

A cell type- and leaf tissue-specific model provides new insights into nitrogen metabolism in the maize leaf.
Abstract
Maize (Zea mays) is an important C4 plant due to its widespread use as a cereal and energy crop. A second-generation genome-scale metabolic model for the maize leaf was created to capture C4 carbon fixation and investigate nitrogen (N) assimilation by modeling the interactions between the bundle sheath and mesophyll cells. The model contains gene-protein-reaction relationships, elemental and charge-balanced reactions, and incorporates experimental evidence pertaining to the biomass composition, compartmentalization, and flux constraints. Condition-specific biomass descriptions were introduced that account for amino acids, fatty acids, soluble sugars, proteins, chlorophyll, lignocellulose, and nucleic acids as experimentally measured biomass constituents. Compartmentalization of the model is based on proteomic/transcriptomic data and literature evidence. With the incorporation of information from the MetaCrop and MaizeCyc databases, this updated model spans 5,824 genes, 8,525 reactions, and 9,153 metabolites, an increase of approximately 4 times the size of the earlier iRS1563 model. Transcriptomic and proteomic data have also been used to introduce regulatory constraints in the model to simulate an N-limited condition and mutants deficient in glutamine synthetase, gln1-3 and gln1-4. Model-predicted results achieved 90% accuracy when comparing the wild type grown under an N-complete condition with the wild type grown under an N-deficient condition.
Maize (Zea mays), also known as corn, is an essential dual-use food and energy crop. Maize production is increasing at the greatest rate among all cereals, with a worldwide trend of 0.06 tons ha−1 year−1 (Leveau et al., 2011) and a record 877 million tons produced in the 2011-2012 fiscal year (International Grains Council, 2013). With the recent completion of the maize genome in 2009 along with the creation and curation of databases such as MaizeGDB in 2011 (Schaeffer et al., 2011), MaizeCyc in 2013 (Monaco et al., 2013), and MetaCrop 2.0 in 2012 (Schreiber et al., 2012), there is a need for an updated genome-scale metabolic model (GSM; Saha et al., 2011) that will integrate all newly available information from diverse sources. The integration of this information with experimental transcriptomic data, proteomic data, and biomass composition measurements obtained with wild-type plants grown under optimal nitrogen (N+ WT) conditions and limited nitrogen (N− WT) conditions (Amiour et al., 2012), as well as two Gln synthetase (GS) mutants grown under optimal nitrogen (N), gln1-3 and gln1-4 (Martin et al., 2006), has provided a more accurate assessment of N metabolism within the maize leaf. Moreover, since integration of transcriptomic, proteomic, and metabolomic data appeared not to be straightforward (Amiour et al., 2012, 2014), the development of a model could help to identify putative candidate genes, proteins, and metabolic pathways contributing to plant growth and development.
Maize is a C4 plant that overcomes the inefficiencies of Rubisco, to capture oxygen over the preferred CO2, by separating the photosynthetic carbon fixation process into two cell types: the bundle sheath and mesophyll cells. In comparison with C3 plants, this separation allows C4 plants to have a lower rate of photorespiration, a higher rate of photosynthesis at high light intensities (under standard air and temperature conditions), and a higher photosynthetic nitrogen use efficiency (NUE; Christin and Osborne, 2013; Driever and Kromdijk, 2013; Peterhansel et al., 2013; Sage, 2014; Wang et al., 2014). A C4-specific maize GSM could provide insight into N metabolism and provide cues for improving NUE (i.e. the vegetative biomass or grain yield produced per unit of N present in the soil). Since N is the major limiting factor in agricultural production among mineral fertilizers (Vitousek et al., 1997; Hirel et al., 2007; Andrews and Lea, 2013; Andrews et al., 2013) and NUE is estimated to be far below 50% in cereal grains (Raun and Johnson, 1999), improving NUE is essential for improving overall productivity in maize (Hirel and Gallais, 2011). Amiour et al. (2012) experimentally determined 150 gene transcripts, 40 proteins, and 89 metabolites that are significantly different between the N+ WT and N− WT conditions during the vegetative stage of growth. N utilization is strongly linked to the GS enzyme, as all N, either in the form of nitrate or ammonium ions, is channeled through the reaction catalyzed by the GS enzyme (Martin et al., 2006; Cañas et al., 2010; Hirel and Gallais, 2011; Andrews et al., 2013). The mesophyll cell-specific GS1-3 isozyme is involved in synthesizing Gln after nitrate reduction from the vegetative state until the plant reaches maturity. Leaf aging induces the synthesis of the bundle sheath-specific GS1-4 isozyme. Consequently, Martin et al. (2006) hypothesized that the GS1-4 isoform is used in the reassimilation of ammonium during protein degradation in senescing leaves. During vegetative growth in the leaf tissue, DNA microarray data revealed that 243 gene transcripts, 46 proteins, and 48 metabolites exhibited significant differences in the gln1-3 mutants and 107 gene transcripts, 14 proteins, and 18 metabolites displayed substantial differences in the gln1-4 mutants (Amiour et al., 2014). In this second-generation maize model, we explore the effect of the computational knockout of genes encoding for GS1-3 and GS1-4 isozymes using flux balance analysis (FBA) to elucidate the role of GS in N metabolism.
FBA of GSMs is used to model organism-specific metabolism by simulating the internal flow of metabolites. The number of GSMs for plants has increased rapidly, with models available for Arabidopsis thaliana (Poolman et al., 2009; de Oliveira Dal’Molin et al., 2010a), barley (Hordeum vulgare) seed (Grafahrend-Belau et al., 2009), maize (de Oliveira Dal’Molin et al., 2010b; Saha et al., 2011), sorghum (Sorghum bicolor; de Oliveira Dal’Molin et al., 2010b), sugarcane (Saccharum officinarum; de Oliveira Dal’Molin et al., 2010b), rapeseed (Brassica napus; Pilalis et al., 2011), and rice (Oryza sativa; Poolman et al., 2013). These models rely on annotation information to assemble comprehensive compilations of all reactions and metabolites known to occur within the organism. Currently, whole-genome sequencing has been completed for approximately 40 vascular plants, including A. thaliana (Arabidopsis Genome Initiative, 2000), Arabidopsis lyrata (Hu et al., 2011), soybean (Glycine max; Schmutz et al., 2010), rice (Goff et al., 2002; Yu et al., 2002), Populus trichocarpa (Tuskan et al., 2006), sorghum (Paterson et al., 2009), Theobroma cacao (Tuskan et al., 2006), and maize (Schnable et al., 2009). Gene annotations of the whole-genome sequences have been used to determine the reactions within an organism and therefore build a GSM. FBA calculates all reaction fluxes in a metabolic network based on the optimization of an objective function (typically the maximization of the biomass yield). A quasi-steady state is assumed, and flux constraints are set based on the specific medium or the reversibility of reactions derived from thermodynamics. Incorporation of omics data into GSMs is achieved through appropriate constraints on fluxes that restrict metabolic flows to only condition-relevant phenotypes.
During the last few years, multiple methods have been developed to integrate omics data into GSMs. Proteomic and transcriptomic data have been used to apply flux constraints on corresponding reactions determined by gene-protein-reaction (GPR) associations. The GIMME (Becker and Palsson, 2008), iMAT (Shlomi et al., 2008), and MADE (Jensen and Papin, 2011) algorithms use a switch approach to turn on/off reactions based on expression levels. The GIMME algorithm turns off reactions based on a user-specified threshold of the expression level. The iMAT algorithm turns on a minimal set of reactions associated with low expression data in order to achieve a user-specified metabolic function. The MADE algorithm incorporates related experimental data sets into the model to activate or repress reactions based on the progression of the experimental conditions. A different class of algorithms, known as the valve approach, was developed to incorporate proteomic and transcriptomic data by constraining the allowable flux ranges of reactions. The E-Flux method incorporates a user-specified function to convert gene expression data to flux constraints (Colijn et al., 2009). Finally, the PROM algorithm (Chandrasekaran and Price, 2010) uses multiple data sets to constrain flux bounds (i.e. allowable flux ranges) based on the probabilities associated with gene activity among all data sets. Lee et al. (2012) integrated gene expression data by minimizing the difference between the predicted flux levels and gene expression data over all reactions with corresponding expression levels. Using the Yeast 5 model (Heavner et al., 2012) for Saccharomyces cerevisiae, Lee et al. (2012) compared the predicted fluxes with experimentally determined exometabolome fluxes using the coefficient of determination r2. The authors achieved r2 values of 0.87 and 0.96 at 75% and 85% of the maximal biomass level, respectively. In comparison, the authors generated a best FBA solution, which maximizes r2 over all feasible solutions generated for FBA, and achieved r2 values of 0.2 and 0.58 at 75% and 85% of the maximal biomass level, respectively. These advancements pertaining to the integration of omics data with GSMs has enabled more accurate model predictions.
In this work, we describe the reconstruction of a second-generation maize leaf model and the incorporation of omics data into the model with the goal of improving the understanding of N metabolism. Both the primary and secondary metabolic pathways of maize are included, by combining information from MetaCrop (Schreiber et al., 2012), MaizeCyc (Monaco et al., 2013), and the earlier iRS1563 (Saha et al., 2011) models. In comparison with the iRS1563 model, this second-generation model spans an additional 4,261 genes and 6,540 reactions. The increased number of genes and reactions enables the inclusion of additional pathways such as fructan biosynthesis, siroheme biosynthesis, and ubiquniol-9 biosynthesis. The model accounts for the two major cell types in the leaf (i.e. the bundle sheath and mesophyll cells). The bundle sheath cell contains seven compartments: the cytosol, mitochondrion, peroxisome, chloroplast stroma, plasma membrane, thylakoid membrane, and vacuole. The mesophyll cell contains six compartments: the cytosol, mitochondrion, chloroplast stroma, plasma membrane, thylakoid membrane, and vacuole. Compartmentalization is based on maize-specific experimental proteomic and transcriptomic measurements (Majeran et al., 2005; Friso et al., 2010; Li et al., 2010; Chang et al., 2012), as opposed to the A. thaliana-based compartmentalization adopted in the previous iRS1563 maize model (Saha et al., 2011). Light reactions have been expanded from an aggregate reaction (as described in the iRS1563 model) to multiple reactions for each complex with the inclusion of a thylakoid membrane compartment. In contrast to the C4GEM maize model (de Oliveira Dal’Molin et al., 2010b), which focuses exclusively on primary metabolism in maize, the developed model also spans secondary metabolism by including all reactions known to occur within the maize leaf tissue. The model includes as many as 763 secondary metabolism reactions (without including duplicate counting due to compartmentalization). Through the incorporation of omics data, regulatory restrictions are introduced in the model to switch-off/on reactions under the N+ WT and N− WT conditions and two GS knockout mutants (gln1-3 and gln1-4) in the vegetative stage, during which the plant absorbs and assimilates N for root and leaf biomass production (Amiour et al., 2012, 2014). Reactions linked to genes or proteins with significantly different expression levels between the N+ WT and N− WT conditions, as well as the gln1-3 and gln1-4 mutants versus the N+ WT condition, are conditionally turned on or off accordingly. The metabolite pool is simulated by maximizing the total flux through a metabolite (i.e. flux sum) as a proxy for the metabolite turnover rate (Chung and Lee, 2009). The directional changes of flux-sum levels between the N− WT condition and the N+ WT condition, as well as the GS mutant conditions and the N+ WT condition, are qualitatively compared with the directional change in experimentally measured concentration levels. These analyses reveal similar trends to the recently developed flux imbalance analysis (Reznik et al., 2013), which makes use of dual variable values associated with metabolite balances to infer the effect of concentration changes on the objective function value.
RESULTS AND DISCUSSION
Effect of N Conditions on Biomass Components
Biomass components were measured in the N+ WT condition as well as for each N background (N− WT, gln1-3, and gln1-4). Table I and Figure 1 display the composition of the classes of biomass metabolites, and Supplemental Table S1 indicates the specific biomass measurements in all modeled conditions. As expected, in the majority of cases, the N− WT condition produced a smaller concentration of biomass components than the N+ WT, gln1-3, and gln1-4 conditions. However, the concentration of amino acids produced was about 5 times higher in the gln1-4 mutant than the gln1-3 mutant, resulting in comparable amino acid concentrations between the gln1-4 mutant and N+ WT as well as between the gln1-3 mutant and N− WT. The similar amino acid concentrations between the gln1-4 mutant and the N+ WT condition in the vegetative stage help to confirm that the GS1-4 isozyme is essential in plant maturity and has a smaller effect compared with the GS1-3 isozyme at the vegetative stage. As expected, the concentration of starch was higher in the N− WT condition than in the N+ WT condition. Under the N− WT condition, the breakdown of starch is limited by the amount of N available (Tercé-Laforgue et al., 2004; Amiour et al., 2012). Due to the limited N available, the starch is stored rather than broken down to produce other biomass components. The stained micrographs depicting the starch visible in the N+ WT, gln1-3 mutant, and gln1-4 mutant conditions are available in Supplemental Figure S1. The condition-specific biomass concentrations have been incorporated in the maize leaf model to more accurately represent metabolism under each condition.
