

Abstract
The use of heat shock protein 90 (Hsp90) inhibitors is an attractive antineoplastic therapy. We wanted to compare the effects of the benzoquinone 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) and the novel isoxazole resorcinol–based Hsp90 inhibitor NVP-AUY922 in a panel of pancreatic and colorectal carcinoma cell lines and in colorectal primary cultures derived from tumors excised to patients. PANC-1, CFPAC-1, and Caco-2 cells were intrinsically resistant to 17-AAG but sensitive to NVP-AUY922. Other cellular models were sensitive to both inhibitors. Human epidermal growth factor receptor receptors and their downstream signaling pathways were downregulated in susceptible cellular models, and concurrently, Hsp70 was induced. Intrinsic resistance to 17-AAG did not correlate with expression of ATP-binding cassette transporters involved in multidrug resistance. Some 17-AAG-resistant, NVP-AUY922–sensitive cell lines lacked NAD(P)H:quinone oxidoreductase 1 (NQO1) enzyme and activity. However, colorectal LoVo cells still responded to both drugs in spite of having undetectable levels and activity of NQO1. Pharmacological and biologic inhibition of NQO1 did not confer resistance to 17-AAG in sensitive cell lines. Therefore, even though 17-AAG sensitivity is related to NQO1 protein levels and enzymatic activity, the absence of NQO1 does not necessarily convey resistance to 17-AAG in these cellular models. Moreover, NVP-AUY922 does not require NQO1 for its action and is a more potent inhibitor than 17-AAG in these cells. More importantly, we show in this report that NVP-AUY922 potentiates the inhibitory effects of chemotherapeutic agents, such as gemcitabine or oxaliplatin, and other drugs that are currently being evaluated in clinical trials as antitumor agents.
Introduction
The heat shock protein 90 (Hsp90) multichaperone complex plays important roles in malignant transformation and therefore is a promising target for cancer therapy. The Hsp90 machinery mediates the folding, maturation, activation, and assembly of various proteins involved in signal transduction, transcriptional regulation, and cell cycle control [1]. Many of these client proteins are oncogenic. Therefore, a great advantage of the use of Hsp90 inhibitors is that multiple key oncogenic proteins can be disrupted simultaneously [2]. The geldanamycin derivative 17-allylamino-17-demethoxygeldanamycin (17-AAG), or tanespimycin, was the first Hsp90 inhibitor that entered clinical trials [3]. There are now about 14 inhibitors of Hsp90 function undergoing clinical trials, which belong to different structural classes [4]. All of them bind to a conserved pocket in the NH2-terminal ATP-binding domain of Hsp90, inhibiting its activity. Geldanamycin and its derivatives belong to the benzoquinone ansamycin class, which was found to inhibit expression of the oncogene c-myc [5] and to cause inactivation [6] and degradation of the tyrosine kinase src [7], human EGFR 2 (HER2)/Neu [8], raf [9], and mutated p53 [10]. However, albeit most of phase I and phase II clinical trials with geldanamycin derivatives have already been completed or terminated due to clinical limitations, these drugs have proved the successful targeting of Hsp90, paving the way for the development of second-generation Hsp90 inhibitors [11], such as synthetic and small molecules, targeted also against the N-terminal ATP-binding site. One class of such small inhibitors is based on the pyrazole or resorcinol subunit, another class on the purine-scaffold, and lastly, novel C-terminal domain–based Hsp90 inhibitors are being developed as well [12]. NVP-AUY922 is a novel resorcinylic isoxazole–based Hsp90 inhibitor that has shown potent preclinical activity in cancer models [13] and in xenografts [14]. In addition, it has shown tolerability in a phase I clinical trial [15]. The Hsp90-client cycle involves the association and dissociation of several cochaperones and is driven by the ATP-binding state of Hsp90 [2]. Thus, Hsp90 participates in two multichaperoning complexes with opposing activities: ATP-bound (mature) and ADP-bound (intermediate). A client protein initially associates with Hsp70/Hsp40 and is loaded onto Hsp90 through p60Hop, forming the ADP-bound intermediate state. When ADP is transformed into ATP, the Hsp90 complex conformation is altered, releasing Hsp70/Hsp40 and p60Hop, allowing other cochaperones such as p23, p50cdc37, and immunophilins to bind Hsp90, forming the mature complex. Then, at this stage, Hsp90-bound ATP is hydrolyzed, and the energy released enables client protein folding. Hsp90 inhibitors such as 17-AAG inhibit the ATPase intrinsic activity of Hsp90, impeding the chaperone to achieve the mature state [16]. Consequently, a ubiquitin ligase is recruited to the intermediate state, and a plethora of protein clients is targeted to degradation (see http://www.picard.ch/downloads for a list of Hsp90 interactors).
Chemoresistance is a common cause of failure to antitumor agents. Resistance to cytotoxic compounds is associated with cross-resistance to different drugs with or without structural similarity to the primary agent. This pleiotropic phenomenon is known as multidrug resistance (MDR) [17]. Although several mechanisms could be involved in the acquisition of this phenotype, the role of P-glycoprotein (Pgp), a member of the ATP-binding cassette (ABC) transporter family, has been well established [18], [19], [20]. Pgp, encoded by the gene MDR1, was first identified as a consequence of its overexpression in multidrug-resistant tumor cells, where it mediates the ATP-dependent efflux of a variety of chemotherapeutic agents [21]. Moreover, high levels of Pgp have been associated with resistance to Hsp90 inhibitors [22]. Other ABC transporters that confer MDR phenotype are MDR-associated protein 1 (MRP1) [23] and breast cancer resistance protein 1 (BCRP1) [24]. The benzoquinone ansamycin class of inhibitors can be reduced to semiquinone and hydroquinone forms through the activity of the two-electron NAD(P)H:quinone oxidoreductase 1 (NQO1)/DT-diaphorase. The hydroquinone forms of 17-AAG and 17-DMAG are more stable and more potent than their quinone partners. Chemoresistance can be intrinsic when existing before the treatment or acquired when it is developed during the treatment. Low levels of NQO1 have been associated to intrinsic resistance to ansamycins [22], [25] and to acquired resistance to 17-AAG [26].
Pancreatic cancer is the fourth leading cause of cancer death in both men and women, with most patients dying within a year [27], and had an increasing incident rate over the last 10 years [28]. Therefore, efforts to find novel therapeutics to fight this disease are challenging. Colorectal carcinoma is the third most prevalent type of cancer in men, the second most frequent type of cancer diagnosed in women [29], and the second leading cause of cancer death [30]. These types of cancer are highly dependent on the epidermal growth factor receptor (EGFR) signaling pathway. Overexpression of EGFR is common in pancreatic adenocarcinoma [31] and novel therapies in metastatic colorectal cancer include antibodies targeted against the EGFR, such as panitumumab and cetuximab [32]. EGFR belongs to the HER family of transmembrane tyrosine kinase receptors, which include HER2 (ErbB2/Neu), HER3 (ErbB3), and HER4 (ErbB4). Upon ligand binding, EGFR undergoes a conformational change that results in homodimerization and/or heterodimerization with the other members of the family [33], [34], which produces activation of the receptor tyrosine kinase, which, in turn, phosphorylates tyrosine residues on several adaptor molecules. Thus, HER receptors transduce a downstream signal either through the extracellular signal–regulated kinase/mitogen-activated kinase (MAPK) pathway or the phosphatidylinositol 3-kinase/Akt pathway, involved in cellular processes such as proliferation and survival [35].
In this report, we sought to determine the effects of 17-AAG and NVP-AUY922 in a panel of pancreatic exocrine adenocarcinoma and colorectal carcinoma cell lines and in colorectal primary cultures derived from tumors excised to patients to find predictive markers of response to such Hsp90 inhibitors, aiming at down-regulation of signaling pathways initiated by HER receptors. We have found some cell lines resistant to 17-AAG but still responsive to NVP-AUY922. We have determined that ABC transporters such as Pgp (Mdr-1), MRP1, and BCRP1 are not involved in 17-AAG resistance and that although the absence of NQO1 is a feature of several pancreatic and colorectal resistant cancer cell lines, its depletion is not enough to generate a resistance phenotype to 17-AAG. Moreover, NQO1 is related to resistance only to 17-AAG but not to other nonbenzoquinone Hsp90 inhibitors such as NVP-AUY922, which is a more potent inhibitor in these cellular models. Indeed, we demonstrate in this report that NVP-AUY922 is able to potentiate the effect of other antitumor drugs in cells that do not respond to these agents.
Materials and Methods
Reagents
17-AAG (tanespimycin), NVP-AUY922, AZD6244, and NVP-BEZ235 were purchased from ChemieTek (Indianapolis, IN) and ES936 and gemcitabine from Tocris Bioscience (Bristol, United Kingdom), and each one of them is dissolved in DMSO or water. Propidium iodide, crystal violet, iodonitrotetrazolium violet, 4-hydroxycoumarin (dicumarol), 2,6-dichlorophenol-indophenol (DCPIP), and oxaliplatin were purchased from Sigma-Aldrich (St Louis, MO).
