

Abstract
Long-range electron transfer (ET) is a crucial step in many energy conversion processes and biological redox reactions in living organisms. We show that newly developed X-ray pulses can directly probe the evolving oxidation states and the electronic structure around selected atoms with detail not available through conventional time-resolved infrared or optical techniques. This is demonstrated in a simulation study of the stimulated X-ray Raman (SXRS) signals in Re-modified azurin, which serves as a benchmark system for photoinduced ET in proteins. Nonlinear SXRS signals offer a direct novel window into the long-range ET mechanism.
Many biological processes including photosynthesis,1 respiration,2 DNA photodamage repair,3 and reducing nucleotides to deoxynucleotides4 involve the controlled transfer of electrons between spatially well-separated groups. Understanding the factors that control ET processes in proteins and DNA5,6 in detail may further help the design of novel sensors and artificial biomimetic light-harvesting devices.7−9 Since the ET rate decreases exponentially with the electron donor–acceptor distance,10 ET in proteins at long distances (e.g., >25 Å) can be drastically accelerated through the sequential mechanism, where an aromatic amino acid such as tryptophan acts as an intermediate for the transferred electron.11−13 Single-step electron tunneling (superexchange) crosses over to a multistep sequential electron hopping mechanism at large distances.12,14 Time-resolved infrared (TRIR) and optical spectroscopy have been widely used to study ET dynamics.11,15 However, measuring the dynamics of each electron hopping step, spectroscopically characterizing of intermediates and identifying different pathways still poses a formidable challenge.
Recently developed state-of-the-art intense X-ray light sources make it possible to carry out nonlinear X-ray spectroscopy experiments that can directly observe electronic and nuclear dynamics in molecules or materials.16−18 Thanks to their broad bandwidth, X-ray pulses can create coherent superpositions of excited states localized near the target atoms. Femtosecond optical pulses introduced in the eighties were able to impulsively probe molecular vibrations. X-ray pulses can achieve the same goal for valence excitations, which are good indicators of the evolving electronic structure in different oxidation states. The atomic pinpoint accuracy of resonant core X-ray transitions can enable the selective detection of the donor, intermediate, and acceptor in an ET system.
Here we show that stimulated X-ray Raman spectroscopy (SXRS) signals19 provide a unique highly sensitive probe for electronic structure around the specific atoms excited by X-ray pulses, which could result in a real-time movie of electron flow in molecules. The resonant core excitations merely serve as a fast atomic-localized trigger for the valence excitations but are not observed directly in this technique. This information is not available from conventional time-resolved IR, optical techniques or transient X-ray absorption spectroscopy (TXAS). TXAS is commonly used for studying photochemical processes.20 However, the short core-excited state lifetime results in very broad peaks compared with valence excitations. By probing valence dynamics via a core-excited intermediate, SXRS combines the spatial selectivity of X-ray spectroscopy with the much narrower linewidths of visible and UV spectra. X-ray diffraction only probes the total electronic density, which does not vary much by the transfer of a single electron. SXRS, in contrast, looks at the structure of valence excitations and their variation with oxidation state, which is an ideal probe for the evolving charge distributions.
Nonlinear X-ray spectroscopy21,22 provides new windows into reaction intermediates. The broad bandwidth (10.9 eV for a 166 as pulse) allows to probe many electronic states in a single snapshot. The attosecond time resolution of the pulses employed in this study is not necessary for the slow electron-transfer processes considered here (100 ps–ns). However, the corresponding bandwidth is crucial for providing a novel window into the intermediate steps.
The interpretation of time-resolved measurements of charge migration in biological complexes is complicated by the numerous pathways and reaction intermediates. Adding artificial groups that can be triggered optically to initiate the process makes it possible to study controlled steps.
