

Abstract
It is hypothesised that Wilms tumour (WT) results from aberrant renal development due to its embryonic morphology, associated undifferentiated precursor lesions (termed nephrogenic rests) and embryonic kidney-like chromatin and gene expression profiles. From the study of overgrowth syndrome-associated WT, germline dysregulation was identified in the imprinted region at 11p15 affecting imprinted genes IGF2 and H19. This is also detected in ~70% sporadic cases, making this the most common somatic molecular aberration in WT. This review summarises the critical discussion at an international workshop held under the auspices of The European Network for Cancer Research in Children and Adolescents (ENCCA) consortium, where the potential for drug development to target IGF2 and the WT epigenome was debated. Here, we consider current cancer treatments which include targeting the IGF pathway and the use of methylation agents alone or in combination with other drugs in clinical trials of paediatric cancers. Finally, we discuss the possibility of the use of these drugs to treat patients with WT.
Keywords: IGF2, IGF signalling pathway, Wilms tumour, targeted therapy, DNA methylation
INTRODUCTION
Wilms tumour (WT) or nephroblastoma is the most frequent renal tumor of childhood affecting one in 10,000 children, with a peak incidence between two and three years of age [1]. With an overall cure rate ≥85%, WT is one of the successes of pediatric oncology. In the Western world, the majority of patients affected by WT are treated within trials conducted by the International Society of Pediatric Oncology (SIOP, Europe) or the Children's Oncology Group (COG, formerly NWTSG, North America) [2]. WTs are frequently large and exhibit radiological, macroscopic and histological heterogeneity. At present, there is evidence of at least three pathways that are associated with WT development: the WNT/β-catenin pathway (15-20% of non-anaplastic WTs), the IGF2 pathway (69% of all WTs) and the p53 pathway (anaplastic WTs, 5-10% of all WTs). At least in part, these may be responsible for the heterogeneity in clinical phenotype and outcome observed in WTs.
Under the auspices of the European Network for Cancer Research in Children and Adolescents (ENCCA) consortium, a European Union Framework 7-funded program (http://www.encca.eu/Pages/home.aspx) a ‘Biology Driven Drug Development Renal Tumors Workshop’ was convened to review the evidence for the genetic pathways disrupted in WT and to develop a translational research strategy. This workshop was part of a wider strategic initiative to involve international experts in the biology of childhood cancers, drug development and early phase clinical trials in children and relevant adult diseases, to accelerate biology-driven drug development and clinical trials across Europe. The outcome of this workshop is being published as two focused reviews. The first discussed the significance of the canonical WNT signaling pathway in a subset of WTs and related therapeutic opportunities [3]. Here, we discuss the subset of WTs with epigenetic aberrations at 11p15 resulting in activation of the IGF signaling pathway and the therapeutic opportunities provided by targeting either the IGF pathway or the epigenome. The IGF pathway is comprised of a complex network of molecules stimulated by insulin-like growth factors (IGFs), which are synthesized by almost any tissue in the body and are important mediators of growth, development, and survival including the embryonic kidney [4]. There are an increasing number of studies highlighting their role in establishing a transformed phenotype in an increasing number of malignancies.
Wilms tumours arise from aberrant renal development and show epigenetic features of embryonic kidney
WT is an embryonic tumour that morphologically resembles foetal kidney with varying proportions of blastemal, epithelial and stromal cells. Immature tubular structures are observed and in some cases skeletal, fat and muscle tissue suggesting a multipotent cell of origin [5]. Concurrent with its embryonal morphology, gene expression profiling has established that WTs express genes corresponding to the earliest stage of metanephric development and under-express genes from later stages of renal development [6]. Furthermore, the cellular composition of WT shows retention of expression patterns reflecting embryonic counterparts [7] Evidence has shown that depending on the location of expression of key renal development gene CITED1, WT cell lines obtain either stem-like properties (cytoplasmic expression, as seen in normal development) or tumourigenic properties (nuclear expression, as seen in WT) [8].
More recent studies have analysed the WT epigenome, which encompasses features that effect gene expression without altering the genetic coding sequence, and found similar results. Epigenetic mechanisms can be broadly classified into chromatin remodelling (by covalent histone modification or protein binding) and DNA cytosine methylation. In detail, researchers identified regions of open chromatin that permit active gene expression (with histone 3 lysine 4 trimethylation [H3K4me3]) and regions of closed chromatin that prevent gene expression (with H3K27me3) in WT, embryonic kidney (EK), embryonic stem cells (ESCs) and normal kidney (NK).
Comparisons between tissues identified open chromatin structures in WT but not NK at genes involved in kidney and mesoderm development which are also highly expressed in foetal kidney. In addition, identification of a network of genes that showed the same “bivalent” chromatin structure (both active and repressive marks simultaneously) and low expression in both WT and ESCs, but were actively expressed in NK, led to the conclusion that developmental arrest occurs in undifferentiated metanephric mesenchyme as the genes required for differentiation (FOXD1 and LHX1 which are involved in stromal and epithelial differentiation, respectively) remained in a poised state awaiting differentiation signals [9]. This evidence suggests that retaining epigenetic features of early renal development is important in the early stages of disease.
Supporting this theory, aberrant epigenetic events have been considered as the earliest events in tumourigenesis whereby epigenetic disruption results in a pool of tumour-progenitor cells. Within these cells, gene-specific epimutations can occur resulting in cellular transformation [10, 11]. Tumours later acquire both epigenetic and genetic plasticity that is proposed to lead to tumour heterogeneity [12]. Therefore, whilst during normal development, epigenetic modifications are remodelled to define embryo patterning and for organ and cell type specification and then upon terminal differentiation is maintained to sustain cell identity, when disrupted (during development or somatically), the epigenome may play a role in cancer initiation and progression, giving the same effect as a “classical” DNA mutation.
Epigenetics of Wilms tumour
Aside from the developmental epigenetic features observed in WT, further aberrant epigenetic events have been observed that are analogous to this “classical” DNA mutation (summarised in Table 1). These occur by aberrant site-specific or global changes in DNA CpG methylation or chromatin structure. In detail, CpG sites are regions of DNA where a cytosine is located next to a guanine nucleotide. In general, gain of DNA methylation at CpG residues can result from the overexpression of DNA (cytosine-5)-methyltransferase 1 (DNMT1) and DNA (cytosine-5)-methyltransferase 3 beta (DNMT3b), which are regulated by TP53 [13, 14]. As well as increase in DNA methylation, trimethylation of histone 3 (H3) at lysine (K) 27 (H3K27me3) also causes gene repression by promoting a closed chromatin structure. Alternatively, loss of DNA methylation, trimethylation of H3K4 or K36, monomethylation of H3K4 or acetylation of H3K36 promote an open chromatin structure, and the binding of transcription factors [9, 15-19]. In cancer, these changes in DNA methylation and chromatin accessibility are associated with the silencing or the overexpression of tumour suppressor genes and oncogenes, respectively (reviewed in [20]).
