

Significance
Unless there are strong constraints, the probability of complex organs originating multiple times through similar trajectories should be vanishingly small. Here, we report that similar light-producing organs (photophores) evolved separately in two squid species, yet each organ expresses similar genes at comparable levels. Gene expression is so similar that overall expression levels alone can predict organ identity, even in separately evolved traits of squid species separated by tens of millions of years. The striking similarity of expression of hundreds of genes in distinct photophores indicates complex trait evolution may sometimes be more constrained and predictable than expected, either because of internal factors, like a limited array of suitable genetic building blocks, or external factors, like natural selection favoring an optimum.
Keywords: novelty, complexity, convergence, parallel evolution, gene expression
Abstract
Despite contingency in life’s history, the similarity of evolutionarily convergent traits may represent predictable solutions to common conditions. However, the extent to which overall gene expression levels (transcriptomes) underlying convergent traits are themselves convergent remains largely unexplored. Here, we show strong statistical support for convergent evolutionary origins and massively parallel evolution of the entire transcriptomes in symbiotic bioluminescent organs (bacterial photophores) from two divergent squid species. The gene expression similarities are so strong that regression models of one species’ photophore can predict organ identity of a distantly related photophore from gene expression levels alone. Our results point to widespread parallel changes in gene expression evolution associated with convergent origins of complex organs. Therefore, predictable solutions may drive not only the evolution of novel, complex organs but also the evolution of overall gene expression levels that underlie them.
Unrelated species often evolve predictably similar features when presented separately with the same environmental or biological challenges (1, 2). For example, the camera eyes of cnidarians, cephalopods, and vertebrates represent convergent and adaptive solutions to visual acuity (3), just as the repeated evolution of electric organs in teleost fish provides solutions for communication, navigation, and defense (4). A long-standing question is to what extent convergent molecular changes, either in protein-coding sequences or in gene expression, are associated with convergent phenotypic traits. Recent studies indicate many parallel genetic changes in protein-coding sequences may sometimes be related to convergent phenotypes, even between distantly related species (5). However, published gene expression studies of convergent traits are mainly limited to one or a few genes, and the transcriptomes underlying convergent traits are poorly understood. Although previous transcriptomic comparisons from long-diverged species indicate that largely conserved gene expression profiles characterize homologous traits (6, 7), the potentially labile nature of gene expression suggests that widespread changes in gene expression could readily produce similar phenotypic outcomes over short evolutionary times (8–10), especially if natural selection acts on a clearly optimal phenotype. In contrast, if gene expression patterns are evolutionarily conserved, perhaps by historical or developmental constraints, then homologous traits should have highly correlated expression, whereas convergent traits should be less correlated because of their different evolutionary origins (11–14). Depending on the extent to which genes are recruited and/or modulated in parallel, expression levels of genes in convergent traits may be more correlated than expression levels of genes in two dissimilar, nonhomologous traits. We tested this prediction using the photophores of bioluminescent squid.
Bioluminescence and complex associated optical structures have originated frequently in the history of life (15, 16), providing numerous instances of convergent evolution. Although bioluminescent structures are found in many cephalopod species (16), photophores harboring bioluminescent bacteria are known only in five genera of sepiolid squid (including Euprymna), and one genus of loliginid squid (Uroteuthis) (17, 18). The bacterial photophore of Euprymna scolopes has served as a model in the field of symbiosis biology (17), but relatively little is known about photophores of Uroteuthis, beyond the harboring of a similar strain of luminescent Vibrionaceae bacteria in an organ bearing a similar morphology to Euprymna’s photophore (19) (SI Appendix, Fig. S1). In addition to using elaborate, eye-like optical devices such as concave reflectors and large crystallin lenses to project emitted light (8–10, 20), these photophores enable E. scolopes to control the intensity of ventrally directed light and to distort the silhouette produced from down-welling light (11, 12, 21, 22). The photophore interior in E. scolopes comprises epithelial crypts capable of mediating bacterial growth and detecting light (15, 16, 19, 23, 24).
Bacterial photophores of squid are evolutionary novelties that may have arisen separately in two squid clades (6, 7, 25), but the details of these evolutionary origins have remained obscure. Although many novelties evolve by modifications to existing structures—such as fishes' electric organs, which are probably modified muscles—bacterial photophores are not obviously related to any structure in nonluminescent squid that might represent a single, direct evolutionary precursor. The best candidate for a trait with historical relationship to photophores is the accessory nidamental gland (ANG), another bacterial symbiotic organ positioned near the photophore in luminous squid (20, 26–28) and also present in nonluminous squid. The ANG consists of a cluster of tubules housing a consortium of bacteria, and was very likely present in the common ancestor of squid (25, 29, 30). The microscopic anatomy, ontogenetic and bacteriogenic properties of the photophore bear striking similarities to those of the ANG (20, 31). Although the photophore does not develop from the ANG, both develop in intimate association with the hindgut–ink sac complex (27, 32). These observations led to the hypothesis that the ANG could have given rise to a second bacteriogenic tubule in the adjacent hindgut–ink sac complex via furcation in sepiolid squid including Euprymna (20, 28, 33). Despite similarities with ANG, bacterial photophores appear to be a complex amalgamation of structural and physiological components, which suggests ANG alone cannot be an evolutionary precursor. For instance, photophores share developmental, phototransduction, and optical components with eyes and share immunological properties with skin, gills, and the ANG (17, 22, 23, 34). As such, we posit that—like mammalian nipples, which are also a complex amalgamation of structural features with different evolutionary histories—bacterial photophores of squid belong to a class of novelty termed “combinations of pleisiomorphic elements” [sensu (35)].
