as exemplified in alkaloid 1.28 However, even today the precise role of the receptors is quite ambiguous since they have been implicated in many therapeutic areas such as cardiology, psychiatry, immunology, and oncology.29 The correlation between castoreum’s affinities to the sigma receptors and its therapeutic effects thus remains obscure at this point.
Conclusions
In summary, an enantiospecific synthesis of Nuphar alkaloids 1 and 2 was achieved from enaminone intermediate 8. The reactions employed were reliable and scalable. The final compounds were obtained in 9 steps from readily available N-Boc-(L)-proline, which renders our synthesis the shortest and most efficient to date. The X-ray analysis validated the syntheses of the two alkaloids as well as synthetic intermediates in an unambiguous manner, which should clarify some of the contradictions in the literature. It is the first time that any biological activity for alkaloid 1 has been reported. In particular, the affinity of compound 1 to the oxytocin receptor is a noteworthy finding, given that castoreum was used in gynecology in ancient days.
Experimental Section
Unless otherwise specified, all reactions were conducted in oven-dried glassware under a nitrogen atmosphere using anhydrous solvents. 3-Furancarboxaldehyde was purified by distillation prior to use. IBX was freshly prepared according to the literature procedure.30 Unless otherwise specified, NMR experiments were conducted at 23 °C. Optical rotations were measured at 23 °C.
(S)-tert-Butyl 2-(2-Oxopropyl)pyrrolidine-1-carboxylate (5)
To a solution of Weinreb amide 4 (5.82 g, 21.4 mmol, 1 equiv) in THF (80 mL) was added slowly MeLi (1.6 M in THF, 33 mL, 53.5 mmol, 2.5 equiv) at −78 °C. After 5 h, additional MeLi (20 mL, 32.1 mmol, 1.5 equiv) was added to the reaction mixture. After 2 h, the reaction was quenched by the addition of 10% H2O in THF (10 mL) and warmed to 0 °C, which was then treated with aqueous HCl (3 M, 30 mL). The partitioned organic layer was washed with aqueous NH4Cl, brine, dried over MgSO4, and concentrated in vacuo. The crude mixture was purified by flash column chromatography (10-50% EtOAc/hexanes) on silica gel and yielded 3.96 g (81%, recovered SM: 8%) of 5 as a clear oil; 1H NMR (400 MHz, CDCl3, 60 °C) δ 4.14 (ddd, J = 11.2, 7.4, 3.5 Hz, 1H), 3.33 (dt, J = 10.2, 5.6 Hz, 2H), 3.00 (s, 1H), 2.41 (dd, J = 15.8, 9.4 Hz, 1H), 2.13 (d, J = 3.1 Hz, 3H), 2.10 – 2.01 (m, 1H), 1.85 – 1.74 (m, 2H), 1.68 – 1.60 (m, 1H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3, 60 °C) δ 207.1, 154.5, 79.6, 53.8, 48.6, 46.6, 31.5, 30.5, 28.7, 23.5; IR (neat, cm−1): 2974, 1692, 1397, 1170, 1103; [α]D −36.9 (c 1.39, CHCl3); HRMS (ESI) calcd for C12H22NO3 (M +H)+ 228.1600, found 228.1603. [Due to the Boc rotamers, the NMR experiments were conducted at a higher temperature.]