Cell Culture
The human pancreatic carcinoma cell lines Hs 766 T, BxPC-3, HPAF-II, CFPAC-1, PANC-1, IMIM-PC-1, IMIM-PC-2, and RWP-1, the human colorectal carcinoma cell lines HT-29, SW620, SW480, HCT-15, HCT 116, LoVo, Caco-2, DLD-1, LS 174 T, and Colo 320 HSR (Colo 320), and the glioblastoma cell line T98G were obtained from the American Type Culture Collection (Manassas, VA) or the IMIM cell line repository (Instituto Hospital del Mar de Investigaciones Médicas (IMIM), Barcelona, Spain). The HGUE-C-1 cell line was kindly donated by Dr Miguel Saceda (Hospital General Universitario de Elche, Elche, Spain). Primary cell culture samples were obtained from colorectal tumors excised to patients at the Hospital Clínico Universitario Virgen de la Arrixaca (Murcia, Spain) or the Hospital General Universitario Santa Lucía (Cartagena, Spain). Surgical samples were digested with 1.5 U/ml dispase, 0.09 mg/ml collagenase II, 0.1 mg/ml pronase E, and 45 U/ml hyaluronidase and incubated at 37°C for 30 minutes. Fragments were incubated with RBC lysis solution (GeneAll Biotechnology, Seoul, Korea) for 10 minutes to eliminate erythrocytes, washed with phosphate-buffered saline (PBS) filtered through a 70-μm mesh, washed with PBS and harvested in Dulbecco's modified Eagle's medium–F12 containing 20% heat-inactivated FBS, 2 mM glutamine, 10 μg/ml insulin–5.5 μg/ml transferrin–6.7 ng/ml selenium, 0.5 μg/ml hydrocortisone, 20 ng/ml epidermal growth factor, 1 mM sodium pyruvate, 10 mM Hepes, 50 units/ml penicillin, 50 mg/ml streptomycin, 2.5 μg/ml amphotericin B, and 50 μg/ml gentamicin. Cell lines were maintained in Dulbecco's modified Eagle's medium and supplemented with 10% heat-inactivated FBS, 2 mM glutamine, 1 mM sodium pyruvate, 10 mM Hepes, 50 units/ml penicillin, and 50 mg/ml streptomycin and incubated at 37°C in a humidified 5% CO2/air atmosphere.
Cell Proliferation Assays
Cells were seeded in 96-well plates at a density of 1500 to 2500 cells per well and treated with vehicle or different concentrations of drugs for 3 days in sextuplicate. Then, cells were washed with PBS, fixed with 4% formaldehyde, and stained with 0.05% crystal violet for 30 minutes at room temperature. Cells were then washed three times with deionized water, and the wells were completely dried for at least 30 minutes. Cells were lysed with 0.1 M HCl and absorbance was determined at 620 nm in a microplate reader (Infinite M200PRO NanoQuant; Tecan Group, Männedorf, Switzerland). Viability of cells was monitored using the trypan blue dye exclusion method.
Colony Formation (Soft Agar Assays)
Cells were suspended in 0.36% agar with appropriate medium in the presence or absence of 17-AAG or NVP-AUY922 and seeded over a 0.6% agar base layer. After 14 days, cells were stained with iodonitrotetrazolium violet and colonies greater than 100 μm were analyzed with a visible light scanner (Image Scanner III; GE Healthcare, Buckinghamshire, United Kingdom) and software Image Quant TL (GE Healthcare Europe GmbH, Freiburg, Germany).
Flow Cytometry
Cells were seeded and treated with 17-AAG or NVP-AUY922 for 24, 48, and 72 hours. Cells were trypsinized, washed with PBS, fixed with 75% cold ethanol at − 20°C for at least 1 hour, treated with 0.5% Triton X-100 and 0.05% RNase A in PBS for 30 minutes, stained with propidium iodide, and analyzed using a flow cytometer (BD FACSCanto; Becton Dickinson & Co, Franklin Lakes, NJ) to determine cell cycle distribution of DNA content.
Western BlotAnalysis
Cells were seeded, treated with DMSO, 17-AAG, or NVP-AUY922, and lysed in a buffer containing 50 mM Tris (pH 7.4), 1% NP-40, 150 mM NaCl, 40 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and 10 μg/ml protease inhibitor cocktail (Sigma-Aldrich). Protein determinations were performed by the Bradford method (Bio-Rad, Richmond, CA). Then, 50 to 80 μg of protein from each lysate was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, blocked and incubated with primary antibodies against EGFR, HER2, HER3, HER4, Akt, Hsp90, Hsp70, Mdr-1, MRP1, BRCP1 and NQO1 from Santa Cruz Biotechnology (Santa Cruz, CA), phospho-ERK1/2, ERK1/2, phospho–ribosomal protein S6 (RPS6), and RPS6 from Cell Signaling Technology (Danvers, MA), or β-actin (Sigma-Aldrich). Then, membranes were incubated with HRP-linked secondary antibodies from GE Healthcare and protein bands were detected using Pierce ECL 2 reagents (Thermo Scientific, Rockford, IL) and scanned with a fluorescent light Typhoon 9410 scanner (GE Healthcare Europe GmbH). Densitometric analyses were performed using Scion Image software or Image Quant TL (GE Healthcare Europe GmbH).
Human Phospho-MAPK Arrays
Cells were seeded and treated with DMSO or 17-AAG (0.5 μM) or NVP-AUY922 (0.1 μM) for 24 hours, lysed, and prepared according to the manufacturer’s instructions of the Human Phospho-MAPK Array Kit (R&D Systems, Minneapolis, MN). Protein concentrations were determined by the Bradford method and 300 μg of each lysate was diluted, mixed with biotinylated phospho-specific detection antibodies, and incubated overnight on nitrocellulose membranes, where capture and control antibodies have been previously spotted in duplicate. After washing and removing unbound material, membranes were incubated with streptavidin conjugated to HRP and washed. Finally, the amount of phosphorylated protein bound in each spot was detected by chemiluminescence. Membranes were incubated with ECL reagents and scanned using a Typhoon 9410 scanner (GE Healthcare Europe GmbH). The levels of phosphorylated proteins were analyzed with the Image Quant TL (GE Healthcare Europe GmbH) software and normalized to the levels of the control spots.
NQO1 Activity Assays
NQO1 specific activity was calculated using the DCPIP reduction rate inhibited by dicumarol in cell extracts [36]. Cells were grown for 72 hours, lysed, and sonicated on ice in a buffer with 25 mM Tris-HCl, pH 7.4, 250 mM sucrose, and 5 μM flavin adenine dinucleotide. Then, the NQO1 activity was measured in 10 μg of protein and diluted in 1 ml with 25 mM Tris-HCl, pH 7.4, 0.7 mg/ml BSA, 200 μM NADH, and 40 μM DCPIP. Reactions were done in the absence and presence of 20 μM dicumarol. The NQO1 activity was determined in cells untreated or treated with 100 nM ES936 for 30 minutes or 4 hours and measured after 2 minutes at 600 nm using a microplate reader (Infinite M200PRO NanoQuant).
Small-interfering (siRNA)-Mediated Inhibition of NQO1 Gene Expression
Cells were seeded and transfected with NQO1 siRNA (Ambion, Life Technologies Corporation, Carlsbad, CA) or control siRNA (scrambled sequence) (Santa Cruz Biotechnology), according to the manufacturer’s instructions for 24 hours, using Opti-MEM I Reduced Serum medium (Gibco, Life Technologies Corporation) and Lipofectamine RNAiMAX (Invitrogen, Life Technologies Corporation). Then, cells were treated with DMSO or 17-AAG for 72 hours and harvested for subsequent experiments.
Clonogenic Assays
Cells were counted and seeded in six-well plates in triplicate and at a density of 1000 cells per well. After plating, cells were grown for 24 hours and some wells were pretreated with ES936 for 30 minutes. Then, cells were washed with PBS and incubated with media containing DMSO (vehicle), ES936, 17-AAG, or ES936 plus 17-AAG, for 4 hours. Media with drugs were removed, cells were washed with PBS again, fresh complete medium was added, and cells were allowed to grow for 14 days. Finally, colonies formed were washed with PBS, fixed with 4% formaldehyde, and stained with 0.05% crystal violet for 30 minutes at room temperature. After that, the wells were washed three times with deionized water and completely dried for at least 30 minutes. The colonies were scanned with a visible light scanner (Image Scanner III, GE Healthcare) and those with areas greater than 100 μm were detected and counted with Image Quant TL software (GE Healthcare Europe GmbH).
Analysis of Interaction between Two Compounds
Cells were seeded in 96-well plates and treated for 3 days using six wells per treatment with suitable vehicle, different concentrations of drugs (gemcitabine, oxaliplatin, AZD6244 (selumetinib), NVP-BEZ235, or in combination with a single suboptimal concentration of NVP-AUY922. Cell proliferation assays were performed as described. The Bliss model [37], [38] was used to determine whether the combination of NVP-AUY922 with other drugs was additive, synergistic, or antagonistic. A theoretical curve (bliss) was calculated by using the following equation: Ebliss = EA + EB − EA × EB, where EA and EB are the effects of drug A and drug B, respectively, expressed as the fractional inhibition between 0 and 1. Eexperimental (Eexp) is the actual result obtained by combination of both drugs. When Ebliss is equal to Eexp, the combination is considered additive. If Ebliss is more than Eexp the combination is synergistic. However, if Ebliss is less than Eexp, the combination is antagonistic.
Statistical Analysis
The experiments were performed with n ≥ 3 and the data are presented as means ± SEM. Statistically significant differences were estimated from P < .05 and evaluated using the Mann-Whitney test. The nonparametric two-tailed Spearman test was used to estimate the correlation between NQO1 enzyme activity and 17-AAG or NVP-AUY922 sensitivity. Statistical analyses were conducted using GraphPad Prism version 4.0 (GraphPad Software Inc., San Diego, CA) or SPSS version 10.0 (SPSS Inc, Chicago, IL).