Azurin is a small (128 residues for the Re-modified azurin) type I blue copper protein (cupredoxin)23 produced by several aerobic bacteria. In the respiration chain, it transfers one electron between cytochrome c-551 and the corresponding oxidase, which helps Pseudomonas oxidase or Pseudomonas nitrite reductase to reduce O2 to H2O, or NO2– to NO.24 Metal-modified azurins have been employed to trigger the ET process optically and explore how electrons flow in real time through the protein backbone.11 Studies so far use optical, infrared, and electron paramagnetic resonance (EPR) probes.25,26
The molecular structure and ET pathway in the Re-modified azurin are shown in Figure 1. The artificial Re metal center excited by a UV photon mimics the electron acceptors in living organisms, and triggers the long-distance electron transfer from the Cu center.11,15 Figure 2 shows the key intermediate states and kinetic pathways. First, a singlet metal-to-ligand charge-transfer (1MLCT) state involving a hole on the Re center and an electron on the dmp (4,7-dimethyl-1,10-phenanthroline) ligand is created by a UV photon. This excited state is rapidly converted into a vibrationally excited triplet state (3*MLCT, state A) through an intersystem crossing process, and the system then relaxes to its lowest vibrational configuration (3MLCT, state B). This state equilibrates with a charge-separated state (3CS, state C), where the hole has migrated to the tryptophan group (Trp122). The 1MLCT state may also rapidly and directly decay to the charge-separated state. Finally an electron from the Cu(I) center fills this hole to form a long-range charge- transfer state (LCT, state D). In this model system a hole is transferred from the Re to the Cu center, while in the respiratory chain of the bacterium Pseudomonas aeruginosa, an electron is transferred from the Cu center to the electron acceptor, cytochrome oxidase.24 Extensive TRIR, transient UV–vis absorption, and fluorescence decay experiments revealed that Trp122 acts as an intermediate and accelerates the electron flow.11 These resulted in the kinetic model
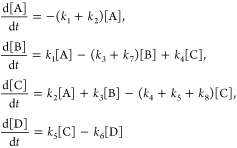 |
1 |
All rate constants k1–8 have been determined by fitting to experiment.11
Figure 1.
Eletron transfer pathway in Re-modified azurin. The red wiggly arrow represents the incident UV pulse that triggers the ET process, and the blue wiggly arrows represent the probe X-ray pulses and the emitted signals. The ET pathway (from the Cu(I) center via the tryptophan group to the Re(I) center) is marked by black arrows. The three X-ray chromophores in the system—Re, Cl, and Cu atoms—are shown in orange, green, and purple, respectively. Ball-and-stick structures represent the electron donor (Cu-complex), intermediate group (Cl-substituted Trp122), and acceptor (Re-complex). Other segments of the protein are represented by solid ribbons.
Figure 2.
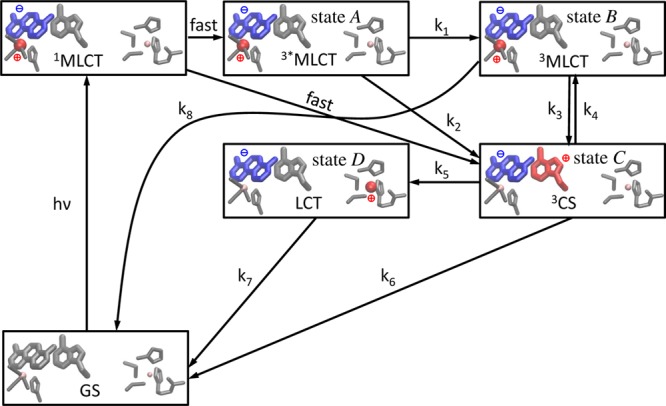
Mechanism of the photoinduced long-range ET in Re-modified azurin (adapted from Figure S1 in ref (11)). The structures of the three molecular fragments in Figure 1 are shown. In each frame, the left fragment is the Re(CO)3(dmp) complex; the middle fragment is the Cl-substituted Trp group; and the right fragment represents the Cu center and the surrounding ligands. Excited species positively or negatively charged compared to the ground state are shown in red and blue, respectively. 1MLCT: singlet metal-to-ligand charge-transfer state; 3*MLCT (state A): vibrationally excited triplet metal-to-ligand charge transfer state; 3MLCT (state B): vibrationally relaxed triplet metal-to-ligand charge transfer state; 3CS (state C): triplet charge-separated state; LCT (state D): long-range charge-transfer state; GS: ground state.