Table I. Experimental content of classes of metabolites in different conditions.
The biomass components were determined experimentally for each of the conditions (N+ WT, N− WT, gln1-3 mutant, and gln1-4 mutant). Values are means of three replicates unless indicated by the asterisk, indicating that two replicate measurements were taken. Biomass measurements for the specific metabolites within each class are displayed in Supplemental Table S1.
| Biomass Components | N+ WT | N− WT | gln1-3 Mutant | gln1-4 Mutant |
|---|---|---|---|---|
| Biomass yield (g dry wt) | 61 ± 3.5 | 15 ± 2 | 64 ± 4.5 | 65 ± 1.5 |
| Soluble amino acid content (mmol g−1 dry wt) | 0.0732 ± 0.0170 | 0.0261 ± 0.0040 | 0.02124 ± 0.00100 | 0.09303 ± 0.00640 |
| Protein content (mg g−1 dry wt) | 132.6 ± 0.7 | 58 ± 3 | 125.25 ± 1.7 | 140.39 ± 5.72 |
| Fatty acid content (mg g−1 dry wt) | 43.3 ± 4.4 | 16.6 ± 2.2 | 45.1* | 16.3 ± 1.1 |
| Starch content (mmol g−1 dry wt) | 0.152 ± 0.005 | 0.199 ± 0.007 | 0.085 ± 0.011 | 0.107 ± 0.006 |
| RNA content (mg g−1 dry wt) | 3.78 ± 0.19 | 0.92 ± 0.10 | 1.05 ± 0.09 | 1.77 ± 0.11 |
| DNA content (mg g−1 dry wt) | 8.315 ± 0.270 | 2.53 ± 0.10 | 9.62 ± 0.22 | 5.48 ± 0.13 |
| Soluble carbohydrate content (mmol g−1 dry wt) | 0.235 ± 0.012 | 0.112 ± 0.013 | 0.198 ± 0.041 | 0.193 ± 0.023 |
| Cell wall carbohydrate content (mg g−1 dry wt) | 0.32 ± 0.03 | 0.26 ± 0.09 | 0.187 ± 0.017 | 0.29 ± 0.07 |
| Chlorophyll content (mg g−1 dry wt) | 1.87 ± 0.16 | 0.69 ± 0.06 | 1.71 ± 0.14 | 1.85 ± 0.08 |
| Total N (% g dry wt) | 4.36 ± 0.08 | 1.80 ± 0.15 | 4.28 ± 0.10 | 4.31 ± 0.12 |
Figure 1.

Weight percentage of biomass components. The weight percentage for each class of metabolites experimentally measured contributing to biomass synthesis is displayed. The composition is displayed for the N+ WT (A), N− WT (B), gln1-3 mutant (C), and gln1-4 mutant (D) conditions. The measurements for specific components within each class of metabolites are shown in Supplemental Table S1. [See online article for color version of this figure.]
Development of the Second-Generation Maize Leaf Model
The second-generation maize leaf model was developed using a combination of gene, protein, and reaction information from the previously developed maize model iRS1563 (Saha et al., 2011), biological databases such as the Kyoto Encyclopedia of Genes and Genomes (Kanehisa et al., 2014), MaizeCyc (Monaco et al., 2013), and MetaCrop (Schreiber et al., 2012), as well as published literature sources. The model contains 5,824 genes and 8,525 reactions, a significant increase from the iRS1563 model, which contained 1,563 genes and 1,985 reactions. The second-generation maize model is split into two cell types (i.e. the bundle sheath and mesophyll cells). The bundle sheath cell is further divided into seven compartments, while the mesophyll cell contains six compartments (Fig. 2). Of the 8,525 reactions in the model, 3,892 reactions are unique, as duplicated counts due to compartmentalization have been disregarded. Of these 3,892 unique reactions, 1,012 reactions were assigned localization information based on transcriptomic and proteomic data (Majeran et al., 2005; Friso et al., 2010; Li et al., 2010; Chang et al., 2012). Light reactions were adjusted to model the flow of protons across the thylakoid membrane to the chloroplast stroma, to represent the pH differential between compartments, and to describe the conversion of light to ATP (Nelson and Cox, 2009). The mitochondrial electron transport chain was similarly updated to include the proton exchange of ATP synthase between the intermembrane space and the mitochondrial matrix (Taiz, 2010). Finally, 303 specific reactions were added to model glycerolipid synthesis, as shown in Supplemental Figure S2 and Supplemental Table S2 (Moore, 1982; Murata, 1983; Murata and Tasaka, 1997; Mekhedov et al., 2000; Bachlava et al., 2009; Li-Beisson et al., 2010; Rolland et al., 2012). To the best of our knowledge, this is the first plant model to include detailed glycerolipid synthesis. Aggregate reactions were included to link specific two-tailed glycerolipids to the experimentally measured single lipids (Supplemental Table S2). Compiling transcriptomic and proteomic compartmentalization data with literature-based pathways yielded a model of 4,103 reactions, leaving 2,880 unique reactions, still with their localizations unknown.
Figure 2.

Number of metabolic and transport reactions distributed between compartments in the bundle sheath and mesophyll cell types. The numbers of metabolic and transport reactions are shown for each compartment. Integral membrane proteins are counted for the compartment in which the main biotransformation occurs. For example, the ATP synthase associated with the mitochondrial electron transport chain is counted as a metabolic reaction in the mitochondrion, not the inner mitochondrial membrane (IMM). [See online article for color version of this figure.]
Once reactions were compartmentalized based on transcriptomic data, proteomic data, and published literature, the reactions were divided into two groups. The first group (core set) includes reactions with known localizations, while the second group (noncore set) spans reactions known to occur within the maize leaf but with no localization evidence. Whenever possible, core reactions were unblocked by first adding reaction(s) from the noncore set to one or multiple compartment(s) and second appending intercellular or intracellular transporters (see “Materials and Methods”). By following this approach, 1,032 unique reactions with previously unknown localizations were assigned to compartments and 729 transporters were added. The remaining 1,848 unique reactions were assigned to compartments based on available pathway information or assigned to the cytosol of both the bundle sheath and mesophyll cells.
With all the reactions assigned to specific compartments, thermodynamically infeasible cycles that were generated due to the overly permissive inclusion of reactions in the model, as well as lack of reaction directionality information, were subsequently identified and eliminated. By first restricting the directionality of reactions and second removing reactions, it was possible to eliminate all thermodynamically infeasible cycles in the model. By this process, we restricted the directionality of 36 reactions and removed 2,055 reactions from the model (Table II). Upon the resolution of thermodynamically infeasible cycles, attempts were made to unblock the remaining blocked core reactions and biomass formation by adding reactions from similar organisms (Krumholz et al., 2012) and model organisms (i.e. rice ssp. japonica, Brachypodium distachyon, sorghum, and A. thaliana). By adding five unique reactions from similar organisms, the flux through three additional reactions known to be in maize was resolved. These reactions were all involved in the formation of Glu from His through urocanic acid. The model is provided in a Microsoft Excel format in Supplemental Table S3 and in Systems Biology Markup Language format in Supplemental Table S4.
Table II. Number of reactions after each model creation and curation step.
The original two data sets are the core set and the noncore set, which combine to form the final model statistics. The total number of metabolic, transport, exchange, and biomass reactions are displayed after each process during model curation. Metabolic reaction totals include duplication from compartmentalization.
| Data Processing | Metabolic Reactions | Transport Reactions | Exchange Reactions | Biomass Reactions |
|---|---|---|---|---|
| Initial data | ||||
| Core set | 3,002 | 418 | 82 | 85 |
| Core set plus manually created pathways | 3,264 | 469 | 285 | 85 |
| Noncore set | 18,951 | 0 | 0 | 0 |
| Processes performed | ||||
| Compartmentalization algorithm | 3,971 | 1,198 | 285 | 85 |
| Manually determined compartmentalization | 9,005 | 1,198 | 285 | 85 |
| Thermodynamically infeasible cycles | 7,033 | 1,115 | 285 | 85 |
| Similar organism GapFill | 7,040 | 1,115 | 285 | 85 |
| Final model | ||||
| Second-generation GSM | 7,040 | 1,115 | 285 | 85 |
Incorporation of Transcriptomic and Proteomic Data in the Model
In order to more accurately model the N+ WT, N− WT, and GS mutant conditions in maize, GPR associations mapped the gene transcripts and proteins that were statistically expressed at a low level to reactions that were turned off in the model. However, no essential reactions to the model, which are required for biomass formation, were altered. For example, the δ-aminolevulinic acid dehydratase reaction was experimentally determined to be higher in the N+ WT condition, suggesting that it should be restricted in the N− WT condition. However, when the flux through the δ-aminolevulinic acid dehydratase reaction is restricted to zero, biomass cannot be formed, as this reaction produces porphobilinogen, a precursor to chlorophyll (Gupta et al., 2013). Due to the incomplete information available in published literature or databases regarding possible alternative routes of the production and degradation of a specific metabolite, regulating reactions that are essential to the model will restrict biomass synthesis. Based on experimental evidence, the fluxes through 83 reactions in the N+ WT condition, 20 in the N− WT condition, 100 in the gln1-3 mutant, and nine in the gln1-4 mutant were restricted. The reactions regulated in the N+ WT condition mainly correspond to reactions known to occur only under stress and are expressed at a low level in comparison with the N− WT and mutant conditions. Reactions that have been down-regulated based on omics data are indicated in the model file (Supplemental Table S3). N perturbations within the leaf tissue were modeled by combining the incorporation of transcriptomic and proteomic data with the unique biomass composition for each condition.
The minimal set of reactions, whose elimination causes a decrease in biomass yield, was determined for the N+ WT, N− WT, gln1-3 mutant, and gln1-4 mutant conditions. There are six reactions across the conditions that encompass the minimal set of reactions, as summarized in Table III. Of the 83 reactions with restricted flux in the N+ WT condition, only two reactions were identified to affect biomass yield. These two reactions are the conversion of ethanol to acetaldehyde through either ethanol oxidoreductase involving NAD+ or a hydrogen peroxide-dependent oxidation of ethanol catalyzed by catalase (Boamfa et al., 2005). These two reactions have a very slight effect on biomass formation, as biomass yield drops by less than 1%. As expected, we find that many of the reactions that correspond to genes that are significantly down-regulated in the N+ WT condition do not hinder biomass formation. In the N− WT condition, none of the reactions have an effect on the biomass yield, suggesting, as expected, that the decreased amount of N is the main limiting factor in biomass yield. In the gln1-3 mutant condition, three of the 100 reactions, which are switched off based on omics data, affect the biomass yield. These three reactions are the glyceraldehyde-3-phosphate dehydrogenase, Fru-bisP aldolase, and Fru-bisphosphatase reactions. The capacity of glyceraldehyde-3-phosphate dehydrogenase to form a multienzyme complex in the chloroplasts for a range of plants is regulated by environmental conditions such as the light/dark transitions (Howard et al., 2011). Glyceraldehyde-3-phosphate is synthesized during carbon fixation in photosynthesis, and 1,3-bisphospho-d-gycerate (i.e. 3-phospho-d-glyceroyl phosphate) can be synthesized from 3-phospho-d-glycerate. ATP is required for the conversion of 3-phospho-d-glycerate to 1,3-bisphospho-d-glycerate catalyzed by 3-phospho-d-glycerate kinase in the bundle sheath chloroplast. This reaction is an important energy-requiring reaction in the Calvin-Benson cycle, as it is essential that the enzyme immediately metabolizes 3-phospho-d-glycerate, the product of the Rubisco reaction. This conclusion is also consistent with the findings that 3-phospho-d-glycerate 1-phosphotransferase is sensitive to changes in energy state (Nakamoto and Edwards, 1987). The Fru-bisP aldolase reaction, which is involved in the Calvin-Benson-Bassham cycle and the glycolysis pathway, can be bypassed using the sedoheptulose 1,7-bisphosphate/d-glyceraldehyde-3-phosphate-lyase reaction, which catalyzes the synthesis of sedoheptulose 1,7-bisphosphate using dihydroxyacetone phosphate (i.e. glycerone phosphate) and d-erythrose 4-phosphate (Lakshmanan et al., 2013). The decreased expression of the cytosolic Fru-bisphosphatase reaction has been shown to decrease the ATP-ADP ratio, lead to the switch from Suc to starch synthesis, and inhibit photosynthesis at high CO2 levels in A. thaliana, resulting in the inhibition of plant growth (Strand et al., 2000). Finally, the regulatory restrictions for the gln1-4 mutant involve only nine reactions, of which one affected the biomass drain (i.e. Rib-5-P isomerase reaction). The lack of the Rib-5-P isomerase reaction has been experimentally shown to cause premature death and affect cellulose synthesis in A. thaliana (Howles et al., 2006; Xiong et al., 2009). A comparison of the number of reactions that affect the GS mutants suggests that at the vegetative stage, the impact of the gln1-4 mutation is less severe than that occurring in the gln1-3 mutant. Such a finding is not surprising, since it has been shown that the gene encoding the GS1-3 isozyme is constitutively expressed irrespective of the leaf development stage and that the expression of the gene encoding the GS1-4 isozyme is much lower and only enhanced at later stages of leaf development (Hirel et al., 2005). Although only a subset of reactions affect the biomass production in the N+ WT, gln1-3 mutant, and gln1-4 mutant conditions, the additional regulation will have an effect on the flux predictions within the model.