Results
17-AAG and NVP-AUY922 Effects in Anchorage-Dependent and -Independent Growth
We pursued the following experiments comparing the effects of 17-AAG and NVP-AUY922. Proliferation of human pancreatic carcinoma cell lines (IMIM-PC-2, RWP-1, BxPC3, Hs 766 T, HPAF-II, and IMIM-PC-1) was inhibited in anchorage-dependent growth assays by 17-AAG. Proliferation of CFPAC-1 and PANC-1 cells was inhibited only 41.3 ± 4.7% and 35.4 ± 4.5%, respectively, even at the maximum concentration used of 2 μM (Figure 1A). However, colorectal carcinoma cell lines were in general more sensitive to 17-AAG. The less 17-AAG-responsive LoVo and Caco-2 colorectal cancer cell lines were growth inhibited only 28.3 ± 0.5% and 28.1 ± 11.9%, respectively, at 0.5 μM but inhibited, respectively, 64.6 ± 10.6% and 54.94 ± 3.9% at higher concentrations (Figure 1B). Colorectal carcinoma cell lines were in general more responsive also to NVP-AUY922 than pancreatic carcinoma cell lines (Figure 1, C and D). Anchorage-independent growth of IMIM-PC-1, HT-29, SW620, and LoVo cells was inhibited after 17-AAG treatment (0.5 μM), whereas PANC-1 and CFPAC-1 were also resistant to 17-AAG in soft agar assays and anchorage-independent growth of Caco-2 cells was only inhibited 50% (Figure 2, A and B). The rest of the cells were also inhibited in anchorage-independent growth assays after NVP-AUY922 treatment (0.1 μM). Primary cultures from colorectal tumors were also inhibited by both Hsp90 inhibitors, even though the half maximal inhibitory concentration (IC50) was higher than the IC50 of the cell lines. Interestingly, the primary culture HCUVA-CC-34 was inhibited only 43.8 ± 4.4% with 17-AAG and 40.4 ± 7.8% with NVP-AUY922 at the maximum concentration used of 10 μM in anchorage-dependent growth assays (Figure 1, E and F). In addition, anchorage-independent growth of the HCUVA-CC-34 primary cell culture was moderately inhibited by 17-AAG and by NVP-AUY922 only at the highest concentration used (Figure 2, C and D).
Figure 1.
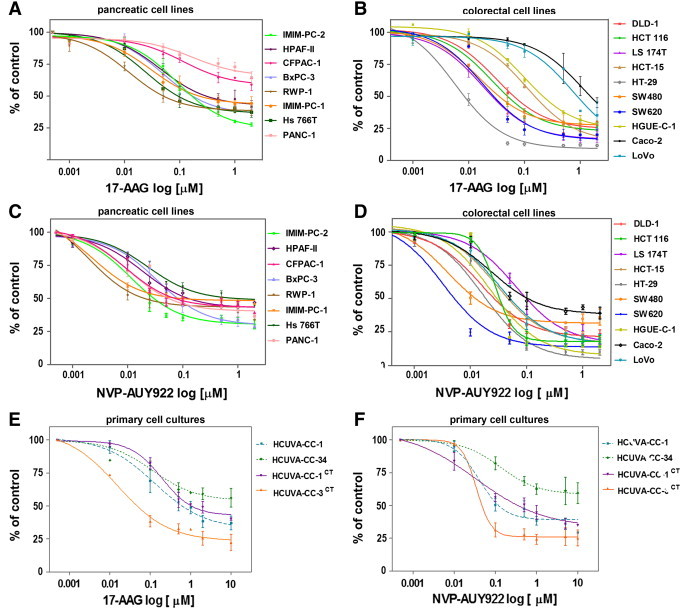
17-AAG and NVP-AUY922 effects on cell proliferation. Pancreatic (A and C) and colorectal (B and D) carcinoma cell lines or primary cell cultures obtained from colorectal tumors excised to patients (E and F) were treated with DMSO or different concentrations of 17-AAG (A, B, and E) or NVP-AUY922 (C, D, and F) for 72 hours. Cells were stained with crystal violet, proliferation rates were determined by colorimetry, and the average of at least four experiments is represented and referred as percentage of control. Error bars are the SEM. Each experiment was performed in sextuplicate (n = 6).
Figure 2.
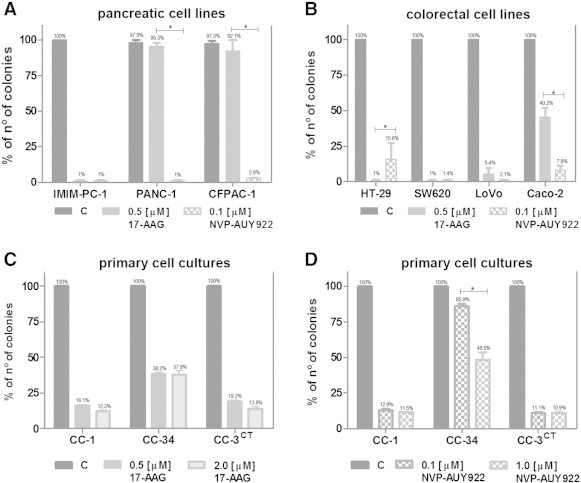
Effects of 17-AAG and NVP-AUY922 on colony formation. Pancreatic cancer cell lines IMIM-PC-1, PANC-1, and CFPAC-1 (A) or colorectal carcinoma cell lines HT-29, SW620, LoVo, and Caco-2 (B) were seeded over agar plates in the presence of DMSO (control), 0.5 μM 17-AAG, or 0.1 μM NVP-AUY922. Colorectal cancer primary cultures HCUVA (C and D) were seeded over agar plates in the presence of DMSO (control), 0.5 μM 17-AAG, 2 μM 17-AAG, 0.1 μM NVP-AUY922, or 1 μM NVP-AUY922. Colonies were stained and counted after 14 days. Each treatment was performed in triplicate and error bars are the SEM. Difference between means was statistically significant (*P < .05, Mann-Whitney test).
17-AAG and NVP-AUY922 Effects on Cell Cycle Distribution of DNA Content
We performed cell cycle analyses and found that pancreatic carcinoma IMIM-PC-2 cells accumulated in the G1 phase of the cell cycle upon 24 hours of 17-AAG or NVP-AUY922 treatment, followed by an accumulation in the sub-G1 phase, indicative of cell death, after 48 or 72 hours of Hsp90 inhibitor treatment (Figure 3, A and C). However, pancreatic carcinoma IMIM-PC-1 cells accumulated in the G2/M phase of the cell cycle, followed by an increase in the sub-G1 phase with both inhibitors (Figure 3, A and C). The pancreatic cell line CFPAC-1 accumulated in the G2/M phase and only slightly in sub-G1, and PANC-1 did not experience any change upon 17-AAG exposure (Figure 3A), suggesting that both CFPAC-1 and PANC-1 cells are unresponsive to 17-AAG but sensitive to NVP-AUY922 treatment. Conversely, when these cells were treated with NVP-AUY922, they accumulated considerably in the G2/M phase of the cell cycle followed by an increase in the sub-G1 phase (Figure 3C). Colorectal carcinoma cell lines HT-29 and SW620 accumulated in the G2/M and sub-G1 phases upon treatment with 17-AAG or NVP-AUY922. Especially, the G2/M arrest induced by 17-AAG treatment was very noticeable in HT29 cells (Figure 3, B and D). LoVo cells mainly accumulated in the sub-G1 phase with both inhibitors, whereas Caco-2 cells barely accumulated in the G2/M phase with 17-AAG but instead were arrested in this phase and also accumulated in sub-G1 after NVP-AUY922 treatment. This indicates that LoVo cells are sensitive to 17-AAG and NVP-AUY922, but Caco-2 cells are practically unresponsive to 17-AAG but sensitive to NVP-AUY922 treatment.
Figure 3.
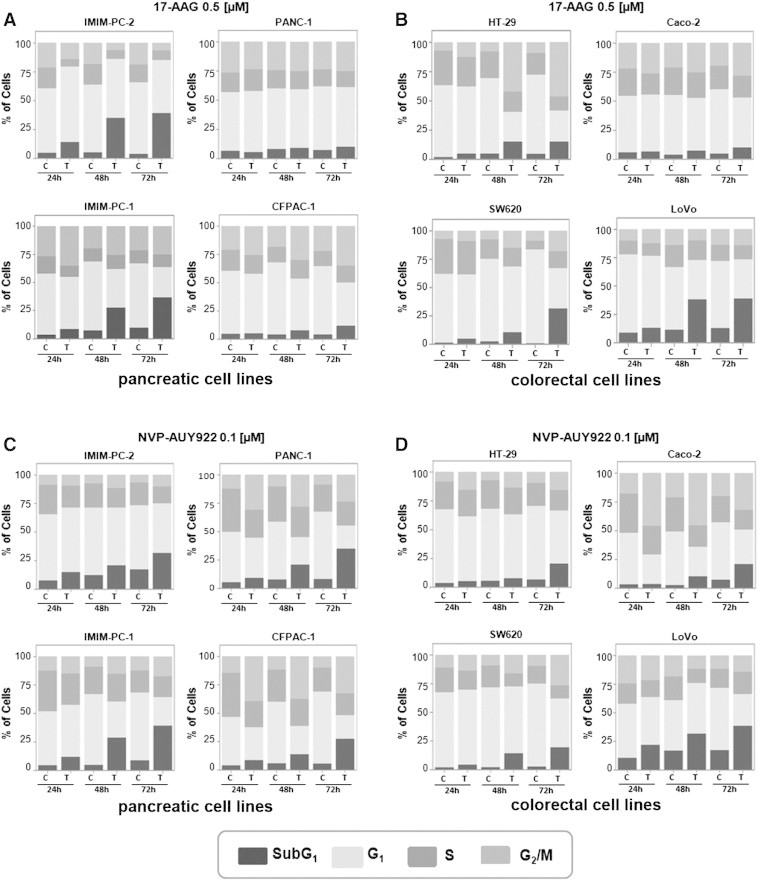
17-AAG and NVP-AUY922 effects on cell death. (A and C) Pancreatic cell lines IMIM-PC-2, IMIM-PC-1, CFPAC-1, and PANC-1 and (B and D) colorectal cell lines HT-29, SW620, LoVo, and Caco-2 were grown and treated with DMSO, 0.5 μM 17-AAG (A and B), or 0.1 μM NVP-AUY922 (C and D) for 24, 48, or 72 hours, and cell cycle distribution of DNA content was determined by flow cytometry. The sub-G1 phase, indicative of cell death, is represented as percentage of control and is the average of at least three separate experiments.