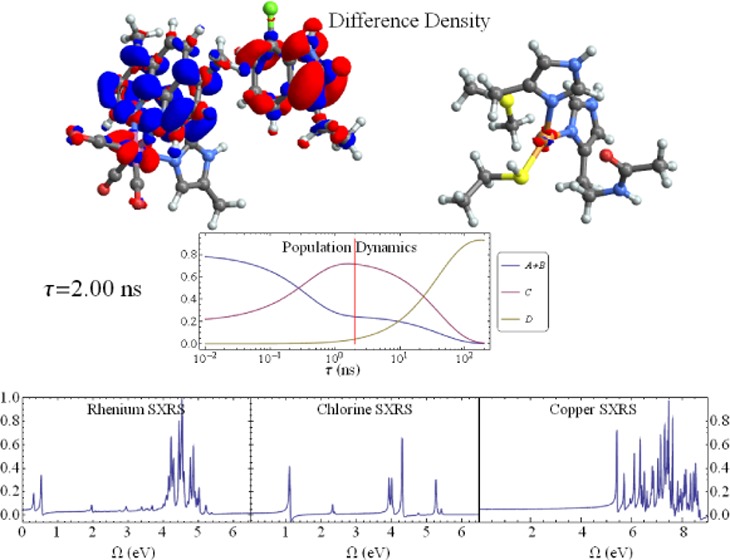
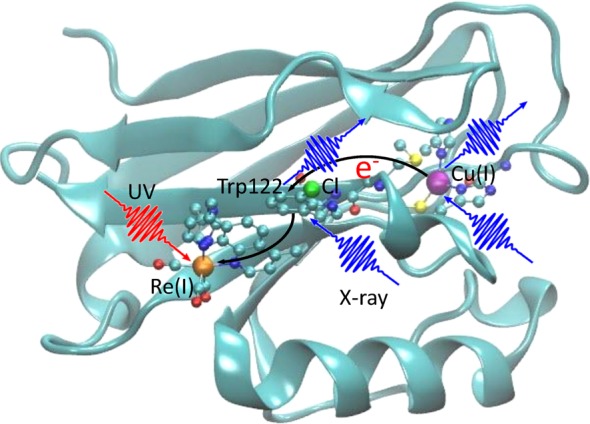
We expect core excitations to be insensitive to vibrational relaxation, therefore the signal contributions from the states A and B (see Figure 2) are lumped together in this study. The time-dependent populations of all states from the kinetic model (eq 1) are shown in Figure 3A. Initially, 3*MLCT and 3MLCT state (A+B) are dominant (population = 0.8), and the 3CS state (C) is small (population = 0.2). With time, a hole is transferred to the Trp122 group, and the population of state C reaches its maximum at ≈1 ns after the excitation. Finally the hole is completely transferred to the Cu center, creating the LCT state D. A movie of the time-dependent electron density differences (excited state density minus ground state density) is given in the Supporting Information. The excitation energy of state A+B is calculated at about 3.1 eV using ΔSCF, which corresponds to the UV pulse (355–400 nm) used in the experiment.15 The TXAS and SXRS signals are calculated at three time snapshots τ = 10 ps, 1 ns, and 200 ns as marked in Figure 3A. These times represent the initial, intermediate and final stages of the ET process, whereby the populations of states A+B, C, and D reach their maximum, respectively.
Figure 3.