Table III. Summary of reactions that affect biomass synthesis.
The minimum set of reactions that are down-regulated as a result of the inclusion of proteomic and transcriptomic data and affect biomass synthesis is displayed. The corresponding condition is displayed for each reaction as well as the role of the reaction.
| Reaction | Condition Affected | Affect of the Reaction |
|---|---|---|
| Ethanol oxidoreductase and ethanol catalase | N+ WT | Produces acetaldehyde, alleviating flux through pyruvate decarboxylase |
| Glyceraldehyde-3-phosphate dehydrogenase | gln1-3 mutant | Participates in glycolysis and carbon fixation but is not required, as 3-phospho-d-glycerate kinase can restore flux to 1,3-bisphospho-d-glycerate |
| Fru-bisP aldolase | gln1-3 mutant | Participates in the Calvin-Benson-Bassham cycle but can be bypassed through the sedoheptulose 1,7-bisphosphate/d-glyceraldehyde-3-phosphate lyase reaction |
| Fru-bisphosphatase | gln1-3 mutant | Decreases ATP-ADP ratio, switches metabolism from Suc to starch synthesis, and inhibits photosynthesis at high CO2 levels in A. thaliana |
| Rib-5-P isomerase | gln1-4 mutant | Affects cellulose synthesis in A. thaliana |
Flux Range Variations among Conditions
The flux range of each reaction was determined in the N+ WT, N− WT, gln1-3 mutant, and gln1-4 mutant conditions under the assumption that biomass is maximized. The flux range of a reaction in the N− WT, gln1-3 mutant, and gln1-4 mutant conditions was compared with the flux range in the N+ WT reference condition to determine reactions with flux ranges that must deviate from the N+ WT flux range. This indicates that the flux through the reaction must change as a result of the limited N or mutation. Overall, the flux through 202 reactions in the N− WT condition is not contained within the flux range of the N+ WT condition, 765 reaction fluxes in the gln1-3 mutant diverge from the N+ WT flux range, and 678 reaction fluxes in the gln1-4 mutant must change from the N+ WT flux range (Supplemental Table S5). In all three N backgrounds (i.e. the N− WT, gln1-3 mutant, and gln1-4 mutant conditions), the flux compared with the N+ WT reference condition decreases under maximum biomass through the chlorophyll cycle, chlorophyllide a biosynthesis, farnesyl diphosphate biosynthesis, methylerythritol phosphate pathway, and tetrapyrrole biosynthesis. Tetrapyrrole biosynthesis, chlorophyllide a biosynthesis, and the chlorophyll cycle link the production of chlorophyll from Glu (Tanaka and Tanaka, 2007; Kim et al., 2013). The methylerythritol phosphate pathway and farnesyl diphosphate biosynthesis lead to a reactant required for the production of chlorophyll a from chlorophyllide a (Lange and Ghassemian, 2003). In both of the GS mutant conditions, the flux through chorismate biosynthesis (Tzin and Galili, 2010), Ser biosynthesis (Ho and Saito, 2001), and the urea cycle (Mérigout et al., 2008) must decrease compared with the N+ WT condition. Choline biosynthesis (McNeil et al., 2001) is decreased in the N− WT condition, increased in the gln1-3 mutant, and decreased in the gln1-4 mutant condition. Flux through Ile and Leu biosynthesis (McCourt and Duggleby, 2006) is lower in the N− WT condition, higher in the gln1-3 mutant condition, and lower in the gln1-4 mutant condition compared with the N+ WT condition, as expected by the proportion of these biomass components in the various conditions. The flux through the glyoxylate cycle (Schnarrenberger and Martin, 2002), stearate biosynthesis (Li-Beisson et al., 2010), and urate degradation (Ramazzina et al., 2006) is higher in the gln1-3 mutant condition compared with the N+ WT condition. Val biosynthesis (McCourt and Duggleby, 2006) is lower in the gln1-3 mutant condition compared with the N+ WT condition. Flux through glutathione biosynthesis/degradation, Trp biosynthesis (Tzin and Galili, 2010), uracil degradation (Zrenner et al., 2006), and Xyl degradation (Penna et al., 2002) is higher in the gln1-4 mutant compared with the N+ WT condition. Glu is converted to glutathione through two ATP-dependent steps requiring the addition of Cys and then Gly. Glutathione is a vitally essential protectant against oxidative stress, heavy metals, and xenobiotics (Noctor et al., 2012; Rahantaniaina et al., 2013). Several routes of glutathione breakdown have been proposed, including the formation of Cys and Gly through cysteinyl-Gly. The Cys is then degraded to form pyruvate, helping to alleviate the gln1-4 mutation. The increased fluxes associated with Xyl (from 1,4-β-d-xylan) and uracil degradation generate a larger pool of xylulose-5-phosphate and β-Ala, respectively. Finally, phenylpropanoid biosynthesis (Vogt, 2010) is lower in the gln1-4 mutant condition compared with the N+ WT condition. The majority of the changes in these pathways are directly related to differences in the proportion of the biomass components between the modeled conditions.
Comparison of Model Predictions with Metabolomic Data
The metabolomic data were compared with flux predictions within the model in each of the various N background conditions. The increasing or decreasing trend of the metabolite concentration, displayed in Figure 3, was qualitatively compared with the change in the flux-sum range determined by the model, as displayed in Figure 4. The flux sum is a measure of the amount of flow through the reactions associated with either the production or consumption of the metabolite. A variability analysis of the flux sum was performed, and flux-sum ranges, normalized by the biomass rate, that do not overlap between the N background condition and the N+ WT condition were analyzed. An increase/decrease in the flux sum (i.e. used as a proxy for the metabolite pool) of a metabolite between the N− WT condition and the N+ WT condition and between the two GS mutants and the N+ WT condition was compared with the metabolite concentration changes. Figure 4 demonstrates the importance of restricting fluxes based on transcriptomic and proteomic data. In the N− WT condition, the accuracy changes from 13% to 90% when the flux constraints based on omics data are incorporated. Without the incorporation of these constraints, all flux-sum ranges normalized by the biomass rate are predicted higher in the N− WT condition. The identified flux-sum levels are included in Supplemental Table S6. The flux-sum variability approach is able to predict the change in metabolite pool sizes more accurately when the flux ranges are similar to the wild-type condition, as in the N− WT condition. Between the N− WT and N+ WT conditions, only approximately 7% of the reactions active in either condition have flux ranges at the maximum biomass that do not overlap. In the gln1-3 and gln1-4 mutant conditions, the fluxes are significantly perturbed, with 49% and 45% of the active reactions at maximum biomass resulting in nonoverlapping ranges compared with the N+ WT condition, respectively. The accuracy of flux sum in the gln1-3 mutant and gln1-4 mutant conditions with omics-based constraints incorporated reaches 53% and 25%, with eight of 15 metabolites predicted correctly and one of four metabolites predicted correctly in the gln1-3 and gln1-4 mutant conditions, respectively. This level of prediction accuracy is far below what was seen for N− WT, suggesting a tenuous connection between concentration changes and gene expression levels when the genetic background changes.
Figure 3.

Number of metabolites in each condition that statistically varied from the N+ WT condition at the vegetative stage. The numbers of metabolites that experimentally significantly increased (up arrows) or decreased (down arrows) in comparison with the N+ WT condition are displayed for each of the N conditions tested (i.e. N− WT, gln1-3 mutant, and gln1-4 mutant conditions). The metabolites are shaded based on whether they are involved in carbon (C), N, or other metabolism. [See online article for color version of this figure.]
Figure 4.

Effect of omics-based regulation on the flux-sum prediction compared with the experimental trend in metabolite concentration. The accuracy in predicting the increasing (up arrows) or decreasing (down arrows) trend in metabolite change between the N background condition and the N+ WT condition is displayed. By restricting the reaction flux based on the transcriptomic and proteomic data, the accuracy of the qualitative trend in metabolite pool size between the N− WT and N+ WT conditions increases. Before adding omics-based constraints, the model was able to correctly predict the direction of change in 13% of the metabolites measured in the N− WT condition compared with the N+ WT condition. The accuracy increases to 90% when omics-based constraints are included. The flux-sum method is not able to accurately represent the gln1-3 and gln1-4 mutant conditions, suggesting that the genetic background affects the ability of the flux-sum method to predict metabolite changes. [See online article for color version of this figure.]
We explored the efficacy of the flux-sum method under different genetic backgrounds for a much more well-studied and data-rich organism (i.e. Escherichia coli) to explore whether the dissonance between gene expression levels and concentrations was maize specific or applied broadly. We applied flux-sum variability to the Ishii et al. (2007) fluxomic and metabolomic data using the iAF1260 (Feist et al., 2007) E. coli model. Two single-gene knockout mutants (i.e. ppsA and glk) were compared with the wild-type condition, and predicting the directional change of the metabolite pool size was met with less than 50% accuracy in each condition. This implies that changes in the genetic background seem to cause concentration changes that are not predictable by gene expression changes alone. In contrast, changes in nutrient availability, as in the N− WT condition, can be captured with 90% accuracy.
We also decided to explore whether the dissonance between gene expression levels and concentration ranges was caused by a deficiency in the proposed flux-sum method. As an alternative, we used flux imbalance analysis (Reznik et al., 2013), which measures the effect of the deviation of a metabolite’s concentration from steady state on the maximum biomass by applying the concept of duality. Flux imbalance analysis examines how the model responds to a deviation from steady state by measuring the effect on biomass when a metabolite is allowed to accumulate or deplete. By determining the change in biomass formation due to the accumulation or depletion of the metabolite, a prediction can be made regarding the change in metabolite levels. Flux imbalance analysis was applied to the model, and the deviation in the maximum biomass was qualitatively compared with the experimental data for the metabolite in each compartment. Only nonoverlapping ranges of the marginal value associated with each compartment-specific metabolite were analyzed. If all compartment-specific metabolites have marginal values that indicate the same trend compared with the N+ WT condition, a prediction was made for the tissue-specific metabolite. The flux imbalance analysis is 66%, 33%, and 78% accurate in the N− WT, gln1-3 mutant, and gln1-4 mutant conditions, respectively, as compared with the N+ WT condition. While flux imbalance analysis makes a prediction for every metabolite in the model, the flux-sum analysis only predicts a direction of change for metabolites whose associated reactions can carry flux. Flux imbalance analysis allows for the prediction of compartment-specific metabolites whose associated reactions do not carry flux under maximum biomass formation. Comparable results between the flux imbalance analysis and flux-sum analysis in the N− WT condition provide independent backing regarding the validity of the flux-sum concept.
CONCLUSION
We have introduced a second-generation model that is specific for the leaf tissue of maize and differentiates between the bundle sheath and mesophyll cell types. By incorporating transcriptomic and proteomic data into the model, we were able to reproduce the metabolomic data with up to 90% accuracy when comparing the N− WT and N+ WT conditions. Ethanol oxidoreductase/catalase, glyceraldehyde-3-phosphate dehydrogenase, Fru-bisP aldolase, Fru-bisphosphatase, and Rib-5-P isomerase were shown to be important genes related to the decrease in biomass formation in the modeled conditions. In order to study the impact of these genes on plant biomass production when optimal N is provided, their functional validation can be undertaken using transgenic technologies, mutagenesis, or association genetics, either at the single-gene or genome-wide level (Simons et al., 2014). The model also predicted a modification of the flux of metabolites formed during glutathione catabolism in the gln1-4 mutant condition compared with the N+ WT condition. This modification is predicted to compensate for the lack of GS1-4 by using the Glu and pyruvate derived from glutathione to produce Ala. Thus, it will be interesting to determine whether the increase in Ala is related to the importance of the enzyme Ala aminotransferase in the improvement of plant productivity in general and NUE in particular (Good and Beatty, 2011; McAllister et al., 2013). In all N background conditions (i.e. N− WT, gln1-3 mutant, and gln1-4 mutant conditions), we find that the flux through chlorophyll biosynthesis, and those pathways directly related to chlorophyll biosynthesis, decrease, confirming the important link between N metabolism and chlorophyll synthesis through the use of its precursor Glu (Forde and Lea, 2007). The leaf model, with the addition of other maize tissue-specific models, can be integrated into a whole-plant genome-scale model for maize. By determining a required metabolic function that is specific to each tissue, tissue-specific models can be created, ensuring that only relevant reactions are included in each tissue.