(2S)-tert-Butyl 2-(4-(Furan-3-yl)-4-hydroxy-2-oxobutyl)pyrrolidine-1-carboxylate (6)
To a solution of methylketone 5 (4.00 g, 17.4 mmol, 1 equiv) and DIPEA (2.58 g, 20.0 mmol, 1.2 equiv) in Et2O (140 mL) was added slowly n-Bu2BOTf (1 M, 18.3 mL, 18.3 mmol, 1.1 equiv) via a syringe pump at −78 °C. After 1 h, 3-furancarboxaldehyde (1.92 g, 20.0 mmol, 1.2 equiv) in Et2O (10 mL) was added dropwise again via a syringe pump over 1h. After 4 h, 7.2 phosphate buffer (20 mL) in MeOH (80 mL) was added to the reaction mixture, which was then warmed to 0 °C, followed by the addition of H2O2 (30%, 20 mL) in MeOH (40 mL). After stirring for 1 h at ambient temperature, the organic layer of the reaction mixture was evaporated under reduced pressure, and Et2O/H2O were added to the solution. The partitioned aqueous layer was extracted with Et2O (×3), and the combined organic layers were washed with aqueous NaHCO3, brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash column chromatography (13-25% EtOAc/hexanes) on silica gel and yielded 5.60 g (quant.) of 6 as a clear oil; 1H NMR (400 MHz, CDCl3, 60 °C) δ 7.38 (dd, J = 9.0, 1.1 Hz, 2H), 6.38 (s, 1H), 5.15 (s, 1H), 4.18 (s, 1H), 3.69 – 3.05 (m, 3H), 3.03 – 2.72 (m, 3H), 2.46 (dd, J = 15.6, 8.3 Hz, 1H), 2.08 (dq, J = 15.9, 8.0 Hz, 1H), 1.82 (d, J = 5.3 Hz, 2H), 1.65 (dt, J = 10.7, 4.9 Hz, 1H), 1.45 (dd, J = 5.0, 4.3 Hz, 9H); 13C NMR (100 MHz, CDCl3, 60 °C) δ 209.0, 154.8, 143.5, 139.2, 128.1, 108.7, 79.8, 63.4, 53.8, 50.9, 48.7, 46.6, 31.7, 28.7, 23.6; IR (neat, cm−1): 3421, 2974, 1690, 1400, 1168, 1121, 1026, 875; [α]D −25 (c 1.1, CHCl3); HRMS (ESI) calcd for C17H26NO5 (M+H)+ 324.1811, found 324.1806. [Despite a number of NMR experiments using different solvents at various temperatures, we were unsuccessful to observe the single isomers of intermediate 6 in the spectra. We found that the best results were obtained in CDCl3.]
(S)-tert-Butyl 2-(4-(Furan-3-yl)-2,4-dioxobutyl)pyrrolidine-1-carboxylate (7)
To a solution of hydroxyketone 6 (2.10 g, 6.5 mmol, 1 equiv) was added freshly prepared IBX (5.5 g, 19.5 mmol, 3 equiv) at room temperature. After heating at 60 °C for 3 h, additional IBX (1.82 g, 6.5 mmol, 1 equiv) was added to the reaction mixture, which was further stirred overnight. Upon Celite filtration, the crude mixture was purified by flash column chromatography (13-17% EtOAc/hexanes) on silica gel and yielded 1.68 g (80%) of 7 as a clear oil; 1H NMR (400 MHz, d4-MeOD) δ 8.23 (s, 1H), 7.61 (s, 1H), 6.81 (s, 1H), 6.07 (d, J = 14.5 Hz, 1H), 4.19 (s, 1H), 3.34 (d, J = 16.0 Hz, 3H), 2.83 (m, 1H), 2.41 (s, 1H), 1.95 (m, 4H), 1.46 (s, 9H); 13C NMR (100 MHz, d4-MeOD, 60 °C) δ 191.7, 182.4, 156.2, 147.7, 145.9, 126.1, 109.0, 99.3, 81.1, 56.5, 47.4, 43.3, 31.5, 28.8, 24.0; IR (neat, cm−1): 2974, 1690, 1619, 1396, 1160, 1119; [α]D −4.7 (c 1.1, CHCl3); HRMS (ESI) calcd for C17H24NO5 (M+H)+ 322.1654, found 322.1651. [Despite a number of NMR experiments using different solvents at various temperatures, we were unsuccessful to observe a single isomer/rotamer of intermediate 7 in the spectra. We found that the best results were obtained in deuterated methanol.]