Here, we present strong statistical support for separate origins of bacterial photophores during cephalopod evolution. Furthermore, we observe significantly higher similarity in transcriptomic profiles among these convergent photophores than between photophores and any other tissues, including one hypothesized precursor structure (ANG), suggesting that extensive parallel evolution of gene expression underlies this morphological convergence.
Results
Strong Phylogenetic Support for Separate Origins of Bacterial Bioluminescent Organs in Squid.
Our phylogenetic analyses significantly support separate evolutionary origins (phylogenetic nonhomology) of bioluminescent organs in two distantly related squid species. We found that three separate analyses of character evolution [maximum parsimony, maximum likelihood (ML), and Bayesian inference] produced congruent inferences of at least two separate gains of bacterial photophores during evolution, with at least one gain before the divergence of the Sepiolinae (including Euprymna) and another gain within the Loliginidae clade (including Uroteuthis) (Fig. 1). Alternatively, a hypothesis of photophore homology requires at least eight losses under parsimony. Under a ML framework, we estimated the marginal likelihood of photophore presence or absence at each node in the ML tree topology under a two-rate Markov model (Fig. 1 and SI Appendix, Fig. S2), which also recovered significant support for nonhomology of photophores. Here, the scenario in which the most recent common ancestor between Euprymna and Uroteuthis lacks a photophore fits the data significantly better. Finally, we also found significant support for nonhomology using a Bayesian, continuous-time Markov chain approach to model presence/absence transitions (36) (SI Appendix, SI Materials and Methods). Here, we conducted a formal Bayes factor test that returns the ratio of the probability of observing our data under the null hypothesis that photophores were gained at most two times, to the probability of the alternative that photophores were gained at least three times. To account for topological uncertainty, we conducted this test integrating over 1,000 bootstrapped ML topologies. Under a wide variety of different priors on photophore gain and loss rates, the log Bayes factor ranged from 10−10 to 0.0002.
Fig. 1.

Evolutionary reconstructions of bacterial photophores on a ML tree of cephalopods. Bacterial photophores are found in species denoted by red tip symbols. Branch color depicts character state evolution inferred by parsimony. Pie charts report marginal likelihood of character states for selected ancestral nodes under an asymmetrical Markov model. Desaturated pie charts identify nodes where neither state significantly improved the model likelihood. A Bayesian test of independent origin (shown in Inset) also strongly favors hypotheses of at least two gains. (Note: branch lengths not to scale for basal lineages.) Right panels of photophores in E. scolopes (Top) and U. edulis (Bottom) depict the organ’s ventral positioning on ink sac, the conspicuous bilobed lensed structure, and internal morphology in cross-section. (b.c., bacterial crypt; i.s., ink sac; l, lens; MRCA, most recent common ancestor; r, reflector layer.)
Convergent Transcriptomes Are Just as Similar as Homologous Transcriptomes.
Although we recover extensive phylogenetic support for separate origins for photophores in squid, we found overall adult gene expression profiles of photophores to be just as similar in our two target species as overall gene expression of homologous organs of those species (Fig. 2). To compare overall gene expression of different organs, we generated 36 separate Illumina transcriptomes from three mature female individuals from each study species (E. scolopes and Uroteuthis edulis). For each of the six animals, we sequenced photophores plus five noncontroversially homologous organs, many of which share physiological or structural similarities to bacterial photophores (eye, brain, gill, skin, ANG). Photophore transcriptomes—even from different species—were statistically more similar to each other than to any other sequenced tissue—including ANG—as evidenced by each of three different distance measures (Spearman rank, cosine, and Bray–Curtis distance; under each measure P < < 0.001). Surprisingly, under the cosine and Bray–Curtis measures, the overall gene expression distance between photophores is indistinguishable from the distance between expression levels of any homologous organ of the two species, on average (Fig. 2 and SI Appendix, Fig. S4). We next used ordination to quantify the amount of tissue-specific signal contained in the transcriptomes. A scree plot of the principal components extracted from the 36 transcriptomes indicates that the amount of variation in gene expression attributable to tissue identity is nearly equivalent to the amount due to species differences (SI Appendix, Fig. S5). Nonmetric multidimensional scaling of the 36 libraries revealed surprisingly close clustering not only within homologous tissue types but also within nonhomologous photophores (SI Appendix, Fig. S5). The considerable degree of shared transcriptomic signal between convergent structures, despite extensive overall gene expression divergence evident between the two species, led us to hypothesize that gene expression profiles of photophores from one species may be sufficient to predict the tissue identity of the phylogenetically nonhomologous photophore transcriptome of another species.