Future efforts will focus on tissue-specific models for the kernel, stalk, tassel, and root tissues. These tissue-specific models will follow community (Zomorrodi and Maranas, 2012; Zomorrodi et al., 2014) and multitissue human model (Duarte et al., 2007; Bordbar et al., 2011; Thiele et al., 2013) reconstruction principles. The tissues can be linked using intertissue transport reactions, with the stalk tissue acting as the central transporter among the various tissues and particularly to the developing ear (Cañas et al., 2012). A whole-plant genome-scale model of maize will help to elucidate the flow of N from the root to the other tissues in the plant, from the shoot to the ear, and within the developing ear (Cañas et al., 2010). By modeling the entire plant, nonintuitive bottlenecks in N metabolism can be determined, which then can be used to suggest genetic interventions through mutagenesis, transgenic technology, or maker-assisted selection to increase the NUE in maize. In addition, the flow of sugars to the kernel tissue can be analyzed to guide the increase of carbohydrate/sugar content of maize kernel by breaking the inverse relationship existing between carbohydrates and proteins (Feil et al., 1990). Apart from its crucial role as a food crop, maize is also used for cellulosic biofuels. To this end, the amount and composition of cell wall polymers is important in developing cellulosic maize. Lignin not only provides rigidity to the maize plant (Vanholme et al., 2008) but also makes the digestion of cellulosic and hemicellulosic sugars difficult during delignification (Li et al., 2008). Recent research endeavors have focused on altering lignin content, since plant viability and fitness are affected by lignin reductions (Li et al., 2008; Bonawitz et al., 2014). Therefore, by utilizing the whole-plant genome-scale model, a system-wide implication of these genetic disruptions can be quantitatively assessed, thus facilitating new strategies for reducing lignin content without affecting the mechanical integrity of the maize plant.
MATERIALS AND METHODS
Plant Material
Maize (Zea mays; genotype B73) wild-type plants and gln1-3 and gln1-4 mutant seeds in the B73 background (for the production, selection, and characterization of the mutants, see Martin et al., 2006) were grown as described by Amiour et al. (2012) in a greenhouse at the Institut National de la Recherche Agronomique (Versailles, France) from May to September 2004. Three individual plants of similar size and of similar developmental stage were selected, corresponding to the three replicates used for the omics experiments. The three youngest fully expanded leaves at the 10- to 11-leaf stage without the midrib were harvested and pooled for the vegetative stage samples to obtain enough homogenous plant material representative of this plant development stage.
Plants were watered daily with a complete nutrient solution containing 10 mm KNO3 as the sole N source in the N+ WT, gln1-3 mutant, and gln1-4 mutant conditions (Coïc and Lesaint, 1971). The N− WT condition was supplied 0.01 mm KNO3. The complete nutrient solution also contained 1.25 mm K+, 0.25 mm Ca2+, 0.25 mm Mg2+, 1.25 mm H2PO4−, 0.75 mm SO42−, 21.5 μm Fe2+ (Sequestrene; Ciba-Geigy), 23 μm B3+, 9 μm Mn2+, 0.3 μm Mo2+, 0.95 μm Cu2+, and 3.5 μm Zn2+.
Yield Components Analysis
Kernel yield, its components, and the N content of different parts of the plant at stages of development from silking to maturity were determined according to the method described by Martin et al. (2005) and corresponded to the data described by Martin et al. (2006) and Amiour et al. (2012).
RNA and DNA Preparation
Total RNA was extracted as described by Verwoerd et al. (1989) from leaves that had been stored at −80°C. Total RNAs (50 μg) for transcriptome and quantitative real-time (qRT)-PCR studies were treated and prepared as described previously by Amiour et al. (2012). Reverse transcription reactions and quantitative first strands were synthesized according to Amiour et al. (2012). Primers for qRT-PCR and reverse transcription-PCR cloning were designed from bacterial artificial chromosome sequences found in the public maize genome databases (Maizesequence.org, PlantGDB, and GenBank). The sequences of the primers used in reverse transcription-PCR and qRT-PCR are presented in Supplemental Table S1.
Gene Expression Profiles Using Maize Complementary DNA Microarrays
Whole-genome leaf transcript profiling was performed using the maize 46K arrays obtained from the maize oligonucleotide array project (http://www.maizecdna.org/outreach/resources.html) as described previously by Amiour et al. (2012). The maize 46K spotted oligonucleotide array contains 46,000 unique probes from maize. Its detailed description, composition, and gene putative annotation can be found at the Gene Expression Omnibus; (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL6438). Transcript abundance in each of the three replicates for vegetative leaves was determined using a mixture of all the samples (18 in total, each with the same mRNA concentration) as a reference. Statistical significance for differentially expressed genes was evaluated through statistical group comparisons performed using multiple testing procedures as described by Amiour et al. (2012). Transcriptomic data were validated by qRT-PCR analysis performed on a selected number of up- or down-regulated gene transcripts.
Statistical Analysis of Maize Complementary DNA Microarray Data
Statistical significance for differentially expressed genes was evaluated with statistical group comparisons using multiple testing procedures. The following two gene selection approaches were applied: the significance analysis of microarrays (Tusher et al., 2001) permutation algorithm and a P value ranking strategy using both z statistics in ArrayStat 1.0 software (Imaging Research) and moderated t statistics using a moderated Student’s t test available in MAnGO tools (http://bioinfome.cgm.cnrs-gif.fr) and BRBArrayTools version 3.2.3 (Korn et al., 2002). For multiple testing corrections, the false discovery rate procedure was used (Benjamini and Hochberg, 1995). Statistical tests were computed and combined for each probe set using the log-transformed data. A significant probe set indicates that an adjusted P value was less than the effective α level (α = 0.05) in at least one of the two gene selection tests. A filtering procedure was used to exclude data points with low signal intensities (average log intensity mean [Amean] < 7.0) that are considered biologically unreliable.
Total Protein Extraction, Solubilization, and Quantification
A TCA/acetone protein precipitation was performed as described by Méchin et al. (2007) from the leaves of optimal N and limited N plants harvested at the vegetative stage of development. The frozen leaf powder was resuspended in acetone with 0.07% (v/v) 2-mercaptoethanol and 10% (w/v) TCA. Proteins were allowed to precipitate for 1 h at −20°C. The pellet was then washed overnight with acetone containing 0.07% (v/v) 2-mercaptoethanol. The supernatant was discarded, and the pellet was dried under vacuum. Protein resolubilization was performed according to Méchin et al. (2007) using 60 μL mg−1 R2D2 buffer [5 m urea, 2 m thiourea, 2% (w/v) CHAPS, 2% (w/v) SB3-10 (i.e. N-decyl-N,N-dimethyl-3-ammonio-1-propane-sulfonate), 20 mm dithiothreitol, 5 mm Tris(2-carboxyethyl)phosphine hydrochloride, and 0.75% (v/v) carrier ampholytes]. After resolubilization, samples were centrifuged and the supernatant was transferred to an Eppendorf tube prior to protein quantification. Total protein content of each sample was evaluated using the 2-D Quant kit (Amersham Biosciences).
Two-Dimensional Electrophoresis, Gel Staining, and Image Analysis
Total protein extraction, solubilization, and quantification were performed as described by Méchin et al. (2007). Solubilized proteins (300 µg) were subjected to two-dimensional gel electrophoresis and identified by liquid chromatography-mass spectrometry as described by Amiour et al. (2012).
Protein Identification by Liquid Chromatography-Tandem Mass Spectrometry
Spot digestion and liquid chromatography-tandem mass spectrometry were performed as described by Martin et al. (2006). In-gel digestion was performed with the Progest system (Genomic Solution). Gel pieces were washed twice by successive separate baths of 10% (v/v) acetic acid, 40% (v/v) ethanol, and acetonitrile (ACN). The pieces were then washed twice with successive baths of 25 mm NH4CO3 and ACN. Digestion was subsequently performed for 6 h at 37°C with 125 ng of modified trypsin (Promega) dissolved in 20% (v/v) methanol and 20 mm NH4CO3. The peptides were extracted successively with 2% (v/v) trifluoroacetic acid and 50% (v/v) ACN and then with ACN. Peptide extracts were dried in a vacuum centrifuge and suspended in 20 mL of 0.05% (v/v) trifluoroacetic acid, 0.05% (v/v) formic acid, and 2% (v/v) ACN. HPLC was performed on an Ultimate liquid chromatography system combined with a Famos autosampler and a Switchos II microcolumn switch system (Dionex). Trypsin digestion was declared with one possible cleavage. Cys carboxyamidomethylation and Met oxidation were set to static and variable modifications, respectively. A multiple-threshold filter was applied at the peptide level: Cross correlation (i.e. Xcorr) magnitudes were up to 1.7, 2.2, 3.3, and 4.3 for peptides with one, two, three, and four isotopic charges, respectively; peptide probability was lower than 0.05, ΔCn > 0.1, with a minimum of two different peptides for an identified protein. Here, ΔCn is the change between the first and second cross correlation. A database search was performed with BioWorks 3.3.1 (Thermo Electron). The Institute for Genomic Research maize gene index database version 16, with 72047*6 (for a total of 72047 EST reads in the six reading frames) EST sequences (http://compbio.dfci.harvard.edu/tgi/), was used.
Metabolite Extraction and Analyses
Lyophilized leaf material was used for metabolite extraction. Approximately 20 mg of the powder was extracted in 1 mL of 80% (v/v) ethanol and 20% (v/v) distilled water for 1 h at 4°C. During extraction, the samples were continuously agitated and then centrifuged for 5 min at 15,000 rpm. The supernatant was removed, and the pellet was subjected to a further extraction in 60% (v/v) ethanol and finally in water at 4°C, as described above. All supernatants were combined to form the aqueous alcohol extract.
Nitrate was determined by the method of Cataldo et al. (1975). Total soluble amino acids were determined by the colorimetric method of Rosen (1957) with Leu as a standard. Chlorophyll was estimated using 10 mg of fresh leaf material (Arnon, 1949). The total N content of 2 mg of lyophilized material was determined in an N elemental analyzer using the combustion method of Dumas (Flash 2000; Thermo Scientific). Starch content was determined as described by Ferrario-Mery et al. (1998).
Total lipids were extracted from frozen leaf material according to Miquel and Browse (1992). Individual lipids were purified from the extracts by one-dimensional thin-layer chromatography on silica gel 60 plates (Lepage, 1967; Ohnishi and Yamada, 1980), which were obtained from Merck-Millipore. Lipids were located by spraying the plates with a solution of 0.001% (w/v) primuline (Sigma) in 80% (v/v) acetone, followed by visualization under UV light. To determine the fatty acid composition and relative amounts of individual lipids, the silica gel for each lipid was transferred to a screw-capped tube with 1 mL of 2.5% (v/v) H2SO4 in methanol and an appropriate amount of C17:0 fatty acid (Sigma) as an internal standard. After heating for 90 min at 80°C, 1 mL of hexane and 1.5 mL of 0.9% (w/v) NaCl2 were added. Fatty acids were extracted in the upper organic phase by shaking and low-speed centrifugation. Samples (1 µL) of the organic phase were separated by gas chromatography on a 30-m × 0.53-mm EC-WAX column (Alltech Associates) and quantified using a flame ionization detector. The gas chromatograph was programmed for an initial temperature of 160°C for 1 min, followed by an increase of 20°C min−1 to 190°C and a ramp of 4°C min−1 to 230°C, with a 9-min hold of the final temperature.
The monosaccharide composition and linkage analysis of polysaccharides were determined as follows: 100 mg (fresh weight) of ground leaf was washed twice in 4 volumes of absolute ethanol for 15 min, then rinsed twice in 4 volumes of acetone at room temperature for 10 min, and left to dry under a fume hood overnight at room temperature. The neutral monosaccharide composition was measured on 5 mg of dried alcohol-insoluble material after hydrolysis in 2.5 m trifluoroacetic acid for 1.5 h at 100°C as described by Harholt et al. (2006). To determine the cellulose content, the residual pellet obtained after the monosaccharide analysis was rinsed twice with 10 volumes of water and hydrolyzed with H2SO4 as described by Updegraff (1969). The released Glc was diluted 500 times and then quantified using high-performance anion-exchange chromatography-pulsed-amperometric detection as described by Harholt et al. (2006).