HER Receptor Down-Regulation and Signaling Pathways Downstream of HER Receptors
To determine whether 17-AAG and NVP-AUY922 were able to downregulate Hsp90 protein clients such as EGFR family members, we performed Western blot analyses and found that indeed EGFR and HER2 down-regulation could be detected within 4 hours of 17-AAG treatment in sensitive cell lines but not in cell lines resistant to 17-AAG (Figure 4A). In addition, EGFR and HER2 receptors were even more efficiently downregulated within 4 hours of NVP-AUY922 exposure (Figure 4A). Moreover, EGFR, HER2, HER3, and HER4 protein steady-state levels were depleted in the 17-AAG–sensitive pancreatic (Figure 4B) and colorectal cell lines (Figure 5A) within 24 hours of 17-AAG treatment and in all cell lines after NVP-AUY922 treatment. The small decrease observed in HER2 or HER3 protein levels in some 17-AAG–resistant cells was not enough to shut down the downstream signaling pathway, as ERK1/2 phosphorylation was unaltered. EGFR, HER4, and Akt protein levels were unaffected in 17-AAG–resistant cells. However, Akt steady-state protein levels and ERK1/2 phosphorylation levels decreased in sensitive cell lines, indicating that signaling pathways downstream of HER receptors were affected by 17-AAG treatment. Likewise, NVP-AUY922 treatment caused depletion of EGFR, HER2 and HER3 receptors, Akt, and ERK1/2 inactivation in all cell lines tested. HER4 receptor was barely downregulated in Caco-2 cells, but still the downstream signaling was interrupted. Hsp70, the hallmark of inhibition of Hsp90 function, was upregulated in 17-AAG–sensitive cell lines in all cell lines within 4 hours of exposure to both drugs (Figure 4A), and only slightly upregulated in some 17-AAG–resistant cell lines at later time points (Figure 5A). In addition, EGFR was downregulated in primary colorectal cell cultures, as Hsp70 levels were augmented, except in the HCUVA-CC-34 primary cell culture after 0.1 μM NVP-AUY922 exposure. ERK1/2 phosphorylation levels decreased after 17-AAG exposure and only in the more NVP-AUY922–sensitive cultures after treatment with this drug (Figure 5B). EGFR protein levels were undetected in SW620 cells and HCUVA-CC-1 primary culture. Hsp90 levels were unaltered upon 17-AAG or NVP-AUY922 treatment (Figure 4, Figure 5). To further determine the effects of Hsp90 inhibitors on the phosphorylation of these and other important signaling molecules downstream of HER receptors, we performed phospho-kinase arrays and found that the phosphorylation levels of the three Akt isoforms decreased after 0.5 μM 17-AAG and 0.1 μM NVP-AUY922 treatment compared to control levels in IMIM-PC-2 cells, except for the Akt2 isoform upon NVP-AUY922 treatment whose phosphorylation levels were unaltered. In addition, the decrease in phosphorylation of ERK1/2 upon exposure to both drugs was confirmed. Interestingly enough, p70S6 kinase (p70S6k) and p90S6 kinase (RSK1) phosphorylation levels also diminished upon 17-AAG and NVP-AUY922 treatment (Figure 6, A and B). The phosphorylation levels of RPS6, the target of p70S6k, which is downstream of Akt, were inhibited in IMIM-PC-2 and HT-29 cells, only slightly in Caco-2 and not affected in PANC-1 cells by 0.5 μM 17-AAG. However, RPS6 phosphorylation levels decreased in all cell lines tested after 0.1 μM NVP-AUY922 treatment (Figure 6C).
Figure 4.
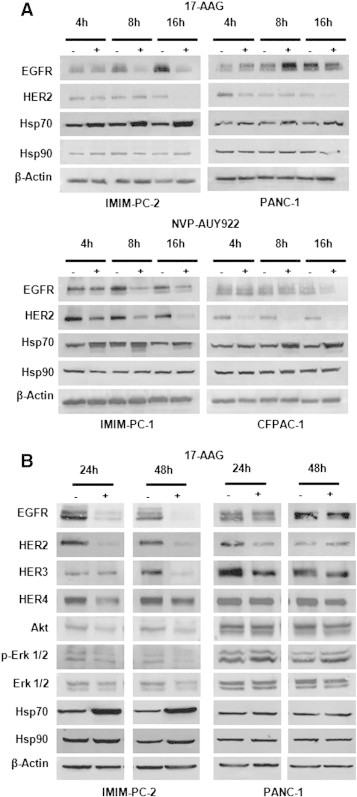
Hsp90 inhibitor effects on Hsp90 client proteins, Hsp70, and Hsp90 in pancreatic cancer cells. (A) Pancreatic cancer IMIM-PC-2 and PANC-1 cells were grown, left untreated (−), or treated (+) with 0.5 μM 17-AAG for 4, 8, or 16 hours. Pancreatic cancer IMIM-PC-1 or CFPAC-1 cells were grown, left untreated (−), or treated (+) with 0.1 μM NVP-AUY922 for 4, 8, or 16 hours. Cell extracts were obtained and analyzed by Western blot using EGFR, HER2, Hsp70, Hsp90, and β-actin antibodies. (B) Pancreatic carcinoma IMIM-PC-2 and PANC-1 cells were grown, left untreated (−), or treated (+) with 0.5 μM 17-AAG for 24 or 48 hours. Cell extracts were obtained and analyzed by Western blot using EGFR, HER2, HER3, HER4, Akt, phospho-ERK1/2, ERK1/2, Hsp70, Hsp90, and β-actin antibodies. In all the experiments, β-actin was used as a loading control.
Figure 5.
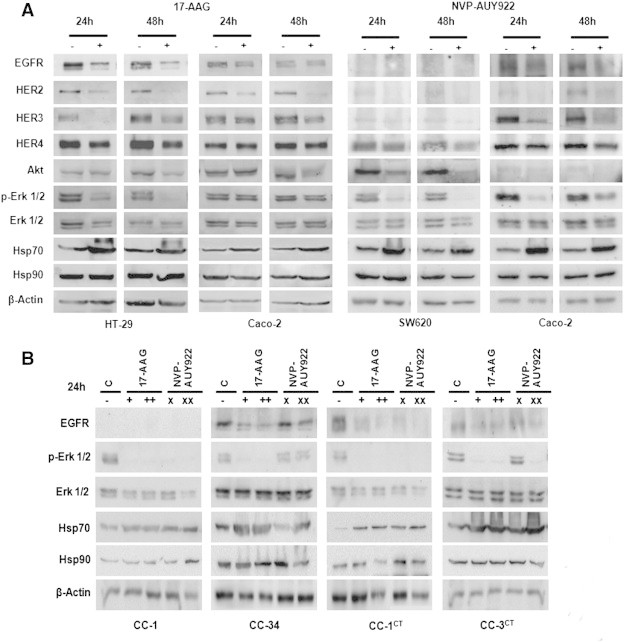
Hsp90 inhibitor effects on Hsp90 client proteins, Hsp70, and Hsp90 in colorectal cancer cells. (A) Colorectal carcinoma HT-29 and Caco-2 cells were grown, left untreated (−), or treated (+) with 0.5 μM 17-AAG for 24 or 48 hours. (A) Colorectal carcinoma SW620 and Caco-2 cells were grown, left untreated (−), or treated (+) with 0.1 μM NVP-AUY922 for 24 or 48 hours. Cell extracts were obtained and analyzed by Western blot using EGFR, HER2, HER3, HER4, Akt, phospho-ERK1/2, ERK1/2, Hsp70, Hsp90, and β-actin antibodies. (B) Primary cultures HCUVA derived from colorectal tumors excised to patients were grown, left untreated (−), or treated with 0.5 μM 17-AAG (+), 2 μM 17-AAG (++), 0.1 μM NVP-AUY922 (×), or 1 μM NVP-AU922 (××). Cells extracts were analyzed using antibodies against EGFR, phospho-ERK1/2, ERK1/2, Hsp70, Hsp90, and β-actin antibodies. β-Actin was used as a loading control.
Figure 6.