Fig. 2.
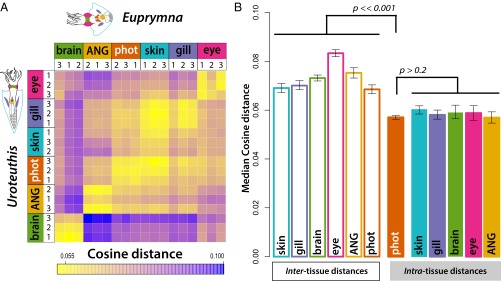
The extent of transcriptomic similarity between convergent photophores is comparable to the similarity between homologous traits. (A) Heat map of cosine distance among the 18 transcriptomes from each species (three replicates of six tissues in either species; yellow: highest similarity between tissue transcriptomes; blue: lowest similarity). (B) Median interspecific cosine distances between transcriptomes of alike tissues (filled bars) and nonalike tissues (open bars). Convergent photophore distances are not significantly different from the distance between any two homologous tissues. However, intraphotophore distances are significantly smaller than all intertissue transcriptome distances. Additional similarity measures are available in SI Appendix, Fig. S4.
Convergent Transcriptomes Predictable in Logistic Regression Models.
We found transcriptomic signal of organ identity to be so strong that a multinomial logistic regression model, a type of generalized linear model (GLM), can accurately predict organ identity from knowledge of gene expression levels alone, even in phylogenetically nonhomologous photophores. Widespread parallel changes to gene expression could occur either through numerous independent regulatory changes or through changes in just a few master control genes. In the case of the latter, distance metrics may overestimate similarity between transcriptomes. With this in mind, we implemented an elastic-net penalization during model training, which reduces overparameterization in the GLM—effectively lessening the number of genes (predictor variables) included in the model. This penalization algorithm addresses the issue of colinearity among gene expression levels that may stem from the expression of master control genes such as transcription factors (37). After training and cross-validating a tissue model from each of the 18 transcriptomes from each of our two study species, we could accurately predict tissue identity for the all transcriptomes of the opposite species (all samples: P < 0.005). We tested the significance of these predictions by generating a set of 10,000 “null transcriptomes” for each tissue (a null transcriptome was produced by randomly sampling genes without respect to tissue identity). We then attempted to predict tissue identity with the GLM models for each null transcriptome. Regardless of which species was used to train the model, all replicate photophore transcriptomes from the other species were accurately predicted as photophores, with significantly better scores than the null distribution (Fig. 3 and SI Appendix, Fig. S6 A and B, and Table S4).
Fig. 3.
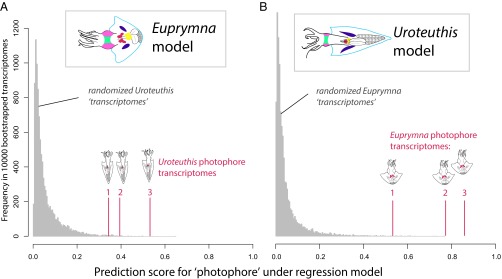
Transcriptomes of convergent traits contain predictable signatures of gene expression. (A) A regression model fit of Euprymna transcriptomes accurately predicts better than chance all three Uroteuthis transcriptomes that originated from independent photophores. Gray distributions show model predictions for null transcriptomes (generated by bootstrapping transcript read counts across six tissues). (B) A reciprocal model fit of Uroteuthis data also predicts which three Euprymna transcriptomes belong to photophores. All real photophore transcriptomes scored prediction values ≥99.5% of null distribution. Colored tissues identify the six sources of transcriptomes used to train each model. Results for homologous tissue prediction are in SI Appendix, Fig. S6 A and B, and Table S4.
Differential Expression in Lens Crystallin, Opsin, and Peroxidase Genes Between Convergent Photophores.
Although expression levels of many genes were indistinguishable in our RNA-seq experiments, we used quantitative PCR (qPCR) on genes previously studied in E. scolopes photophore to corroborate and to assess robustness of single-gene estimates of differential expression from RNA-seq experiments. These qPCR tests confirmed significant differences in expression levels of four important candidate loci (SI Appendix, Fig. S3) between photophores in adults of a representative sepiolid (E. scolopes) and loliginid (U. edulis) species.
Using qPCR, we first compared opsin expression. Although E. scolopes uses an opsin phototransduction pathway in the light-sensitive epithelial lining that harbors bacterial symbionts (24), we detected no opsin expression in U. edulis outside the retina. We next compared halide peroxidase expression, because E. scolopes relies on this enzyme to regulate symbiont populations within the photophore (38). We found that E. scolopes expressed nearly 800 times as much halide peroxidase in the photophores as U. edulis (Mann–Whitney, P = 0.048), suggesting that the Uroteuthis photophore may rely on alternate mechanisms to mediate symbiont growth and may be under reduced oxidative stress compared with Euprymna. Third, we compared expression of lens crystallin genes. Although the lenses of squid eyes are largely composed of S-crystallins (derived from glutathione S-transferases), the photophore of the squid E. scolopes uses an unrelated Ω-crystallin (derived from aldehyde dehydrogenase) (39, 40). We found that, although both S- and Ω-crystallins are detectable in bioluminescent organs, U. edulis expresses predominantly S-crystallins, whereas E. scolopes expresses predominantly Ω-crystallins (Mann–Whitney, P < 0.003). Because the recruitment of Ω-crystallins may be related to adaptation to high levels of oxidative stress in E. scolopes bioluminescent organs (41), the dominance of S-crystallins in U. edulis organs also suggests reduced oxidative stress compared with Euprymna.