For lignin quantification, 100 mg (fresh weight) of ground leaf was washed twice in 4 volumes of absolute ethanol for 15 min and twice with 4 volumes of water at room temperature, then rinsed twice in 4 volumes of acetone at room temperature for 10 min, and left to dry under a fume hood overnight at room temperature. The following protocol is adapted from Fukushima and Hatfield (2001). Lignins from the prepared cell wall residue were solubilized in 1 mL of acetyl bromide solution (acetyl bromide:acetic acid, 1:3 [v/v]) in a glass vial at 55°C for 2.5 h under shaking. Samples were then allowed to cool to room temperature, and 1.2 mL of 2 m NaOH:acetic acid (9:50 v/v) was added in the vial. One hundred microliters of this sample was transferred in 300 µL of 0.5 m hydroxylamine chlorhydrate and mixed with 1.4 mL of acetic acid. The A280 of the samples was measured. The lignin content was calculated using the following formula:
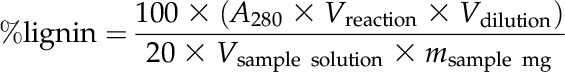 |
(1) |
Metabolome Analysis
All steps were adapted from the original protocol described by Fiehn (2006) following the procedure described by Amiour et al. (2012).
Model Development and Curation
Figure 5 outlines the work flow used for model development. Our previously developed maize model, iRS1563 (Saha et al., 2011), and biological databases such as MetaCrop (downloaded in December 2012; Schreiber et al., 2012) and MaizeCyc (version 2.0.2; Monaco et al., 2013) provided information pertaining to the genes, proteins, reactions, and metabolites used to reconstruct the second-generation maize leaf genome-scale model. In addition, available proteomic and transcriptomic data, maize-specific biological databases, namely MetaCrop and MaizeCyc, and published literature were used to assign cellular (i.e. bundle sheath or mesophyll) and intracellular organelle specificity to the curated reactions.
Figure 5.

Model development and curation schematic. The work flow for the second-generation genome-scale metabolic model of the maize leaf is displayed. The data sources give three types of retrieved data (i.e. raw reaction data, reaction directionality, and compartmentalization) that are then manipulated as shown to create the final model. [See online article for color version of this figure.]
When the gene expression level was reported in reads per kilobase per million mapped reads (RPKM; Li et al., 2010; Chang et al., 2012), the cell specificity of any gene i can be calculated as:
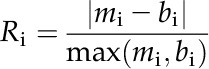 |
(2) |
Here, mi and bi are the RPKM abundance of gene i in the mesophyll and bundle sheath cells, respectively (Chang et al., 2012). A gene that is only expressed in one cell type will have an Ri of 1, while a gene that is equally expressed in both cell types will have an Ri of 0. As suggested by Chang et al. (2012), a threshold of 0.8 or a 5-fold abundance difference is adopted to assign gene cell type specificity. In the absence of RPKM information, an adjusted spectral count (adjSPC) along with the fold change difference between the mesophyll and bundle sheath cells was used to determine gene cell type specificity (Friso et al., 2010). The adjSPC is the number of mass spectra identified for a protein normalized by the number of unique spectral counts. Since low counts are not statistically informative, a cutoff of 10 was used for adjSPC (Zybailov et al., 2008; Kim et al., 2009). Similar to the threshold used for RPKM data, a 5-fold difference between the mesophyll and bundle sheath cell type normalized spectral abundance factor was used to determine the cellular specificity of any gene (Friso et al., 2010). The normalized spectral abundance factor is a weighted adjSPC based on the number of theoretical tryptic peptides with a relevant length (Ehleringer et al., 1997; Friso et al., 2010). Additional intracellular compartmentalization was carried out based on the MetaCrop database (Schreiber et al., 2012), the MaizeCyc database (Monaco et al., 2013), and primary literature sources (Chang et al., 2012; Zhao et al., 2013).
The intracellular compartmentalization was determined based first on the MetaCrop database (Schreiber et al., 2012), literature sources (Friso et al., 2010; Chang et al., 2012), compartmentalization information in the MaizeCyc database, and finally the Plant Proteomics Database (Sun et al., 2009). An original set of intercellular and intracellular transporters was determined based on literature evidence (Alberte and Thornber, 1977; Leegood, 1985; Stitt and Heldt, 1985; Furbank et al., 1989; Weiner and Heldt, 1992; Doulis et al., 1997; Burgener et al., 1998; Taniguchi et al., 2004; Sowiński et al., 2008; Friso et al., 2010). In the subsequent standardization step, the MetRxn knowledgebase (Kumar et al., 2012) as well as manual curation were used to standardize the description of metabolites and reactions such as fixing stoichiometric errors (i.e. elemental or charge imbalances) and incomplete atomistic details (e.g. absence of stereospecificity and presence of unspecified side chains). Reactions and metabolites were given the Kyoto Encyclopedia of Genes and Genomes identifiers where available or were otherwise given new identifiers (in the form of MR or MC, respectively). Reaction directionality was adopted from the manually curated MetaCrop database, as available, and from the MaizeCyc database for the remaining reactions.
In the next step of model development, all reactions (including metabolic, intracellular, and extracellular transport reactions) were divided into two categories based on the evidence of their intercellular and intracellular compartmental specificity. The core set contains all metabolic reactions with experimental or literature-backed evidence of intracellular or intercellular compartmentalization as well as known intracellular and intercellular transporters. The noncore set contains reactions with partial or completely absent localization information. Barring any conflicting evidence, these reactions were provisionally placed in all compartments. An optimization formulation (as shown below) was developed by imposing flow though the maximal number of core reactions while including minimal intracellular and intercellular transporters and minimal participation of noncore reactions in various compartments. A parsimony criterion was used to apportion noncore functions so that core functions could be restored. Furthermore, in order to restore a core function, the resolution strategy was prioritized in the following order: (1) apportion noncore reaction(s) in one/multiple compartment(s); (2) add intracellular transporter(s); and (3) add intercellular transporter(s). To this end, an objective function was formulated by taking the weighted sum of the number of noncore reactions and intracellular and intercellular transporters by providing weights of 1, 104, and 106, respectively, for these three groups of reactions. However, it is important to ensure that any resolution strategy does not cause thermodynamically infeasible cycles. Therefore, each of these solutions was further checked, and those reactions that result in the formation of a cycle were rejected. For each core reaction, multiple solutions were determined, and the solution that fixes the largest number of core reactions was accepted. When required, manual curation was used to delineate between multiple solutions. This approach is analogous to the one proposed by Mintz-Oron et al. (2012) but does not rely on a complicated scoring system. It is also computationally less taxing, as it activates one core reaction at a time. Furthermore, in contrast to the approach of Mintz-Oron et al. (2012), the method proposed here allows for the minimal number of transporters added, rather than potentially minimizing the flux through many transporters. The process of minimally adding the number of reactions and transporters to the model is similar to that used by the model SEED (Henry et al., 2010). In order to allow flux through all reactions in the core set C = {1,…,c}, we minimized the addition of reactions from the noncore set NC = {c + 1,…,g}, intracellular transporter set T = {g + 1,…,t}, and intercellular transporter set IC = {t + 1,…,m}. This encompasses an overall set of reactions M = {1,…,m} and a set of metabolites N = {1,…,n}. In addition, binary variable yj is defined as:
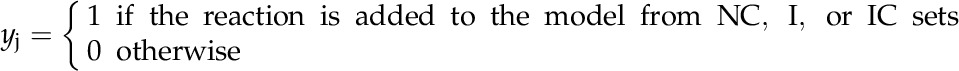 |
The task of identifying the minimal set of additional reactions that enable flux through a core reaction j* is posed as the following mixed-integer linear programming problem:
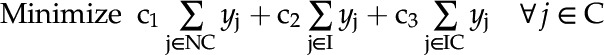 |
(3) |
subject to:
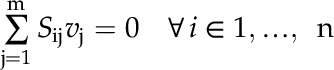 |
(4) |
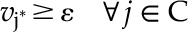 |
(5) |
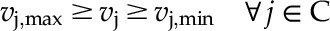 |
(6) |
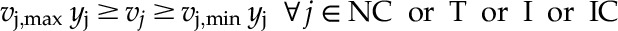 |
(7) |
Here, Sij is the stoichiometric coefficient of metabolite i in reaction j and vj is the flux value of reaction j. Parameters vj,min and vj,max denote the minimum and maximum allowable fluxes for reaction j, respectively. vj* represents the core reaction flux that is currently being unblocked, and ε is a small value to ensure a threshold amount of flux through each core reaction. c1, c2, and c3 represent weights associated with each set of reactions (i.e. noncore set, intracellular transporter set, and intercellular transporter set, respectively). In this formulation, the objective function 3 above minimizes the number of added reactions (from three reaction sets as mentioned earlier) so as to restore flux through reaction j*. We chose values of 1, 104, and 106 for c1, c2, and c3, respectively, so that metabolic reactions without experimental or literature evidence for compartmental specificity are added to specific compartment(s) before including additional transport reactions with no literature evidence. Constraint set 4 above represents the pseudo-steady-state assumption, while constraint 5 determines the threshold amount of flux necessary through j*. Bounds on core reaction fluxes are imposed by constraint set 6, while constraint set 7 ensures that only reactions from those three sets having nonzero flow are added to the model. This algorithm is repeated for each core reaction j* to ensure flux and, hence, provides compartmentalization assignments for 431 metabolic reactions by assigning them to at least one compartment, adding 1,032 total metabolic reactions to the model, as shown in Table II.
The reactions identified by the above-mentioned algorithm plus the reactions from the core set constituted two new sets, a set of reactions with resolved compartmental information and a set whose location still needs resolution, as shown in Figure 5. Reactions from the latter set that are known to occur within the maize leaf tissue but were not in the initial model were added to intracellular/intercellular compartments manually based on pathway localization or simply added to the cytosol of bundle sheath and/or mesophyll cells. Thermodynamically infeasible cycles were resolved by changing the minimum number of reaction directionalities possible and eliminating the smallest number of reactions from the model (Schellenberger et al., 2011) while conserving biomass formation. An optimization procedure was iteratively run for each reaction in a thermodynamically infeasible cycle to determine the minimum number of directionality changes or reaction removals required to fix the cycle. These results were then compared for each reaction to determine the changes that resolve the largest number of reactions participating in thermodynamically infeasible cycles. The solutions found were manually inspected before the changes were applied to the model. The application of this optimization procedure led to restricting the directionality for 507 reactions that prevented 889 reactions from carrying unbounded fluxes, thus eliminating the corresponding thermodynamically infeasible cycles.
In the final step, as shown in Figure 5, the GapFind/GapFill (Kumar et al., 2007) procedure was applied to identify blocked/dead-end metabolites and subsequently restore their connectivity. A gap-filling database of reactions was created by combining reactions from phylogenetically close/model plant species (i.e. rice [Oryza sativa ssp. japonica], Brachypodium distachyon, sorghum [Sorghum bicolor], and Arabidopsis thaliana), noncore reactions without compartmental specificity (not identified by our aforementioned algorithm), and all possible intracellular/intercellular transporters. The gap-filling procedure was modified by prioritizing the addition of reactions from closely related/model plant species or noncore reactions over transporters to unblock the flow-through metabolites while ensuring that no new thermodynamically infeasible cycles are created. After completing this step, we added five reactions from closely related/model plant species, changed the directionality of 14 reactions, and added eight intracellular transporters.