Table 1. Epigenetics alterations found in Wilms tumours.
| Aberration | Finding within Wilms tumour |
|---|---|
| CASP8 hypermethylation | Frequency of 19% and associated with RASSF1A methylation [88] |
| GLIPR1/RTVP hypomethylation | Frequency of 87.5%, results in overexpression [89] |
| Global hypomethylation | Results in genome instability in tumour cells [90, 91]. |
| Gain of methylation at 6p22.1, 6p21.32 and 11q13.5 | Biomarker for WT, can be detected in the circulation of patients [92] |
| HACE1 hypermethylation | Frequency of 73% [93] |
| Hypermethylation of a CTCF binding site downstream of WT1 | Correlated with high WT1 expression [94] |
| Hypermethylation of protocadherin cluster at 5q31 | Results in expression loss of these proteins at the cell surface [95] |
| LOI 11p15 | Frequency of 69%, results in overexpression of IGF2 and down-regulation of H19 [29, 36, 96] |
| P16 hypermethylation | Frequency of 23% [97] |
| RASSF1 hypermethylation | Frequency of 54% [98] |
| WT1-antisense transcript hypomethylation | Results in biallelic expression [99] |
Of the known epimutations in WT, epigenetic aberration at 11p15 has received the most attention due to its association with Beckwith-Wiedemann Syndrome (BWS), a paediatric overgrowth disorder with germline gain of methylation at 11p15 and functional relationship with expression of imprinted genes IGF2 and H19 [21, 22]. There are over 40 human imprinted genes that show parental allele-specific expression [23]. This monoallelic expression tightly controls the levels of the proteins encoded by imprinted genes, usually important factors of embryonic growth, placental growth or adult metabolism [24]. The regulation of imprinted genes is largely dependent on DNA methylation marks, which are laid down during embryological development of germ cells. Once in place, the methylation status of precise chromosomal regions, imprinting control regions (ICRs), is read by either of two mechanisms, chromatin barrier formation or untranslated RNAs, thereby ensuring that only the maternal or paternal allele is expressed [25, 26]. Each imprinted gene is classified as maternal or paternal according to the expressed allele. Misregulation of imprinted gene expression (loss of imprinting [LOI]) is seen frequently in a large variety of human tumours [27]. Specifically, LOI of IGF2 and H19 is seen in ~69% WT either by gain of methylation at the H19-ICR (37%) or by paternal UPD (32%) [28, 29]. Around 10-20% WT patients have constitutional LOI at this locus [30, 31].
Evidence that the IGF pathway is disrupted in Wilms tumour
The H19-ICR (which regulates expression of paternally imprinted IGF2 and maternally imprinted H19) contains differentially methylated domains (DMD) and is located between IGF2 and H19 [32]. The ICR comprises CTCF (CCCTC-binding factor, zinc finger protein) binding sites and acts by regulating interactions between both gene promoters and their shared enhancers downstream of H19 [33]. CTCF protects the maternal H19-ICR from de novo methylation in normal tissue [34]; however aberrant gain of methylation at this allele results in silencing of H19 expression and transcription of IGF2 replicating the paternal allele.
Clinically, WT with LOI at 11p15 are associated with perilobar nephrogenic rests (PLNRs), lesions of retained embryonic renal tissue found towards the periphery of the renal lobe, and with blastemal or epithelial-predominant WTs [35, 36] and show increased expression of IGF2. Increased expression of IGF2 results in activation of the insulin signalling pathway. Binding of insulin, IGF1 or IGF2 to the extracellular portion of the insulin receptor (IR), IGF1R or hybrid receptor leads to autophosphorylation of the β-subunit tyrosine kinase, followed by the phosphorylation of additional tyrosine residues. This leads to recruitment of insulin receptor substrates (IRS)1 to IRS4 and other proteins, allowing activation of the PI3K and mitogen activated protein kinase (MAPK) signalling pathways which lead to unregulated protein synthesis, cell cycle progression and cell growth, and prevention of apoptosis [37]. Therefore, in WT, LOI is driving IGF2 overexpression and oncogenic pathway activation in the cell.
Although constitutional 11p15 abnormalities are found in a small proportion (3%) of patients with sporadic WT (31)and in those with asymmetric overgrowth, including BWS patients, not all develop WT, adding evidence that a second aberrant event is required. Supporting this hypothesis, a strong association between pUPD at 11p15 and WT1 mutation as well as both mechanisms of LOI at 11p15 and WTX mutation are found [29, 30, 36]. Other genes that are commonly mutated in around 30% of sporadic WT cases include WTX, CTNNB1, HACE1, LIN28A, DROSHA and FBXW7 although the total number of known mutated genes in WT is limited [38-43].
Further supporting IGF pathway disruption in WT, low level copy number increase of IGF1R resulting in aberrant mRNA and protein levels was found in WT, particularly in those with blastemal predominance [44]. Subsequently a mouse model was developed whereby upregulation of Igf2 was achieved through IGF1R signalling transduced via pIRS1 and pERK1/2. This upregulation alone was insufficient to cause WT, however in combination with WT1 mutation (present in around 15% WT), 64% of engineered mice developed WT [45]. The resulting WT is probably due to the alteration in normal differentiation of the induced nephrogenic mesenchyme caused by WT1 mutation together with increased IGF signalling driving proliferation of these cells. This showed that although 11p15 defects are one of the most common aberrations in WT, additional events are necessary for WT development.
Relating this epigenetic aberration to patient treatment, in chemotherapy-naïve tumours, the stage I favourable histology WT weighing less than 550g in children younger than 24 months of age treated with surgery alone showed a significant association between LOH at 11p15, WT1 mutation and relapse [46]. This finding is being tested in the current COG clinical trial (AREN0532) and if validated, 11p15 methylation analysis may be used to select patients with stage I favourable histology WT who do not require adjuvant therapy [47]. In chemotherapy treated tumours, the relationship between 11p15 disruption and patient outcome remains to be addressed.
Potential for IGF2 pathway inhibition
Having establishing that the majority of WT, independent of tumour histology [29, 36], are dependent on the IGF signalling pathway it follows that treatment strategies aimed at its inhibition may be useful in the clinic. Currently there are no clinically available drugs which target IGF2 directly although two small molecules are currently under development, m-Cresol and Myristic acid.
Instead, the IGF2 receptor, IGF1R, seems to be the most viable therapeutic target due to its redundancy in normal growth and its role in tumorigenesis and growth in cancer [48]. Approaches to targeting IGF1R activity include impeding expression, blocking IGF1R interaction with its ligands or preventing receptor activation (Figure 1). There is also novel evidence suggesting that reduction in dietary protein intake may inhibit tumour growth possibly through the inhibition of the IGF/AKT/mTOR pathway [49].
Figure 1. A summary of agents that target either the IGF pathway or the epigenome.
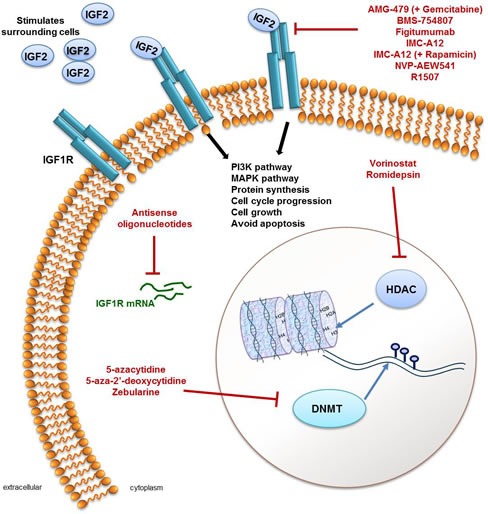
IGF signalling occurs through stimulation of insulin-like growth factor 1 receptors (IGF1R; shown in blue in the cell membrane) by IGF2 and results in activation of the PI3K and MAPK signalling pathways, an increase in protein synthesis, cell cycle progression, cell growth and avoiding apoptosis. Activation of the IGF pathway through overexpression of IGF2 is frequently seen in Wilms tumour (WT) and is therefore a viable therapeutic target. IGF pathway inhibitors include antisense oligonucleotides which target IGF1R mRNA (green) to prevent their translation into functional protein or molecules that target the IGF1R, which include AMG-479 in combination with Gemcitabine, BMS-754807, Figitumumab, IMC-A12 (alone or with Gemcitabine), NVP-AEW541 and R1507. As frequent epigenomic aberrations are also found in WT (see main text and Table 1) targeting enzymes that regulate DNA methylation (DNA methyal transferases; DNMT) or that regulate histone modifications (histone deacetylases; HDAC) may be a viable therapy for patients with WT. In terms of targeting the epigenome, 5-azacytidine, 5-aza-2'-deoxycytidine and Zebularine are DNMT inhibitors and Vorinostat and Romidepsin are both HDAC inhibitors, preventing normal enzymatic activity.