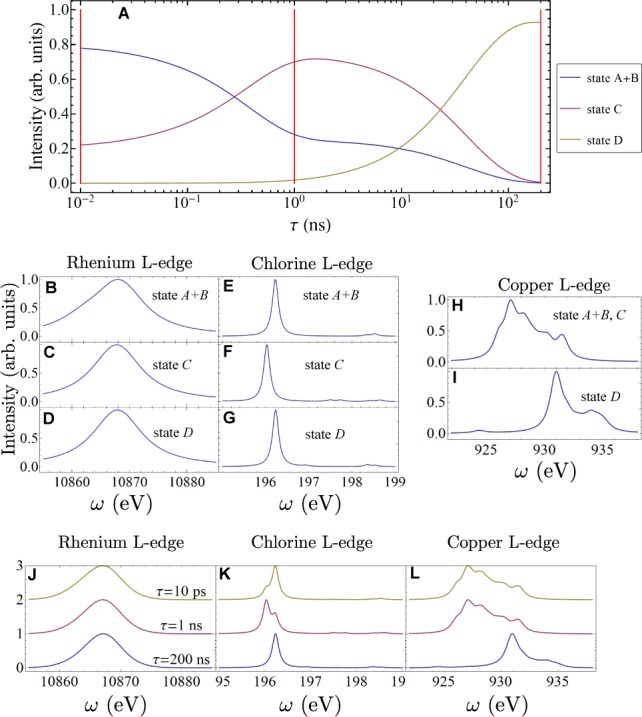
Time-dependent populations and simulated X-ray absorption signals. See the spectroscopy signal calculation section of the Supporting Information for X-ray parameters. (A) Time-dependent populations for different states in the ET process calculated from eq 1 and with the initial conditions [A]0 = 0.8, [B]0 = 0.0, [C]0 = 0.2 and [D]0 = 0.0. See Figure 2 for explanations of different states. TXAS and SXRS signals are calculated at the three selected times marked with vertical red lines, at τ = 10 ps, 1 ns, and 200 ns. Middle panels: XANES spectra of different species. (B) Simulated Re L-edge XANES spectrum for state A+B. (C) Simulated Re L-edge XANES spectrum for state C. (D) Simulated Re L-edge XANES spectrum for state D. (E) Simulated Cl L-edge XANES spectrum for state A+B. (F) Simulated Cl L-edge XANES spectrum for state C. (G) Simulated Cl L-edge XANES spectrum for state D. (H) Simulated Cu L-edge XANES spectrum for state A+B and C. (I) Simulated Cu L-edge XANES spectrum for state D. Bottom panels: Time-dependent XANES. (J) TXAS of a 200 as pulse tuned to Re L-edge with interpulse delays set to 10 ps, 1 and 200 ns. (K) Same as (J), but the Cl L-edge TXAS signals are shown. (L) Same as (J), but the Cu L-edge TXAS signals are shown.
We used three pruned molecular fragments to represent the electron donor (Cu-complex), the tryptophan intermediate group (Trp122) and the acceptor (Re-complex) (see the ball-and-stick structures in Figures 1 and S5). To make Trp122 unambiguously detectable by X-rays and distinguish it from other tryptophans, we had substituted it with a chlorine atom, which is a soft X-ray chromophore. This strategy is analogous to isotope labeling in infrared spectroscopy.27 Test calculations show that the added chlorine atom does not change the electronic structure of tryptophan. The electron density changes on the group may be probed by variation of the X-ray absorption or Raman signals at the Cl L-edge. L-edge excitations can be also used for pump and probe at the Re or Cu centers.
Quantum chemistry calculation details for the three molecular fragments shown in Figure S5 are given in the Computational Details section in the Supporting Information. The resulting X-ray absorption near edge structure (XANES) signals for different states are displayed in Figure 3B–I. Only the copper signals significantly change for different oxidation states, with the strongest peak shifting from 927.0 (Cu (I), state A+B, C) to 931.1 eV (Cu (II), state D). The rhenium signals show very broad peaks for all states. The dominant chlorine XANES peaks of the neutral and positively charged tryptophan differ only by about 0.2 eV, and all other features are weak. This can be rationalized by the large electronegativity of chlorine. The positive charge on the tryptophan group avoids this atom and so that it has little impact on the localized Cl core excitations. The chlorine atom serves as a good chromophore that does not perturb the electron flow.
In the pump–probe (transient absorption) experiment, a UV actinic pulse first excites the system at time zero, initiating the hole transfer. A two-dimensional time-resolved signal is obtained by recording the frequency-dispersed transmission of a broadband X-ray pulse k2 versus the delay time τ:
| 2 |
where the summation extends
over the set of
valence-excited (core-excited) states, the a (x) states, and Pa(τ) is the time-dependent population of state a obtained from eq 1. Vxa and ωxa are the dipole moment and frequency for the core transition
from state a to x, 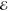 2(ω) is the X-ray pulse
spectral envelope, and Γx is the
lifetime broadening of state x.
2(ω) is the X-ray pulse
spectral envelope, and Γx is the
lifetime broadening of state x.