(S)-5-(Furan-3-yl)-2,3,8,8a-tetrahydroindolizin-7(1H)-one (8)
To a cold flask in an ice-bath, containing 7 (1.43 g, 4.46 mmol, 1 equiv) was added cold (0 °C) formic acid (45 mL). After the addition, the reaction mixture was allowed to reach room temperature and was stirred for 6 h at ambient temperature, after which the reaction mixture was concentrated in vacuo. Next, MeOH (45 mL) was added to the residue and cooled to 0 °C. After addition of K2CO3 (1.23 g, 8.91 mmol, 2 equiv), the ice bath was removed and the reaction mixture was stirred overnight. Upon evaporation of MeOH, resulting crude mixture was extracted with CH2Cl2 (×3). The extraction was concentrated, subjected to flash column chromatography (2-3% MeOH/CH2Cl2) on silica gel, and yielded 845 mg (93%) of 8 as a white solid; mp: 107.6-108.5 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.46 (d, J = 1.6 Hz, 1H), 6.53 (d, J = 1.0 Hz, 1H), 5.19 (s, 1H), 4.03 – 3.91 (m, 1H), 3.71 – 3.63 (m, 1H), 3.56 – 3.48 (m, 1H), 2.44 (s, 1H), 2.41 (d, J = 2.7 Hz, 1H), 2.31 (m, 1H), 2.15 – 2.04 (m, 1H), 1.94 (m, 1H), 1.77 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 192.0, 154.3, 143.6, 142.7, 121.9, 110.1, 98.9, 59.3, 49.6, 41.6, 32.0, 24.7; IR (neat, cm−1): 3117, 2968, 2879, 1622, 1507, 1266, 1162, 1045; [α]D −739 (c 0.590, CHCl3); HRMS (ESI) calcd for C12H14NO2 (M+H)+ 204.1025, found 204.1020.
(8S,8aS)-Methyl 5-(Furan-3-yl)-7-oxo-1,2,3,7,8,8a-hexahydroindolizine-8-carboxylate (9)
To a solution of diisopropylamine (263 mg, 2.6 mmol, 1.3 equiv) in THF (10 mL) at −78 °C was slowly added n-BuLi (1.6 M, 1.63 mL, 2.6 mmol, 1.3 equiv). The solution was warmed to 0 °C and stirred for 10 min to generate LDA, and cooled again to −78 °C. Enaminone 8 (408 mg, 2.0 mmol, 1.0 equiv) in THF (5 mL) was added dropwise to the LDA solution, which was stirred for 1 h. Methyl cyanoformate (Mander’s reagent, 272 mg, 3.2 mmol, 1.6 equiv) was added to the reaction mixture. After 1 h, the reaction mixture was poured into ice and extracted with EtOAc (×3). The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by recrystallization (EtOAc/hexanes, twice, 1st crop and 2nd crop were combined) and yielded 468 mg (90%) of 9 as a yellow solid; mp: 140.2-141.1 °C; 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 7.47 (t, J = 1.6 Hz, 1H), 6.53 (d, J = 1.0 Hz, 1H), 5.25 (s, 1H), 4.29 (dt, J = 14.9, 7.3 Hz, 1H), 3.82 (s, 3H), 3.76 – 3.68 (m, 1H), 3.54 (dt, J = 10.6, 7.6 Hz, 1H), 3.41 (d, J = 15.2 Hz, 1H), 2.32 (m, 1H), 2.16 – 2.04 (m, 1H), 1.96 (m, 1H), 1.76 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 186.1, 170.1, 154.2, 143.8, 142.9, 121.5, 110.1, 98.5, 60.7, 56.7, 52.5, 50.0, 30.9, 24.5; IR (neat, cm−1): 1737, 1624, 1510, 1255, 1160; [α]D −729 (c 0.650, CHCl3); HRMS (ESI) calcd for C14H16NO4 (M +H)+ 262.1079, found 262.1074.