Impeding expression involves targeting IGF1R mRNA using antisense oligonucleotides complementary to the translational start site. Expression of such oligonucleotides in glioblastoma cell lines inhibited IGF1 mediated growth in culture [50] and showed a pronounced growth-blocking effect in murine and human cancer cell lines [51, 52]. In Ewing's sarcoma cell lines, after oligonucleotide delivery by plasmid transfection, an increased response due to an increased sensitivity to Doxorubicin was observed, suggesting a potential use in combination therapy [53].
An alternative approach aims to block the interaction between IGF1R and its ligands either with small molecule inhibitors or using monoclonal antibodies. There are several fully humanised anti-IGF1R monoclonal antibodies and small molecule inhibitors of IGF1R in various stages of clinical development, including in the Pediatric Preclinical Testing Program (PPTP). The IGF1R inhibitor BMS-754807 is an ATP-competitive small molecule that was tested in vivo using xenograft models of several paediatric tumours (including WT) and demonstrated significant growth delay [54]. The anti-IGF1R monoclonal antibody IMC-A12 was also used to treat the same WT models but showed very limited efficacy as a single agent [55]. The data is insufficient to draw firm conclusions as neither study evaluated IGF1R network status in the cells. In WT cell lines growing in the kidney environment, the use of the IGF1R inhibitor NVP-AEW541 resulted in growth inhibition associated with down-regulation of PI3K and MAPK pathways and down-regulation of cell cycle control genes CCNA2 and CCNB1, with drug efficacy dependent on the levels of phosphorylated IGF1R [56]. Two anti-IGF1R antibodies, Figitumumab (CP-751,871) and R1507, have shown success in patients with Ewing's sarcoma and thymoma but have not yet been tested in WT patients or animal models [57, 58]. As well as its role in the IGF pathway as a tyrosine kinase, evidence shows that IGF1R also has kinase-independent activity suggesting that combination targeting using both antibodies that target the receptor as well as small molecule inhibitors may be more effective [59].
The lack of response in some IGF2-overexpressing WT may be due to the cancer cells overcoming their dependence on IGF signalling, or that the IGF pathway is not the sole dominant driver, thus suggesting that combination therapies may be more effective. In fact, in vivo studies of IGF1R antibody AMG-479 with gemcitabine showed additive inhibitory activity in pancreatic carcinoma [60]. Combined inhibition of the mTOR and IGF1R signalling pathways is an attractive and biologically rational approach to development of anticancer therapy for sarcomas and other solid tumours. Separately, both pathways appear to be relevant in multiple forms of cancer, and preclinical models suggest synergistic antitumor activity with combined inhibition of these pathways. The PPTP has demonstrated that the combination of IGF1R inhibitor IMC-A12 and mTORC1 signalling inhibitor rapamicin have a superior tumour response compared to either agent alone in several models of paediatric cancer, however testing has not been carried out in the WT model [61]. In human WTs, overexpression of IGF2, through allele loss of 11p15.5 or loss of imprinting, frequently co-exists with WT1 mutation. The same combination of critical genetic events proved essential for tumour formation in a genetically engineered mouse model [45]. ERK1/2 phosphorylation was upregulated in WTs in both species, suggesting a further target for therapy.
Unfortunately, some drugs that target the IGF pathway, when tested in clinical trials have not given encouraging results. Amgen (Amgen Inc. CA, USA) recently terminated ganitumab (AMC-479 [62]), an antibody against IGF1R, for a late-stage pancreatic cancer trial [63]. Earlier in 2010, Pfizer abandoned figitumumab after trials failed to show clinical efficacy in adult cancers (lung carcinoma and myeloma). Several other drug manufacturers have discontinued their own earlier-stage programs directed at the same target. However, Merck are still developing an IGF1R inhibitor dalotuzumab, with an emphasis on the identification of predictive biomarkers for subgroups that benefit most from this drug and Eli Lilly has an IGF1R-targeted antibody, cixutumumab, in phase 2 trials. COG recently carried out a randomised phase 2 study testing cixutumumab in adolescents and children with refractory solid tumours including ten patients with WT. The drug was well tolerated in patients and prolonged stable disease was seen in 15% patients [64]. Although no response in patients with refractory WT was observed, this was a single-agent trial and drugs that only target IGF1R may be non-efficacious as they fail to target the insulin receptor, another tyrosine kinase receptor that is activated by IGF1 and IGF2 in addition to insulin itself. Following this idea, Astellas Pharma developed a drug, linsitinib which is undergoing phase 2 trials for several tumour types, that targets both receptors. The observed poor response may be due to the complexity of the IGF1R/insulin receptor system and parallel growth and survival pathways. However, IGF1R does remain a valid target for selected tumour types and agents targeting IGF1R are attractive therapeutic targets for WT.
An alternative approach to overcome the potential side effects of inhibiting the whole IGF pathway is the inhibition of specific arms of the pathway important for tumour development. The monoclonal antibody mAb 1/41 targets the metalloproteinase, pregnancy-associated plasma protein-A (PAPP-A) resulting in indirect inhibition of IGF signalling [65]. Tumours with increased PAPP-A activity will benefit from the use of mAb 1/41 with the advantage of local and indirect targeting of the IGF-IR, rather than global targeting which lacks specificity and is likely to interfere with metabolism [66], however there is no information in the literature about PAPP-A activity in WT.
Therapeutic options based on the WT epigenome
It is now clear that cancer development depends not only on genetic alterations but also on epigenetic changes that convey heritable gene expression patterns critical for neoplastic initiation and progression [67]. However, unlike genetic mutations which are permanent, the discovery that these epigenetic marks can be reversed by compounds targeting aberrant transcription factor/co-activator/co-repressor interactions and histone-modifying activities, suggests the possibility that the epigenome of cancer cells may be manipulated with potential therapeutic benefits [68]. Given the proven dysregulation of epigenetic architecture in WT and the pattern, interactions and clinical associations of molecular events in WT there is clear potential for targeting the epigenome to treat WT. A significant and growing number of pharmaceutical companies have research programs that aim to define proteins that target the epigenome. Drugs currently FDA approved or in the pipeline for approval for this purpose include the DNA methyltransferase inhibitors 5-azacytidine (Vidaza and AZA), 5-aza-2′-deoxycytidine (Decitabine, Dacogen and DAC), as well as two histone remodelling inhibitors: suberanilohydroxamic acid (Vorinostat, Zolinza) and depsipeptide (Romidepsin, Istodax) [69], shown in Figure 1. More epigenetic drugs are entering into pre-clinical development every year [70].