The calculated transient absorptions signals for the reacting ET system at the three selected times are shown in Figures 3J–L. The signal calculation protocol is explained in the Computational Details section in the Supporting Information. The copper TXAS signal changes at the last stage of the ET process, when the copper center changes its oxidation state from I to II. Since the dominant Cl absorption peak only shows a minor shift during the ET process, it is hard to resolve the ET dynamics from the tryptophan group to the Re center from the Cl TXAS signals. The Re signals are also not very informative about ET. The Cu signals only reveal the last step of the ET process. These simulations show that the linear TXAS is not a sensitive probe of the two-step electron hopping mechanism. Below we demonstrate that nonlinear SXRS signals, in contrast, do vary significantly with the ET process.
The two-pulse stimulated X-ray Raman spectroscopy19,28 signal is represented diagrammatically in Figure S1 in Supporting Information. As in TXAS, ET dynamics are initiated by the actinic UV pump pulse k1 at time zero. Following the delay time τ, the molecule now interacts with two Raman pulses k2 and k3 separated by delay time T. Each pulse interacts with the system twice: a core–electron is excited into an unoccupied orbital, the excited system then evolves for a short period before a second interaction with the same pulse stimulates a down transition, eliminating the core-hole, emitting an X-ray photon, and creating a valence electronic state.
The SXRS signal is given by28
| 3 |
where
| 4 |
is the imaginary
part of the effective isotropic
polarizability,29 averaged over the spectral
envelope of the jth pulse, 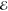 j, and θ(T) is the
Heaviside step function. The summation in eq 3 runs over the valence-excited states, and x denotes
core-excitations. The broad pulse envelope
j, and θ(T) is the
Heaviside step function. The summation in eq 3 runs over the valence-excited states, and x denotes
core-excitations. The broad pulse envelope 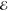 (ω)
controls the active valence frequencies
ωab accessible by the technique.
The core edges considered here, the L edges of rhenium, chlorine,
and copper, have lifetimes of 0.1, 6.5, and 1.0 fs,30 respectively. Auger processes are expected to fill the
core hole and ionize the molecule, bringing it out of resonance with
the probe pulse. We can thus safely ignore the short-lived contributions
to the signal from core-excited populations created by the first X-ray
pulse.
(ω)
controls the active valence frequencies
ωab accessible by the technique.
The core edges considered here, the L edges of rhenium, chlorine,
and copper, have lifetimes of 0.1, 6.5, and 1.0 fs,30 respectively. Auger processes are expected to fill the
core hole and ionize the molecule, bringing it out of resonance with
the probe pulse. We can thus safely ignore the short-lived contributions
to the signal from core-excited populations created by the first X-ray
pulse.
The SXRS signal is collected in the time domain by varying the delay T between the Raman pump k2 and probe k3 pulses. In this technique the k2 pulse will necessarily perturb the ET dynamics which we seek to measure. It is therefore important that the sampling time window T be short compared with the ET time scale and long enough to allow good spectral resolution. The ET time scale varies from tens of picoseconds to hundreds of nanoseconds. We varied T between 0 and 200 fs, and collected the signal in 200-as timesteps. This finite collection period provides an adequate spectral resolution of the Fourier transform signal, as shown in the Supporting Information.
Unlike the TXAS signals shown in Figures 3B–I, the SXRS spectra for the various ET states in Figures 4A–H are much more clearly distinct (see the Computational Details section in the Supporting Information for the signal calculation protocol). Characteristic peak patterns can be identified for all oxidation states of the various species. In particular, we see that low-frequency peaks appear on a given atoms SXRS signal when the hole is on that fragment. The rhenium SXRS signal from the MLCT state (state A+B) shows strong peaks at Ω = 0.3, 0.55, and 1.98 eV which do not show up in the Re signal from the CS or LCT states. These represent excitations into that hole from other valence orbitals. These intravalence excitations have energies lower than the highest occupied molecular orbital to lowest unoccupied molecular orbital (HOMO–LUMO) gap and are therefore indicative of excited state SXRS signals. The Re signals from these states, states C and D, respectively, are similar to each other showing a cluster of peaks between 4 and 5.5 eV. The signal from state D features a peak at 2.90 eV that is not present in the MLCT state.
Figure 4.