(8S,8aS)-5-(Furan-3-yl)-8-methyl-2,3,8,8a-tetrahydroindolizin-7(1H)-one (10a)
To a solution of enaminone 9 (501 mg, 1.92 mmol, 1.0 equiv) in THF (19 mL) at −78 °C was added NaHMDS (1.0 M, 2.10 mL, 2.1 mmol, 2.1 equiv). After 1 h, MeI (817 mg, 5.75 mmol, 3.0 equiv) was added to the reaction mixture. Again the reaction flask was stirred for 1 h, and warmed to 0 °C. After 1 h, the reaction was quenched with aqueous. NaHCO3 and then extracted with EtOAc (×3). The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The crude mixture was purified by flash column chromatography (1-3% MeOH/CH2Cl2), and two inseparable diastereomers were obtained (yellow oil).
To the resulting diastereomeric mixtures in DMSO (19 mL) was added NaCN (466 mg, 9.5 mmol, 5.0 equiv) and H2O (171 mg, 9.5 mmol, 5.0 equiv). This reaction mixture was heated to 160 °C using a microwave for 1.5 h. EtOAc and water were added to the reaction mixture, which was extracted with EtOAc (×3). The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The crude products were purified by flash column chromatography (25-33% EtOAc/hexanes for the major diastereomer, 1-3% MeOH/CH2Cl2 for the minor diastereomer) on silica gel to give each product. Colorless solid (303 mg, 73%, major diastereomer); mp: 128.7-129.6 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 – 7.66 (m, 1H), 7.45 (t, J = 1.7 Hz, 1H), 6.53 (dd, J = 1.8, 0.8 Hz, 1H), 5.21 (s, 1H), 3.77 – 3.67 (m, 1H), 3.58 – 3.45 (m, 2H), 2.33 (dtd, J = 12.6, 6.5, 3.2 Hz, 1H), 2.23 (dq, J = 13.7, 6.8 Hz, 1H), 2.09 (m, 1H), 1.99 – 1.87 (m, 1H), 1.84 – 1.73 (m, 1H), 1.15 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 194.2, 153.5, 143.5, 142.5, 121.8, 110.2, 98.6, 65.2, 49.8, 43.7, 31.7, 24.5, 11.8; IR (neat, cm−1): 3117, 2967, 2869, 1613, 1538, 1302, 1231, 1027; [α]D −688 (c 0.590, CHCl3); HRMS (ESI) calcd for C13H16NO2 (M+H)+ 218.1181, found 218.1180.
(8R,8aS)-5-(Furan-3-yl)-8-methyl-2,3,8,8a-tetrahydroindolizin-7(1H)-one (10b)
Colorless solid (88.2 mg, 21%, minor diastereomer); mp: 78.8-80.3 °C; 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 7.46 (t, J = 1.6 Hz, 1H), 6.56 – 6.53 (m, 1H), 5.12 (s, 1H), 4.04 (dd, J = 11.6, 7.4 Hz, 1H), 3.74 – 3.67 (m, 1H), 3.54 (dt, J = 10.7, 6.9 Hz, 1H), 2.31 – 2.23 (m, 1H), 2.14 – 2.03 (m, 2H), 2.01 – 1.90 (m, 2H), 1.04 (d, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 196.9, 153.3, 143.6, 142.6, 121.6, 110.1, 97.0, 62.4, 49.9, 42.5, 27.1, 25.0, 11.0; IR (neat, cm−1): 2967, 1619, 1584, 1511, 1474, 1258, 1161; [α]D −455 (c 0.840, CHCl3); HRMS (ESI) calcd for C13H16NO2 (M +H)+ 218.1181, found 218.1171.