Histone deacetylases (HDACs) are critical in the control of gene expression. Molecules that interfere with histone remodelling, for example histone deacetylase inhibitors (HDACi) are an emerging class of therapeutics with potential as anticancer drugs with promising results in clinical trials [71]. To date, two HDACi have received FDA approval for the treatment of cutaneous T-cell lymphoma: Vorinostat and Romidepsin. Clinical trials assessing the effects of various HDACi on haematological and solid malignancies are currently being conducted [72]. Despite the proven anticancer effects of particular HDACi, a detailed understanding of how HDACi with different pharmacological properties affect biological functions in vitro and in vivo is still missing [73]. Therefore while drugs that target histone remodelling proteins exist and have been tested in both haematological and solid malignancies [74], the potential efficacy of such inhibitors in WT is unknown and unpredictable as no studies have revealed a particular dependence on HDACs in WTs. In addition, the potential downside of drugs that target global chromatin modifications is their broad effect on fundamental cellular mechanisms. They can cause multiple side effects - anorexia, nausea, vomiting, suppression of white blood cells and thrombocytopenia, and profound fatigue [75].
DNA methyl transferase (DNMT) 1, 3a and 3b are enzymes that transfer a methyl group to cytosine residues in DNA. DNMT1 maintains methylation during DNA synthesis whereas 3a and 3b act during embryonic development [76, 77]. A number of different types of DNMT inhibitors are known including non-nucleoside analogues which are antisense oligonucleotides targeting the DNMT family of enzymes, resulting in DNA demethylation. One such inhibitor underwent phase I and phase II trials for treatment of renal cancer but failed due to a lack of efficacy [78].
The more common inhibitors for DNMT are nucleoside analogues that become phosphorylated and are incorporated into DNA during replication also resulting in global DNA demethylation. The DNMT inhibitors Vidaza and Decitabine are the most successful epigenetic drugs to date and are still the most widely used as epigenetic modulators, even though their application for oncological diseases is restricted by their relative toxicity and poor chemical stability. Decitabine was approved for the treatment of myelodysplastic syndrome (MDS) in 2006 and shows anti-leukemic activity against acute myeloid leukemia (AML). Its clinical activity against solid tumours is under investigation, but initial studies in patients with solid tumours have demonstrated only limited responses. However, as there is a close correlation between the concentration of Decitabine, exposure time, demethylating effects and thus anti-tumour activity, these patients potentially received a suboptimal exposure to the drug compared to that expected from in vitro and in vivo animal studies [79]. Zebularine (1-(β-D-ribofuranosyl)-1,2-dihydropyrimidin-2-one), a more stable and less toxic cytidine analog, is another DNMT inhibitor which also shows inhibitory activity towards cytidine deaminase [80]. Zebularine preferentially targets cancer cells and exhibits low toxicity toward normal cells and mice [81, 82]. Zebularine induces apoptosis and decreases clonogenic capacity of acute lymphoblastic leukemia cell lines in a dose-dependent manner [83]. Although it is orally available and more stable than FDA-approved Vidaza and Decitabine, its clinical benefit is yet to be evaluated.
As WT shows a triphasic morphology, demethylating agents may show different activity in each cell type however, as no evidence currently exists as to whether one cell type may respond better, this would also need to be assessed.
Finally, enhanced expression of EZH2 in WT, a key polycomb group protein that functions to repress transcription through critical histone 3 methylation markers, has been identified to correlate with WT progression [84]. Agents targeting EZH2 such as GSK126 have entered pre-clinical and clinical development [85, 86].
CONCLUSIONS AND CHALLENGES
Here, we have discussed a wide range of agents that target the IGF signalling pathway or the epigenome (summarised in Table 2). It is clear that epigenetic alterations play a key role in a significant fraction of WTs and result in LOI at H19/IGF2 [33]. This epimutation has also been observed in the associated nephrogenic rests (precursor lesions) and surrounding normal kidney of patients with WT. Furthermore, other epigenetic aberrations have been found within the tumour tissue and it appears that the founding event in a large subset of WTs is the failure to complete nephrogenesis, thus leaving lesions with an epigenetically unstable composition.
Table 2. A summary of the agents discussed that target the IGF pathway or epigenome.
| Mechanism | Compound | Type | Status | Tumour (patients enrolled) |
| IGF2 inhibitor | m-Cresol | small molecule | developed | not tested in vivo |
| Myristic acid | small molecule | developed | not tested in vivo | |
| IGF1R inhibitor | BMS-754807 | ATP-competitive small molecule | developed | xenograft models |
| IMC-A12 | monoclonal antibody | phase III | hepatocellular carcinoma (n=24) | |
| NVP-AEW541 | small molecule inhibitor | pre-clinical | Ewing sarcoma and neuroblastoma | |
| Figitumumab (CP-751,871) | monoclonal antibody | phases II/III | Ewing's sarcoma (n=138) | |
| R1507 | monoclonal antibody | phases I/II | Ewing's sarcoma and thymoma (n=29) | |
| AMG-479 | monoclonal antibody | phases III | pancreatic carcinoma (n=800) | |
| gemcitabine | monoclonal antibody | phases III | pancreatic carcinoma (n=800) | |
| ganitumab | monoclonal antibody | phases III | pancreatic carcinoma (n=800) | |
| figitumumab | monoclonal antibody | phases I | myeloma (n=47) | |
| dalotuzumab | monoclonal antibody | phase I | paediatric solid tumours (n=21) | |
| cixutumumab | monoclonal antibody | phases II | refractory solid tumours (n=10 patients with WT) | |
| linsitinib | small molecule inhibitor | phases II/III | non-small Cell Lung Cancer (n=205), ovarian cancer (n=79), adrenocortical carcinoma (n=139) | |
| DNA methyltransferase inhibitors | 5-azacytidine | analogue of cytidine | FDA approved for use | myelodysplastic syndrome, acute myeloid leukemia |
| 5-aza-2′-deoxycytidine | analogue of cytidine | FDA approved for use | myelodysplastic syndrome, acute myeloid leukemia | |
| MG98 | anti-sense nucleotides | phase I and II, terminated | metastatic renal cancer (n=17) | |
| Zebularine | analog of cytidine | developed | acute lymphoblastic leukemia | |
| GSK126 | small-molecule inhibitor of EZH2 methyltransferase activity | pre-clinical | EZH2 mutated B-cell lymphoma and follicular lymphoma | |
| histone remodelling inhibitors | suberanilohydroxamic acid | histone deacetylase inhibitors | FDA approved for use | cutaneous T-cell lymphoma |
| depsipeptide | amino acid-containing small molecule or chain | FDA approved for use | cutaneous T-cell lymphoma | |
| Romidepsin | histone deacetylase inhibitors | FDA approved for use | cutaneous T-cell lymphoma |
Considering this evidence, WT seems at present to be an ideal model for epigenetic therapy. While global demethylators can be assessed for use as adjuvant therapy, most of the epigenetic drugs under development require effective cell proliferation, suggesting that they may only be effective in a subset of tumours.
We have discussed the use of targeting IGF signalling in WT as a possible therapeutic strategy. Most importantly, the assessment of IGF signalling disruption in clinical trials may help to identify the patient subgroup most likely to benefit from treatment with pathway inhibitors. Moreover, the COG study AREN0532, which is currently analysing the relationship between 11p15 methylation in very low risk WT and patient outcome, may support previous evidence that patients with 11p15 LOH are more likely to relapse which would aid future patient stratification and treatment decision planning. 11p15 disruption and its association with outcome remains to be addressed in pre-treated tumours.