Static and time-dependent SXRS signals. See the spectroscopy signal calculation section of the Supporting Information for X-ray parameters. (A) SXRS signal taken with 100 as pulses tuned to Re L-edge for state A+B. (B) Re L-edge SXRS signal for state C. (C) Re L-edge SXRS signal for state D. (D) Cl L-edge SXRS signal for state A+B. (E) Cl L-edge SXRS signal for state C. (F) Cl L-edge SXRS signal for state D. (G) Cu L-edge SXRS signal for state A+B and C. (H) Cu L-edge SXRS signal for state D. (I) Time-dependent Re L-edge SXRS signals for the three snapshots indicated in the figure. (J) Same as panel I, but for the Cl L-edge SXRS signals. (K) Same as panel I, but for the Cu L-edge SXRS signals. (L) Three snapshots, with the delay between the initiation and the Raman pulses set to 10 ps, 1 ns, and 200 ns from top to bottom, of the evolving electron density difference of the system (excited state density minus ground state density). Red denotes a negative sign (hole) and blue denotes a positive sign (electron). Rhenium is in pink, chlorine is in green, copper is in orange, nitrogen is in blue, oxygen is in red, carbon is in gray, and hydrogen is in white.
The chlorine and the copper signals show a similar trend to the rhenium. When the tryptophan fragment is neutral in the MLCT and LCT states (states A+B and D), the chlorine signal’s largest peak is at Ω = 5.28 eV. When the positive charge is on the tryptophan in state C, this peak is absent and in its place are low-energy peaks at 1.1, 2.3, 3.9, and 4.3 eV. The SXRS signal from the neutral Cu-complex shows a great number of peaks between 4.5 and 8.5 eV. These peaks disappear when the hole migrates to this fragment in state D, and the signal shows a cluster of peaks between 0 and 4 eV with prominent peaks at 0.7 and 1.1 eV.
By averaging the spectra of the various species over the distribution of populations (Figure 3A) obtained from the kinetic equations, we obtained the time-dependent SXRS signals shown in Figure 4I–K at three selected times. SXRS signals resonant with different atoms are complementary to each other and together provide a full picture of the sequential electron-hopping process. The decay of the Re SXRS peak around 0.3 and 0.55 eV and the enhancement of the Cl SXRS peaks around 1.1 and 4.0–4.3 eV can jointly reveal the mechanism of the first electron hopping step (electron moving from Trp122 to Re). The second step is revealed by the decay of the high-energy peaks, between 4.5 and 8.5 eV, in the Cu signal and the rise of the low-energy peaks.
Using a chlorine substitution of the tryptophan intermediate we obtained three distinct spatial regions that can be selectively probed by tuning the frequency of the broadband X-ray pulses. Our simulations reveal clear spectral signature of charge transfer, via the presence of low-frequency valence excitations not present in the neutral species. The rhenium, chlorine, and copper SXRS signals provide windows onto the sequential ET process. By comparing the time-evolving amplitudes of low- and high-energy peaks, it should be possible to extract the ET kinetic rates from experiment.
X-ray Raman spectroscopy may be used to study several fast initial steps (see Figure 2), which may not be resolved by conventional time-resolved optical absorption.11 For example, the sub-femtosecond time-resolution of broadband X-ray pulse could be used to observe the intersystem crossing from the singlet to triplet state. Multireference methods such as complete active space self-consistent field (CASSCF) must then be used to describe the open-shell MLCT singlet state, and spin–orbit coupling which governs the singlet-to-triplet transition dynamics can be estimated by time-dependent density-functional theory (TDDFT) or DFT/MRCI (multireference configuration interaction) methods. This will be an interesting future extension of the present work. When using X-ray Raman spectroscopy to monitor dynamics that are faster than the collection period for SXRS (200 fs in this report), it should be possible to employ a combination of broad and narrow beams to obtain the signal in one shot, as is done in conventional vibrational Raman.31 In the Supporting Information we show that the Raman spectrum obtained this way is equivalent to the SXRS spectra reported here.
Recently, Weninger et al. used nontransform-limited pulses with 7 eV bandwidth and 40 fs duration to collect Raman signals from neon gas.32 The effect of such pulses, with noiselike power spectra, on the two-pulse SXRS signal is an interesting and open question. We expect the experiment proposed in this study could be conducted in the near future.
Acknowledgments
The support of the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy is gratefully acknowledged. We also gratefully acknowledge the support of the National Science Foundation (Grant CHE-1361516), and the National Institutes of Health (Grant GM-59230). A portion of the research was performed using EMSL, a national scientific user facility sponsored by the Department of Energy’s Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory. The authors wish to thank Prof. Harry B. Gray for most useful discussions.
