

Abstract
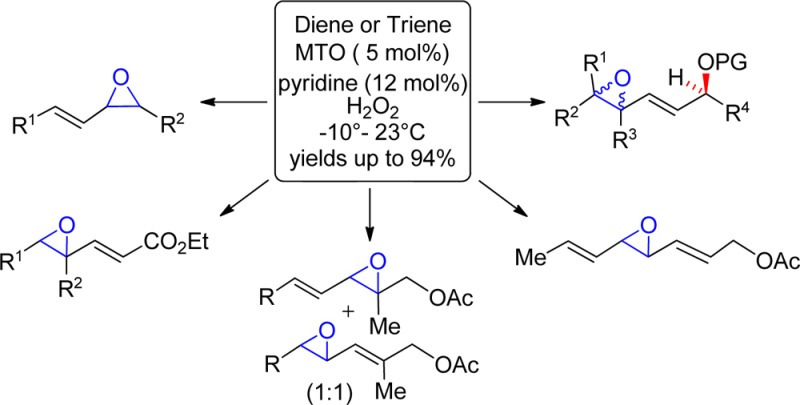
Methyltrioxorhenium (MTO) complexed with pyridine was shown to be a highly effective catalyst for the regioselective monoepoxidation of conjugated di- and trienes using 30% H2O2 at or below room temperature. The resultant allylic epoxides, and the triols derived from them, are versatile synthetic intermediates as well as substructures present in many bioactive natural products. The site of epoxidation was dependent upon olefin substitution, olefin geometry (Z vs E), and the presence of electron-withdrawing substituents on adjacent carbons. For 1-acyl(silyl)oxypenta-2,4-dienes, epoxidation of the distal olefin was generally favored in contrast to the adjacent regioselectivity characteristic of Sharpless, peracid, and other directed epoxidations of hydroxylated dienes.
Introduction
An array1 of protocols is available for the preparation of epoxides as befits their prominence as versatile synthetic intermediates2 and as substructures in numerous bioactive compounds.2,3 The most common and generally economic synthetic approach is the direct, catalytic epoxidation of olefins.4 The task is more problematic for the monoepoxidation of 1,3-conjugated dienes and higher homologues.5 Of the few reagents that have been studied, inter alia, Mo(CO)6,6 OTi(tetraphenylporphyrin),7 Mn(tetraphenylporphyrin),8 transition metal salens,9 and dimethyldioxirane,10 most have one or more limitations such as modest yields, variable regioselectivities, low cis-/trans-selectivity, polyoxidation, stereoisomerization, and/or instability of the allylic epoxide product under the reaction conditions. A prominent exception is the Shi fructose-based dioxirane reagents,11 although the strict reaction regimen and catalyst availability can be a deterrence.
 |
1 |
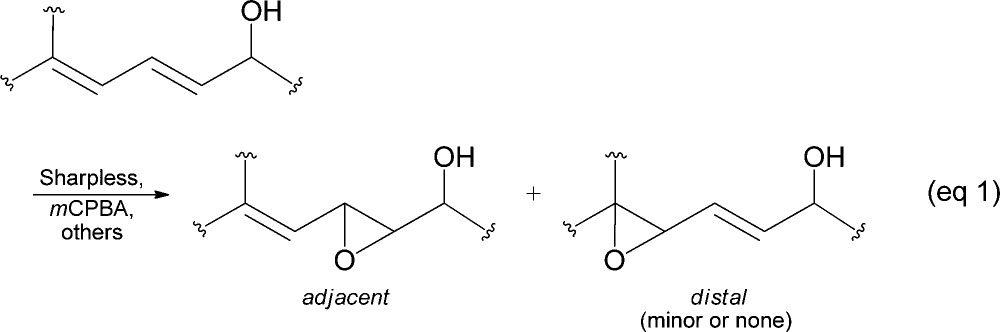
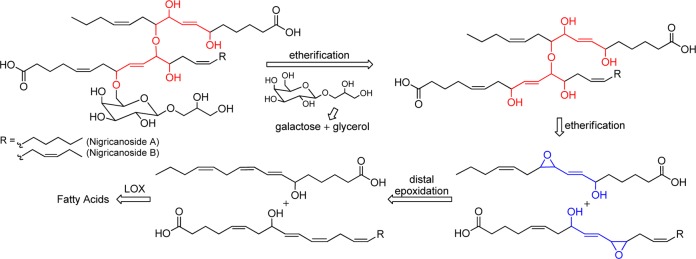
The epoxidation of the 2,4-pentadien-1-ol substructure is of particular interest to many laboratories. In addition to being useful synthetic building blocks,5 the resultant allylic epoxyols12 and their chemically or enzymatically derived allylic triols are well-represented among natural products of current interest (Figure 1).13 Functional group directed epoxidations, exemplified by peracid, Sharpless,14 and related catalytic reagents,15 generally offer an excellent level of stereocontrol, but they predominately epoxidize the olefin adjacent to the hydroxyl and not the distal olefin (eq 1).16 We were, thus, motivated to develop an inexpensive and direct distal-selective, catalytic epoxidation of conjugated buta-1,3-dienes/penta-2,4-dien-1-ols and exploit this methodology as a key transformation in a biogenetically inspired total synthesis17 of the potent antimitotic marine natural products nigricanoside A/B18 and their analogues (Scheme 1).
Figure 1.
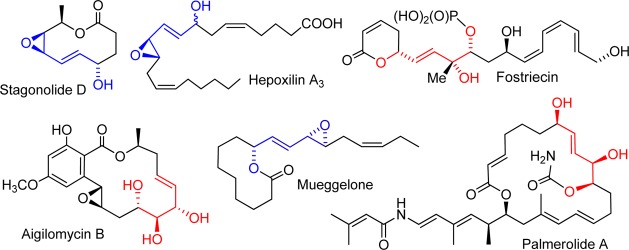
Representative allylic epoxyol and triol natural products.
Scheme 1. Retrosynthetic Analysis of Nigricanosides A/B.
Results and Discussion
A wide variety of catalysts and oxidants were surveyed for distal-selective epoxidation of the penta-2,4-diene-ol moiety. A sampling is compiled in Table 1. Methyl ester 1 was selected as the model substrate because it is readily available in high stereochemical purity via incubation of linoleic acid with soybean lipoxygenase19 on a multigram scale and also provides a stereochemical vantage point to monitor the influence of an adjacent chiral center on the course of the epoxidation. Initially, epoxidations were conducted with the C(13)-hydroxyl unprotected; in many cases, however, the hydroxyl underwent oxidation and/or any epoxide product decomposed under the experimental conditions. Hence, future screenings were conducted with the hydroxyl protected as its acetate.
Table 1. Survey of Catalysts for Distal-Selective Epoxidation of Diene 1.