Discussion
We report very strong statistical phylogenetic evidence that bacterial photophores of two cephalopod species are evolutionarily convergent, having originated separately in some loliginids (including Uroteuthis) and some sepiolids (including Euprymna). At the same time, we show that overall gene expression patterns within the convergent photophores are themselves convergent, to the point that photophore identity is predictable from gene expression models constructed from a convergent photophore. The striking similarity of expression of hundreds of genes in evolutionarily distinct photophores could indicate complex trait evolution may sometimes be more constrained and predictable than expected, either because of internal factors, like developmental constraints, or external factors, like natural selection favoring an optimum. However, there are also important alternatives to consider.
A first alternative to separate origins and massively parallel evolution of gene expression is that bacterial photophores are homologous and were lost numerous times in species that lack them. Parsimony would require eight losses, a number that cannot be evaluated statistically, leading to our Bayesian phylogenetic test. The test quantifies different probabilities to indicate—given our standard discrete state Markov model—the observed data are approximately and conservatively 5,000 times less likely to have arisen from an evolutionary history with less than three gains of photophores than from an evolutionary history with three or more photophore gains (SI Appendix, Table S2). Although dependent on the underlying homogeneous model of evolution, this is extraordinarily strong evidence for separate origins of bacterial bioluminescent organs in squid. To put these results in the context of classical t tests, Bayes factors of such absolute magnitude lead to rejection of the null hypothesis with a significance threshold much lower than 0.005 (36).
A second alternative to massively parallel evolution of gene expression accompanying separate origins of photophores is that a homologous precursor trait evolved in parallel in at least two instances into photophores, while maintaining very similar gene expression patterns. Here, much of the similarity in gene expression we observe could be attributable to homology, whereas the differences in expression in a few genes could indicate evolutionary divergence. Some of our results do not support this alternative. Previous morphological and developmental data (20, 27, 28, 31, 32) suggest the best candidate for such an evolutionary precursor of bacterial photophores is the ANG, even though similarities also exist between photophores and other organs like eyes, skin, and gills. If the ANG was the “proto-photophore” and the cause of our observed transcriptome similarities between convergent photophores, we would also expect greater transcriptome similarities between each species’ photophore and its own ANG than between the photophores of different species because—as shown for the homologous organs we studied—traits related by evolutionary descent should share greater transcriptomic similarity than unrelated traits (see also refs. 7, 42, and 43). In contrast, we observe significantly greater profile similarity between photophores than between photophores and ANGs (SI Appendix, Fig. S7). In addition, photophores share physiological and developmental features with other organs besides ANG, notably gills and eyes (24, 34, 38, 39). The convergent photophores are also more similar to each other in gene expression than to eye or gill expression. As such, we posit that—like mammalian nipples, which are also a complex amalgamation of structural features with different evolutionary histories—bacterial photophores of squid belong to a class of novelty termed “combinations of pleisiomorphic elements” [sensu (35)]. This origin of a photophore by amalgamation may have occurred at least two times during the history of squid in surprisingly similar ways, leading to predictable similarity in gene expression.
Regardless of the evolutionary significance of the bacteriogenic ANG for the origins of photophores, the gene expression profiles in these two focal species suggest an important role for bricolage or “tinkering”—the separate recruitment of similar genetic modules recruited from other tissues into each evolving photophore (44–46). Bricolage could help explain the chimeric nature of photophores, which share some developmental and structural features with eyes, and also immunological properties with organs like the ANG and gill (24, 34, 38, 39). The evolution of photophores in each lineage probably required extensive co-option of preexisting homologous genetic modules enabling symbiosis with luminous bacteria and eventually light modulation at both the cellular and behavioral levels (17, 22, 47). However, we also found RNA-seq evidence, corroborated by qPCR, supporting differential gene use in each photophore. At least four genes with established roles in the Euprymna photophore (opsin, halide peroxidase, ω-crystallin, and S-crystallin) are expressed at substantially different levels in Uroteuthis (SI Appendix, Fig. S3). These genes, in contrast with the overall pattern of similarity, highlight how bricolage may lead to specific differences during the evolutionary courses of these organs. For example, although Euprymna expresses predominantly L-crystallin in its lens, Uroteuthis has co-opted the unrelated gene S-crystallin, known for its role in cephalopod ocular lenses (48). Significant gene expression differences are consistent with divergent evolution in the photophores of each species, and the incongruity between some individual genes and full expression profiles underscores the importance of analyzing not just a few candidate genes, but rather expression across entire transcriptomes.