At present, targeting IGF signalling has been tested on WT cell lines and xenograft models which showed significant growth delay. Given IGF2 overexpression in WT, targeting IGF1R seems plausible. However, it must be considered that tumours can overcome IGF pathway dependence and therefore combination therapy might be a more effective option, though the effective pathways to be co-inhibited have not yet been defined. Furthermore, the use of new epigenetic therapies needs to be established before we can consider assessing efficacy in clinical trials. Should this therapeutic approach be considered in the future, the status of 11p15 imprinting would have to be assessed at diagnosis to determine whether IGF-targeted therapy would be beneficial for a patient, perhaps in the same way that assessing ERBB2 status impacted on breast cancer treatment. This can be done by methylation-specific MLPA (MS-MLPA), a simple method based on digestion of genomic DNA-probe hybrid complexes combined with methylation-sensitive endonucleases. As a quantitative method, MS-MLPA discriminates between methylation of one, both or none of the alleles making it a powerful screening tool [87]. Further clinical trials assessing IGF1R inhibitors in vivo as well as mRNA therapy blocking IGF1R expression are potential avenues for further consideration.
Acknowledgments
The authors thank Professor Riccardo Riccardi, Director of Pediatrics, Universita’ Cattolica, Rome, for hosting the workshop. This work has received funding from the European Union's Seventh Framework Programme under the European Network for Cancer Research in Children and Adolescents project (FP7-HEALTH-F2-2011 Grant no. 261474). K Pritchard-Jones is supported by program grant funding from Cancer Research UK (C1188/A11859), ENCCA (FP7-HEALTH-F2-2011 Grant no. 261474) and by the National Institute for Health Research Great Ormond Street Hospital UCL Biomedical Research Centre award and Great Ormond Street Hospital for Children's Charitable funds. Daniela Perotti and Paolo Radice are sponsored by Associazione Bianca Garavaglia, Busto Arsizio, Varese, Italy, and the Italian Association for Cancer Research (AIRC). The other authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
REFERENCES
- 1.Breslow N, Olshan A, Beckwith JB, Green DM. Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993;21(3):172–181. doi: 10.1002/mpo.2950210305. [DOI] [PubMed] [Google Scholar]
- 2.Pritchard-Jones K, Pieters R, Reaman GH, Hjorth L, Downie P, Calaminus G, Naafs-Wilstra MC, Steliarova-Foucher E. Sustaining innovation and improvement in the treatment of childhood cancer: lessons from high-income countries. Lancet Oncol. 2013;14(3):e95–e103. doi: 10.1016/S1470-2045(13)70010-X. [DOI] [PubMed] [Google Scholar]
- 3.Perotti D, Hohenstein P, Bongarzone I, Maschietto M, Weeks M, Radice P, Pritchard-Jones K. Is Wilms tumor a candidate neoplasia for treatment with WNT/β-catenin pathway modulators?--A report from the renal tumors biology-driven drug development workshop. Mol Cancer Ther. 2013;12(12):2619–2627. doi: 10.1158/1535-7163.MCT-13-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott J, Cowell J, Robertson ME, Priestley LM, Wadey R, Hopkins B, Pritchard J, Bell GI, Rall LB, Graham CF, et al. Insulin-like growth factor-II gene expression in Wilms’ tumour and embryonic tissues. Nature. 1985;317(6034):260–262. doi: 10.1038/317260a0. [DOI] [PubMed] [Google Scholar]
- 5.Beckwith JB, Kiviat NB, Bonadio JF. Nephrogenic Rests, Nephroblastomatosis, and the Pathogenesis of Wilms’ Tumor. Fetal & Pediatric Pathology. 1990;10(1-2):1–36. doi: 10.3109/15513819009067094. [DOI] [PubMed] [Google Scholar]
- 6.Li CM, Guo M, Borczuk A, Powell CA, Wei M, Thaker HM, Friedman R, Klein U, Tycko B. Gene expression in Wilms’ tumor mimics the earliest committed stage in the metanephric mesenchymal-epithelial transition. Am J Pathol. 2002;160(6):2181–2190. doi: 10.1016/S0002-9440(10)61166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maschietto M, de Camargo B, Brentani H, Grundy P, Sredni ST, Torres C, Mota LD, Cunha IW, Patrao DF, Costa CM, Soares FA, Brentani RR, Carraro DM. Molecular profiling of isolated histological components of wilms tumor implicates a common role for the Wnt signaling pathway in kidney and tumor development. Oncology. 2008;75(1-2):81–91. doi: 10.1159/000155210. [DOI] [PubMed] [Google Scholar]
- 8.Murphy AJ, Pierce J, de Caestecker C, Ayers GD, Zhao A, Krebs JR, Saito-Diaz VK, Lee E, Perantoni AO, de Caestecker MP, Lovvorn HN., 3rd CITED1 confers stemness to Wilms tumor and enhances tumorigenic responses when enriched in the nucleus. Oncotarget. 2014;5(2):386–402. doi: 10.18632/oncotarget.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aiden AP, Rivera MN, Rheinbay E, Ku M, Coffman EJ, Truong TT, Vargas SO, Lander ES, Haber DA, Bernstein BE. Wilms tumor chromatin profiles highlight stem cell properties and a renal developmental network. Cell Stem Cell. 2010;6(6):591–602. doi: 10.1016/j.stem.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7(1):21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 11.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 13.Park IY, Sohn BH, Choo JH, Joe CO, Seong JK, Lee YI, Chung JH. Deregulation of DNA methyltransferases and loss of parental methylation at the insulin-like growth factor II (Igf2)/H19 loci in p53 knockout mice prior to tumor development. Journal of cellular biochemistry. 2005;94(3):585–596. doi: 10.1002/jcb.20263. [DOI] [PubMed] [Google Scholar]
- 14.Biniszkiewicz D, Gribnau J, Ramsahoye B, Gaudet F, Eggan K, Humpherys D, Mastrangelo MA, Jun Z, Walter J, Jaenisch R. Dnmt1 overexpression causes genomic hypermethylation, loss of imprinting, and embryonic lethality. Mol Cell Biol. 2002;22(7):2124–2135. doi: 10.1128/MCB.22.7.2124-2135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences. 2010 doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, Lee W, Mendenhall E, O/'Donovan A, Presser A, Russ C, Xie X, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. Whole-Genome Analysis of Histone H3 Lysine 4 and Lysine 27 Methylation in Human Embryonic Stem Cells. Cell Stem Cell. 2007;1(3):299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81(4):303–311. doi: 10.1111/j.1399-0004.2011.01809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown KW, Villar AJ, Bickmore W, Clayton-Smith J, Catchpoole D, Maher ER, Reik W. Imprinting mutation in the Beckwith-Wiedemann syndrome leads to biallelic IGF2 expression through an H19-independent pathway. Human molecular genetics. 1996;5(12):2027–2032. doi: 10.1093/hmg/5.12.2027. [DOI] [PubMed] [Google Scholar]
- 22.Cerrato F, Sparago A, Verde G, De Crescenzo A, Citro V, Cubellis MV, Rinaldi MM, Boccuto L, Neri G, Magnani C, D'Angelo P, Collini P, Perotti D, Sebastio G, Maher ER, Riccio A. Different mechanisms cause imprinting defects at the IGF2/H19 locus in Beckwith-Wiedemann syndrome and Wilms’ tumour. Human molecular genetics. 2008;17(10):1427–1435. doi: 10.1093/hmg/ddn031. [DOI] [PubMed] [Google Scholar]