(5S,8R,8aS)-5-(Furan-3-yl)-8-methyloctahydroindolizine (1)
To a solution of enaminone 10a (150 mg, 0.692 mmol, 1.0 equiv) in CH2Cl2 (7 mL) at −78 °C was added Tf2O (390 mg, 1.38 mmol, 2.0 equiv). The reaction mixture was warmed to ambient temperature and stirred for 3 h, which was then cooled to −78 °C. LAH (2.0 M, 0.69 mL, 1.38 mmol, 2.0 equiv) was added to the solution, which was warmed to ambient temperature. After 1 h, the reaction was quenched by the addition of saturated Rochelle salt solution (2 mL) at −78 °C. The partitioned aqueous layer was extracted with CH2Cl2, and the combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The crude product was carried to the next step without further purification. Pd/C (10%, 370 mg, 0.35 mmol, 0.5 equiv) was added to the crude triflate in MeOH (7 mL). H2 was initially bubbled through a needle to the reaction mixture for 5 min, which was then stirred overnight under a H2 atmosphere using a balloon. Upon Celite filtration, MeOH was evaporated and the crude mixture was re-dissolved in CH2Cl2, which was washed with aq. NaHCO3, dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash column chromatography on Al2O3 (10-20% Et2O/hexanes) and yielded 106 mg (75%) of 1 as a clear oil (upon cooling at 0 °C, the title compound formed colorless crystals); 1H NMR (400 MHz, CDCl3) δ 7.34 (m, 2H), 6.44 (s, 1H), 2.95 – 2.84 (m, 2H), 1.93 (m, 2H), 1.83 – 1.36 (m, 8H), 1.14 – 1.00 (m, 1H), 0.91 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 142.8, 139.5, 128.4, 109.9, 71.6, 59.9, 53.3, 36.6, 34.3, 34.1, 29.2, 20.3, 19.0; IR (neat, cm−1): 2953, 2926, 2781, 1501, 1458, 1166, 1023; [α]D −121 (c 1.17, CHCl3); HRMS (ESI) calcd for C13H20NO (M +H)+ 206.1545, found 206.1541.
(5S,8S,8aS)-5-(Furan-3-yl)-8-methyloctahydroindolizine (2)
To a solution of enaminone 10b (31 mg, 0.14 mmol, 1.0 equiv) in CH2Cl2 (0.8 mL) at −78 °C was added Tf2O (79 mg, 0.28 mmol, 2.0 equiv). The reaction mixture was warmed to 0 °C and stirred for 3 h, which was then cooled to −78 °C. LAH (2.0 M, 0.14 mL, 0.28 mmol, 2.0 equiv) was added to the solution, which was warmed to ambient temperature. The crude triflate as well as the title compound were obtained using the same protocol as described in the previous procedure, and yielded 7.6 mg (26%) of 2 as a clear oil; 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.30 (m, 2H), 6.43 (s, 1H), 2.90 – 2.83 (m, 2H), 2.14 – 2.06 (m, 1H), 1.93 (td, J = 6.8, 2.8 Hz, 1H), 1.87 – 1.75 (m, 2H), 1.70 – 1.53 (m, 6H), 1.52 – 1.45 (m, 1H), 1.04 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 142.7, 139.2, 129.0, 109.9, 67.7, 61.1, 53.6, 32.3, 29.6, 29.1, 27.0, 20.3, 12.4; IR (neat, cm−1): 2925, 1459, 1158, 1023, 873, 784; [α]D −96 (c 0.52, CHCl3); HRMS (ESI) calcd for C13H20NO (M +H)+ 206.1545, found 206.1535. [For the concentration of the eluent with the rotary evaporator, a temperature of 25 °C was used for water bath due to the low boiling point of the product.]
(5S,8S,8aS)-5-(Furan-3-yl)-8-methylhexahydroindolizin-7(1H)-one (11).