Irrespective of whether photophores have repeatedly originated as homonomous structures to the ANG or whether they represent morphological novelties (35) through bricolage and amalgamation, the extent of gene expression similarity we observe in convergent photophores is surprising because of the expectation for convergent traits to evolve by distinct genetic mechanisms (11–14, 48, 49). The finding of widespread coincident changes in convergent photophores indicates that transcriptome evolution in complex novelties may be more predictable than previously suspected.
Materials and Methods
Sample Collection.
We collected mature female specimens of both species at night and killed them the following morning, before preserving in RNAlater (Ambion) for 1 mo. Local fisherman in Saga Prefecture, Japan, caught Uroteuthis edulis individuals. We used a dip net in Honolulu, Hawaii, to collect Euprymna scolopes. We dissected eyes, brains, skin samples, distal gill samples, ANGs, and photophores.
Transcriptome Sequencing.
We prepared samples of all six organs from three individuals from each species. We first extracted RNA from the resulting 36 samples using the RiboPure kit (Ambion) and then prepared libraries for Illumina sequencing on the Hi-Seq platform at University of California, Berkeley, using the TruSeq kit (Illumina).
Phylogenetic Analysis.
We estimated the ML topology of 70 cephalopod species in RAxML, version 7.6.8 (50), under a GTRGAMMA model using concatenated nucleotide sequences for up to 13 loci (12S, 16S, 18S, aldolase, ATP synthetase B, COI, cytB, EF1a, histone3a, octopine dehydrogenase, opsin, Pax6) from previously published genes and published or newly generated transcriptome datasets (see SI Appendix, Table S1, for accessions). In particular, species belonging to Sepiolid and Loliginid families were heavily targeted for sequence sampling. The final taxon sample of 70 cephalopods included representatives from all (10 out of 15) sepiolid and all (9 out of 11) loliginid genera for which sequence data are accessible. Sepiolid sampling covered 25 of ∼91 nominal species. We scored species as present or absent for bacterial photophores (summarized in ref. 25). We estimated ancestral character states using the ML topology under parsimony in Mesquite (51) and under a two-rate Markov model in the R package “corHMM” (52). To account for phylogenetic uncertainty in cephalopod relationships and to estimate the minimum number of times photophores originated, we used a continuous-time Markov chain model and Bayes factor test on the set of 1,000 RAxML-bootstrapped ML topologies. [The Bayes factor test can be implemented in a new R package (53) (“indorigin”), with additional details on independent origins test in SI Appendix, SI Materials and Methods.] Briefly, these Bayes factors (54) are calculated from the probability of the data conditioned on a null and alternative hypothesis specifying a minimum number (m) of photophore gains occurring on a set of trees:
Quantitative PCR on Candidate Loci.
We used qPCR to test whether the relative expression levels differ in U. edulis and E. scolopes for the following genes, which have been previously shown to play important roles in the physiology of the E. scolopes photophore: opsin, cryptochrome1, cryptochrome2, s-crystallin, Ω-crystallin, NFkappaB, and peroxidase. RNA from 10 individuals from each species was isolated from photophores as described earlier for sequencing library preparation. qPCRs were prepared using GoTaq qPCR Master Mix (Promega), and quantification was measured on a Bio-Rad iCycler with a 54 °C annealing temperature. β-Actin serves as a reference gene for comparing expression levels between species. We used a Mann–Whitney U test to determine if log-fold changes were significant between species. Primer sequences are available in SI Appendix, Table S2.
Transcriptome Raw Read Processing.
We used FastQC (55) to assess read qualities and identify sequencing artifacts. We then trimmed residual adapters and low-quality end via sliding window using FastX Toolkit (56) until all reads scored above 20 at each position. We identified overrepresented sequences by FastQC removed by TagDust (57). All six read libraries constructed from each individual were pooled for assembly using Trinity (05-2012 release) (58). To remove potential bacterial contamination, any assembled contigs receiving a tBLASTn match (e < 1e-50) to the genomes of Vibrio fischeri ES114 (GenBank: NC_00684[0-2]0.2) or Photobacterium leiognathi (NZ_DF093[593-603]0.1) were restricted from further analysis. Reads from each tissue library were then mapped back to the individual’s single assembly using Bowtie (59) with the RSEM package (60). The original unaltered reads have been deposited in the National Center for Biotechnology Information Short Read Archive under BioProject PRJNA257113.
Transcriptome Ortholog Clustering.
To assign orthology among genes in the six Trinity assemblies, we used an iterative clustering method using CD-HIT software (61). Redundancy within each of the six individual assemblies was reduced by clustering sequences at 100% similarity. Intraspecies assembly clustering was then performed to further cluster sequences at 90% similarity. The longest sequence from each cluster was translated using Trinity’s ORFpredictor tool and then used in an interspecific reciprocal BLASTp to identify orthologous sequences.
Quantification of Transcriptome Similarity.
Transcriptome saturation was verified by rarefaction analysis using the “vegan” R package (62). Any orthologous gene represented by fewer than three reads in a given library was considered absent in that library. Differences in library size were accounted for by adjusting RSEM’s expected read counts for each library using TMM-normalization factors in EdgeR (63). To prevent artifacts stemming from the separate de novo read assemblies performed on each individual, we only retained orthologs from the analysis in which more than one individual had recovered a minimum of 20 mappable reads across their libraries.