- 23.Morison IM, Paton CJ, Cleverley SD. The imprinted gene and parent-of-origin effect database. Nucleic Acids Res. 2001;29(1):275–276. doi: 10.1093/nar/29.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tycko B, Morison IM. Physiological functions of imprinted genes. J Cell Physiol. 2002;192(3):245–258. doi: 10.1002/jcp.10129. [DOI] [PubMed] [Google Scholar]
- 25.Lewis A, Murrell A. Genomic imprinting: CTCF protects the boundaries. Current biology : CB. 2004;14(7):R284–286. doi: 10.1016/j.cub.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 26.Jelinic P, Shaw P. Loss of imprinting and cancer. The Journal of Pathology. 2007;211(3):261–268. doi: 10.1002/path.2116. [DOI] [PubMed] [Google Scholar]
- 27.Rainier S, Johnson LA, Dobry CJ, Ping AJ, Grundy PE, Feinberg AP. Relaxation of imprinted genes in human cancer. Nature. 1993;362(6422):747–749. doi: 10.1038/362747a0. [DOI] [PubMed] [Google Scholar]
- 28.Jelinic P, Shaw P. Loss of imprinting and cancer. The Journal of pathology. 2007;211(3):261–268. doi: 10.1002/path.2116. [DOI] [PubMed] [Google Scholar]
- 29.Scott RH, Murray A, Baskcomb L, Turnbull C, Loveday C, Al-Saadi R, Williams R, Breatnach F, Gerrard M, Hale J, Kohler J, Lapunzina P, Levitt GA, Picton S, Pizer B, Ronghe MD, et al. Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget. 2012;3(3):327–335. doi: 10.18632/oncotarget.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Segers H, Kersseboom R, Alders M, Pieters R, Wagner A, van den Heuvel-Eibrink MM. Frequency of WT1 and 11p15 constitutional aberrations and phenotypic correlation in childhood Wilms tumour patients. European journal of cancer (Oxford, England : 1990) 2012;48(17):3249–3256. doi: 10.1016/j.ejca.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Scott RH, Douglas J, Baskcomb L, Huxter N, Barker K, Hanks S, Craft A, Gerrard M, Kohler JA, Levitt GA, Picton S, Pizer B, Ronghe MD, Williams D, Cook JA, Pujol P, et al. Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nature genetics. 2008;40(11):1329–1334. doi: 10.1038/ng.243. [DOI] [PubMed] [Google Scholar]
- 32.Li YM, Franklin G, Cui HM, Svensson K, He XB, Adam G, Ohlsson R, Pfeifer S. The H19 transcript is associated with polysomes and may regulate IGF2 expression in trans. J Biol Chem. 1998;273(43):28247–28252. doi: 10.1074/jbc.273.43.28247. [DOI] [PubMed] [Google Scholar]
- 33.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137(7):1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fedoriw AM, Stein P, Svoboda P, Schultz RM, Bartolomei MS. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science (New York, NY) 2004;303(5655):238–240. doi: 10.1126/science.1090934. [DOI] [PubMed] [Google Scholar]
- 35.Vuononvirta R, Sebire NJ, Dallosso AR, Reis-Filho JS, Williams RD, Mackay A, Fenwick K, Grigoriadis A, Ashworth A, Pritchard-Jones K, Brown KW, Vujanic GM, Jones C. Perilobar nephrogenic rests are nonobligate molecular genetic precursor lesions of insulin-like growth factor-II-associated Wilms tumors. Clin Cancer Res. 2008;14(23):7635–7644. doi: 10.1158/1078-0432.CCR-08-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gadd S, Huff V, Huang CC, Ruteshouser EC, Dome JS, Grundy PE, Breslow N, Jennings L, Green DM, Beckwith JB, Perlman EJ. Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: a Children's Oncology Group Study. Neoplasia. 2012;14(8):742–756. doi: 10.1593/neo.12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallagher EJ, LeRoith D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends in endocrinology and metabolism: TEM. 2010;21(10):610–618. doi: 10.1016/j.tem.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maiti S, Alam R, Amos CI, Huff V. Frequent Association of β-Catenin and WT1 Mutations in Wilms Tumors. Cancer Research. 2000;60(22):6288–6292. [PubMed] [Google Scholar]
- 39.Perotti D, Gamba B, Sardella M, Spreafico F, Terenziani M, Collini P, Pession A, Nantron M, Fossati-Bellani F, Radice P. Functional inactivation of the WTX gene is not a frequent event in Wilms’ tumors. Oncogene. 2008;27(33):4625–4632. doi: 10.1038/onc.2008.93. [DOI] [PubMed] [Google Scholar]
- 40.Rivera MN, Kim WJ, Wells J, Driscoll DR, Brannigan BW, Han M, Kim JC, Feinberg AP, Gerald WL, Vargas SO, Chin L, Iafrate AJ, Bell DW, Haber DA. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science (New York, NY) 2007;315(5812):642–645. doi: 10.1126/science.1137509. [DOI] [PubMed] [Google Scholar]
- 41.Slade I, Stephens P, Douglas J, Barker K, Stebbings L, Abbaszadeh F, Pritchard-Jones K, Cole R, Pizer B, Stiller C, Vujanic G, Scott RH, Stratton MR, Rahman N. Constitutional translocation breakpoint mapping by genome-wide paired-end sequencing identifies HACE1 as a putative Wilms tumour susceptibility gene. Journal of medical genetics. 2010;47(5):342–347. doi: 10.1136/jmg.2009.072983. [DOI] [PubMed] [Google Scholar]
- 42.Urbach A, Yermalovich A, Zhang J, Spina CS, Zhu H, Perez-Atayde AR, Shukrun R, Charlton J, Sebire N, Mifsud W, Dekel B, Pritchard-Jones K, Daley GQ. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes & development. 2014;28(9):971–982. doi: 10.1101/gad.237149.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torrezan GT, Ferreira EN, Nakahata AM, Barros BD, Castro MT, Correa BR, Krepischi AC, Olivieri EH, Cunha IW, Tabori U, Grundy PE, Costa CM, de Camargo B, Galante PA, Carraro DM. Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nature communications. 2014;5:4039. doi: 10.1038/ncomms5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Natrajan R, Reis-Filho JS, Little SE, Messahel B, Brundler MA, Dome JS, Grundy PE, Vujanic GM, Pritchard-Jones K, Jones C. Blastemal expression of type I insulin-like growth factor receptor in Wilms’ tumors is driven by increased copy number and correlates with relapse. Cancer research. 2006;66(23):11148–11155. doi: 10.1158/0008-5472.CAN-06-1931. [DOI] [PubMed] [Google Scholar]
- 45.Hu Q, Gao F, Tian W, Ruteshouser EC, Wang Y, Lazar A, Stewart J, Strong LC, Behringer RR, Huff V. Wt1 ablation and Igf2 upregulation in mice result in Wilms tumors with elevated ERK1/2 phosphorylation. J Clin Invest. 2011;121(1):174–183. doi: 10.1172/JCI43772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perlman EJ, Grundy PE, Anderson JR, Jennings LJ, Green DM, Dome JS, Shamberger RC, Ruteshouser EC, Huff V. WT1 mutation and 11P15 loss of heterozygosity predict relapse in very low-risk wilms tumors treated with surgery alone: a children's oncology group study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(6):698–703. doi: 10.1200/JCO.2010.31.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dome JS, Perlman EJ, Graf N. Risk stratification for wilms tumor: current approach and future directions. American Society of Clinical Oncology educational book / ASCO American Society of Clinical Oncology Meeting. 2014;34:215–223. doi: 10.14694/EdBook_AM.2014.34.215. [DOI] [PubMed] [Google Scholar]