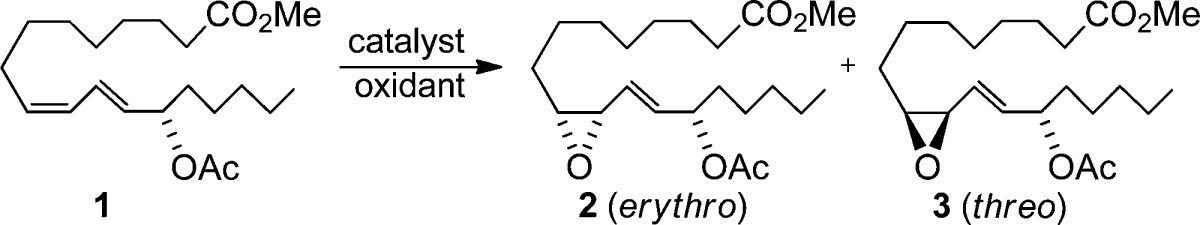
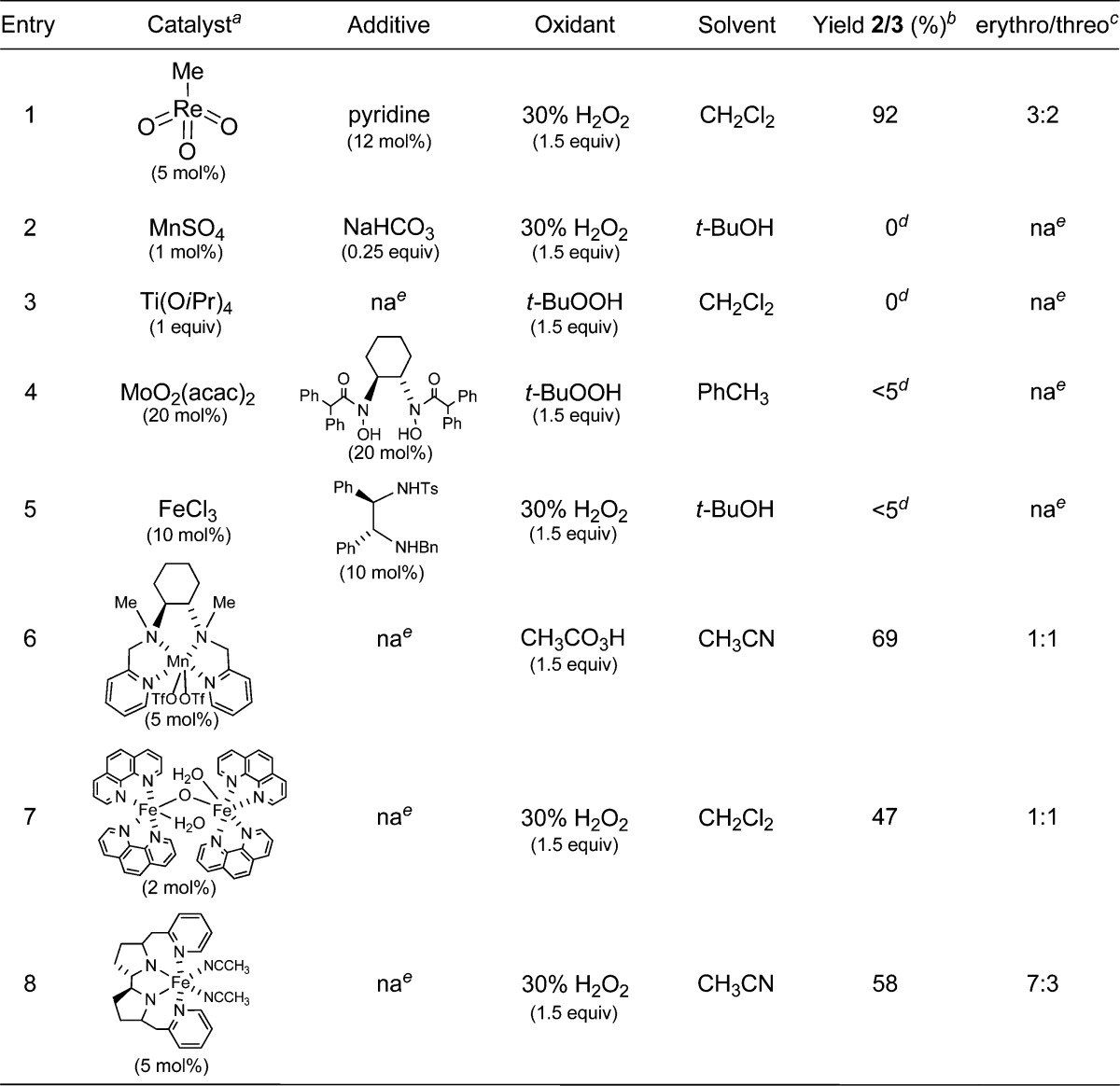
It was evident that methyltrioxorhenium20 (MTO) (entry 1) in CH2Cl2 was the most efficacious for distal epoxidation, although the product was generated as a mixture of diastereomers 2 and 3.28,29 Yields were diminished somewhat in CH3CN and CH3NO2, and the dr (2/3) was unchanged. Other common reagents (entries 2–5) were ineffective or gave minor amounts of epoxide. Interestingly, Mn25 (entry 6) and Fe26,27 (entries 7 and 8) complexed with chiral ligands were also distal-selective but still produced mixtures of 2 and 3. To modulate MTO’s Lewis acidity, pyridine was added, as recommended by Sharpless;20a however, increasing the level of pyridine beyond 2.4 equiv with respect to MTO did not improve either the yield or dr. Replacement of the pyridine with other ligands (Table 2) had some effect on yield but, disappointingly, little influence on the dr even when using chiral pyridines and amines (entries 9–14).30 The latter likely reflects the weak coordination of the chiral bases with the metal center.31
Table 2. MTO Ligand Screeninga.
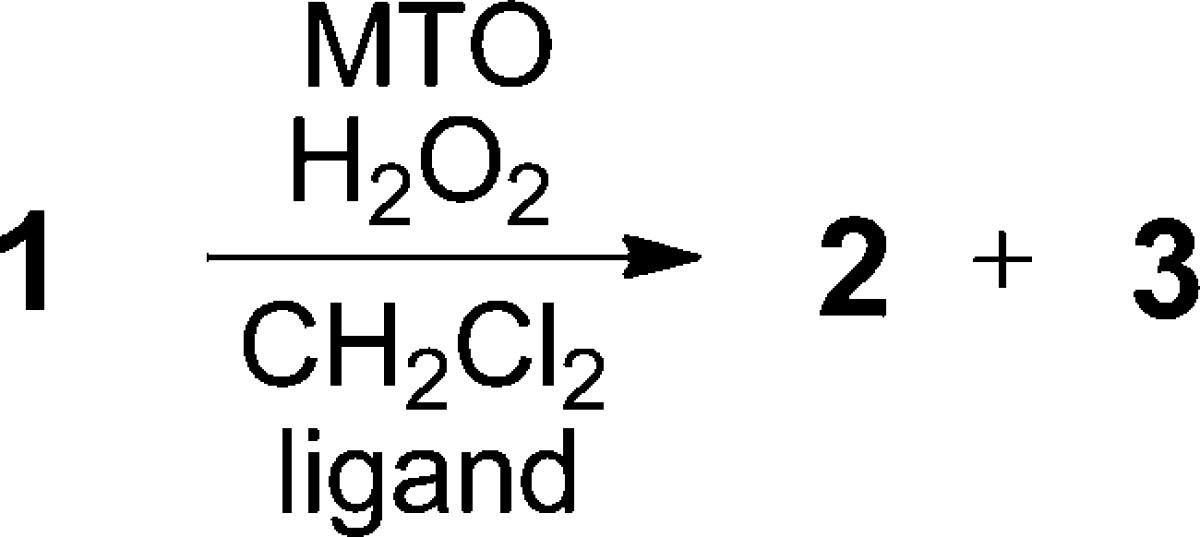
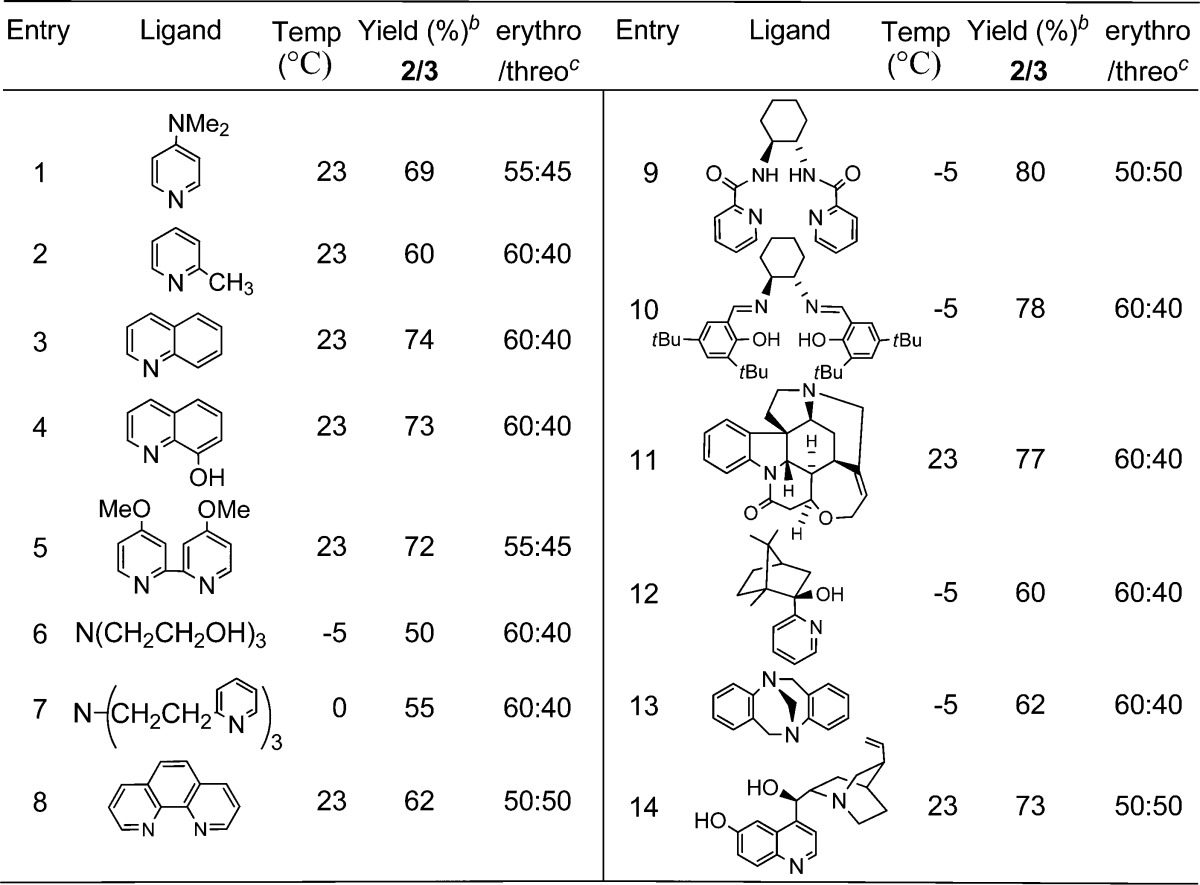
12 mol % each MTO and ligand in CH2Cl2.