Supporting Information Available
Additional notes about the computational details, the loop diagrams for the SXRS signal, the molecular fragment model of the Re-modified azurin system, sampling time and SXRS spectral resolution, X-ray Raman spectroscopy with a broadband and a narrowband pulse; and a movie to show the correlation between the electronic density and the SXRS signals. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Y.Z. and J.D.B. contributed equally to this study.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Moore G. F.; Brudvig G. W. Energy Conversion in Photosynthesis: A Paradigm for Solar Fuel Production. Annu. Rev. Condens. Matter Phys. 2011, 2, 303303–303327. [Google Scholar]
- Kaila V. R. I.; Verkhovsky M. I.; Wikström M. Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem. Rev. 2010, 110, 7062–7081. [DOI] [PubMed] [Google Scholar]
- Sancar A. Structure and Function of DNA Photolyase and Cryptochrome Blue-Light Photoreceptors. Chem. Rev. 2003, 103, 2203–2238. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; Nocera D. G.; Yee C. S.; Chang M. C. Y. Radical Initiation in the Class I Ribonucleotide Reductase: Long-Range Proton-Coupled Electron Transfer?. Chem. Rev. 2003, 103, 2167–2202. [DOI] [PubMed] [Google Scholar]
- Murphy C. J.; Arkin M. R.; Jenkins Y.; Ghatlia N. D.; Bossmann S. H.; Turro N. J.; Barton J. K. Long-Range Photoinduced Electron Transfer Through a DNA Helix. Science 1993, 262, 1025–1029. [DOI] [PubMed] [Google Scholar]
- Bixon M.; Giese B.; Wessely S.; Langenbacher T.; Michel-Beyerle M. E.; Jortner J. Long-Range Charge Hopping in DNA. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 11713–11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J. J.; Morgan D. A.; Wrathmell C. L.; Axford D. N.; Zhao J.; Wang N. Molecular Bioelectronics. J. Mater. Chem. 2005, 15, 2160–2174. [Google Scholar]
- Ron I.; Sepunaru L.; Itzhakov S.; Belenkova T.; Friedman N.; Pecht I.; Sheves M.; Cahen D. Proteins as Electronic Materials: Electron Transport through Solid-State Protein Monolayer Junctions. J. Am. Chem. Soc. 2010, 132, 4131–4140. [DOI] [PubMed] [Google Scholar]
- Patil A.; Davis J. Molecular Scale Bioelectrochemistry. Coord. Chem. Rev. 2011, 255, 1970–1980. [Google Scholar]
- Marcus R. A.; Sutin N. Electron Transfers in Chemistry and Biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar]
- Shih C.; Museth A. K.; Abrahamsson M.; Blanco-Rodriguez A. M.; Bilio A. J. D.; Sudhamsu J.; Crane B. R.; Ronayne K. L.; Towrie M.; Vlc̆ek A. Jr.; et al. Tryptophan-Accelerated Electron Flow through Proteins. Science 2008, 320, 1760–1762. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Ener M. E.; Vlc̆ek A. Jr.; Winkler J. R.; Gray H. B. Electron Hopping through Proteins. Coord. Chem. Rev. 2012, 256, 2478–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B. Long-Range Electron Tunneling. J. Am. Chem. Soc. 2014, 136, 2930–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada A.; Chernyak V.; Mukamel S. Solvent Reorganization in Long-Range Electron Transfer: Density Matrix Approach. J. Phys. Chem. A 1998, 102, 1241–1251. [Google Scholar]
- Blanco-Rodríguez A. M.; Di Bilio A. J.; Shih C.; Museth A. K.; Clark I. P.; Towrie M.; Cannizzo A.; Sudhamsu J.; Crane B. R.; Sýkora J.; et al. Phototriggering Electron Flow through Re(I)-Modified Pseudomonas aeruginosa Azurins. Chem.—Eur. J. 2011, 17, 5350–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutman A. A.; Coffee R.; Ding Y.; Huang Z.; Krzywinski J.; Maxwell T.; Messerschmidt M.; Nuhn H.-D. Experimental Demonstration of Femtosecond Two-Color X-ray Free-Electron Lasers. Phys. Rev. Lett. 2013, 110, 134801. [DOI] [PubMed] [Google Scholar]