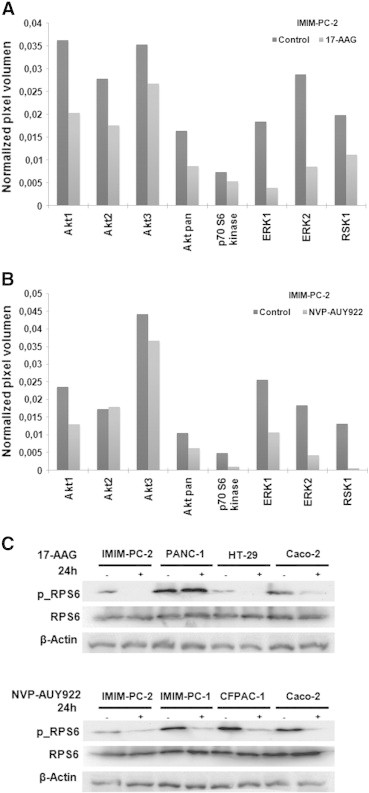
Hsp90 inhibitor effects on different kinases and RPS6 phosphorylation levels. Phospho-MAPK array (A) IMIM-PC-2 pancreatic cells were grown and treated with DMSO (control) or 0.5 μM 17-AAG. Cell extracts were subjected to the human phospho-MAPK array, as described in the Materials and Methods section. The graph represents relative phosphorylation levels of the most relevant kinases: Akt1 Ser473), Akt2 (Ser474), Akt3 (Ser472), Akt pan (Ser473, Ser474, and Ser472), p70S6k (Thr421/Ser424), ERK1 (Thr202/Tyr204), ERK2 (Thr185/Tyr187), and RSK1 (Ser380) in treated cells compared to nontreated cells (control). (B) IMIM-PC-2 pancreatic cells were grown and treated with DMSO (control) or 0.1 μM NVP-AUY922 for 24 hours and analyzed using the human phospho-MAPK array. The graph represents relative phosphorylation levels of relevant kinases as in (A). RPS6 phosphorylation levels. (C) Pancreatic cancer IMIM-PC-2 and PANC-1 and colorectal carcinoma HT-29 and Caco-2 were grown, left untreated (−), or treated (+) with 0.5 μM 17-AAG. In the same way, pancreatic cancer IMIM-PC-2, IMIM-PC-1, and CFPAC-1 cells and colorectal carcinoma Caco-2 cells were grown, left untreated (−), or treated (+) with 0.1 μM NVP-AUY922 for 24 hours. Cell extracts were obtained and analyzed by Western blot using phospho-RPS6, RPS6, and β-actin antibodies. β-Actin was used as a loading control.
Role of ABC Transporters in 17-AAG Intrinsic Resistance
Since MDR is frequently associated with overexpression of ABC transporters, we wanted to determine whether these ABC transporters were involved in the intrinsic resistance to 17-AAG observed in these cell lines. We found that none of the pancreatic carcinoma cell lines used expressed Mdr-1 (Pgp) protein (Figure 7). However, the colorectal carcinoma cell lines HCT-15, DLD-1, LS 174 T, and LoVo cells that express Mdr-1 are growth inhibited by 17-AAG. We used Colo 320 cells as a positive control for Mdr-1 expression. MRP1 expression could be barely detected only in DLD-1 cells, which respond to 17-AAG. T98G cells were used as a positive control. On the contrary, BCRP1 expression was detected mainly in the sensitive Hs 766 T pancreatic carcinoma cells and to a lesser extent in several colorectal carcinoma cell lines: DLD-1, SW480, LS 174 T, SW620, HCT-15, and HGUE-C-1 sensitive to 17-AAG and in Caco-2 cells resistant to 17-AAG. The 17-AAG–resistant PANC-1 and CFPAC-1 cells do not express any of the ABC transporters used in our study.
Figure 7.
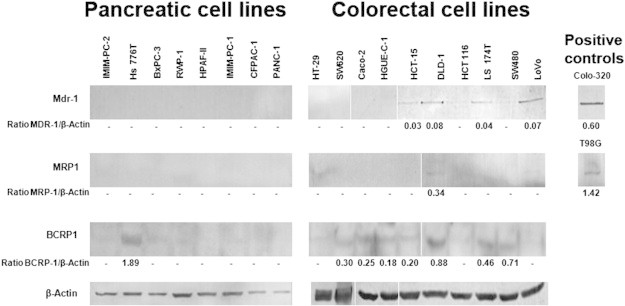
Expression of ABC transporters. Pancreatic and colorectal cells were grown, lysed, and subjected to Western blot analyses to detect basal levels of Mdr-1, MRP1, BRCP1, or β-actin, using specific antibodies. Colo 320 and T98G cell extracts were used as positive controls. β-Actin was used as a loading control. Densitometric values of the ratio between ABC transporters and β-actin are indicated.
NQO1/DT-Diaphorase Expression and Enzymatic Activity
We wanted to confirm whether NQO1 was involved in the intrinsic resistance to 17-AAG found by others in pancreatic cancer cell lines [39] and to determine its role in our panel of pancreatic and colorectal carcinoma cell lines and primary tumor cell cultures. The protein NQO1 levels and enzymatic activity were undetectable in the 17-AAG–resistant CFPAC-1 and PANC-1 pancreatic carcinoma cells and in Caco-2 colorectal cells, which are 17-AAG–resistant (Figure 8, A and B). In fact, there was a negative correlation between the IC50 for 17-AAG after 72 hours of drug exposure and NQO1 activity in the pancreatic and colorectal carcinoma cells studied (Figure 8C). In addition, the primary cell cultures derived from colorectal tumors express different levels of NQO1 and Hsp70 (Figure 8A). Interestingly, NQO1 protein levels were relatively high in the less sensitive primary culture to both 17-AAG and NVP-AUY922, HCUVA-CC-34. As expected, there was no correlation between the IC50 for NVP-AUY922 and NQO1 enzymatic activity in the pancreatic and colorectal carcinoma cell lines studied (Figure 8C).
Figure 8.
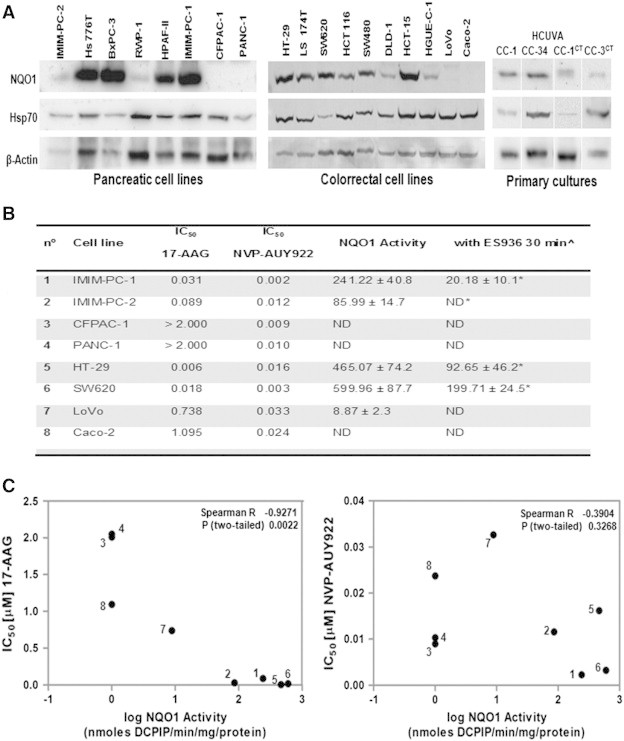
Comparison of NQO1 protein levels and activity. (A) Western blot analyses were performed with total cellular extracts to determine basal NQO1, Hsp70, or β-actin levels in the cell lines or primary cultures indicated. NQO1 activity assays. (B) NQO1 specific activity was calculated using the dicumarol-inhibitable rate of DCPIP reduction in cell extracts. IC50 (μM) of growth inhibition of cell lines for 17-AAG and NVP-AUY922 is indicated. The inhibition of NQO1 activity by ES936 was also determined. ^Cells were treated with 0.1 μM ES936 for 30 minutes. The average of at least three experiments is represented. *Reduction of NQO1 after treatment with ES936 was statistically significant (P < .05, Mann-Whitney test). ND, nondetectable (< 5 nmol DCPIP/min per mg protein); Spearman correlation test. (C) The IC50 values (50% of growth inhibition) after 72 hours of treatment for 17-AAG and for NVP-AUY922 in the different cell lines were plotted against NQO1 activity. Each number corresponds to the cell line represented in (B). R and P values are indicated.
Pharmacological Inhibition of NQO1
To determine the role of NQO1 in sensitivity to 17-AAG, we performed cell proliferation assays in 17-AAG–sensitive cell lines in the presence of the NQO1-specific inhibitor ES936 [5-methoxy-1,2-dimethyl-3-[(4-nitrophenoxy)methyl]indole-4,7-dione], which was added 30 minutes before exposure to 17-AAG and sustained throughout 17-AAG treatment for 72 hours. In spite of significantly reducing NQO1 activity (Figure 8B), this inhibitor was unable to confer 17-AAG resistance to sensitive cells (Figure 9A). As expected, no effect was observed in cell lines devoid of NQO1 protein or enzymatic activity, such as CFPAC-1, PANC-1, or Caco-2 cells (data not shown). Then, we wanted to determine the effects of NQO1 ablation in long-term clonogenic assays. First, we determined that after 4 fours of treatment with ES936, NQO1 activity was still inhibited (Figure 9D). Then, we performed clonogenic experiments after incubating HT-29 cells for 4 hours with 17-AAG or a combination of the specific inhibitor ES936 and 17-AAG and found that clonogenic survival of cells was only slightly recovered after the combination treatment (Figure 9, B and C).
Figure 9.