Combined, isolated yield.
Determined by NMR.
In addition to offering the best combined yield of 2/3, there is much to recommend the MTO/H2O2 system versus other catalysts.32 It is commercially available, inexpensive, air stable, reacts at room temperature or below, uses environmentally friendly H2O2 or H2O2–urea adduct33 instead of more corrosive oxidants, generates water as the only byproduct, and is operationally simple. Careful optimization of the reaction conditions showed that best results could be obtained with 5 mol % of MTO and 12 mol % of pyridine. Importantly, this methodology was also amenable to the multigram conversion of 1 into a mixture of 2 and 3 in an 84% combined yield.
Early optimization studies of the MTO/H2O2 catalyzed epoxidation of unprotected 4 (PG = H) found that the yields were somewhat compromised by the formation of ketone and other uncharacterized products (Table 3, entry a), so a brief survey of commonly used protecting groups (PGs) was initiated. This revealed bulky (entry b), aryl (entry c), and aliphatic (entry d) esters, ethoxycarbonyl (entry e), and t-butyldiphenylsilyl ether (entry f) were all well-tolerated and afforded good yields of epoxides 5/6, but they showed little variation in the dr. In concert with acetate 1, there was a slight preference in favor of the erythro diastereomer 5. All epoxides were identified by comparisons with authentic standards.29
Table 3. Effect of Alcohol Protecting Groupa.
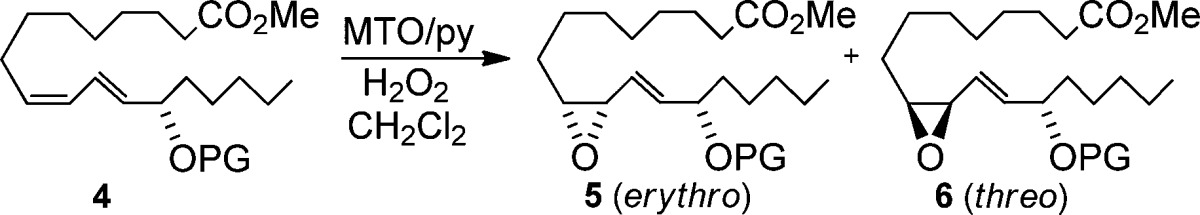
| Entry | PG | Time (h) | Yield 5/6 (%)b | erythro/threoc |
|---|---|---|---|---|
| a | H | 6d | 56 | 60:40 |
| b | C(O)tBu | 3 | 80 | 60:40 |
| c | C(O)Ph | 4 | 78 | 60:40 |
| d | C(O)CH2Ph | 3 | 73 | 55:45 |
| e | C(O)OEt | 3 | 79 | 55:45 |
| f | SiPh2tBu | 5 | 82 | 60:40 |
5 mol % MTO, 12 mol % pyridine, and 2 equiv H2O2 at rt.
Combined, isolated yield.
Determined by NMR.
Conducted at −10 °C. Remaining material balance mainly ketone or decomposition.
To elucidate the scope34 of MTO-mediated epoxidations of di/trienes, a panel of representative substrates was subjected to the standard epoxidation conditions (Table 4). Acetate 7 (entry 1) and carbonate 9 (entry 2), both derived from the soybean lipoxidase metabolite35 of linolenic acid, smoothly led to distal epoxides 8 and 10, respectively, in good yields at −5 °C; at room temperature, however, ∼10–15% of the Δ15,16-olefin of 10 was also epoxidized. Exposure of the structurally related natural fatty acid 11 to the standard reaction conditions revealed a modest 7:3 regioselectivity favoring the Z-olefin 12 (entry 3). This is consistent with inductive and/or steric contributions of the acyloxy group to the observed regioselectivity in the preceding examples (cf., 1, 4, 7, and 9). As a testimony to the mildness of the reaction conditions, 1,4-diphenyl-1(E),3(E)-butadiene (14), despite its well-known proclivity toward polymerization and isomerization,8a was well-behaved and gave the somewhat sensitive allylic-styrenyl epoxide 15 (entry 4) in good yield. As anticipated, substrate bias33b led to α-epoxides 17 (entry 5) and 19 (entry 6) from cholest-4,6-dienes 16 and 18, respectively. The reduced yield for 18 suggests the α-acetyloxy partially blocks the bottom face. Simple 1-acetyloxy-E,E-dienes 20, 22, and 24 reacted similarly to their Z,E-counterparts, but they required lower temperatures to optimize the yields of 21 (entry 7), 23 (entry 8), and 25 (entry 9), respectively, with the latter two produced as diastereomeric mixtures. Notably, an increase in the level of substitution on the allylic olefin induced a change in oxidation regioselectivity and gave rise to a 1:1 mixture of 27 and 28 (entry 10). Increasing the substitution level of the distal olefin, e.g., trialkyl (entries 11 and 12), cyclic trialkyl (entry 13), and cyclic tetraalkyl (entry 14), was well-tolerated and uneventfully afforded 30, 32, 34, and 36, respectively. Unexpectedly, 2,4,6-triene 37 was converted to bis-allylic epoxide 38 as the only mono-oxidation product (entry 15). Both conjugated dienyl esters 39 and 41 underwent epoxidation at the terminal olefin, albeit slowly. Control experiments with both 39 and 41 confirmed that MTO was required for epoxidation.
Table 4. MTO Epoxidation of Representative Conjugated Dienes/Trienesa.
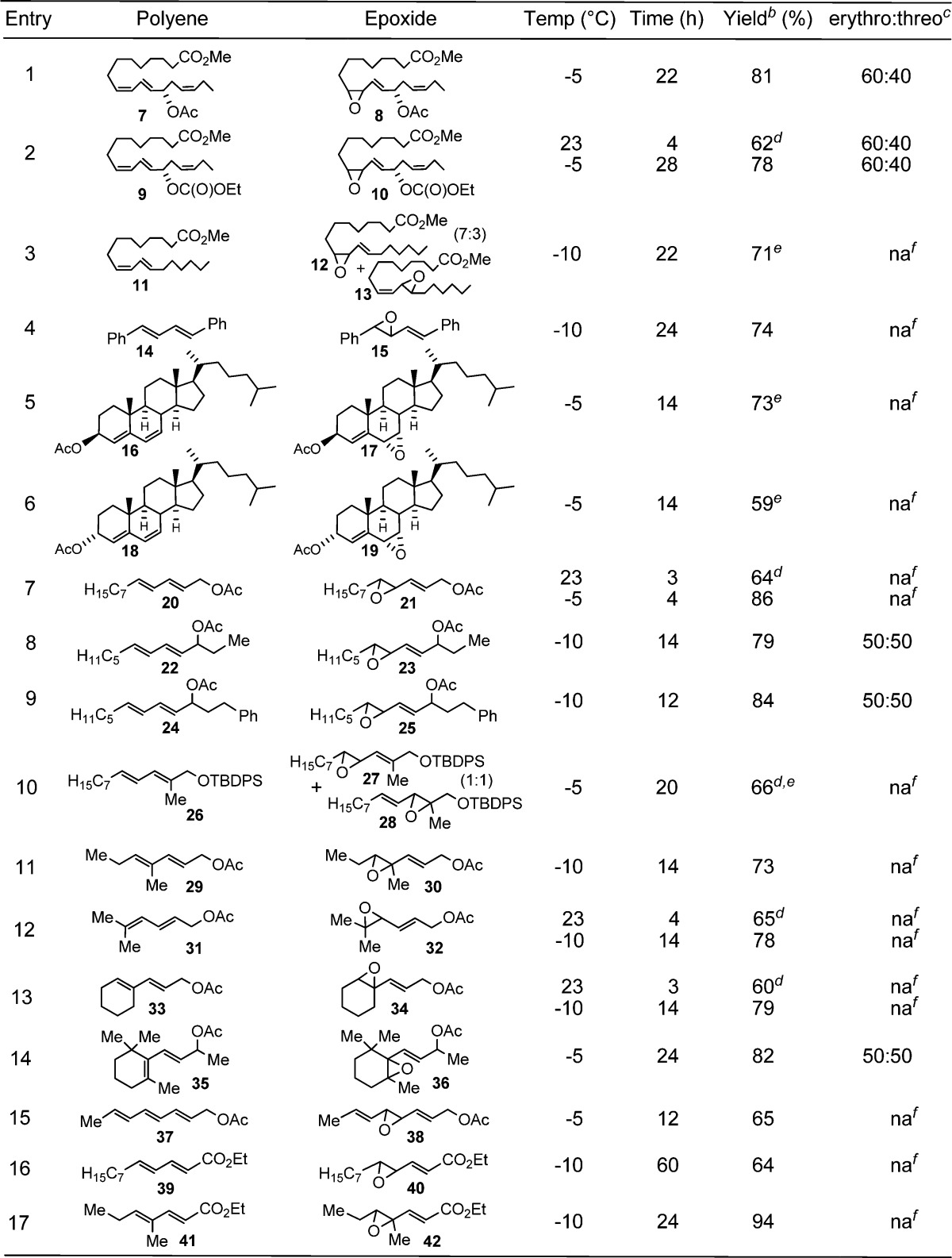
5 mol % MTO, 12 mol % pyridine, and 2 equiv H2O2 in CH2Cl2.