Quantifying similarity requires considering the caveats inherent to similarity/distance measures. To ensure that our choice of similarity measure alone did not bias our conclusions, we report results using three different measures. Cosine similarity (an information retrieval metric similar to Pearson’s correlation in that it assesses shared variance but is less sensitive to zeros), Spearman rank correlation coefficient (although similar to Pearson’s, is less sensitive to outliers and more conservative if violating distribution assumptions), and Bray–Curtis similarity (an ecological index that is less influenced by presence of low-count variables) were evaluated across the pairwise tissue comparisons on the log-transformed normalized read count libraries using R packages “lsa,” “bioDist,” and “fossil.” We evaluated whether the interspecific similarity among convergent tissues (photophores) (i) differs from the similarity between nonhomologous tissues or (ii) differs from the similarity observed between homologous tissues. Because the choice of similarity measure could influence the outcome of significance tests, we conducted all tests with the same three measures as described earlier. We used a two-way test of independence (permutation test) in the “coin” R package to determine significance (64).
Multinominal Logistic Regression Model.
To determine whether the gene expression signature of a given tissue is sufficient to accurately predict a sample of the same tissue type in another species, we fit a multinomial logistic regression model for each species using the “glmnet” R package (65), using a hybrid elastic-net penalization on variable coefficients to minimize the number of genes effectively included in the model. For each species, we fit the model using all 18 normalized log-transformed read-count libraries and then used the model to predict the tissue identities of the other species’ libraries. To test whether these model predictions were significantly better than chance, we created a null distribution of read-count data matrices by bootstrapping the read counts of each gene, creating bootstrapped tissue libraries with randomized gene abundances. For each species, we created 10,000 sets of these 18 bootstrapped libraries. These bootstrapped datasets were then subjected to the GLM model fit by the original data from the opposite species. The resulting tissue predictions were used as a null distribution with which we tested the significance of the model predictions on real data.
Supplementary Material
Acknowledgments
We are grateful to M. Matsuyama, G. Nishihara, M. J. McFall-Ngai, and the Kewalo Marine Laboratory at the University of Hawaii for facilitating animal collections. We thank P. Weakliem for providing computational resources. We thank D. Stern, S. Haddock, S. Hodges, and T. DeTomaso for manuscript comments. This work was funded by National Science Foundation (NSF) Grants CNS-0960316 awarded to University of California, Santa Barbara Center for Scientific Computing, NSF Grants DEB-1210673 (to M.S.P. and T.H.O.) and DEB-1146337 (to T.H.O.), and NSF Grants DMS-0856099 and DMS-1264153 and NIH Grant R01-AI107034 (to V.N.M. and M.A.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper has been deposited in the in National Center for Biotechnology Information Short Read Archive, www.ncbi.nlm.nih.gov/sra (BioProject PRJNA257113).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1416574111/-/DCSupplemental.
References
- 1.Conway-Morris S. Life's Solution: Inevitable Humans in a Lonely Universe. Cambridge Univ Press; Cambridge, UK: 2003. [Google Scholar]
- 2.Stern DL. The genetic causes of convergent evolution. Nat Rev Genet. 2013;14(11):751–764. doi: 10.1038/nrg3483. [DOI] [PubMed] [Google Scholar]
- 3.Land M, Nilsson D-E. Animal Eyes. Oxford Univ Press; Oxford: 2002. [Google Scholar]
- 4.Gallant JR, et al. Nonhuman genetics. Genomic basis for the convergent evolution of electric organs. Science. 2014;344(6191):1522–1525. doi: 10.1126/science.1254432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Liu Z, Shi P, Zhang J. The hearing gene Prestin unites echolocating bats and whales. Curr Biol. 2010;20(2):R55–R56. doi: 10.1016/j.cub.2009.11.042. [DOI] [PubMed] [Google Scholar]
- 6.Khaitovich P, Enard W, Lachmann M, Pääbo S. Evolution of primate gene expression. Nat Rev Genet. 2006;7(9):693–702. doi: 10.1038/nrg1940. [DOI] [PubMed] [Google Scholar]
- 7.Chan ET, et al. Conservation of core gene expression in vertebrate tissues. J Biol. 2009;8(3):33. doi: 10.1186/jbiol130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones FC, et al. Broad Institute Genome Sequencing Platform and Whole Genome Assembly Team The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012;484(7392):55–61. doi: 10.1038/nature10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whittall JB, Voelckel C, Kliebenstein DJ, Hodges SA. Convergence, constraint and the role of gene expression during adaptive radiation: Floral anthocyanins in Aquilegia. Mol Ecol. 2006;15(14):4645–4657. doi: 10.1111/j.1365-294X.2006.03114.x. [DOI] [PubMed] [Google Scholar]