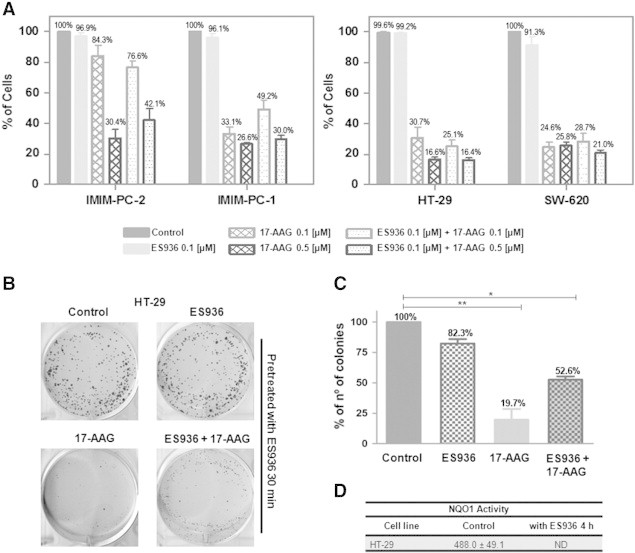
Pharmacological NQO1 inhibition. Cell proliferation assays. (A) Pancreatic and colorectal carcinoma cells were treated with ES936 for 30 minutes before treatment with DMSO (control), 0.1 μM or 0.5 μM 17-AAG for 72 hours, and cell proliferation was determined. The average of at least three experiments is represented as percentage of control. Error bars are the SEM. Clonogenic assays. HT-29 cells were pretreated with 0.1 μM ES936 during 30 minutes and then treated with DMSO (control), 0.1 μM ES936 (ES936), 0.5 μM 17-AAG (17-AAG), or 0.1 μM ES936 plus 0.5 μM 17-AAG (ES936 + 17-AAG) for 4 hours. Then, cells were seeded at a very low density, allowed to grow for 14 days, fixed, stained, and visualized and colonies formed were counted. (B) Scanned images showing colonies of HT-29 cells. (C) Graphic representation of the average of three independent experiments. Error bars are the SEM. Difference between means was statistically significant (*P < .05, **P < .01, Mann-Whitney test). NQO1 activity assays. (D) NQO1 specific activity was calculated using the dicumarol-inhibitable rate of DCPIP reduction in cell extracts of HT-29 cells, treated with DMSO (control) or 0.1 μM ES936 for 4 hours. The average of at least three experiments is indicated. ND, nondetectable (< 5 nmol DCPIP/min per mg protein).
Biologic Inhibition of NQO1
Since reexpression of NQO1 in PANC-1 cells devoid of this enzyme has already been performed [39], we wanted to determine whether the silencing of NQO1 would render resistance to sensitive cells. To do that, we used a biologic approach to knock down NQO1 by using a specific siRNA targeted against NQO1 in the 17-AAG–sensitive IMIM-PC-2 cells. Both NQO1 protein levels and enzymatic activity were abolished (Figure 10, A and C), and still the proliferation of IMIM-PC-2 cells was inhibited by 17-AAG to the same extent as in the nontransfected cells or in the cells transfected with scrambled siRNA (Figure 10B), indicating that 17-AAG is effective even in the absence of NQO1. Furthermore, Hsp70 protein levels increased and EGFR protein levels decreased on 17-AAG treatment in IMIM-PC-2 cells devoid of NQO1 (Figure 10A).
Figure 10.
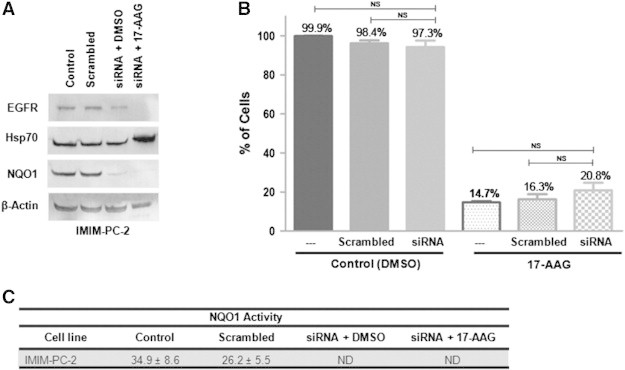
Biologic NQO1 inhibition. IMIM-PC-2 cells were nontransfected (control) or transfected with a control siRNA (scrambled sequence) or a specific siRNA against NQO1, before treatment with DMSO or 0.5 μM 17-AAG for 72 hours. (A) EGFR, Hsp70, NQO1, and β-actin protein levels were analyzed by Western blot. (B) Cell proliferation rates were determined after transfection and 17-AAG treatment and represented as percentage of control. Error bars are the SEM. Difference between means was nonsignificant (NS, Mann-Whitney test). (C) NQO1 specific activity was calculated using the dicumarol-inhibitable rate of DCPIP reduction in cell extracts after transfection. Reduction of NQO1 was nondetectable (ND; < 5 nmol DCPIP/min per mg protein).
Combination of NVP-AUY922 with Other Drugs
After demonstrating that NVP-AUY922 is more efficacious than 17-AAG in our cellular models, we set out to determine whether this drug was able to potentiate the effects of chemotherapeutic drugs currently in the clinic, such as gemcitabine for pancreatic cancer and oxaliplatin for colorectal cancer. Additionally, we tested the effect of combining NVP-AUY922 with the Mek inhibitor AZD-6244 (selumetinib) and with the dual phosphatidylinositol 3-kinase/mammalian target of Rapamycin inhibitor NVP-BEZ235. The effects of various concentrations of these antitumor drugs with a single concentration of NVP-AUY922 are depicted in Figure 11. We selected cell lines that were nonresponsive to such drugs, according to unpublished data from our laboratory. Suboptimal concentrations of NVP-AUY922 were synergistic (Ebliss > Eexp) with gemcitabine in the CFPAC-1 (Figure 11A) and PANC-1 (Figure 11B) pancreatic cancer cells, with oxaliplatin in the HCT-15 colorectal cancer cells (Figure 11C), with AZD6244 in DLD-1 colorectal cancer cells (Figure 11D), and with NVP-BEZ235 in the pancreatic carcinoma PANC-1 cells (Figure 11E).
Figure 11.
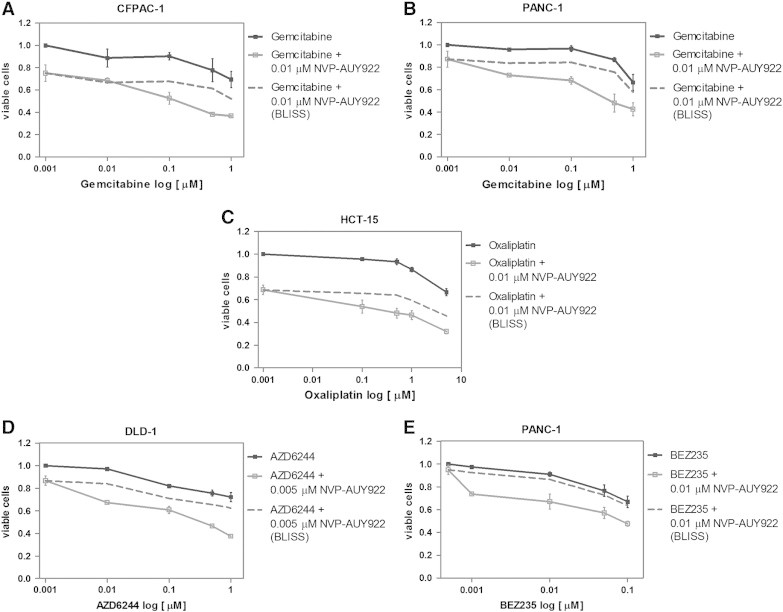
Combinatory effects of NVP-AUY922 with other drugs. (A) CFPAC-1 and (B) PANC-1 pancreatic cancer cells were treated with different concentrations of gemcitabine (0.01, 0.1, 0.5, and 1 μM) in the absence or presence of 0.01 μM NVP-AUY922 for 72 hours and cell proliferation was determined. Curves for gemcitabine alone, experimental curves for gemcitabine and NVP-AUY922, and theoretical (Ebliss) curves for gemcitabine and NVP-AUY922 are represented. (C) HCT-15 colorectal carcinoma cells were treated with various concentrations of oxaliplatin (0.1, 0.5, 1, and 5 μM) in the absence or presence of 0.01 μM NVP-AUY922. Curve for oxaliplatin alone, experimental curves for oxaliplatin and NVP-AUY922, and theoretical (Ebliss) curves for oxaliplatin and NVP-AUY922 are represented. (D) DLD-1 colorectal carcinoma cells were treated with varying concentrations of AZD6244 (0.01, 0.1, 0.5, and 1 μM) in the absence or presence of 0.005 μM NVP-AUY922. Curve for AZD6244 alone, experimental curves for AZD6244 and NVP-AUY922, and theoretical (Ebliss) curves for AZD6244 and NVP-AUY922 are represented. (E) PANC-1 pancreatic carcinoma cells were treated with different concentrations of NVP-BEZ235 (BEZ235; 0.001, 0.01, 0.05, and 0.1 μM) in the absence or presence of 0.01 μM NVP-AUY922. Curve for BEZ235 alone, experimental curves for BEZ235 and NVP-AUY922, and theoretical (Ebliss) curves for BEZ235 and NVP-AUY922 are represented. In all graphs, results represent the average of three independent experiments and error bars are the SEM. Each experiment was performed in sextuplicate (n = 6).