Isolated yield.
Determined by 1H/13C NMR or chiral-phase HPLC.
10–15% bis-epoxide also formed.
Combined, isolated yield.
na, not applicable.
The mechanism of MTO-mediated epoxidation has been well-studied.33 Hydrogen bonding between the substrate and peroxyrhenium intermediate in the transition state has been invoked33a to explain stereospecificity, but this does not apply in the examples in Table 4. Steric factors have also been found to effect stereospecificity.33 The faster reaction rate for Z-olefins versus E-olefins20a is also observed in conjugated dienes (e.g., entry 3). When present, acyloxy groups inductively deactivate the adjacent olefin of the diene, thus directing epoxidation to the distal olefin regardless of olefin configuration (entries 1, 2, and 7); however, this can be overcome, at least partially, by greater olefin substitution (entry 10).
Conclusions
MTO complexed with pyridine was shown to be a highly effective catalyst for the regioselective monoepoxidation of conjugated di- and trienes. The site of epoxidation was dependent upon the olefin substitution, olefin geometry (Z vs E), and the presence of electron-withdrawing substituents on adjacent carbons. For the special case of 1-acyloxypenta-2,4-dienes, the regioselectivity was complementary to that achieved in Sharpless and other directed epoxidations of 1-hydroxypenta-2,4-dienes.
Experimental Section
General Methods and Materials
Proton and carbon nuclear magnetic resonance spectra (1H and 13C NMR) were recorded at 500 and 126 MHz, respectively, or at 400 and 101 MHz, respectively, in CDCl3 with TMS as internal standard, unless otherwise stated. 1H NMR data are reported as follows: chemical shift (ppm), multiplicity (s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, app q = apparent quartet, qn = quintet, app qn = apparent quintet, m = multiplet), and coupling constant (Hz). High-resolution mass spectra (HRMS) were obtained using a TOF mass spectrometer, whereas infrared (IR) spectra were obtained using a Fourier transform infrared spectrometer. Melting points were measured using an automated melting point apparatus and are uncorrected. Analytical thin-layer chromatography (TLC) used EMD Chemicals TLC silica gel 60 F254 plates (0.040–0.063 mm) with visualization by UV light and/or KMnO4 or phosphomolybdic acid (PMA) solution followed by heating. Chromatographic purifications utilized Et3N or t-BuNH2 basified preparative TLC or flash chromatography using prepacked SiO2 columns on an automated medium-pressure chromatograph with eluents containing 0.5–2% t-BuNH2. Determinations of diastereomeric ratios (dr) were conducted by 1H and 13C NMR or chiral phase-HPLC as specified in the experimental. Unless otherwise noted, yields refer to isolated, purified material with spectral data consistent with assigned structures or, if known, were in agreement with published data. All reactions were conducted under an argon atmosphere in oven-dried glassware with magnetic stirring. Reagents were purchased at the highest commercial quality and used without further purification. Dichloromethane (CH2Cl2) and tetrahydrofuran (THF) were dried by passage through a column of activated, neutral alumina under argon and stored under argon until use.
General Epoxidation Procedure
Aqueous 30% H2O2 (1.5–2.0 equiv) was added to a stirring, 0 °C solution of polyene, methyltrioxorhenium (MTO, 5 mol %), and pyridine (12 mol %) in CH2Cl2. The yellow reaction mixture was stirred at the specified temperature for the indicated time and then quenched with 10% tetrasodium EDTA solution. The colorless solution was extracted with CH2Cl2 (3–4 times), and the combined extracts were washed with water and brine and dried over Na2SO4. Evaporation of all volatiles and purification of the residue by flash chromatography using 0.5–2% t-butylamine or 1% Et3N in EtOAc/hexane afforded the allylic epoxide in the indicated yield.
Methyl 13(S)-Acetyloxyoctadeca-9(Z),11(E)-dienoate36 (1)
Acetic anhydride (50 μL, 0.58 mmol), pyridine (50 μL, 0.58 mmol), and a catalytic amount of DMAP (1 mg) were added to a 0 °C solution of 4a(37) (150 mg, 0.48 mmol) in CH2Cl2 (10 mL). After stirring at rt for 3 h, the reaction mixture was washed with 1 N aq. HCl (2 mL) and water (2 mL) and dried over anhydrous Na2SO4, and all volatiles were evaporated in vacuo. Purification of the residue via silica gel column chromatography using 5–15% ethyl acetate/hexane as eluent gave 1 (155 mg, 91%) as a clear oil. TLC: Rf ≈ 0.6 (20% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 6.48 (dd, J = 11.2, 15.2 Hz, 1H), 5.92 (t, J = 11.2 Hz, 1H), 5.54 (dd, J = 7.6, 15.2 Hz, 1H), 5.48–5.40 (m, 1H), 5.26 (dt, J = 7.2, 14 Hz, 1H), 3.65 (s, 3H), 2.28 (t, J = 7.2 Hz, 2H), 2.15 (dt, J = 7.2, 14.4 Hz, 2H), 2.03 (s, 3H), 1.64–1.58 (m, 4H), 1.36–1.26 (m, 14 H), 0.86 (t, J = 6.8 Hz, 3H).
Methyl 13(S)-Acetyloxyoctadeca-9(R*),10(S*)-epoxy-11(E)-enoate (2/3)
Following the general epoxidation procedure, 1 (2.4 g, 6.81 mmol), MTO (84 mg, 5 mol %), pyridine (66 μL, 12 mol %), and 30% H2O2 (1.30 mL, 10.2 mmol) were stirred in dry CH2Cl2 (70 mL) at −5 °C for 16 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (2.31 g, 92%, ∼3:2 mixture of diastereomers).29 TLC: Rf ≈ 0.5 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 5.85–5.78 (m, 1H), 5.60–5.52 (m, 1H), 5.27–5.23 (m, 1H), 3.66 (s, 3H), 3.39–3.37 (m, 1H), 3.07–3.04 (m, 1H), 2.29 (t, J = 6.0 Hz, 2H), 2.05 (s, 3H), 1.66–1.27 (m, 20 H), 0.87 (t, J = 4.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.4, 170.41, 170.38, 134.7, 134.6, 127.4, 127.1, 74.1, 73.9, 59.1, 59.0, 56.4, 56.3, 51.6, 34.5, 34.4, 34.2, 31.7, 29.42, 29.39, 29.37, 29.35, 29.2, 27.88, 27.87, 26.5, 26.4, 25.1, 24.93, 24.91, 22.7, 21.5, 21.4, 14.2. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C21H37O5, 369.2642; found, 369.2638.
Methyl 13(S)-Hydroxyoctadeca-9(R*),10(S*)-epoxy-11(E)-enoate (5a/6a)
Following the general epoxidation procedure, 4a (100 mg, 0.32 mmol), MTO (4 mg, 5 mol %), pyridine (4 μL, 12 mol %), and 30% H2O2 (72 μL, 0.64 mmol) were stirred in dry CH2Cl2 (3 mL) at −10 °C for 6 h. Chromatographic purification of the crude product by silica gel column using a gradient of 50–70% EtOAc/hexanes + 2% t-BuNH2 as eluent afforded the known38 diastereomeric epoxides 5/6 as a colorless oil (58 mg, 56%, ∼3:2 mixture).29 TLC: Rf ≈ 0.3 (40% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.80–5.74 (m, 1H), 5.58–5.50 (m, 1H), 3.89–3.83 (m, 1H), 3.31 (s, 3H), 3.21–3.19 (m, 1H), 2.82–2.79 (m, 1H), 2.05 (t, J = 7.5 Hz, 2H), 1.49–1.28 (m, 8H), 1.19–1.07 (m, 12H), 0.82 (t, J = 8.0 Hz, 3H).