To a solution of the crude triflate (175 mg, 0.5 mmol, 1 equiv) obtained from 10a (109 mg, 0.5 mmol) in 1,4-dioxane (3 mL) and MeOH (1.5 mL) was added aqueous NaOH solution (1 M, 1.5 mL, 1.5 mmol, 3 equiv). After 6 h, MeOH was evaporated and EtOAc, and water were added to the crude mixture. The partitioned aqueous layer was extracted with EtOAc (×3), and the combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash column chromatography (1-3% MeOH/CH2Cl2) on silica gel and yielded 103 mg (95%) of 11 as a white solid; mp: 94.8-95.6 °C; 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.32 (m, 2H), 6.47 (s, 1H), 3.34 (dd, J = 11.9, 3.2 Hz, 1H), 2.94 (ddd, J = 9.0, 9.0, 2.0 Hz, 1H), 2.76 – 2.65 (ddd, J = 13.1, 13.0, 1.0 Hz, 1H), 2.50 – 2.39 (m, 2H), 2.09 – 1.97 (m, 3H), 1.92 – 1.79 (m, 1H), 1.79 – 1.58 (m, 2H), 1.04 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 209.8, 143.6, 139.6, 126.8, 109.1, 70.5, 58.1, 52.2, 50.5, 48.6, 30.5, 21.5, 10.7; IR (neat, cm−1): 3140, 2966, 2787, 1710, 1504, 1359, 1171, 1019; [α]D −97.6 (c 0.505, CHCl3); HRMS (ESI) calcd for C13H18NO2 (M+H)+ 220.1338, found 220.1341.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM081267) and the University of Minnesota through the Vince and McKnight Endowed Chairs. CNS target screen and Ki determination were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. Contract # HHSN-271-2008-025C. We thank Dr. Subhashree Francis for mass spectrometry assistance, Dr. Sean Murray for GC/MS assistance, Dr. Victor Young, Jr. for X-ray analysis, Dr. Defeng Tian for the cytotoxic assay, and David Huang for the attempt to isolate the natural product. We acknowledge helpful NMR discussion with Dr. Thomas R. Hoye, Dr. Franklin A. Davis, Dr. Letitia Yao, Alex Stoye, and Dr. David VanderVelde. The authors thank Mariko Seki for the drawing in the TOC.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.xxxxxxxxx.
Crystallographic data for compound 8, 9, and 11 have been deposited (CCDC 853380 – 853382). GC/MS data for compound 1 and 2, and characterization data and copies of the 1H NMR and 13C NMR spectra for all new compounds.
References
- [1].Arima J, Takahashi B. Nippon Kagaku Kaishi (1921-47) 1931;52:815–817. [Google Scholar]
- [2].(a) Arata Y. Chem. Pharm. Bull. 1964;12:1394–1395. doi: 10.1248/cpb.12.1394. [DOI] [PubMed] [Google Scholar]; (b) LaLonde RT, Wong CF, Cullen WP. Tetrahedron Lett. 1970:4477–4480. [Google Scholar]; (c) LaLonde RT, Wong CF. Phytochemistry. 1972;11:3305–3306. [Google Scholar]
- [3].(a) Matsuda H, Shimoda H, Yoshikawa M. Bioorg. Med. Chem. 2001;9:1031–1035. doi: 10.1016/s0968-0896(00)00327-8. [DOI] [PubMed] [Google Scholar]; (b) Matsuda H, Yoshida K, Miyagawa K, Nemoto Y, Asao Y, Yoshikawa M. Bioorg. Med. Chem. Lett. 2006;16:1567–1573. doi: 10.1016/j.bmcl.2005.12.032. [DOI] [PubMed] [Google Scholar]; (c) Matsuda H, Morikawa T, Oda M, Asao Y, Yoshikawa M. Bioorg. Med. Chem. Lett. 2003;13:4445–4449. doi: 10.1016/j.bmcl.2003.09.019. [DOI] [PubMed] [Google Scholar]
- [4].(a) Muller-Schwarze D, Houlihan PW. J. Chem. Ecol. 1991;17:715–734. doi: 10.1007/BF00994195. [DOI] [PubMed] [Google Scholar]; (b) Schulte BA, Muller-Schwarze D, Tang R, Webster FX. J. Chem. Ecol. 1995;21:941–957. doi: 10.1007/BF02033800. [DOI] [PubMed] [Google Scholar]