Discussion
The use of Hsp90 inhibitors has emerged as a potential antitumor therapy. Exocrine pancreatic adenocarcinoma has a very poor prognosis. In addition, colorectal carcinoma is one of the most common types of cancer. HER receptors and their downstream signaling are very important in these types of cancer. Therefore, we were interested in using Hsp90 inhibitors able to downregulate HER receptors as a therapeutic strategy in pancreatic and colorectal carcinomas. As a matter of fact, 17-AAG is being studied in phase I and phase II clinical trials for a variety of solid tumors with promising results, especially in HER2 + breast cancer and multiple myeloma [3] and in combination with therapeutic agents such as Herceptin and Vortezomib [40]. Furthermore, although numerous clinical trials with 17-AAG have already been completed or terminated, IPI-504 (retaspimycin), the water-soluble hydroquinone derivative of 17-AAG [41], is currently being evaluated in several clinical trials (see http://www.clinicaltrials.gov/ct2/results?term=17-AAG&Search=Search). However, the isoxazole derivative NVP-AUY922 is able to deplete HER2 in breast cancer cells [13] and EGFR in non–small lung cancer cells [42] and is also under clinical evaluation for the treatment of various solid tumors (see http://www.clinicaltrials.gov/ct2/results?term=AUY922&Search=Search). Other Hsp90 small molecule inhibitors under current clinical evaluation include AT13387, STA9090, and MPC3100. In particular, STA-9090 (ganetespib) is being evaluated over 25 clinical trials, including breast, lung, colorectal, and hematologic tumors (http://www.clinicaltrials.gov/ct2/results?term=ganetespib&pg=1). In this report, we have used a panel of pancreatic and colorectal carcinoma cell lines and primary cultures derived from human tumors to test the effects of 17-AAG and NVP-AUY922. In addition, we were interested in finding molecular determinants of sensitivity or resistance to these drugs. We have determined that pancreatic carcinoma PANC-1 and CFPAC-1 cells were resistant to 17-AAG both in anchorage-dependent and -independent growth assays (Figure 1, Figure 2). The colorectal carcinoma cell line Caco-2 was also resistant to 17-AAG (Figure 1). Pancreatic and colorectal sensitive cell lines underwent cell death upon 17-AAG treatment, as indicated by an increase in the sub-G1 phase of the cell cycle, whereas resistant cell lines did not (Figure 3). However, all cell lines were sensitive to NVP-AUY922. A previous report has shown that NVP-AUY922 is able to inhibit migratory and invasive properties of pancreatic cancer cells [43]. However, when we performed anchorage-dependent and -independent growth assays in primary cultures obtained from colorectal tumors, we found that the HCUVA-CC-34 was not very responsive to 17-AAG and even less responsive to NVP-AUY922. We have demonstrated in this report that EGFR, HER2, HER3, and HER4 are Hsp90 client proteins that are depleted upon 17-AAG treatment in sensitive pancreatic and colorectal cell lines such as IMIM-PC-1, IMIM-PC-2, SW620, or HT-29 but not in resistant PANC-1, CFPAC-1, or Caco-2 cells within 4 or 8 hours (Figure 4, Figure 5 and data not shown). Not only HER receptors but also the signaling pathways downstream this class of tyrosine kinase receptors were also downregulated in sensitive cell lines, since Akt protein levels, Akt, RSK1, p70S6k, RPS6, and ERK2 phosphorylation levels diminished upon 17-AAG treatment (Figure 4, Figure 5, Figure 6). Albeit HER2 and HER3 protein levels were partially downregulated by 17-AAG in some of the resistant cells, the signaling pathways in these cells were unaltered. NVP-AUY922 was also able to deplete HER receptors in all cell lines tested within 4 or 8 hours (Figure 4, Figure 5 and data not shown). The induction of Hsp70 was observed in sensitive cell lines to 17-AAG very rapidly, within 4 or 8 hours of treatment. The slight increase in Hsp70 and down-regulation of some protein clients that we observed in some cases in resistant cell lines could be due to the fact that 17-AAG binds poorly to the Hsp90 complex and has some inhibitory capacity that is not translated into cell growth inhibition or cell death. As all cell lines respond to NVP-AUY922, the increase in Hsp70 is very significant and occurs rapidly. In the HCUVA-CC-34 primary culture however, EGFR depletion, ERK1/2 phosphorylation, and Hsp70 up-regulation are not very dramatic, which explain the moderate effects of this drug in anchorage-dependent and anchorage-independent growth assays. Experiments are underway to try to identify a possible mechanism of resistance of HCUVA-CC-34 and other colorectal cellular models to NVP-AUY922.
Since all our cellular models, apart from the exception just mentioned, were sensitive to NVP-AUY922, we sought to find markers of sensitivity/resistance to 17-AAG. In fact, phospho-kinase arrays were performed in 17-AAG–sensitive as well as in 17-AAG–resistant cell lines with the intention to find putative markers. However, we could not clearly associate differences found between cell lines to resistance to this drug. As it has been suggested that ABC transporters may play a role in resistance to Hsp90 inhibitors, we analyzed Mdr-1, MRP1, and BRCP1 protein levels in these cell lines and found that none of the 17-AAG–resistant pancreatic and colorectal carcinoma cell lines expressed these transporters, with the exception of Caco-2 cells that express very low levels of BRCP1. However, many of the 17-AAG–sensitive cell lines express some of these ABC transporters (Figure 7). Therefore, we can rule out the role of these ABC transporters in 17-AAG resistance. In addition to Pgp (Mdr-1), it has been suggested in several reports that NQO1/DT-diaphorase is necessary for benzoquinone ansamycin function. This enzyme is able to metabolize quinones to the corresponding hydroquinones, which are more stable and bind Hsp90 with greater affinity. We have found that the 17-AAG–resistant pancreatic carcinoma PANC-1 and CFPAC-1 cells lack NQO1 protein and activity (Figure 8), confirming the results previously reported by Siegel et al. [39]. The 17-AAG–resistant Caco-2 cells also lack NQO1 protein and enzymatic activity. However, LoVo cells, which are also devoid of NQO1 enzyme (Figure 8), are still responsive to 17-AAG, as demonstrated especially in soft agar assays and cell cycle analyses (Figure 2, Figure 3). We speculate that other reductases, albeit with less potency, may be able to reduce 17-AAG to 17-AAGH2 in these cells. Another possibility is that although less potent, the nonreduced benzoquinones may also have an activity and be able to exert the same effects as their reduced counterparts at higher concentrations. When we blocked NQO1 activity in 17-AAG–sensitive cell lines with ES936, these cells were still growth inhibited by 17-AAG (Figure 9). Furthermore, knocking down NQO1 with siRNA did not confer 17-AAG resistance to the pancreatic carcinoma IMIM-PC-2 (Figure 10), IMIM-PC-1 cells, and the colorectal carcinoma HT-29 and SW620 cells (data not shown), indicating that NQO1 activity is dispensable in these cellular models for 17-AAG action. Increased levels of pro-survival chaperones such as Hsp27 [44] and Hsp70 due to elevation in the heat shock response [45] have been proposed as possible NQO1-unrelated causes of resistance to benzoquinone ansamycins [46]. In our system however, Hsp70 protein levels were not significantly induced after 17-AAG treatment in resistant cells. Inaccessibility of Hsp90 inhibitors to the Hsp90 isoforms located in mitochondria [47], which contribute to apoptosis inhibition, may be another plausible cause of resistance. Furthermore, mutations or alterations in posttranslational modifications in the Hsp90 itself may contribute to Hsp90 inhibitor resistance [46]. Our cellular models, however, were sensitive to NVP-AUY922, which is based on resorcinol and not structurally related to benzoquinones [14]. This inhibitor is not dependent on the presence of NQO1 and we have demonstrated that NVP-AUY922 sensitivity does not correlate with NQO1 activity (Figure 8C). In a clinical setting, it is more useful to use NVP-AUY922 that offers several advantages over benzoquinones: no liver toxicity and no NQO1 or other reductase requirement for its function. Furthermore, we have shown in this report for the first time that this novel Hsp90 inhibitor is very potent in combination with other drugs such as gemcitabine, oxaliplatin, AZD6244, or NVP-BEZ235 in cell lines that are not very responsive to these drugs (Figure 11). Moreover, it has been shown that NVP-AUY922 is able to sensitize prostate cancer cell to radiation [48]. Therefore, NVP-AUY922 has a great potential to be used not only as a single agent but also in combination with chemotherapy or radiation therapy, even when these agents are not very effective when used alone.
Conclusions
NVP-AUY922 is a more potent inhibitor than 17-AAG in pancreatic and colorectal cellular models, as demonstrated by inhibition of cell proliferation and colony formation, cell death induction, HER receptor depletion, and inhibition of ERK and Akt signaling pathways. Some of these models show resistance to 17-AAG, especially pancreatic carcinoma cell lines. The ABC transporters examined are not involved in resistance to the Hsp90 inhibitors 17-AAG and NVP-AUY922. The use of NQO1 as a biomarker of response to Hsp90 inhibitors is limited only to 17-AAG and not to NVP-AUY922 and is dependent on the cellular context. Moreover, we show that rather than a marker of response to 17-AAG, NQO1 is a marker of sensitivity, as cells devoid of this enzyme can still respond to 17-AAG. Therefore, the utilization of non-benzoquinone compounds such as NVP-AUY922 is more appropriate. In fact, NVP-AUY922 is being clinically evaluated both as a single agent and in combination with other antineoplastic drugs or radiation and represents a promising novel therapeutic strategy for the treatment of solid malignancies such as pancreatic and colorectal carcinomas.
Acknowledgments
We are especially grateful to M. Angeles Ros Roca for technical assistance. We thank Rosario Martinez and all the Biobank personnel for their help. We are also grateful to our laboratory members for helpful comments. Conflicts of interest: The authors declare no conflict of interest.
Footnotes
This article has been funded by a grant from the Asociación Española Contra el Cancer to support M.A. Ros Roca, L.M.-L., E.T., and I.M.-L. and by a Seneca Foundation grant and Instituto de Salud Carlos III grant FIS PI10/01123 to I.M.-L.