Methyl 13(S)-(Pivaloyloxy)octadeca-9(Z),11(E)-dienoate (4b)
Following the acylation procedure above, 4a (R = H) (1.0 g, 3.2 mmol) was treated with pivaloyl chloride (0.77 mL, 6.4 mmol), pyridine (0.38 mL, 4.8 mmol), and DMAP (20 mg) in CH2Cl2 (30 mL) at rt for 12 h. Chromatographic purification of the crude product using 10–20% EtOAc/hexanes as eluent afforded 4b (1.20 g, 98%) as a clear oil. TLC: Rf ≈ 0.6 (15% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 6.46 (dd, 1H, J = 11.2, 15.0 Hz), 5.93 (t, 1H, J = 10.5 Hz), 5.56 (dd, 1H, J = 6.5, 15.0 Hz), 5.46–5.40 (m, 1H), 5.27 (dt, 1H, J = 6.5, 13.5 Hz), 3.66 (s, 3H), 2.29 (t, 2H, J = 7.2 Hz), 2.14 (dt, 2H, J = 7.6, 14.4 Hz), 1.63–1.56 (m, 4H), 1.35–1.16 (m, 23H), 0.87 (t, 3H, J = 6 Hz). 13C NMR (125 MHz, CDCl3) δ 177.8, 174.4, 133.3, 131.4, 127.8, 127.1, 74.2, 51.6, 38.9, 34.7, 34.2, 31.7, 29.6, 29.3, 29.2, 29.1, 27.8, 27.3, 25.0, 24.9, 22.6, 14.1. HRMS (ESI-TOF) calcd for C24H42O4Na m/z [M + Na]+, 417.2983; found, 417.2975.
Methyl 13(S)-(Pivaloyloxy)octadeca-9(R*),10(S*)-epoxy-11(E)-enoate (5b/6b)
Following the general epoxidation procedure, 4b (50 mg, 0.13 mmol), MTO (2 mg, 5 mol %), pyridine (1.5 μL, 12 mol %), and 30% H2O2 (22 μL, 0.19 mmol) were stirred in dry CH2Cl2 (3 mL) at 0 °C to rt for 3 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (42 mg, 80%, ∼3:2 mixture of diastereomers). TLC: Rf ≈ 0.5 (30% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.87–5.80 (m, 1H), 5.58–5.52 (m, 1H), 5.25 (dt, J = 6.5, 12.5 Hz, 1H), 3.66 (s, 3H), 3.38 (t, J = 6.5 Hz, 1H), 3.09–3.04 (m, 1H), 2.29 (t, J = 8.0 Hz, 2H), 1.63–1.27 (m, 20 H), 1.19 (s, 9H), 0.87 (t, J = 4.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 177.8, 177.7, 174.4, 134.88, 134.85, 126.8, 126.2, 73.5, 73.3, 59.0, 56.4, 51.6, 39.02, 39.01, 34.50, 34.47, 34.3, 31.7, 29.5, 29.42, 29.38, 29.37, 29.2, 27.89, 27.88, 27.4, 27.3, 26.2, 25.10, 25.09, 24.9, 24.8, 22.7, 14.2. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C24H43O5, 411.3111; found, 411.3105.
Methyl 13(S)-(Benzoyloxy)octadeca-9(R*),10(S*)-epoxy-11(E)-enoate (5c/6c)
Following the general epoxidation procedure, 4c(39) (50 mg, 0.12 mmol), MTO (1.5 mg, 5 mol %), pyridine (1.5 μL, 12 mol %), and 30% H2O2 (21 μL, 0.18 mmol) were stirred in dry CH2Cl2 (3 mL) at rt for 4 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (41 mg, 78%, ∼3:2 mixture of diastereomers). TLC: Rf ≈ 0.4 (20% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 8.05 (d, J = 7.5 Hz, 2H), 7.57 (t, J = 8.0 Hz, 1H), 7.47–7.43 (m, 2H), 5.99–5.92 (m, 1H), 5.72–5.64 (m, 1H), 5.54 (dt, J = 6.5, 13.5 Hz, 1H), 3.67 (s, 3H), 3.43–3.40 (m, 1H), 3.08–3.05 (m, 1H), 2.29 (t, J = 7.5 Hz, 2H), 1.83–1.69 (m, 2H), 1.61–1.24 (m, 18H), 0.89 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.47, 174.46, 165.9, 134.6, 134.5, 133.14, 133.12, 130.73, 130.67, 129.8, 128.6, 127.74, 127.71, 74.7, 74.4, 59.2, 59.1, 56.42, 56.35, 51.68, 51.67, 34.6, 34.55, 34.3, 31.8, 29.9, 29.5, 29.42, 29.41, 29.33, 29.26, 29.24, 29.22, 27.91, 27.87, 26.5, 26.4, 25.12, 25.11, 25.1, 25.02, 25.0, 22.8, 14.23, 14.20. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C26H39O5, 431.2798; found, 431.2792.
Methyl 13(S)-(2-Phenylacetyloxy)octadeca-9(Z),11(E)-dienoate (4d)
Following the acylation procedure above, 4a (100 mg, 0.32 mmol) was treated with phenylacetyl chloride (70 μL, 0.48 mmol) and pyridine (52 μL, 0.64 mmol) in CH2Cl2 (5 mL) at rt for 12 h. Chromatographic purification of the crude product using 10–20% EtOAc/hexanes as eluent afforded the title compound 4d (130 mg, 91%) as a clear oil. TLC: Rf ≈ 0.6 (10% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.34–7.27 (m, 5H), 6.44 (dd, J = 11.0, 15.5 Hz, 1H), 5.92 (t, J = 10.5 Hz, 1H), 5.56 (dd, J = 7.5, 15.0 Hz, 1H), 5.44 (dt, J = 8.0, 18.2 Hz, 1H), 5.31 (dt, J = 7.0, 13.5 Hz, 1H), 3.67 (s, 3H), 3.62 (s, 2H), 2.31 (t, J = 7.5 Hz, 2H), 2.14–2.09 (m, 2H), 1.62–1.56 (m, 3H), 1.35–1.24 (m, 15H), 0.86 (t, J = 5.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.3, 170.8, 134.2, 133.6, 130.8, 129.2, 128.5, 127.8, 127.5, 126.9, 75.1, 51.4, 41.7, 34.5, 34.1, 31.5, 29.4, 29.10, 29.08, 29.05, 29.03, 29.1, 27.7, 24.9, 24.7, 22.5, 14.0. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C27H40O4, 451.2819; found, 451.2825.
Methyl 13(S)-(2-Phenylacetyloxy)octadeca-9(R*),10(S*)-epoxy-11(E)-enoate (5d/6d)
Following the general epoxidation procedure, 4d (50 mg, 0.12 mmol), MTO (1.5 mg, 5 mol %), pyridine (1.3 μL, 12 mol %), and 30% H2O2 (21 μL, 0.18 mmol) were stirred in dry CH2Cl2 (2 mL) at rt for 3 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (38 mg, 73%, ∼55:45 mixture of diastereomers).29 TLC: Rf ≈ 0.5 (30% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.33–7.25 (m, 5H), 5.85–5.77 (m, 1H), 5.53–5.45 (m, 1H), 5.30–5.26 (m, 1H), 3.67 (s, 3H), 3.61 (s, 2H), 3.37–3.34 (m, 1H), 3.07–3.02 (m, 1H), 2.31 (t, J = 7.5 Hz, 2H), 1.63–1.23 (m, 20H), 0.87–0.85 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 174.5, 171.0, 170.9, 134.51, 134.46, 134.33, 134.31, 129.45, 129.44, 128.77, 128.76, 127.4, 127.3, 126.9, 74.5, 74.3, 59.2, 59.12, 59.10, 56.43, 56.41, 51.71, 51.70, 41.94, 41.93, 34.5, 34.3, 31.7, 29.5, 29.44, 29.43, 29.3, 26.5, 25.2, 24.84, 24.82, 22.72, 22.71, 14.20. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C27H41O5, 445.2955; found, 445.2949.
Methyl 13(S)-[(Ethoxycarbonyl)oxy]octadeca-9(Z),11(E)-dienoate (4e)
Following the acylation procedure above, 4a (200 mg, 0.65 mmol) was treated with ethyl chloroformate (0.18 mL, 1.9 mmol) and pyridine (0.15 mL, 1.9 mmol) in CH2Cl2 (10 mL) at rt for 3 h. Chromatographic purification of the crude product using 10–20% EtOAc/hexanes as eluent afforded the title compound (226 mg, 92%) as a clear oil. TLC: Rf ≈ 0.6 (20% EtOAc/hexanes); [α]D25 = +0.14 (c 0.014, CHCl3). 1H NMR (400 MHz, CDCl3) δ 6.53 (dd, J = 10.8, 15.2 Hz, 1H), 5.93 (t, J = 10.8 Hz, 1H), 5.56 (dd, J = 8.0, 15.2 Hz, 1H), 5.49–5.42 (m, 1H), 5.08 (dt, J = 7.2, 14.0 Hz, 1H), 4.19–4.14 (m, 2H), 3.65 (s, 3H), 2.29 (t, J = 7.6 Hz, 2H), 2.15 (dt, J = 7.2, 14.4 Hz, 2H), 1.72–1.54 (m, 4H), 1.36–1.24 (m, 17 H), 0.87 (t, J = 3.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.4, 154.9, 134.2, 130.5, 128.8, 127.7, 79.2, 63.9, 51.6, 34.8, 34.3, 31.7, 29.7, 29.31, 29.27, 29.2, 28.0, 25.1, 25.0, 22.7, 14.5, 14.2. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C22H39O5, 383.2798; found, 383.2794.