- 10.Reed RD, et al. optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science. 2011;333(6046):1137–1141. doi: 10.1126/science.1208227. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka K, Barmina O, Kopp A. Distinct developmental mechanisms underlie the evolutionary diversification of Drosophila sex combs. Proc Natl Acad Sci USA. 2009;106(12):4764–4769. doi: 10.1073/pnas.0807875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voelckel C, Borevitz JO, Kramer EM, Hodges SA. Within and between whorls: Comparative transcriptional profiling of Aquilegia and Arabidopsis. PLoS One. 2010;5(3):e9735. doi: 10.1371/journal.pone.0009735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prud’homme B, et al. Repeated morphological evolution through cis-regulatory changes in a pleiotropic gene. Nature. 2006;440(7087):1050–1053. doi: 10.1038/nature04597. [DOI] [PubMed] [Google Scholar]
- 14.Emera D, et al. Convergent evolution of endometrial prolactin expression in primates, mice, and elephants through the independent recruitment of transposable elements. Mol Biol Evol. 2012;29(1):239–247. doi: 10.1093/molbev/msr189. [DOI] [PubMed] [Google Scholar]
- 15.Haddock SHD, Moline MA, Case JF. Bioluminescence in the sea. Annu Rev Mar Sci. 2010;2:443–493. doi: 10.1146/annurev-marine-120308-081028. [DOI] [PubMed] [Google Scholar]
- 16.Herring PJ. In: Biology of Cephalopods. Symposia of the Zoological Society of London No. 38. Nixon M, Messenger J, editors. Academic; London: 1977. [Google Scholar]
- 17.McFall-Ngai MJ. Consequences of evolving with bacterial symbionts: Insights from the squid-vibrio associations. Annu Rev Ecol Syst. 1999;30:235–256. [Google Scholar]
- 18.Anderson FE, Bergman A, Cheng SH, Pankey MS, Valinassab T. Lights out: The evolution of bacterial bioluminescence in Loliginidae. Hydrobiologia. 2013;725(1):189–203. [Google Scholar]
- 19.Guerrero-Ferreira RC, Nishiguchi MK. Ultrastructure of light organs of loliginid squids and their bacterial symbionts: A novel model system for the study of marine symbioses. Vie Milieu Paris. 2009;59(3-4):307–313. [PMC free article] [PubMed] [Google Scholar]
- 20.Montgomery M, McFall-Ngai MJ. Embryonic development of the light organ of the sepiolid squid Euprymna scolopes Berry. Biol Bull. 1993;184:296–308. doi: 10.2307/1542448. [DOI] [PubMed] [Google Scholar]
- 21.Young RE. Intensity regulation of bioluminescence during countershading in living midwater animals. Fish Bull. 1977;75:239–252. [Google Scholar]
- 22.Jones B, Nishiguchi MK. Counterillumination in the Hawaiian bobtail squid, Euprymna scolopes Berry (Mollusca: Cephalopoda) Mar Biol. 2004;144:1151–1155. [Google Scholar]
- 23.Pringgenies D, Jorgensen JM. Morphology of the luminous organ of the squid Loligo duvauceli D'Orbigny 1839. Acta Zoologica. 1994;75:305–309. [Google Scholar]
- 24.Tong D, et al. Evidence for light perception in a bioluminescent organ. Proc Natl Acad Sci USA. 2009;106(24):9836–9841. doi: 10.1073/pnas.0904571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindgren AR, Pankey MS, Hochberg FG, Oakley TH. A multi-gene phylogeny of Cephalopoda supports convergent morphological evolution in association with multiple habitat shifts in the marine environment. BMC Evol Biol. 2012;12:129. doi: 10.1186/1471-2148-12-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naef A. Die Cephalopoden, I. Teil. R. Friedländer & Sohn; Berlin: 1921. [Google Scholar]
- 27.Boletzky SV. On the presence of light organs in Semirossia Streenstrup, 1887 (Mollusca: Cephalopoda) Bull Mar Sci. 1970;20:374. [Google Scholar]
- 28.Nishiguchi MK, Lopez JE, Boletzky Sv. Enlightenment of old ideas from new investigations: More questions regarding the evolution of bacteriogenic light organs in squids. Evol Dev. 2004;6(1):41–49. doi: 10.1111/j.1525-142x.2004.04009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bloodgood RA. The squid accessory nidamental gland: Ultrastructure and association with bacteria. Tissue Cell. 1977;9(2):197–208. doi: 10.1016/0040-8166(77)90016-7. [DOI] [PubMed] [Google Scholar]
- 30.Collins AJ, et al. Diversity and partitioning of bacterial populations within the accessory nidamental gland of the squid Euprymna scolopes. Appl Environ Microbiol. 2012;78(12):4200–4208. doi: 10.1128/AEM.07437-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herring PJ. Luminescent Organs. Academic; London: 1988. [Google Scholar]
- 32.Naef A. Die Cephalopoden. Embryologie. Fauna Flora Golf Neapel. 1928;35:1–357. [Google Scholar]
- 33.Gould SJ, Vrba ES. Exaptation—a missing term in the science of form. Paleobiology. 1982;8(1):4–15. [Google Scholar]