Incorporation of Transcriptomic, Proteomic, and Metabolomic Data
Significantly different gene transcripts and proteins were incorporated into the model by switching off corresponding reactions under the N+ WT, N− WT (Amiour et al., 2012), gln1-3 mutant, and gln1-4 mutant (Martin et al., 2006) conditions. The number of proteins, gene transcripts, and metabolites with abundances that are statistically differentially expressed in the various conditions are listed in Table IV. Reactions with GPRs associated with significantly lowered transcriptomic and proteomic expression are switched off under the corresponding conditions. Metabolite turnover rates were determined based on the flux-sum analysis method (Chung and Lee, 2009) and compared with the metabolomic data. The range of the flux sum or the flow through of each metabolite with experimental measurements was maximized/minimized as follows:
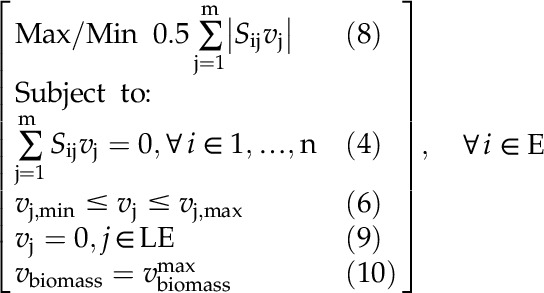 |
Here, set E represents the set of metabolites with experimental measurements and set LE represents reactions with statistically lower expression of gene transcripts and/or proteins. The formulation was run in an iterative manner for each metabolite with experimental measurements. The formulation was also repeated for each individual condition, ensuring that the proper nutrients and simulated knockouts were considered. By linearizing the objective function, the resulting formulation is a mixed-integer linear programming problem similar to the description by Chung and Lee (2009). Therefore, the basic idea is to determine the range of the flux sum of a metabolite (for which metabolomic data are available) under a given condition by switching off reaction fluxes corresponding to gene transcripts and/or proteins with lower expression levels (i.e. constraint 9). The flux-sum ranges were determined at the maximum biomass for the condition as displayed in constraint 10. Predictions were made only when the flux-sum ranges did not overlap between the N background condition and the N+ WT condition and when the direction of change in all compartments was consistent. In this way, the compartment-specific predictions of the flux-sum ranges were compared with tissue-specific experimental measurements. The flux-sum levels in the N− WT, gln1-3 mutant, and gln1-4 mutant conditions were compared with the reference N+ WT condition to find the qualitative trend in the change of metabolite pool size between the conditions.
Table IV. Number of gene transcripts, proteins, and metabolites that vary significantly.
The wild-type condition for each study was combined to create one uniform N+ WT condition. The numbers of gene transcripts, proteins, and metabolites that statistically vary are displayed.
| Type of Data | N+ WT Condition | N− WT Condition | gln1-3 Mutant | gln1-4 Mutant |
|---|---|---|---|---|
| Transcriptomic | 256 | 76 | 102 | 53 |
| Proteomic | 38 | 14 | 29 | – |
| Metabolomic | 83 | 20 | 31 | 13 |
Flux variability analysis was used to determine the flux range of each reaction under maximum biomass by subsequently maximizing and minimizing the flux through each reaction. The flux range of each reaction for the N− WT, gln1-3 mutant, and gln1-4 mutant conditions was compared with the reference N+ WT condition. Flux ranges that did not overlap between one of the N background conditions and the reference condition were further analyzed. These are reactions that must change in response to the limited amount of N or the mutant conditions. Finally, we determined for each condition the minimum number of reactions that, when not regulated, will restore the biomass to the yield obtained when no omics-based regulation is applied. This was done by identifying the minimal set of reactions, included in the omics-based regulation, that when active would allow for a biomass yield equivalent to the yield under no omics-based regulation. This set of reactions represent the reactions whose restriction affects the biomass yield.
The CPLEX solver (version 12.3 IBM ILOG) was used in the GAMS environment (version 23.3.3; GAMS Development) to solve the optimization problems. The Python programming language was also used during model development (mainly for scripting and data analysis). All computations were carried out on Intel Xeon X5675 Six-Core 3.06 GHz processors constituting the Lion-XF cluster, which was built and operated by the Research Computing and Cyberinfrastructure Group of Pennsylvania State University.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. Stained micrograph of starch in the N+ WT, gln1-3 mutant, and gln1-4 mutant conditions.
Supplemental Figure S2. Glycerolipid synthesis in maize.
Supplemental Table S1. Experimental biomass measurements.
Supplemental Table S2. Glycerolipid synthesis reactions included in the model.
Supplemental Table S3. Second-generation leaf model in Excel format.
Supplemental Table S4. Second-generation leaf model in Systems Biology Markup Language format.
Supplemental Table S5. Metabolic reactions with fluxes that must change in an N background condition compared with the N+ WT condition.
Supplemental Table S6. Flux-sum levels in each condition.
Supplementary Material
Acknowledgments
We thank Isabelle Quilleré, François Gosse, and Michel Lebrusq for technical help.
Glossary
- GSM
genome-scale metabolic model
- N+ WT
wild-type plants grown under optimal nitrogen conditions
- N− WT
wild-type plants grown under limited nitrogen conditions
- N
nitrogen
- NUE
nitrogen use efficiency
- FBA
flux balance analysis
- GPR
gene-protein-reaction
- qRT
quantitative real-time
- ACN
acetonitrile
- RPKM
reads per kilobase per million mapped reads
- adjSPC
adjusted spectral count
Footnotes
This work was supported by the U.S. Department of Energy (grant no. DE–FG02–05ER25684) and by the Agence National pour la Recherche Genoplante program Maize and Yield (grant no. GNP05015G).
Some figures in this article are displayed in color online but in black and white in the print edition.
The online version of this article contains Web-only data.
Articles can be viewed online without a subscription.
References
- Alberte RS, Thornber JP. (1977) Water stress effects on the content and organization of chlorophyll in mesophyll and bundle sheath chloroplasts of maize. Plant Physiol 59: 351–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiour N, Imbaud S, Clément G, Agier N, Zivy M, Valot B, Balliau T, Armengaud P, Quilleré I, Cañas R, et al. (2012) The use of metabolomics integrated with transcriptomic and proteomic studies for identifying key steps involved in the control of nitrogen metabolism in crops such as maize. J Exp Bot 63: 5017–5033 [DOI] [PubMed] [Google Scholar]
- Amiour N, Imbaud S, Clément G, Agier N, Zivy M, Valot B, Balliau B, Quilleré I, Tercé-Laforgue T, Dargel-Graffin C, et al. (2014) An integrated “omics” approach to the characterization of maize (Zea mays L.) mutants deficient in the expression of two genes encoding cytosolic glutamine synthetase. BMC Genomics (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews M, Lea PJ. (2013) Our nitrogen ‘footprint’: the need for increased crop nitrogen use efficiency. Ann Appl Biol 163: 165–169 [Google Scholar]
- Andrews M, Raven JA, Lea PJ. (2013) Do plants need nitrate? The mechanisms by which nitrogen form affects plants. Ann Appl Biol 163: 174–199 [Google Scholar]
- Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815 [DOI] [PubMed] [Google Scholar]
- Arnon DI. (1949) Copper enzymes in isolated chloroplasts: polyphenoloxidase in Beta vulgaris. Plant Physiol 24: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachlava E, Dewey R, Burton J, Cardinal AJ. (2009) Mapping candidate genes for oleate biosynthesis and their association with unsaturated fatty acid seed content in soybean. Mol Breed 23: 337–347 [Google Scholar]
- Becker SA, Palsson BO. (2008) Context-specific metabolic networks are consistent with experiments. PLoS Comput Biol 4: e1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc 57: 289–300 [Google Scholar]
- Boamfa EI, Veres AH, Ram PC, Jackson MB, Reuss J, Harren FJ. (2005) Kinetics of ethanol and acetaldehyde release suggest a role for acetaldehyde production in tolerance of rice seedlings to micro-aerobic conditions. Ann Bot (Lond) 96: 727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonawitz ND, Kim JI, Tobimatsu Y, Ciesielski PN, Anderson NA, Ximenes E, Maeda J, Ralph J, Donohoe BS, Ladisch M, et al. (2014) Disruption of Mediator rescues the stunted growth of a lignin-deficient Arabidopsis mutant. Nature 509: 376–380 [DOI] [PubMed] [Google Scholar]
- Bordbar A, Feist AM, Usaite-Black R, Woodcock J, Palsson BO, Famili I. (2011) A multi-tissue type genome-scale metabolic network for analysis of whole-body systems physiology. BMC Syst Biol 5: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgener M, Suter M, Jones S, Brunold C. (1998) Cyst(e)ine is the transport metabolite of assimilated sulfur from bundle-sheath to mesophyll cells in maize leaves. Plant Physiol 116: 1315–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cañas RA, Quilleré I, Gallais A, Hirel B. (2012) Can genetic variability for nitrogen metabolism in the developing ear of maize be exploited to improve yield? New Phytol 194: 440–452 [DOI] [PubMed] [Google Scholar]
- Cañas RA, Quilleré I, Lea PJ, Hirel B. (2010) Analysis of amino acid metabolism in the ear of maize mutants deficient in two cytosolic glutamine synthetase isoenzymes highlights the importance of asparagine for nitrogen translocation within sink organs. Plant Biotechnol J 8: 966–978 [DOI] [PubMed] [Google Scholar]
- Cataldo DA, Haroon M, Schrader LE, Youngs VL. (1975) Rapid colorimetric determination of nitrate in plant-tissue by nitration of salicylic-acid. Commun Soil Sci Plant Anal 6: 71–80 [Google Scholar]
- Chandrasekaran S, Price ND. (2010) Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc Natl Acad Sci USA 107: 17845–17850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YM, Liu WY, Shih ACC, Shen MN, Lu CH, Lu MYJ, Yang HW, Wang TY, Chen SCC, Chen SM, et al. (2012) Characterizing regulatory and functional differentiation between maize mesophyll and bundle sheath cells by transcriptomic analysis. Plant Physiol 160: 165–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christin PA, Osborne CP. (2013) The recurrent assembly of C4 photosynthesis, an evolutionary tale. Photosynth Res 117: 163–175 [DOI] [PubMed] [Google Scholar]
- Chung BK, Lee DY. (2009) Flux-sum analysis: a metabolite-centric approach for understanding the metabolic network. BMC Syst Biol 3: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coïc Y, Lesaint C. (1971) Comment assurer une bonne nutrition en eau et en ions minéraux en horticulture. Hortic Francaise 8: 11–14 [Google Scholar]
- Colijn C, Brandes A, Zucker J, Lun DS, Weiner B, Farhat MR, Cheng TY, Moody DB, Murray M, Galagan JE. (2009) Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput Biol 5: e1000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Dal’Molin CG, Quek LE, Palfreyman RW, Brumbley SM, Nielsen LK. (2010a) AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol 152: 579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Dal’Molin CG, Quek LE, Palfreyman RW, Brumbley SM, Nielsen LK. (2010b) C4GEM, a genome-scale metabolic model to study C4 plant metabolism. Plant Physiol 154: 1871–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulis AG, Debian N, Kingston-Smith AH, Foyer CH. (1997) Differential localization of antioxidants in maize leaves. Plant Physiol 114: 1031–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driever SM, Kromdijk J. (2013) Will C3 crops enhanced with the C4 CO2-concentrating mechanism live up to their full potential (yield)? J Exp Bot 64: 3925–3935 [DOI] [PubMed] [Google Scholar]
- Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BO. (2007) Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci USA 104: 1777–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehleringer JR, Cerling TE, Helliker BR. (1997) C4 photosynthesis, atmospheric CO2, and climate. Oecologia 112: 285–299 [DOI] [PubMed] [Google Scholar]
- Feil B, Thiraporn R, Geisler G, Stamp P. (1990) Genotype variation in grain nutrient concentration in tropical maize grown during a rainy and a dry season. Agronomie 10: 717–725 [Google Scholar]
- Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BO. (2007) A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol 3: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario-Mery S, Valadier MH, Foyer CH. (1998) Overexpression of nitrate reductase in tobacco delays drought-induced decreases in nitrate reductase activity and mRNA. Plant Physiol 117: 293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O. (2006) Metabolite profiling in Arabidopsis. Methods Mol Biol 323: 439–447 [DOI] [PubMed] [Google Scholar]
- Forde BG, Lea PJ. (2007) Glutamate in plants: metabolism, regulation, and signalling. J Exp Bot 58: 2339–2358 [DOI] [PubMed] [Google Scholar]
- Friso G, Majeran W, Huang M, Sun Q, van Wijk KJ. (2010) Reconstruction of metabolic pathways, protein expression, and homeostasis machineries across maize bundle sheath and mesophyll chloroplasts: large-scale quantitative proteomics using the first maize genome assembly. Plant Physiol 152: 1219–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima RS, Hatfield RD. (2001) Extraction and isolation of lignin for utilization as a standard to determine lignin concentration using the acetyl bromide spectrophotometric method. J Agric Food Chem 49: 3133–3139 [DOI] [PubMed] [Google Scholar]
- Furbank RT, Jenkins CLD, Hatch MD. (1989) CO2 concentrating mechanism of C4 photosynthesis: permeability of isolated bundle sheath-cells to inorganic carbon. Plant Physiol 91: 1364–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, et al. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296: 92–100 [DOI] [PubMed] [Google Scholar]
- Good AG, Beatty PH (2011) Biotechnological approaches to improving nitrogen use efficiency in plants: alanine aminotransferase as a case study. In MJ Hawkesford, P Barraclough, eds, The Molecular and Physiological Basis of Nutrient Use Efficiency in Crops. Wiley-Blackwell, Sussex, UK, pp 165–191 [Google Scholar]
- Grafahrend-Belau E, Schreiber F, Koschützki D, Junker BH. (2009) Flux balance analysis of barley seeds: a computational approach to study systemic properties of central metabolism. Plant Physiol 149: 585–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Jain M, Sarangthem J, Gadre R. (2013) Inhibition of 5-aminolevulinic acid dehydratase by mercury in excised greening maize leaf segments. Plant Physiol Biochem 62: 63–69 [DOI] [PubMed] [Google Scholar]
- Harholt J, Jensen JK, Sørensen SO, Orfila C, Pauly M, Scheller HV. (2006) ARABINAN DEFICIENT 1 is a putative arabinosyltransferase involved in biosynthesis of pectic arabinan in Arabidopsis. Plant Physiol 140: 49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heavner BD, Smallbone K, Barker B, Mendes P, Walker LP. (2012) Yeast 5: an expanded reconstruction of the Saccharomyces cerevisiae metabolic network. BMC Syst Biol 6: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. (2010) High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol 28: 977–982 [DOI] [PubMed] [Google Scholar]
- Hirel B, Gallais A (2011) Nitrogen use efficiency: physiological, molecular and genetic investigations towards crop improvement. In JL Prioul, C Thévenot, T Molnar, eds, Advances in Maize, Vol 3. Society for Experimental Biology, London, pp 285–310 [Google Scholar]
- Hirel B, Le Gouis J, Ney B, Gallais A. (2007) The challenge of improving nitrogen use efficiency in crop plants: towards a more central role for genetic variability and quantitative genetics within integrated approaches. J Exp Bot 58: 2369–2387 [DOI] [PubMed] [Google Scholar]
- Hirel B, Martin A, Terce-Laforgue T, Gonzalez-Moro MB, Estavillo JM. (2005) Physiology of maize: I. A comprehensive and integrated view of nitrogen metabolism in a C4 plant. Physiol Plant 124: 167–177 [Google Scholar]
- Ho CL, Saito K. (2001) Molecular biology of the plastidic phosphorylated serine biosynthetic pathway in Arabidopsis thaliana. Amino Acids 20: 243–259 [DOI] [PubMed] [Google Scholar]
- Howard TP, Lloyd JC, Raines CA. (2011) Inter-species variation in the oligomeric states of the higher plant Calvin cycle enzymes glyceraldehyde-3-phosphate dehydrogenase and phosphoribulokinase. J Exp Bot 62: 3799–3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howles PA, Birch RJ, Collings DA, Gebbie LK, Hurley UA, Hocart CH, Arioli T, Williamson RE. (2006) A mutation in an Arabidopsis ribose 5-phosphate isomerase reduces cellulose synthesis and is rescued by exogenous uridine. Plant J 48: 606–618 [DOI] [PubMed] [Google Scholar]
- Hu TT, Pattyn P, Bakker EG, Cao J, Cheng JF, Clark RM, Fahlgren N, Fawcett JA, Grimwood J, Gundlach H, et al. (2011) The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet 43: 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Grains Council (2013) Report for fiscal year 2011/12. International Grains Council, London [Google Scholar]
- Ishii N, Nakahigashi K, Baba T, Robert M, Soga T, Kanai A, Hirasawa T, Naba M, Hirai K, Hoque A, et al. (2007) Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science 316: 593–597 [DOI] [PubMed] [Google Scholar]
- Jensen PA, Papin JA. (2011) Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 27: 541–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. (2014) Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42: D199–D205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Rudella A, Ramirez Rodriguez V, Zybailov B, Olinares PDB, van Wijk KJ. (2009) Subunits of the plastid ClpPR protease complex have differential contributions to embryogenesis, plastid biogenesis, and plant development in Arabidopsis. Plant Cell 21: 1669–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Schlicke H, Van Ree K, Karvonen K, Subramaniam A, Richter A, Grimm B, Braam J. (2013) Arabidopsis chlorophyll biosynthesis: an essential balance between the methylerythritol phosphate and tetrapyrrole pathways. Plant Cell 25: 4984–4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn EL, McShane LM, Troendle JF, Rosenwald A, Simon R. (2002) Identifying pre-post chemotherapy differences in gene expression in breast tumours: a statistical method appropriate for this aim. Br J Cancer 86: 1093–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumholz EW, Yang H, Weisenhorn P, Henry CS, Libourel IG. (2012) Genome-wide metabolic network reconstruction of the picoalga Ostreococcus. J Exp Bot 63: 2353–2362 [DOI] [PubMed] [Google Scholar]
- Kumar A, Suthers PF, Maranas CD. (2012) MetRxn: a knowledgebase of metabolites and reactions spanning metabolic models and databases. BMC Bioinformatics 13: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar VS, Dasika MS, Maranas CD. (2007) Optimization based automated curation of metabolic reconstructions. BMC Bioinformatics 8: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan M, Zhang Z, Mohanty B, Kwon JY, Choi HY, Nam HJ, Kim DI, Lee DY. (2013) Elucidating rice cell metabolism under flooding and drought stresses using flux-based modeling and analysis. Plant Physiol 162: 2140–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange BM, Ghassemian M. (2003) Genome organization in Arabidopsis thaliana: a survey for genes involved in isoprenoid and chlorophyll metabolism. Plant Mol Biol 51: 925–948 [DOI] [PubMed] [Google Scholar]
- Lee D, Smallbone K, Dunn WB, Murabito E, Winder CL, Kell DB, Mendes P, Swainston N. (2012) Improving metabolic flux predictions using absolute gene expression data. BMC Syst Biol 6: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leegood RC. (1985) The intercellular compartmentation of metabolites in leaves of Zea mays L. Planta 164: 163–171 [DOI] [PubMed] [Google Scholar]
- Lepage M. (1967) Identification and composition of turnip root lipids. Lipids 2: 244–250 [DOI] [PubMed] [Google Scholar]
- Leveau V, Lorgeou J, Prioul JL (2011) Maize in the world economy: a challenge for scientific research–how to produce more cheaper! In JL Prioul, C Thévenot, C Molnar, eds, Advances in Maize, Vol 3. Society for Experimental Biology, London, pp 509–534 [Google Scholar]
- Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR, et al. (2010) The developmental dynamics of the maize leaf transcriptome. Nat Genet 42: 1060–1067 [DOI] [PubMed] [Google Scholar]
- Li X, Weng JK, Chapple C. (2008) Improvement of biomass through lignin modification. Plant J 54: 569–581 [DOI] [PubMed] [Google Scholar]
- Li-Beisson Y, Shorrosh B, Beisson F, Andersson MX, Arondel V, Bates PD, Baud S, Bird D, Debono A, Durrett TP, et al. (2010) Acyl-lipid metabolism. The Arabidopsis Book 8: e0133, /10.1199/tab.0133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeran W, Cai Y, Sun Q, van Wijk KJ. (2005) Functional differentiation of bundle sheath and mesophyll maize chloroplasts determined by comparative proteomics. Plant Cell 17: 3111–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Belastegui-Macadam X, Quilleré I, Floriot M, Valadier MH, Pommel B, Andrieu B, Donnison I, Hirel B. (2005) Nitrogen management and senescence in two maize hybrids differing in the persistence of leaf greenness: agronomic, physiological and molecular aspects. New Phytol 167: 483–492 [DOI] [PubMed] [Google Scholar]
- Martin A, Lee J, Kichey T, Gerentes D, Zivy M, Tatout C, Dubois F, Balliau T, Valot B, Davanture M, et al. (2006) Two cytosolic glutamine synthetase isoforms of maize are specifically involved in the control of grain production. Plant Cell 18: 3252–3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister CH, Facette M, Holt A, Good AG. (2013) Analysis of the enzymatic properties of a broad family of alanine aminotransferases. PLoS ONE 8: e55032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCourt JA, Duggleby RG. (2006) Acetohydroxyacid synthase and its role in the biosynthetic pathway for branched-chain amino acids. Amino Acids 31: 173–210 [DOI] [PubMed] [Google Scholar]
- McNeil SD, Nuccio ML, Ziemak MJ, Hanson AD. (2001) Enhanced synthesis of choline and glycine betaine in transgenic tobacco plants that overexpress phosphoethanolamine N-methyltransferase. Proc Natl Acad Sci USA 98: 10001–10005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méchin V, Thévenot C, Le Guilloux M, Prioul JL, Damerval C. (2007) Developmental analysis of maize endosperm proteome suggests a pivotal role for pyruvate orthophosphate dikinase. Plant Physiol 143: 1203–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekhedov S, de Ilárduya OM, Ohlrogge J. (2000) Toward a functional catalog of the plant genome: a survey of genes for lipid biosynthesis. Plant Physiol 122: 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mérigout P, Lelandais M, Bitton F, Renou JP, Briand X, Meyer C, Daniel-Vedele F. (2008) Physiological and transcriptomic aspects of urea uptake and assimilation in Arabidopsis plants. Plant Physiol 147: 1225–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz-Oron S, Meir S, Malitsky S, Ruppin E, Aharoni A, Shlomi T. (2012) Reconstruction of Arabidopsis metabolic network models accounting for subcellular compartmentalization and tissue-specificity. Proc Natl Acad Sci USA 109: 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquel M, Browse J. (1992) Arabidopsis mutants deficient in polyunsaturated fatty acid synthesis: biochemical and genetic characterization of a plant oleoyl-phosphatidylcholine desaturase. J Biol Chem 267: 1502–1509 [PubMed] [Google Scholar]