Methyl 13(S)-((Ethoxycarbonyl)oxy)octadeca-9(R*),10(S*)-epoxy-11(E)-enoate (5e/6e)
Following the general epoxidation procedure, 4e (50 mg, 0.13 mmol), MTO (2 mg, 5 mol %), pyridine (1.5 μL, 12 mol %), and 30% H2O2 (23 μL, 0.19 mmol) were stirred in dry CH2Cl2 (3 mL) at rt for 3 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (41 mg, 79%, ∼55:45 mixture of diastereomers).29 TLC: Rf ≈ 0.5 (30% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.85 (dd, J = 7.0, 15.5 Hz, 1H), 5.67–5.60 (m, 1H), 5.10 (dt, J = 6.5, 13.5 Hz, 1H), 4.18 (q, J = 7.5 Hz, 2H), 3.67 (s, 3H), 3.41–3.38 (m, 1H), 3.07 (br s, 1H), 2.31 (t, J = 7.5 Hz, 2H), 1.73–1.26 (m, 23H), 0.88 (t, J = 6.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.42, 174.41, 154.73, 154.72, 134.1, 133.9, 128.1, 127.9, 78.1, 78.0, 64.0, 59.1, 59.0, 56.4, 56.2, 51.6, 34.6, 34.4, 34.2, 31.70, 31.69, 29.41, 29.38, 29.3, 29.23, 29.2, 26.5, 25.10, 25.09, 24.9, 24.8, 22.8, 22.7, 14.47, 14.46, 14.1. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C22H38O6Na, 421.2568; found, 421.2561.
Methyl 13(S)-(tert-Butyldiphenylsilyloxy)octadeca-9(R*),10(S*)-epoxy-11(E)-enoate (5f/6f)
Following the general epoxidation procedure, 4f(40) (539 mg, 0.98 mmol), MTO (12 mg, 5 mol %), pyridine (10 μL, 12 mol %), and 30% H2O2 (220 μL, 1.96 mmol) were stirred in dry CH2Cl2 (10 mL) at rt for 5h. Chromatographic purification of the crude product afforded the title product as a colorless oil (440 mg, 82%, ∼3:2 mixture of diastereomers).29 TLC: Rf ≈ 0.4 (30% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 7.70–7.62 (m, 4H), 7.50–7.27 (m, 6H), 5.98–5.71 (m, 1H), 5.40 (dd, J = 15.6, 7.8 Hz, 0.4H), 5.28 (dd, J = 15.5, 7.5 Hz, 0 6H), 4.22–4.19 (m, 1H), 3.66 (d, J = 2.9 Hz, 3H), 3.33 (dd, J = 7.9, 4.3 Hz, 0.40H), 3.29 (dd, J = 7.6, 4.3 Hz, 0.6H), 3.00 (dt, J = 15.1, 5.3 Hz, 1H), 2.30 (q, J = 8.2 Hz, 2H), 1.72–1.54 (m, 2H), 1.50–1.12 (m, 18H), 1.10 (s, 9H), 0.83–0.79 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 174.2, 139.3, 135.9, 135.85, 135.83, 134.3, 134.1, 134.0, 129.6, 129.5, 129.4, 127.5, 127.4, 127.3, 124.2, 124.0, 73.6, 73.3, 58.8, 58.7, 56.6, 56.5, 51.4, 37.54, 37.47, 34.05, 34.04, 31.7, 29.3, 29.23, 29.18, 29.17, 29.05, 29.02, 27.9, 27.8, 27.0, 26.3, 24.90, 24.88, 24.0, 23.9, 22.5, 19.3, 13.99, 13.98. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C35H52O4Si, 587.3527; found, 587.3539.
Methyl 13(S)-Acetyloxyoctadeca-9(Z),11(E),15(Z)-trienoate (7)
Following the acylation procedure above, methyl 13(S)-hydroxyoctadeca-9(Z),11(E),15(Z)-trienoate41 (200 mg, 0.65 mmol) was treated with acetic anhydride (80 μL, 0.78 mmol) and pyridine (68 μL, 0.84 mmol) in CH2Cl2 (10 mL) at rt for 6 h. Chromatographic purification of the crude product using 10–20% EtOAc/hexanes as eluent afforded the title compound 7 (210 mg, 93%) as a clear oil. TLC: Rf ≈ 0.6 (20% EtOAc/hexanes); [α]D25 = −0.181 (c 0.016, CHCl3). 1H NMR (500 MHz, CDCl3) δ 6.52 (dd, J = 11.5, 15.0 Hz, 1H), 5.94 (app. t, J = 10.5 Hz, 1H), 5.60 (dd, J = 7.5, 15.5 Hz, 1H), 5.54–5.44 (m, 2H), 5.34–5.27 (m, 2H), 3.67 (s, 3H), 2.46–2.34 (m, 2H), 2.31 (t, J = 7.5 Hz, 2H), 2.18–2.12 (m, 2H), 2.08–2.02 (m, 2H), 2.06 (s, 3H), 1.66–1.58 (m, 2H), 1.38–1.30 (m, 8H), 0.96 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.5, 170.5, 134.9, 134.1, 130.5, 128.3, 127.7, 123.2, 74.5, 51.7, 34.3, 32.6, 29.7, 29.33, 29.30, 29.2, 28.0, 25.1, 21.5, 20.9, 14.4. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C21H34O4Na, 373.2357; found, 373.2349.
Methyl 13(S)-Acetyloxyoctadeca-9(R*),10(S*)-epoxy-11(E),15(Z)-dienoate (8)
Following the general epoxidation procedure, 7 (150 mg, 0.43 mmol), MTO (5.0 mg, 5 mol %), pyridine (4 μL, 12 mol %), and 30% H2O2 (72 μL, 0.64 mmol) were stirred in dry CH2Cl2 (3 mL) at −5 °C for 22 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (126 mg, 81%, ∼3:2 mixture of diastereomers).29 TLC: Rf ≈ 0.6 (30% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.88–5.82 (m, 1H), 5.66–5.54 (m, 1H), 5.53–5.47 (m, 1H), 5.31–5.24 (m, 2H), 3.66 (s, 3H), 3.39–3.36 (m, 1H), 3.07–3.05 (m, 1H), 2.44–2.34 (m, 2H), 2.30 (t, J = 7.5 Hz, 2H), 2.06–1.99 (m, 2H), 2.04 (s, 3H), 1.63–1.29 (m, 12H), 0.96 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.4, 170.3, 135.03, 135.02, 134.03, 133.98, 127.6, 127.2, 122.80, 122.76, 73.5, 73.3, 59.1, 59.0, 56.34, 56.27, 51.6, 34.2, 32.3, 32.2, 29.5, 29.32, 29.30, 29.2, 27.8, 26.4, 26.3, 25.0, 21.4, 21.3, 20.8, 14.3. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C21H35O5, 367.2485; found, 367.2479.
Methyl 13(S)-((Ethoxycarbonyl)oxy)octadeca-9(Z),11(E),15(Z)-trienoate (9)
Following the acylation procedure above, methyl 13(S)-hydroxyoctadeca-9(Z),11(E),15(Z)-trienoate34a (350 mg, 1.13 mmol) was treated with ethyl chloroformate (161 μL, 1.70 mmol) and pyridine (180 μL, 2.20 mmol) in CH2Cl2 (10 mL) at rt for 12 h. Chromatographic purification of the crude product using 10–20% EtOAc/hexanes as eluent afforded the title compound 9 (210 mg, 93%) as a clear oil. TLC: Rf ≈ 0.6 (20% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 6.54 (dd, 1H, J = 11.2, 15.5 Hz), 5.93 (t, 1H, J = 11.2 Hz), 5.59 (dd, 1H, J = 7.6, 15.2 Hz), 5.53–5.42 (m, 1H), 5.33–5.25 (m, 1H), 5.11 (dt, 1H, J = 7.2, 14.4 Hz), 4.16 (q, 2H, J = 7.2 Hz), 3.65 (s, 3H), 2.53–2.32 (m, 2H), 2.28 (t, 2H, J = 7.6 Hz), 2.15 (dt, 2H, J = 6.8, 14.0 Hz), 2.07–1.98 (m, 2H), 1.60 (t, 3H, J = 7.2 Hz), 1.37–1.25 (m, 10 H), 0.94 (t, 3H, J = 7.6 Hz). 13C NMR (100 MHz, CDCl3) δ 174.3, 154.7, 135.0, 134.3, 129.9, 128.8, 127.6, 122.8, 78.4, 63.8, 51.5, 34.2, 32.6, 29.6, 29.3, 29.2, 29.1, 27.9, 25.1, 20.8, 14.4, 14.2. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C22H36O5Na, 403.2463; found, 403.2458.