- [5].King H. Hippocrates’ Woman: Reading the Female Body in Ancient Greece. Routledge; London: 1998. [Google Scholar]
- [6].Thomas R. Herodotus in Context. Cambridge University Press; New York: 2000. [Google Scholar]
- [7].Lucretius T. On the Nature of Things. 6 [Google Scholar]
- [8].Pliny E. Natural History. XXXII [Google Scholar]
- [9].Riddle JM. Contraception and abortion from the ancient world to the Renaissance. Harvard University Press; Cambridge: 1992. [Google Scholar]
- [10].King AH. Ph.D. thesis. Indiana University; Ann Arbor: 2007. H.S. thanks Dr. Anya. H. King for providing a useful reference. [Google Scholar]
- [11].Burdock GA. Int. J. Toxicol. 2007;26:51–55. doi: 10.1080/10915810601120145. [DOI] [PubMed] [Google Scholar]
- [12].Maurer B, Ohloff G. Helv. Chim. Acta. 1976;59:1169–1185. [Google Scholar]
- [13].(a) Joseph S, Comins DL. Curr. Opin. Drug Discovery Dev. 2002;5:870–880. [PubMed] [Google Scholar]; (b) Comins DL. J. Heterocycl. Chem. 1999;36:1491–1500. [Google Scholar]; (c) Comins DL, Joseph SP. Adv. Nitrogen Heterocycl. 1996;2:251–294. [Google Scholar]
- [14].(a) Turunen BJ, Georg GI. J. Am. Chem. Soc. 2006;128:8702–8703. doi: 10.1021/ja0609046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Niphakis MJ, Turunen BJ, Georg GI. J. Org. Chem. 2010;75:6793–6805. doi: 10.1021/jo100907u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Seki H, Georg GI. J. Am. Chem. Soc. 2010;132:15512–15513. doi: 10.1021/ja107329k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Seki H, Georg GI. Org. Lett. 2011;13:2147–2149. doi: 10.1021/ol200358h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Niphakis MJ, Georg GI. J. Org. Chem. 2010;75:6019–6022. doi: 10.1021/jo101051w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Leighty MW, Georg GI. ACS Med. Chem. Lett. 2011;2:313–315. doi: 10.1021/ml1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Niphakis MJ, Georg GI. Org. Lett. 2011;13:196–199. doi: 10.1021/ol1023954. [DOI] [PubMed] [Google Scholar]
- [15].(a) LaLonde RT, Muhammad N, Wong CF, Sturiale ER. J. Org. Chem. 1980;45:3664–3671. [Google Scholar]; (b) Ohnuma T, Tabe M, Shiiya K, Ban Y, Date T. Tetrahedron Lett. 1983;24:4249–4252. [Google Scholar]; (c) Tufariello JJ, Dyszlewski AD. J. Chem. Soc., Chem. Commun. 1987:1138–1140. [Google Scholar]; (d) Kurihara T, Matsubara Y, Osaki H, Harusawa S, Yoneda R. Heterocycles. 1990;30:885–896. [Google Scholar]; (e) Clive DLJ, Bergstra RJ. J. Org. Chem. 1991;56:4976–4977. [Google Scholar]; (f) Bates RW, Lim CJ. Synlett. 2010:866–868. [Google Scholar]
- [16].(a) Barluenga J, Aznar F, Ribas C, Valdes C. J. Org. Chem. 1999;64:3736–3740. doi: 10.1021/jo9823240. [DOI] [PubMed] [Google Scholar]; (b) Davis FA, Santhanaraman M. J. Org. Chem. 2006;71:4222–4226. doi: 10.1021/jo060371j. [DOI] [PubMed] [Google Scholar]; (c) Stoye A, Quandt G, Brunnhofer B, Kapatsina E, Baron J, Fischer A, Weymann M, Kunz H. Angew. Chem., Int. Ed. 2009;48:2228–2230. doi: 10.1002/anie.200805606. [DOI] [PubMed] [Google Scholar]
- [17].We thank Dr. Horst Kunz for providing us his GC/MS spectra of alkaloid 1 and 2.