- 48.Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. International journal of cancer Journal international du cancer. 2003;107(6):873–877. doi: 10.1002/ijc.11487. [DOI] [PubMed] [Google Scholar]
- 49.Fontana L, Adelaiye RM, Rastelli AL, Miles KM, Ciamporcero E, Longo VD, Nguyen H, Vessella R, Pili R. Dietary protein restriction inhibits tumor growth in human xenograft models. Oncotarget. 2013;4(12):2451–2461. doi: 10.18632/oncotarget.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Resnicoff M, Sell C, Rubini M, Coppola D, Ambrose D, Baserga R, Rubin R. Rat Glioblastoma Cells Expressing an Antisense RNA to the Insulin-like Growth Factor-1 (IGF-1) Receptor Are Nontumorigenic and Induce Regression of Wild-Type Tumors. Cancer Research. 1994;54(8):2218–2222. [PubMed] [Google Scholar]
- 51.Chernicky CL, Tan H, Yi L, Loret de Mola JR, Ilan J. Treatment of murine breast cancer cells with antisense RNA to the type I insulin-like growth factor receptor decreases the level of plasminogen activator transcripts, inhibits cell growth in vitro, and reduces tumorigenesis in vivo. Mol Pathol. 2002;55(2):102–109. doi: 10.1136/mp.55.2.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pavelic J, Pavelic L, Karadza J, Krizanac S, Unesic J, Spaventi S, Pavelic K. Insulin-like growth factor family and combined antisense approach in therapy of lung carcinoma. Mol Med. 2002;8(3):149–157. [PMC free article] [PubMed] [Google Scholar]
- 53.Scotlandi K, Maini C, Manara MC, Benini S, Serra M, Cerisano V, Strammiello R, Baldini N, Lollini PL, Nanni P, Nicoletti G, Picci P. Effectiveness of insulin-like growth factor I receptor antisense strategy against Ewing's sarcoma cells. Cancer Gene Ther. 2002;9(3):296–307. doi: 10.1038/sj.cgt.7700442. [DOI] [PubMed] [Google Scholar]
- 54.Kolb EA, Gorlick R, Lock R, Carol H, Morton CL, Keir ST, Reynolds CP, Kang MH, Maris JM, Billups C, Smith MA, Houghton PJ. Initial testing (stage 1) of the IGF-1 receptor inhibitor BMS-754807 by the pediatric preclinical testing program. Pediatric blood & cancer. 2011;56(4):595–603. doi: 10.1002/pbc.22741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Houghton PJ, Morton CL, Gorlick R, Kolb EA, Keir ST, Reynolds CP, Kang MH, Maris JM, Wu J, Smith MA. Initial testing of a monoclonal antibody (IMC-A12) against IGF-1R by the Pediatric Preclinical Testing Program. Pediatric blood & cancer. 2010;54(7):921–926. doi: 10.1002/pbc.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bielen A, Box G, Perryman L, Bjerke L, Popov S, Jamin Y, Jury A, Valenti M, Brandon AdH, Martins V, Romanet V, Jeay S, Raynaud FI, Hofmann F, Robinson SP, Eccles SA, et al. Dependence of Wilms tumor cells on signaling through insulin-like growth factor 1 in an orthotopic xenograft model targetable by specific receptor inhibition. Proceedings of the National Academy of Sciences. 2012;109(20):E1267–E1276. doi: 10.1073/pnas.1105034109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, Batzel GN, Yin D, Pritchard-Jones K, Judson I, Worden FP, Gualberto A, Scurr M, de Bono JS, Haluska P. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing's sarcoma: a phase 1 expansion cohort study. The Lancet Oncology. 2010;11(2):129–135. doi: 10.1016/S1470-2045(09)70354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurzrock R, Patnaik A, Aisner J, Warren T, Leong S, Benjamin R, Eckhardt SG, Eid JE, Greig G, Habben K, McCarthy CD, Gore L. A Phase I Study of Weekly R1507, A Human Monoclonal Antibody Insulin-like Growth Factor-I Receptor Antagonist, in Patients with Advanced Solid Tumors. Clinical Cancer Research. 2010;16(8):2458–2465. doi: 10.1158/1078-0432.CCR-09-3220. [DOI] [PubMed] [Google Scholar]
- 59.Janku F, Huang HJ, Angelo LS, Kurzrock R. A kinase-independent biological activity for insulin growth factor-1 receptor (IGF-1R) : implications for inhibition of the IGF-1R signal. Oncotarget. 2013;4(3):463–473. doi: 10.18632/oncotarget.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beltran PJ, Mitchell P, Chung Y-A, Cajulis E, Lu J, Belmontes B, Ho J, Tsai MM, Zhu M, Vonderfecht S, Baserga R, Kendall R, Radinsky R, Calzone FJ. AMG 479, a fully human anti–insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Molecular Cancer Therapeutics. 2009;8(5):1095–1105. doi: 10.1158/1535-7163.MCT-08-1171. [DOI] [PubMed] [Google Scholar]
- 61.Kolb EA, Gorlick R, Maris JM, Keir ST, Morton CL, Wu J, Wozniak AW, Smith MA, Houghton PJ. Combination testing (Stage 2) of the Anti-IGF-1 receptor antibody IMC-A12 with rapamycin by the pediatric preclinical testing program. Pediatric blood & cancer. 2012;58(5):729–735. doi: 10.1002/pbc.23157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. http://www.amgen.com/media/media_pr_detail.jsp?releaseID=1723925
- 63.McCaffery I, Tudor Y, Deng H, Tang R, Suzuki S, Badola S, Kindler HL, Fuchs CS, Loh E, Patterson SD, Chen L, Gansert JL. Putative predictive biomarkers of survival in patients with metastatic pancreatic adenocarcinoma treated with gemcitabine and ganitumab, an IGF1R inhibitor. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(15):4282–4289. doi: 10.1158/1078-0432.CCR-12-1840. [DOI] [PubMed] [Google Scholar]
- 64.Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, Krailo M, Villaluna D, Adamson PC, Blaney SM. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children's Oncology Group. Pediatric blood & cancer. 2014;61(3):452–456. doi: 10.1002/pbc.24605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mikkelsen JH, Resch ZT, Kalra B, Savjani G, Kumar A, Conover CA, Oxvig C. Indirect targeting of IGF receptor signaling in vivo by substrate-selective inhibition of PAPP-A proteolytic activity. Oncotarget. 2014;5(4):1014–1025. doi: 10.18632/oncotarget.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Conover CA, Bale LK, Overgaard MT, Johnstone EW, Laursen UH, Fuchtbauer EM, Oxvig C, van Deursen J. Metalloproteinase pregnancy-associated plasma protein A is a critical growth regulatory factor during fetal development. Development. 2004;131(5):1187–1194. doi: 10.1242/dev.00997. [DOI] [PubMed] [Google Scholar]
- 67.Ting AH, McGarvey KM, Baylin SB. The cancer epigenome - components and functional correlates. Genes & development. 2006;20(23):3215–3231. doi: 10.1101/gad.1464906. [DOI] [PubMed] [Google Scholar]
- 68.Rice KL, Hormaeche I, Licht JD. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene. 2007;26(47):6697–6714. doi: 10.1038/sj.onc.1210755. [DOI] [PubMed] [Google Scholar]
- 69.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nature reviews Cancer. 2011;11(10):726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Di Costanzo A, Del Gaudio N, Migliaccio A, Altucci L. Epigenetic drugs against cancer: an evolving landscape. Arch Toxicol. 2014 doi: 10.1007/s00204-014-1315-6. [DOI] [PubMed] [Google Scholar]