Methyl 13(S)-((Ethoxycarbonyl)oxy)octadeca-9(R*),10(S*)-epoxy-11(E),15(Z)-dienoate (10)
Following the general epoxidation procedure, 9 (100 mg, 0.26 mmol), MTO (3.2 mg, 5 mol %), pyridine (3 μL, 12 mol %), and 30% H2O2 (45 μL, 0.39 mmol) were stirred in dry CH2Cl2 (3 mL) at −5 °C for 28 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (81 mg, 78%, ∼3:2 mixture of diastereomers).29 TLC: Rf ≈ 0.6 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 5.87 (dd, J = 6.4, 15.2 Hz, 1H), 5.67–5.59 (m, 1H), 5.55–5.47 (m, 1H), 5.11 (dt, J = 6.8, 13.2 Hz, 1H), 4.17 (q, J = 7.2 Hz, 2H), 3.66 (s, 3H), 3.38 (dd, J = 4.4, 7.2 Hz, 1H), 3.08–3.03 (m, 1H), 2.53–2.34 (m, 2H), 2.29 (t, J = 7.6 Hz, 2H), 2.07–2.01 (m, 2H), 1.63–1.24 (m, 12H), 0.96 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 174.4, 154.64, 154.63, 135.41, 135.37, 133.5, 133.3, 128.3, 128.0, 122.5, 122.4, 77.5, 77.3, 64.1, 59.13, 59.06, 56.4, 56.2, 51.7, 34.3, 32.43, 32.36, 29.43, 29.40, 29.2, 27.91, 27.86, 26.49, 26.45, 25.1, 20.9, 14.5, 14.3. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C22H36O6Na, 419.2412; found, 419.2406.
Methyl Octadeca-cis-9,10-epoxy-11(E)-enoate (12) and Methyl Octadeca-trans-11,12-epoxy-9(Z)-enoate (13)
Following the general epoxidation procedure, commercial methyl conjugated linoleate34a (11; Me CLA) (40 mg, 0.14 mmol), MTO (2 mg, 5 mol %), pyridine (2 μL, 12 mol %), and 30% H2O2 (16 μL, 0.14 mmol) were stirred in dry CH2Cl2 (2 mL) at −10 °C for 4 h. Chromatographic purification of the crude product afforded the title products as a colorless oil (30 mg, 71%, 7:3 mixture of regioisomers) whose spectral data were in accord with literature values.43
2-Phenyl-3-(2-phenyleth-(E)-en)-2,3-(E)-oxirane (15)
Following the general epoxidation procedure, commercial 4-phenyl-1(E),3(E)-butadienyl]benzene (14) (200 mg, 1.00 mmol), MTO (12 mg, 5 mol %), pyridine (10 μL, 12 mol %), and 30% H2O2 (226 μL, 2.0 mmol) were stirred in dry CH2Cl2 (10 mL) at −10 °C for 24 h. Chromatographic purification of the crude product afforded the title product44 as a colorless oil (169 mg, 74%). TLC: Rf ≈ 0.5 (20% EtOAc/hexanes).
3β-Acetyloxy-α-6,7-epoxy-4-cholestene (17)
Following the general epoxidation procedure, 16(45) (330 mg, 0.78 mmol), MTO (10 mg, 5 mol %), pyridine (8 μL, 12 mol %), and 30% H2O2 (180 μL, 1.56 mmol) were stirred in dry CH2Cl2 (10 mL) at −5 °C for 14 h. Chromatographic purification of the crude product afforded the title product as a white solid (250 mg, 73%), mp 108–110 °C. TLC: Rf ≈ 0.3 (40% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.79 (d, J = 2.0 Hz, 1H), 5.51–4.99 (m, 1H), 3.37 (d, J = 3.8 Hz, 1H), 3.23 (d, J = 3.8 Hz, 1H), 2.07 (s, 3H), 1.99–1.91 (m, 2H), 1.87–1.65 (m, 2H), 1.61–1.53 (m, 2H), 1.40–1.15 (m, 9H), 1.21–1.05 (m, 6H), 0.98 (s, 3H), 0.91 (d, 3H, J = 6.4 Hz), 0.87 (d, 3H, J = 2.5 Hz), 0.86 (d, 3H, J = 2.4 Hz), 0.71 (s, 3H), 0.59 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 170.9, 143.6, 129.9, 70.7, 56.0, 54.94, 53.2, 51.6, 43.3, 42.6, 39.71, 39.65, 36.4, 36.1, 35.3, 34.6, 33.3, 28.6, 28.2, 25.2, 24.1, 23.8, 23.1, 22.8, 21.6, 20.2, 19.1, 18.9, 12.1. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C29H46O3Na, 465.3339; found, 465.3345.
3α-Acetyloxy-α-6,7-epoxy-4-cholestene (19)
Following the general epoxidation procedure, 18(46) (145 mg, 0.34 mmol), MTO (5 mg, 5 mol %), pyridine (4 μL, 12 mol %), and 30% H2O2 (77 μL, 0.68 mmol) were stirred in dry CH2Cl2 (5 mL) at −5 °C for 14 h. Chromatographic purification of the crude product afforded the title product as a white solid (91 mg, 59%), mp 96–98 °C. TLC: Rf ≈ 0.4 (40% EtOAc/hexanes). 1H NMR (500 MHz, C6D6) δ 5.83 (d, J = 2.4 Hz, 1H), 5.47–5.43 (m,1H) 3.14 (d, J = 3.8 Hz, 1H), 2.91 (d, J = 3.7 Hz, 1H), 1.96–1.74 (m, 6H), 1.71 (s, 3H), 1.58–1.50 (m, 2H), 1.45–1.35 (m, 4H), 1.30–1.15 (m, 8H), 1.12–0.92 (m, 4H), 0.96 (d, J = 6.4 Hz, 3H), 0.93 (d, J = 2.5 Hz, 3H), 0.91 (d, J = 2.4 Hz, 3H), 0.75 (s, 3H), 0.59 (s, 3H). 13C NMR (126 MHz, C6D6) δ 169.7, 143.5, 129.8, 70.37, 70.35, 56.0, 53.9, 52.6, 51.6, 43.1, 42.3, 39.80, 39.77, 36.4, 36.1, 35.3, 34.4, 33.1, 28.6, 28.3, 25.3, 24.2, 23.8, 23.0, 22.7, 20.8, 20.1, 18.7, 11.9. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C29H46O3Na, 465.3339; found, 465.3343.
Dodeca-2(E),4(E)-dien-1-yl Acetate (20)
Following the acylation procedure above, dodeca-2(E),4(E)-dien-1-ol47 (2.2 g, 12 mmol) was treated with acetic anhydride (1.4 mL, 14.5 mmol) and pyridine (1.45 mL, 18 mmol) in CH2Cl2 (30 mL) at rt for 3 h. Chromatographic purification of the crude product using 10–20% EtOAc/hexanes as eluent afforded 20 (2.5 g, 93%) as a clear oil. TLC: Rf ≈ 0.6 (10% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 6.25 (dd, J = 10.4, 15.2 Hz, 1H), 6.02 (dd, J = 10.4, 15.2 Hz, 1H), 5.74 (dt, J = 6.8, 14.4 Hz, 1H), 5.63 (dt, J = 6.8, 14.4 Hz, 1H), 4.59 (d, J = 6.8 Hz, 2H), 2.10–2.03 (m, 2H), 2.06 (s, 3H), 1.40–1.25 (m, 10 H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.7, 136.9, 135.1, 129.2, 123.9, 65.0, 32.7, 31.9, 29.3, 29.2, 22.7, 21.0, 14.2. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C14H24O2Na, 247.1676; found, 247.1669.
(E)-3-(3-Heptyloxiran-2-yl)allyl Acetate (21)
Following the general epoxidation procedure, 20 (100 mg, 0.45 mmol), MTO (6 mg, 5 mol %), pyridine (5 μL, 12 mol %), and 30% H2O2 (100 μL, 0.90 mmol) were stirred in dry CH2Cl2 (10 mL) at −5 °C for 4 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (93 mg, 86%). TLC: Rf ≈ 0.5 (10% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.98 (dt, J = 6.0, 15.5 Hz, 1H), 5.52 (dd, J = 7.5, 15.5 Hz, 1H), 4.58 (d, J = 6.5 Hz, 2H), 3.10 (dd, J = 2.0, 7.5 Hz, 1H), 2.84–2.80 (m, 1H), 2.08 (s, 3H), 1.59–1.54 (m, 2 H), 1.47–1.38 (m, 2H), 1.31–1.25 (m, 8H), 0.89 (t, J = 6.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 170.9, 132.2, 128.7, 64.1, 60.9, 57.8, 32.2, 32.0, 29.6, 29.4, 26.1, 22.9, 21.1, 14.3. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C14H24O3, 263.1625; found, 263.1619.