- Beye M.; Schreck S.; Sorgenfrei F.; Trabant C.; Pontius N.; Schüßler-Langeheine C.; Wurth W.; Föhlisch A. Stimulated X-ray Emission for Materials Science. Nature 2013, 501, 191–194. [DOI] [PubMed] [Google Scholar]
- Miller R. J. D. Femtosecond Crystallography with Ultrabright Electrons and X-rays: Capturing Chemistry in Action. Science 2014, 343, 1108–1116. [DOI] [PubMed] [Google Scholar]
- Biggs J. D.; Zhang Y.; Healion D.; Mukamel S. Two-Dimensional Stimulated Resonance Raman Spectroscopy of Molecules with Broadband X-ray Pulses. J. Chem. Phys. 2012, 136, 174117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. X.; Zhang X. Photochemical Processes Revealed by X-ray Transient Absorption Spectroscopy. J. Phys. Chem. Lett. 2013, 4, 4000–4013. [Google Scholar]
- Barty A.; Küpper J.; Chapman H. N. Molecular Imaging Using X-ray Free-Electron Lasers. Annu. Rev. Phys. Chem. 2013, 64, 415–435. [DOI] [PubMed] [Google Scholar]
- Emma P.; Akre R.; Arthur J.; Bionta R.; Bostedt C.; Bozek J.; Brachmann A.; Bucksbaum P.; Coffee R.; Decker F.-J.; et al. First Lasing and Operation of An Ångstrom-Wavelength Free-Electron Laser. Nat. Photonics 2010, 4, 641–647. [Google Scholar]
- Kolczak U.; Dennison C.; Messerschmidt A.; Canters G. W.. Handbook of Metalloproteins; Wiley: New York, 2001; Vol. 2, pp 1170–1794. [Google Scholar]
- Silvestrini M. C.; Tordi M. G.; Colosimo A.; Antonini E.; Brunori M. The Kinetics of Electron Transfer Between Pseudomonas aeruginosa Cytochrome c-551 and Its Oxidase. Biochem. J. 1982, 203, 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takematsu K.; Williamson H.; Blanco-Rodríguez A. M.; Sokolová L.; Nikolovski P.; Kaiser J. T.; Towrie M.; Clark I. P.; Vlc̆ek A.; Winkler J. R.; et al. Tryptophan-Accelerated Electron Flow Across a Protein–Protein Interface. J. Am. Chem. Soc. 2013, 135, 15515–15525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bilio A. J.; Crane B. R.; Wehbi W. A.; Kiser C. N.; Abu-Omar M. M.; Carlos R. M.; Richards J. H.; Winkler J. R.; Gray H. B. Properties of Photogenerated Tryptophan and Tyrosyl Radicals in Structurally Characterized Proteins Containing Rhenium (I) Tricarbonyl Diimines. J. Am. Chem. Soc. 2001, 123, 3181–3182. [DOI] [PubMed] [Google Scholar]
- Kim Y. S.; Hochstrasser R. M. Applications of 2D IR Spectroscopy to Peptides, Proteins, and Hydrogen-Bond Dynamics. J. Phys. Chem. B 2009, 113, 8231–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs J. D.; Zhang Y.; Healion D.; Mukamel S. Multidimensional X-ray Spectroscopy of Valence and Core Excitations in Cysteine. J. Chem. Phys. 2013, 138, 144303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamel S.; Healion D.; Zhang Y.; Biggs J. D. Multidimensional Attosecond Resonant X-ray Spectroscopy of Molecules: Lessons from the Optical Regime. Annu. Rev. Phys. Chem. 2013, 64, 101–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschornack G.Handbook of X-ray Data; Springer-Verlag: Berlin Heidelberg, 2007. [Google Scholar]
- Kukura P.; McCamant D. W.; Mathies R. A. Femtosecond Stimulated Raman Spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. [DOI] [PubMed] [Google Scholar]
- Weninger C.; Purvis M.; Ryan D.; London R. A.; Bozek J. D.; Bostedt C.; Graf A.; Brown G.; Rocca J. J.; Rohringer N. Stimulated Electronic X-ray Raman Scattering. Phys. Rev. Lett. 2013, 111, 233902. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.