- 34.Peyer SM, Pankey MS, Oakley TH, McFall-Ngai MJ. ScienceDirect. Mech Dev. 2013;131:111–116. doi: 10.1016/j.mod.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Müller GB, Wagner GP. Novelty in evolution: Restructuring the concept. Annu Rev Ecol Syst. 1991;22:229–256. [Google Scholar]
- 36.Minin VN, Suchard MA. Counting labeled transitions in continuous-time Markov models of evolution. J Math Biol. 2008;56(3):391–412. doi: 10.1007/s00285-007-0120-8. [DOI] [PubMed] [Google Scholar]
- 37.Michaut L, et al. Analysis of the eye developmental pathway in Drosophila using DNA microarrays. Proc Natl Acad Sci USA. 2003;100(7):4024–4029. doi: 10.1073/pnas.0630561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Small AL, McFall-Ngai MJ. Halide peroxidase in tissues that interact with bacteria in the host squid Euprymna scolopes. J Cell Biochem. 1999;72(4):445–457. [PubMed] [Google Scholar]
- 39.Montgomery MK, McFall-Ngai MJ. The muscle-derived lens of a squid bioluminescent organ is biochemically convergent with the ocular lens. Evidence for recruitment of aldehyde dehydrogenase as a predominant structural protein. J Biol Chem. 1992;267(29):20999–21003. [PubMed] [Google Scholar]
- 40.Tomarev SI, Zinovieva RD, Piatigorsky J. Characterization of squid crystallin genes. Comparison with mammalian glutathione S-transferase genes. J Biol Chem. 1992;267(12):8604–8612. [PubMed] [Google Scholar]
- 41.Zinovieva RD, Tomarev SI, Piatigorsky J. Aldehyde dehydrogenase-derived omega-crystallins of squid and octopus. Specialization for lens expression. J Biol Chem. 1993;268(15):11449–11455. [PubMed] [Google Scholar]
- 42.Wang Z, Young RL, Xue H, Wagner GP. Transcriptomic analysis of avian digits reveals conserved and derived digit identities in birds. Nature. 2011;477(7366):583–586. doi: 10.1038/nature10391. [DOI] [PubMed] [Google Scholar]
- 43.Brawand D, et al. The evolution of gene expression levels in mammalian organs. Nature. 2011;478(7369):343–348. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- 44.Jacob F. Evolution and tinkering. Science. 1977;196(4295):1161–1166. doi: 10.1126/science.860134. [DOI] [PubMed] [Google Scholar]
- 45.Wagner GP. Homologues, natural kinds and the evolution of modularity. Integr Comp Biol. 1996;36:36–43. [Google Scholar]
- 46.von Dassow G, Munro E. Modularity in animal development and evolution: Elements of a conceptual framework for EvoDevo. J Exp Zool. 1999;285(4):307–325. [PubMed] [Google Scholar]
- 47.Ruby EG, McFall-Ngai MJ. Oxygen-utilizing reactions and symbiotic colonization of the squid light organ by Vibrio fischeri. Trends Microbiol. 1999;7(10):414–420. doi: 10.1016/s0966-842x(99)01588-7. [DOI] [PubMed] [Google Scholar]
- 48.Tomarev SI, Zinovieva RD. Squid major lens polypeptides are homologous to glutathione S-transferases subunits. Nature. 1988;336(6194):86–88. doi: 10.1038/336086a0. [DOI] [PubMed] [Google Scholar]
- 49.Hoffmann FG, Opazo JC, Storz JF. Gene cooption and convergent evolution of oxygen transport hemoglobins in jawed and jawless vertebrates. Proc Natl Acad Sci USA. 2010;107(32):14274–14279. doi: 10.1073/pnas.1006756107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 51.Maddison W, Maddison D. 2008. Mesquite: A modular system for evolutionary analysis. Version 2.5. Available at mesquiteproject.org. Accessed October 9, 2014.
- 52.Beaulieu JM, O’Meara BC, Donoghue MJ. Identifying hidden rate changes in the evolution of a binary morphological character: The evolution of plant habit in campanulid angiosperms. Syst Biol. 2013;62(5):725–737. doi: 10.1093/sysbio/syt034. [DOI] [PubMed] [Google Scholar]
- 53.Minin VN, Suchard MA, Imholte GC. 2014. indorigin: Testing how many times a trait of interest was regained during evolution (R package). Available at github.com/vnminin/indorigin. Accessed October 9, 2014.
- 54.Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc. 1995;90:773–795. [Google Scholar]
- 55.Andrews S. 2010 FastQC: A quality control tool for high throughput sequence data. Available at www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed October 9, 2014.
- 56.Hannon G. 2009. FASTX Toolkit. Available at hannonlab.cshl.edu/fastx_toolkit/. Accessed October 9, 2014.
- 57.Lassmann T, Hayashizaki Y, Daub CO. TagDust—a program to eliminate artifacts from next generation sequencing data. Bioinformatics. 2009;25(21):2839–2840. doi: 10.1093/bioinformatics/btp527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dixon P. VEGAN, a package of R functions for community ecology. Journal of Vegetation Science. 2003;14:927–930. [Google Scholar]
- 63.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hothorn T, Hornik K, van de Wiel MA, Zeileis A. A Lego System for Conditional Inference. Am Stat. 2006;60:257–263. [Google Scholar]
- 65.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.