- Monaco MK, Sen TZ, Dharmawardhana PD, Ren L, Schaeffer M, Naithani S, Amarasinghe V, Thomason J, Harper L, Gardiner J, et al. (2013) Maize metabolic network construction and transcriptome analysis. Plant Genome 6: 1–12 [Google Scholar]
- Moore TS. (1982) Phospholipid biosynthesis. Annu Rev Plant Physiol Plant Mol Biol 33: 235–259 [Google Scholar]
- Murata N. (1983) Molecular-species composition of phosphatidylglycerols from chilling-sensitive and chilling-resistant plants. Plant Cell Physiol 24: 81–86 [Google Scholar]
- Murata N, Tasaka Y. (1997) Glycerol-3-phosphate acyltransferase in plants. Biochim Biophys Acta 1348: 10–16 [DOI] [PubMed] [Google Scholar]
- Nakamoto H, Edwards GE. (1987) Effect of adenine-nucleotides on the reaction catalyzed by pyruvate, ortho-phosphate dikinase in maize. Biochim Biophys Acta 924: 360–368 [Google Scholar]
- Nelson DL, Cox MM (2009) Oxidative phosphorylation and photophosphorylation. In K Ahr, ed, Lehninger Principles of Biochemistry, Ed 5. WH Freeman, New York, pp 707–772 [Google Scholar]
- Noctor G, Mhamdi A, Chaouch S, Han Y, Neukermans J, Marquez-Garcia B, Queval G, Foyer CH. (2012) Glutathione in plants: an integrated overview. Plant Cell Environ 35: 454–484 [DOI] [PubMed] [Google Scholar]
- Ohnishi J, Yamada M. (1980) Glycerolipid synthesis in Avena leaves during greening of etiolated seedlings. II. Alpha-linolenic acid synthesis. Plant Cell Physiol 21: 1607–1618 [DOI] [PubMed] [Google Scholar]
- Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, et al. (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457: 551–556 [DOI] [PubMed] [Google Scholar]
- Penna S, Sági L, Swennen R. (2002) Positive selectable marker genes for routine plant transformation. In Vitro Cell Dev Biol Plant 38: 125–128 [Google Scholar]
- Peterhansel C, Krause K, Braun HP, Espie GS, Fernie AR, Hanson DT, Keech O, Maurino VG, Mielewczik M, Sage RF. (2013) Engineering photorespiration: current state and future possibilities. Plant Biol (Stuttg) 15: 754–758 [DOI] [PubMed] [Google Scholar]
- Pilalis E, Chatziioannou A, Thomasset B, Kolisis F. (2011) An in silico compartmentalized metabolic model of Brassica napus enables the systemic study of regulatory aspects of plant central metabolism. Biotechnol Bioeng 108: 1673–1682 [DOI] [PubMed] [Google Scholar]
- Poolman MG, Kundu S, Shaw R, Fell DA. (2013) Responses to light intensity in a genome-scale model of rice metabolism. Plant Physiol 162: 1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolman MG, Miguet L, Sweetlove LJ, Fell DA. (2009) A genome-scale metabolic model of Arabidopsis and some of its properties. Plant Physiol 151: 1570–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahantaniaina MS, Tuzet A, Mhamdi A, Noctor G. (2013) Missing links in understanding redox signaling via thiol/disulfide modulation: how is glutathione oxidized in plants? Front Plant Sci 4: 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramazzina I, Folli C, Secchi A, Berni R, Percudani R. (2006) Completing the uric acid degradation pathway through phylogenetic comparison of whole genomes. Nat Chem Biol 2: 144–148 [DOI] [PubMed] [Google Scholar]
- Raun WR, Johnson GV. (1999) Improving nitrogen use efficiency for cereal production. Agron J 91: 357–363 [Google Scholar]
- Reznik E, Mehta P, Segrè D. (2013) Flux imbalance analysis and the sensitivity of cellular growth to changes in metabolite pools. PLoS Comput Biol 9: e1003195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland N, Curien G, Finazzi G, Kuntz M, Maréchal E, Matringe M, Ravanel S, Seigneurin-Berny D. (2012) The biosynthetic capacities of the plastids and integration between cytoplasmic and chloroplast processes. Annu Rev Genet 46: 233–264 [DOI] [PubMed] [Google Scholar]
- Rosen H. (1957) A modified ninhydrin colorimetric analysis for amino acids. Arch Biochem Biophys 67: 10–15 [DOI] [PubMed] [Google Scholar]
- Sage RF. (2014) Stopping the leaks: new insights into C4 photosynthesis at low light. Plant Cell Environ 37: 1037–1041 [DOI] [PubMed] [Google Scholar]
- Saha R, Suthers PF, Maranas CD. (2011) Zea mays iRS1563: a comprehensive genome-scale metabolic reconstruction of maize metabolism. PLoS ONE 6: e21784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangwan RS, Bourgeois Y, Brown S, Vasseur G, Sangwan-Norreel B. (1992) Characterization of competent cells and early events of Agrobacterium-mediated genetic transformation in Arabidopsis thaliana. Planta 188: 439–456 [DOI] [PubMed] [Google Scholar]
- Schaeffer ML, Harper LC, Gardiner JM, Andorf CM, Campbell DA, Cannon EK, Sen TZ, Lawrence CJ. (2011) MaizeGDB: curation and outreach go hand-in-hand. Database (Oxford) 2011: bar022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger J, Lewis NE, Palsson BO. (2011) Elimination of thermodynamically infeasible loops in steady-state metabolic models (vol 100, pg 544, 2010). Biophys J 100: 1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, et al. (2010) Genome sequence of the palaeopolyploid soybean. Nature 463: 178–183 [DOI] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112–1115 [DOI] [PubMed] [Google Scholar]
- Schnarrenberger C, Martin W. (2002) Evolution of the enzymes of the citric acid cycle and the glyoxylate cycle of higher plants: a case study of endosymbiotic gene transfer. Eur J Biochem 269: 868–883 [DOI] [PubMed] [Google Scholar]
- Schreiber F, Colmsee C, Czauderna T, Grafahrend-Belau E, Hartmann A, Junker A, Junker BH, Klapperstück M, Scholz U, Weise S. (2012) MetaCrop 2.0: managing and exploring information about crop plant metabolism. Nucleic Acids Res 40: D1173–D1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomi T, Cabili MN, Herrgård MJ, Palsson BO, Ruppin E. (2008) Network-based prediction of human tissue-specific metabolism. Nat Biotechnol 26: 1003–1010 [DOI] [PubMed] [Google Scholar]
- Simons M, Saha R, Guillard L, Clément G, Armengaud P, Cañas R, Maranas CD, Lea PJ, Hirel B. (2014) Nitrogen-use efficiency in maize (Zea mays L.): from ‘omics’ studies to metabolic modelling. J Exp Bot 65: 5657–5671 [DOI] [PubMed] [Google Scholar]
- Sowiński P, Szczepanik J, Minchin PEH. (2008) On the mechanism of C4 photosynthesis intermediate exchange between Kranz mesophyll and bundle sheath cells in grasses. J Exp Bot 59: 1137–1147 [DOI] [PubMed] [Google Scholar]
- Stitt M, Heldt HW. (1985) Generation and maintenance of concentration gradients between the mesophyll and bundle sheath in maize leaves. Biochim Biophys Acta 808: 400–414 [Google Scholar]
- Strand A, Zrenner R, Trevanion S, Stitt M, Gustafsson P, Gardeström P. (2000) Decreased expression of two key enzymes in the sucrose biosynthesis pathway, cytosolic fructose-1,6-bisphosphatase and sucrose phosphate synthase, has remarkably different consequences for photosynthetic carbon metabolism in transgenic Arabidopsis thaliana. Plant J 23: 759–770 [DOI] [PubMed] [Google Scholar]
- Sun Q, Zybailov B, Majeran W, Friso G, Olinares PDB, van Wijk KJ. (2009) PPDB, the Plant Proteomics Database at Cornell. Nucleic Acids Res 37: D969–D974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiz L (2010) Plant Physiology, Ed 5. Sinauer Associates, Sunderland, MA [Google Scholar]
- Tanaka R, Tanaka A. (2007) Tetrapyrrole biosynthesis in higher plants. Annu Rev Plant Biol 58: 321–346 [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Nagasaki J, Kawasaki M, Miyake H, Sugiyama T, Taniguchi M. (2004) Differentiation of dicarboxylate transporters in mesophyll and bundle sheath chloroplasts of maize. Plant Cell Physiol 45: 187–200 [DOI] [PubMed] [Google Scholar]
- Tercé-Laforgue T, Mack G, Hirel B. (2004) New insights towards the function of glutamate dehydrogenase revealed during source-sink transition of tobacco (Nicotiana tabacum) plants grown under different nitrogen regimes. Physiol Plant 120: 220–228 [DOI] [PubMed] [Google Scholar]
- Thiele I, Swainston N, Fleming RM, Hoppe A, Sahoo S, Aurich MK, Haraldsdottir H, Mo ML, Rolfsson O, Stobbe MD, et al. (2013) A community-driven global reconstruction of human metabolism. Nat Biotechnol 31: 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, et al. (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313: 1596–1604 [DOI] [PubMed] [Google Scholar]
- Tzin V, Galili G. (2010) The biosynthetic pathways for shikimate and aromatic amino acids in Arabidopsis thaliana. The Arabidopsis Book 8: e0132, /10.1199/tab.0132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Updegraff DM. (1969) Semimicro determination of cellulose in biological materials. Anal Biochem 32: 420–424 [DOI] [PubMed] [Google Scholar]
- Vanholme R, Morreel K, Ralph J, Boerjan W. (2008) Lignin engineering. Curr Opin Plant Biol 11: 278–285 [DOI] [PubMed] [Google Scholar]
- Verwoerd TC, Dekker BMM, Hoekema A. (1989) A small-scale procedure for the rapid isolation of plant RNAs. Nucleic Acids Res 17: 2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitousek PM, Aber JD, Howarth RW, Likens GE, Matson PA, Schindler DW, Schlesinger WH, Tilman DG. (1997) Human alteration of the global nitrogen cycle: sources and consequences. Ecol Appl 7: 737–750 [Google Scholar]
- Vogt T. (2010) Phenylpropanoid biosynthesis. Mol Plant 3: 2–20 [DOI] [PubMed] [Google Scholar]
- Wang Y, Suzek T, Zhang J, Wang J, He S, Cheng T, Shoemaker BA, Gindulyte A, Bryant SH. (2014) PubChem BioAssay: 2014 update. Nucleic Acids Res 42: D1075–D1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner H, Heldt HW. (1992) Inter- and intracellular distribution of amino acids and other metabolites in maize (Zea mays L.) leaves. Planta 187: 242–246 [DOI] [PubMed] [Google Scholar]
- Xiong Y, DeFraia C, Williams D, Zhang X, Mou Z. (2009) Deficiency in a cytosolic ribose-5-phosphate isomerase causes chloroplast dysfunction, late flowering and premature cell death in Arabidopsis. Physiol Plant 137: 249–263 [DOI] [PubMed] [Google Scholar]
- Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, et al. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296: 79–92 [DOI] [PubMed] [Google Scholar]
- Zhao Q, Chen S, Dai S. (2013) C4 photosynthetic machinery: insights from maize chloroplast proteomics. Front Plant Sci 4: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomorrodi AR, Islam MM, Maranas CD. (2014) d-OptCom: dynamic multi-level and multi-objective metabolic modeling of microbial communities. ACS Synth Biol 3: 247–257 [DOI] [PubMed] [Google Scholar]
- Zomorrodi AR, Maranas CD. (2012) OptCom: a multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput Biol 8: e1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zrenner R, Stitt M, Sonnewald U, Boldt R. (2006) Pyrimidine and purine biosynthesis and degradation in plants. Annu Rev Plant Biol 57: 805–836 [DOI] [PubMed] [Google Scholar]
- Zybailov B, Rutschow H, Friso G, Rudella A, Emanuelsson O, Sun Q, van Wijk KJ. (2008) Sorting signals, N-terminal modifications and abundance of the chloroplast proteome. PLoS ONE 3: e1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
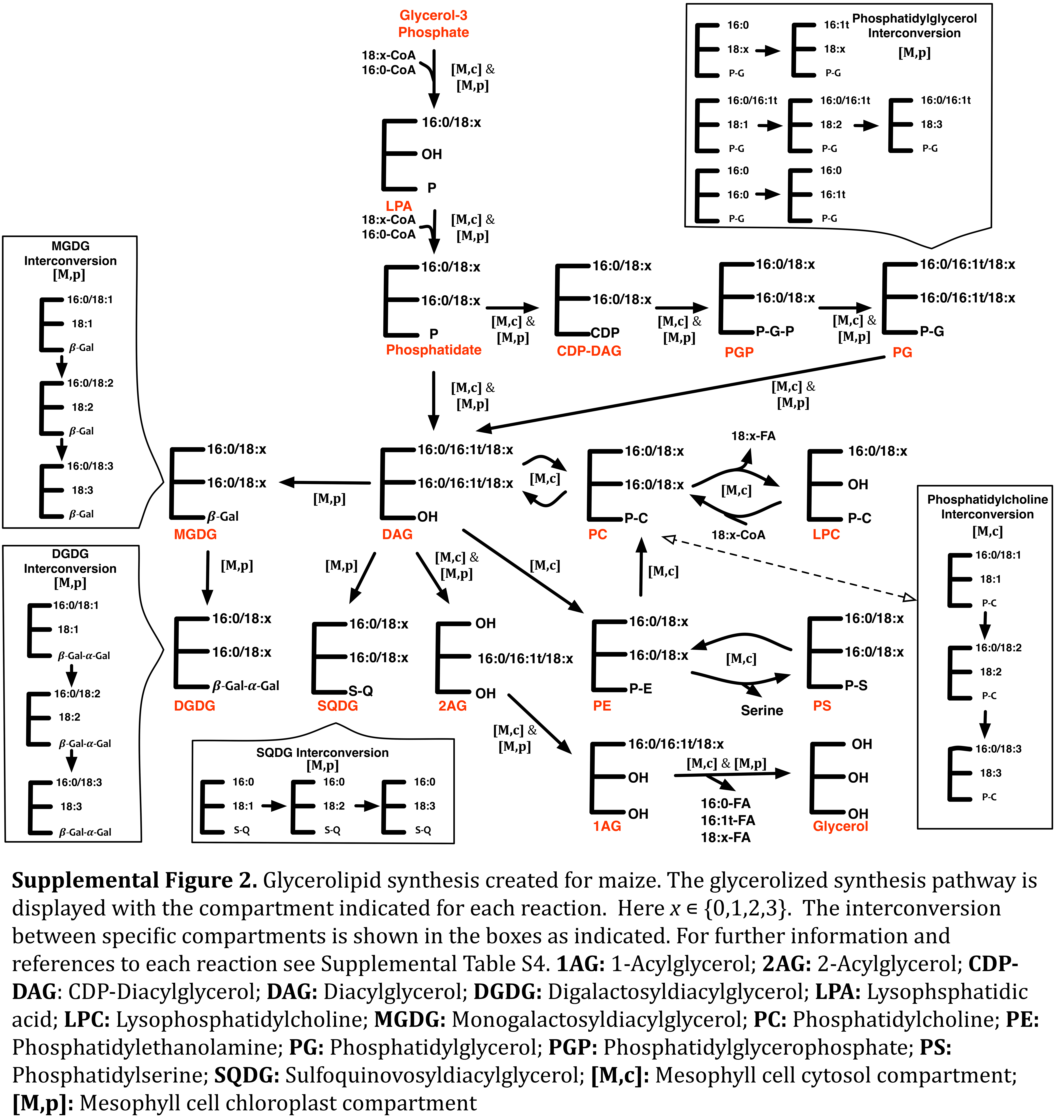{kind=link}