References
- 1.Kamal A, Boehm MF, Burrows FJ. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med. 2004;10:283–290. doi: 10.1016/j.molmed.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharp S, Workman P. Inhibitors of the HSP90 molecular chaperone: current status. Adv Cancer Res. 2006;95:323–348. doi: 10.1016/S0065-230X(06)95009-X. [DOI] [PubMed] [Google Scholar]
- 3.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter JR, Fritz CC, Depew KM. Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy. Curr Opin Chem Biol. 2010;14:412–420. doi: 10.1016/j.cbpa.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 5.Yamaki H, Iguchi-Ariga SM, Ariga H. Inhibition of c-myc gene expression in murine lymphoblastoma cells by geldanamycin and herbimycin, antibiotics of benzoquinoid ansamycin group. J Antibiot (Tokyo) 1989;42:604–610. doi: 10.7164/antibiotics.42.604. [DOI] [PubMed] [Google Scholar]
- 6.Uehara Y, Hori M, Takeuchi T, Umezawa H. Phenotypic change from transformed to normal induced by benzoquinonoid ansamycins accompanies inactivation of p60src in rat kidney cells infected with Rous sarcoma virus. Mol Cell Biol. 1986;6:2198–2206. doi: 10.1128/mcb.6.6.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller P, DiOrio C, Moyer M, Schnur RC, Bruskin A, Cullen W, Moyer JD. Depletion of the erbB-2 gene product p185 by benzoquinoid ansamycins. Cancer Res. 1994;54:2724–2730. [PubMed] [Google Scholar]
- 9.Schulte TW, Blagosklonny MV, Ingui C, Neckers L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J Biol Chem. 1995;270:24585–24588. doi: 10.1074/jbc.270.41.24585. [DOI] [PubMed] [Google Scholar]
- 10.Blagosklonny MV, Toretsky J, Neckers L. Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene. 1995;11:933–939. [PubMed] [Google Scholar]
- 11.Jhaveri K, Ochiana SO, Dunphy MP, Gerecitano JF, Corben AD, Peter RI, Janjigian YY, Gomes-DaGama EM, Koren J, III, Modi S. Heat shock protein 90 inhibitors in the treatment of cancer: current status and future directions. Expert Opin Investig Drugs. 2014;23:611–628. doi: 10.1517/13543784.2014.902442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R. Targeting the molecular chaperone heat shock protein 90 (HSP90): lessons learned and future directions. Cancer Treat Rev. 2013;39:375–387. doi: 10.1016/j.ctrv.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Jensen MR, Schoepfer J, Radimerski T, Massey A, Guy CT, Brueggen J, Quadt C, Buckler A, Cozens R, Drysdale MJ. NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008;10:R33. doi: 10.1186/bcr1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven BA, Gowan S, Boxall F. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68:2850–2860. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 15.Sessa C, Shapiro GI, Bhalla KN, Britten C, Jacks KS, Mita M, Papadimitrakopoulou V, Pluard T, Samuel TA, Akimov M. First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res. 2013;19:3671–3680. doi: 10.1158/1078-0432.CCR-12-3404. [DOI] [PubMed] [Google Scholar]
- 16.Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55–S61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 17.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 18.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 19.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 20.Tan B, Piwnica-Worms D, Ratner L. Multidrug resistance transporters and modulation. Curr Opin Oncol. 2000;12:450–458. doi: 10.1097/00001622-200009000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Johnstone RW, Ruefli AA, Smyth MJ. Multiple physiological functions for multidrug transporter P-glycoprotein? Trends Biochem Sci. 2000;25:1–6. doi: 10.1016/s0968-0004(99)01493-0. [DOI] [PubMed] [Google Scholar]
- 22.Kelland LR, Sharp SY, Rogers PM, Myers TG, Workman P. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst. 1999;91:1940–1949. doi: 10.1093/jnci/91.22.1940. [DOI] [PubMed] [Google Scholar]
- 23.He SM, Li R, Kanwar JR, Zhou SF. Structural and functional properties of human multidrug resistance protein 1 (MRP1/ABCC1) Curr Med Chem. 2011;18:439–481. doi: 10.2174/092986711794839197. [DOI] [PubMed] [Google Scholar]
- 24.Mo W, Zhang JT. Human ABCG2: structure, function, and its role in multidrug resistance. Int J Biochem Mol Biol. 2012;3:1–27. [PMC free article] [PubMed] [Google Scholar]
- 25.Guo W, Reigan P, Siegel D, Zirrolli J, Gustafson D, Ross D. Formation of 17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:quinone oxidoreductase 1: role of 17-AAG hydroquinone in heat shock protein 90 inhibition. Cancer Res. 2005;65:10006–10015. doi: 10.1158/0008-5472.CAN-05-2029. [DOI] [PubMed] [Google Scholar]
- 26.Gaspar N, Sharp SY, Pacey S, Jones C, Walton M, Vassal G, Eccles S, Pearson A, Workman P. Acquired resistance to 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) in glioblastoma cells. Cancer Res. 2009;69:1966–1975. doi: 10.1158/0008-5472.CAN-08-3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 28.Simard EP, Ward EM, Siegel R, Jemal A. Cancers with increasing incidence trends in the United States: 1999 through 2008. CA Cancer J Clin. 2012;62:118–128. doi: 10.3322/caac.20141. [DOI] [PubMed] [Google Scholar]
- 29.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 30.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 31.Papageorgio C, Perry MC. Epidermal growth factor receptor-targeted therapy for pancreatic cancer. Cancer Invest. 2007;25:647–657. doi: 10.1080/07357900701522653. [DOI] [PubMed] [Google Scholar]
- 32.Ballestrero A, Garuti A, Cirmena G, Rocco I, Palermo C, Nencioni A, Scabini S, Zoppoli G, Parodi S, Patrone F. Patient-tailored treatments with anti-EGFR monoclonal antibodies in advanced colorectal cancer: KRAS and beyond. Curr Cancer Drug Targets. 2012;12:316–328. doi: 10.2174/156800912800190956. [DOI] [PubMed] [Google Scholar]
- 33.Earp HS, Dawson TL, Li X, Yu H. Heterodimerization and functional interaction between EGF receptor family members: a new signaling paradigm with implications for breast cancer research. Breast Cancer Res Treat. 1995;35:115–132. doi: 10.1007/BF00694752. [DOI] [PubMed] [Google Scholar]
- 34.Riese DJ, Stern DF. Specificity within the EGF family/ErbB receptor family signaling network. Bioessays. 1998;20:41–48. doi: 10.1002/(SICI)1521-1878(199801)20:1<41::AID-BIES7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 35.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 36.Benson AM, Hunkeler MJ, Talalay P. Increase of NAD(P)H:quinone reductase by dietary antioxidants: possible role in protection against carcinogenesis and toxicity. Proc Natl Acad Sci U S A. 1980;77:5216–5220. doi: 10.1073/pnas.77.9.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berenbaum MC. Criteria for analyzing interactions between biologically active agents. Adv Cancer Res. 1981;35:269–335. doi: 10.1016/s0065-230x(08)60912-4. [DOI] [PubMed] [Google Scholar]
- 38.Buck E, Eyzaguirre A, Brown E, Petti F, McCormack S, Haley JD, Iwata KK, Gibson NW, Griffin G. Rapamycin synergizes with the epidermal growth factor receptor inhibitor erlotinib in non–small-cell lung, pancreatic, colon, and breast tumors. Mol Cancer Ther. 2006;5:2676–2684. doi: 10.1158/1535-7163.MCT-06-0166. [DOI] [PubMed] [Google Scholar]
- 39.Siegel D, Shieh B, Yan C, Kepa JK, Ross D. Role for NAD(P)H:quinone oxidoreductase 1 and manganese-dependent superoxide dismutase in 17-(allylamino)-17-demethoxygeldanamycin-induced heat shock protein 90 inhibition in pancreatic cancer cells. J Pharmacol Exp Ther. 2011;336:874–880. doi: 10.1124/jpet.110.176438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sydor JR, Normant E, Pien CS, Porter JR, Ge J, Grenier L, Pak RH, Ali JA, Dembski MS, Hudak J. Development of 17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci U S A. 2006;103:17408–17413. doi: 10.1073/pnas.0608372103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garon EB, Finn RS, Hamidi H, Dering J, Pitts S, Kamranpour N, Desai AJ, Hosmer W, Ide S, Avsar E. The HSP90 inhibitor NVP-AUY922 potently inhibits non–small cell lung cancer growth. Mol Cancer Ther. 2013;12:890–900. doi: 10.1158/1535-7163.MCT-12-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moser C, Lang SA, Hackl C, Wagner C, Scheiffert E, Schlitt HJ, Geissler EK, Stoeltzing O. Targeting HSP90 by the novel inhibitor NVP-AUY922 reduces growth and angiogenesis of pancreatic cancer. Anticancer Res. 2012;32:2551–2561. [PubMed] [Google Scholar]
- 44.McCollum AK, TenEyck CJ, Sauer BM, Toft DO, Erlichman C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res. 2006;66:10967–10975. doi: 10.1158/0008-5472.CAN-06-1629. [DOI] [PubMed] [Google Scholar]
- 45.McCollum AK, TenEyck CJ, Stensgard B, Morlan BW, Ballman KV, Jenkins RB, Toft DO, Erlichman C. P-glycoprotein–mediated resistance to Hsp90-directed therapy is eclipsed by the heat shock response. Cancer Res. 2008;68:7419–7427. doi: 10.1158/0008-5472.CAN-07-5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Millson SH, Prodromou C, Piper PW. A simple yeast-based system for analyzing inhibitor resistance in the human cancer drug targets Hsp90α/β. Biochem Pharmacol. 2010;79:1581–1588. doi: 10.1016/j.bcp.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 47.Kang BH, Altieri DC. Compartmentalized cancer drug discovery targeting mitochondrial Hsp90 chaperones. Oncogene. 2009;28:3681–3688. doi: 10.1038/onc.2009.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gandhi N, Wild AT, Chettiar ST, Aziz K, Kato Y, Gajula RP, Williams RD, Cades JA, Annadanam A, Song D. Novel Hsp90 inhibitor NVP-AUY922 radiosensitizes prostate cancer cells. Cancer Biol Ther. 2013;14:347–356. doi: 10.4161/cbt.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]