- [18].We thank Drs. José Barluenga and Carlos Valdés for providing the 1H NMR spectrum of alkaloid 1.
- [19].See supporting information for the details.
- [20].Evans DA, Cote B, Coleman PJ, Connell BT. J. Am. Chem. Soc. 2003;125:10893–10898. doi: 10.1021/ja027640h. [DOI] [PubMed] [Google Scholar]
- [21].More JD, Finney NS. Org. Lett. 2002;4:3001–3003. doi: 10.1021/ol026427n. [DOI] [PubMed] [Google Scholar]
- [22].Krapcho AP, Glynn GA, Grenon BJ. Tetrahedron Lett. 1967:215–217. doi: 10.1016/s0040-4039(00)90519-7. [DOI] [PubMed] [Google Scholar]
- [23].For the similar example using amides: Pelletier G, Bechara WS, Charette AB. J. Am. Chem. Soc. 2010;132:12817–12819. doi: 10.1021/ja105194s.
- [24].For prior CNS studies: Suzuki Y, Taguchi K, Hagiwara Y, Kajiyama K, Ikeda T. Jpn J. Pharmacol. 1981;31:653–659. doi: 10.1254/jjp.31.653.
- [25].Chini B, Manning M. Biochem. Soc. Trans. 2007;35:737–741. doi: 10.1042/BST0350737. [DOI] [PubMed] [Google Scholar]
- [26].(a) Gimpl G, Fahrenholz F. Physiol. Rev. 2001;81:629–683. doi: 10.1152/physrev.2001.81.2.629. [DOI] [PubMed] [Google Scholar]; (b) Zingg HH, Laporte SA. Trends Endocrinol. Metab. 2003;14:222–227. doi: 10.1016/s1043-2760(03)00080-8. [DOI] [PubMed] [Google Scholar]
- [27].(a) Ring Robert H, Schechter Lee E, Leonard Sarah K, Dwyer Jason M, Platt Brian J, Graf R, Grauer S, Pulicicchio C, Resnick L, Rahman Z, Sukoff Rizzo Stacey J, Luo B, Beyer Chad E, Logue Sheree F, Marquis Karen L, Hughes Zoe A, Rosenzweig-Lipson S. Neuropharmacology. 2010;58:69–77. doi: 10.1016/j.neuropharm.2009.07.016. [DOI] [PubMed] [Google Scholar]; (b) Chaviaras S, Mak P, Ralph D, Krishnan L, Broadbear JH. Psychopharmacology. 2010;210:35–43. doi: 10.1007/s00213-010-1815-x. [DOI] [PubMed] [Google Scholar]
- [28].Guitart X, Codony X, Monroy X. Psychopharmacology. 2004;174:301–319. doi: 10.1007/s00213-004-1920-9. [DOI] [PubMed] [Google Scholar]
- [29].(a) Collier TL, Waterhouse RN, Kassiou M. Curr. Pharm. Des. 2007;13:51–72. doi: 10.2174/138161207779313740. [DOI] [PubMed] [Google Scholar]; (b) Monassier L, Bousquet P. Fundam. Clin. Pharmacol. 2002;16:1–8. doi: 10.1046/j.1472-8206.2002.00063.x. [DOI] [PubMed] [Google Scholar]
- [30].Frigerio M, Santagostino M, Sputore S. J. Org. Chem. 1999;64:4537–4538. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.