- 71.Stankov MV, El Khatib M, Kumar Thakur B, Heitmann K, Panayotova-Dimitrova D, Schoening J, Bourquin JP, Schweitzer N, Leverkus M, Welte K, Reinhardt D, Li Z, Orkin SH, Behrens GM, Klusmann JH. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2013 doi: 10.1038/leu.2013.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics : targets & therapy. 2013;7:47–60. doi: 10.2147/BTT.S29965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, Ortwine DF, Gunzner J, Modrusan Z, Neumann L, Koth CM, Lupardus PJ, et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. The Journal of biological chemistry. 2013;288(37):26926–26943. doi: 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wagner JM, Hackanson B, Lubbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clinical epigenetics. 2010;1(3-4):117–136. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dimond PF. Epigenetics Targets May Enable Discovery of Novel Drugs. Genetic Engineering & Biotechnology News. 2013 http://www.genengnews.com/insight-and-intelligenceand153/epigenetics-targets-may-enable-discovery-of-novel-drugs/77899928/: Genetic Engineering & Biotechnology News website [Google Scholar]
- 76.Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proceedings of the National Academy of Sciences. 2000;97(10):5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 78.Graham JS, Kaye SB, Brown R. The promises and pitfalls of epigenetic therapies in solid tumours. European journal of cancer (Oxford, England : 1990) 2009;45(7):1129–1136. doi: 10.1016/j.ejca.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 79.Karahoca M, Momparler RL. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2'-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clinical epigenetics. 2013;5(1):3. doi: 10.1186/1868-7083-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gnyszka A, Jastrzebski Z, Flis S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer research. 2013;33(8):2989–2996. [PubMed] [Google Scholar]
- 81.Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, Jones PA, Selker EU. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. Journal of the National Cancer Institute. 2003;95(5):399–409. doi: 10.1093/jnci/95.5.399. [DOI] [PubMed] [Google Scholar]
- 82.Cheng JC, Yoo CB, Weisenberger DJ, Chuang J, Wozniak C, Liang G, Marquez VE, Greer S, Orntoft TF, Thykjaer T, Jones PA. Preferential response of cancer cells to zebularine. Cancer cell. 2004;6(2):151–158. doi: 10.1016/j.ccr.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 83.Andrade AF, Borges KS, Castro-Gamero AM, Silveira VS, Suazo VK, Oliveira JC, Moreno DA, de Paula Queiroz RG, Scrideli CA, Tone LG. Zebularine induces chemosensitization to methotrexate and efficiently decreases AhR gene methylation in childhood acute lymphoblastic leukemia cells. Anticancer Drugs. 2014;25(1):72–81. doi: 10.1097/CAD.0000000000000028. [DOI] [PubMed] [Google Scholar]
- 84.Zirn B, Hartmann O, Samans B, Krause M, Wittmann S, Mertens F, Graf N, Eilers M, Gessler M. Expression profiling of Wilms tumors reveals new candidate genes for different clinical parameters. Int J Cancer. 2006;118(8):1954–1962. doi: 10.1002/ijc.21564. [DOI] [PubMed] [Google Scholar]
- 85.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(19):7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 87.Nygren AO, Ameziane N, Duarte HM, Vijzelaar RN, Waisfisz Q, Hess CJ, Schouten JP, Errami A. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Research. 2005;33(14):e128. doi: 10.1093/nar/gni127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harada K, Toyooka S, Shivapurkar N, Maitra A, Reddy JL, Matta H, Miyajima K, Timmons CF, Tomlinson GE, Mastrangelo D, Hay RJ, Chaudhary PM, Gazdar AF. Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer research. 2002;62(20):5897–5901. [PubMed] [Google Scholar]
- 89.Chilukamarri L, Hancock AL, Malik S, Zabkiewicz J, Baker JA, Greenhough A, Dallosso AR, Huang TH, Royer-Pokora B, Brown KW, Malik K. Hypomethylation and aberrant expression of the glioma pathogenesis-related 1 gene in Wilms tumors. Neoplasia. 2007;9(11):970–978. doi: 10.1593/neo.07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ehrlich M, Hopkins NE, Jiang G, Dome JS, Yu MC, Woods CB, Tomlinson GE, Chintagumpala M, Champagne M, Dillerg L, Parham DM, Sawyer J. Satellite DNA hypomethylation in karyotyped Wilms tumors. Cancer Genet Cytogenet. 2003;141(2):97–105. doi: 10.1016/s0165-4608(02)00668-4. [DOI] [PubMed] [Google Scholar]
- 91.Ludgate JL, Le Mee G, Fukuzawa R, Rodger EJ, Weeks RJ, Reeve AE, Morison IM. Global demethylation in loss of imprinting subtype of wilms tumor. Genes Chromosomes Cancer. 2012 doi: 10.1002/gcc.22017. [DOI] [PubMed] [Google Scholar]
- 92.Charlton J, et al. Methylome analysis identifies a Wilms tumor epigenetic biomarker detectable in blood. Genome Biology. 2014 doi: 10.1186/s13059-014-0434-y. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang L, Anglesio MS, O'Sullivan M, Zhang F, Yang G, Sarao R, Mai PN, Cronin S, Hara H, Melnyk N, Li L, Wada T, Liu PP, Farrar J, Arceci RJ, Sorensen PH, et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med. 2007;13(9):1060–1069. doi: 10.1038/nm1621. [DOI] [PubMed] [Google Scholar]
- 94.Zitzmann F, Mayr D, Berger M, Stehr M, von Schweinitz D, Kappler R, Hubertus J. Frequent hypermethylation of a CTCF binding site influences Wilms tumor 1 expression in Wilms tumors. Oncology reports. 2014;31(4):1871–1876. doi: 10.3892/or.2014.3019. [DOI] [PubMed] [Google Scholar]
- 95.Dallosso AR, Hancock AL, Szemes M, Moorwood K, Chilukamarri L, Tsai HH, Sarkar A, Barasch J, Vuononvirta R, Jones C, Pritchard-Jones K, Royer-Pokora B, Lee SB, Owen C, Malik S, Feng Y, et al. Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms’ tumor. PLoS Genet. 2009;5(11):e1000745. doi: 10.1371/journal.pgen.1000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haruta M, Arai Y, Sugawara W, Watanabe N, Honda S, Ohshima J, Soejima H, Nakadate H, Okita H, Hata J, Fukuzawa M, Kaneko Y. Duplication of paternal IGF2 or loss of maternal IGF2 imprinting occurs in half of Wilms tumors with various structural WT1 abnormalities. Genes, chromosomes & cancer. 2008;47(8):712–727. doi: 10.1002/gcc.20572. [DOI] [PubMed] [Google Scholar]
- 97.Arcellana-Panlilio MY, Egeler RM, Ujack E, Pinto A, Demetrick DJ, Robbins SM, Coppes MJ. Decreased expression of the INK4 family of cyclin-dependent kinase inhibitors in Wilms tumor. Genes, Chromosomes and Cancer. 2000;29(1):63–69. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1006>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 98.Wagner KJ, Cooper WN, Grundy RG, Caldwell G, Jones C, Wadey RB, Morton D, Schofield PN, Reik W, Latif F, Maher ER. Frequent RASSF1A tumour suppressor gene promoter methylation in Wilms’ tumour and colorectal cancer. Oncogene. 2002;21(47):7277–7282. doi: 10.1038/sj.onc.1205922. [DOI] [PubMed] [Google Scholar]
- 99.Malik K, Salpekar A, Hancock A, Moorwood K, Jackson S, Charles A, Brown KW. Identification of Differential Methylation of the WT1 Antisense Regulatory Region and Relaxation of Imprinting in Wilms’ Tumor. Cancer Res. 2000;60(9):2356–2360. [PubMed] [Google Scholar]