Dodeca-4(E),6(E)-dien-3-yl Acetate (22)
Ethyl magnesium bromide (2.6 mL, 7.9 mmol, 3 M in THF) was added over 10 min to a 0 °C solution of E,E-2,4-decadienal (1.0 g, 6.6 mmol) in dry THF (60 mL). After 3 h, the reaction was quenched with 10% aq. NH4Cl (20 mL), the THF was removed under reduced pressure, and the reaction mixture was extracted with EtOAc (2 × 80 mL). The combined organic extracts were washed with water (2 × 40 mL) and brine (30 mL) and dried, and the residue purified by flash chromatography to provide dodeca-4(E),6(E)-dien-3-ol (1.0 g, 84%) as a colorless liquid. TLC: Rf ≈ 0.5 (20% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 6.19 (dd, J = 10.5, 15 Hz, 1H), 6.03 (dd, J = 10.5, 15.5 Hz, 1H), 5.71 (dt, J = 7.0, 14.5 Hz, 1H), 5.57 (dd, J = 7.5, 15.0 Hz, 1H), 4.05 (dt, J = 6.5, 13.5 Hz, 1H), 2.08 (q, J = 7.0 Hz, 2H), 1.63–1.49 (m, 3H), 1.42–1.36 (m, 2H), 1.39–1.25 (m, 4H), 0.90–0.87 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 135.7, 133.4, 131.4, 129.6, 77.4, 32.8, 31.6, 30.3, 29.1, 22.7, 14.3, 9.9. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C12H22ONa, 205.1571; found, 205.1567.
Following the acylation procedure above, dodeca-4(E),6(E)-dien-3-ol (500 mg, 2.7 mmol) was treated with acetic anhydride (0.3 mL, 3.2 mmol) and pyridine (0.30 mL, 3.5 mmol) in CH2Cl2 (15 mL) at rt for 3 h. Chromatographic purification of the crude product using 5–10% EtOAc/hexanes as eluent afforded the title compound 22 (580 mg, 94%) as a clear oil. TLC: Rf ≈ 0.5 (10% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 6.21 (dd, J = 10.5, 15.0 Hz, 1H), 6.00 (dd, J = 10.4, 15.0 Hz, 1H), 5.72 (dt, J = 7.5, 14.5 Hz, 1H), 5.47 (dd, J = 7.5, 15.5 Hz, 1H), 5.17 (dt, J = 7.5, 14.0 Hz, 1H), 2.07 (app q, J = 7.0 Hz, 2H), 2.05 (s, 3H), 1.70–1.58 (m, 3H), 1.41–1.34 (m, 2H), 1.33–1.24 (m, 3H), 0.90–0.87 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 170.6, 136.6, 133.4, 129.3, 128.5, 76.1, 32.8, 31.6, 29.0, 27.7, 22.7, 21.5, 14.2, 9.7. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C14H24O2Na, 247.1676; found, 247.1669.
(E)-1-(3-Pentyloxiran-2-yl)pent-1-en-3-yl Acetate (23)
Following the general epoxidation procedure, 22 (50 mg, 0.22 mmol), MTO (3 mg, 5 mol %), pyridine (2.2 μL, 12 mol %), and 30% H2O2 (38 μL, 0.33 mmol) were stirred in dry CH2Cl2 (3 mL) at −10 °C for 14 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (42 mg, 79%, ∼1:1 mixture of diastereomers). TLC: Rf ≈ 0.4 (10% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.86–5.78 (m, 1H), 5.48–5.41 (m, 1H), 5.20 (dt, J = 6.0, 12.5 Hz, 1H), 3.08–3.06 (m, 1H), 2.84–2.80 (m, 1H), 2.06 (s, 3H), 1.68–1.53 (m, 5H), 1.49–1.41 (m, 2H), 1.34–1.29 (m, 4H), 0.90 (t, J = 7.5 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 170.43, 170.41, 132.9, 132.7, 130.79, 130.1, 75.0, 74.7, 60.83, 60.81, 57.84, 57.82, 32.04, 32.04, 32.02, 31.7, 27.43, 27.37, 25.7, 22.7, 21.4, 21.3, 14.1, 9.6, 9.5. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C14H25O3, 241.1804; found, 241.1798.
1-Phenyldodeca-4(E),6(E)-dien-3-yl Acetate (24)
Following the procedure above, addition of phenylethyl Grignard to 2(E),4(E)-decadienal gave 1-phenyldodeca-4(E),6(E)-dien-3-ol. 1H NMR (500 MHz, CDCl3) δ 7.31–7.27 (m, 2H), 7.22–7.20 (m, 3H), 6.20 (dd, J = 15.2, 10.4 Hz, 1H), 6.05 (dd, J = 15.2, 10.4 Hz, 1H), 5.73 (dt, J = 14.6, 7.0 Hz, 1H), 5.62 (dd, J = 15.2, 7.1 Hz, 1H), 4.46–3.94 (m, 1H), 2.89–2.53 (m, 2H), 2.09 (app q, J = 7.3 Hz, 2H), 1.91–1.81 (m, 2H), 1.50 (br s, 1H), 1.43–1.33 (m, 2H), 1.31–1.29 (m, 4H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 141.9, 135.8, 133.1, 131.3, 129.4, 128.5, 128.44, 128.35, 125.8, 72.1, 38.8, 32.6, 31.7, 31.4, 28.9, 22.5, 14.1. HRMS (ESI-TOF) m/z [M – OH]+ calcd for C18H25, 241.1951; found, 241.1951.
Following the acylation procedure above, 1-phenyldodeca-4(E),6(E)-dien-3-ol (600 mg, 2.40 mmol) was treated with acetic anhydride (0.3 mL, 2.81 mmol) and pyridine (0.30 mL, 3.51 mmol) in CH2Cl2 (15 mL) at rt for 3 h. Chromatographic purification of the crude product using 5–10% EtOAc/hexanes as eluent afforded the title compound 24 (610 mg, 87%) as a clear oil. TLC: Rf ≈ 0.5 (10% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.45–7.22 (m, 2H), 7.21–7.19 (m, 3H), 6.25 (dd, J = 15.2, 10.4 Hz, 1H), 6.12–5.92 (m, 1H), 5.75 (dt, J = 14.7, 6.9 Hz, 1H), 5.53 (dd, J = 15.3, 7.4 Hz, 1H), 5.40–5.17 (m, 1H), 2.68–2.63 (m, 2H), 2.17–2.06 (m, 2H), 2.05 (s, 3H), 2.04–1.81 (m, 2H), 1.53–1.34 (m, 2H), 1.31–1.29 (m, 4H), 0.91 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.3, 141.4, 136.7, 133.5, 129.1, 128.4, 128.3, 128.2, 125.9, 74.5, 36.1, 32.7, 31.6, 31.4, 28.8, 22.5, 14.1. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C20H28O2Na, 323.1982; found, 323.1995.
(E)-1-(3-Pentyloxiran-2-yl)-5-phenylpent-1-en-3-yl Acetate (25)
Following the general epoxidation procedure, 24 (300 mg, 1.00 mmol), MTO (10 mg, 5 mol %), pyridine (10 μL, 12 mol %), and 30% H2O2 (225 μL, 2.0 mmol) were stirred in dry CH2Cl2 (10 mL) at −10 °C for 12 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (269 mg, 84%, ∼1:1 mixture of diastereomers). TLC: Rf ≈ 0.4 (20% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.29–7.25 (m, 2H), 7.20–7.14 (m, 3H), 5.84 (dd, J = 15.6, 6.8 Hz, 1H), 5.61–5.31 (m, 1H), 5.41–5.09 (m, 1H), 3.07 (dd, J = 7.6, 2.1 Hz, 1H), 2.79 (td, J = 5.5, 2.0 Hz, 1H), 2.66–2.61(m, 2H), 2.04 (s, 3H), 2.07–1.79 (m, 4H), 1.46–1.42 (m, 2H), 1.52–1.13 (m, 4H), 1.13–0.65 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 170.2, 141.11, 141.08, 132.6, 132.4, 130.8, 130.2, 128.4, 128.3, 126.0, 73.2, 72.9, 60.74, 60.73, 57.62, 57.59, 35.78, 35.75, 31.88, 31.86, 31.6, 31.4, 31.3, 25.5, 22.5, 21.2, 14.0. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C20H28O3, 339.1931; found, 339.1929.
tert-Butyl(((2E,4E)-2-methyldodeca-2,4-dien-1-yl)oxy)diphenylsilane (26)
To a solution of 2-methyldodeca-2(E),4(E)-dien-1-ol48 (80 mg, 0.40 mmol) and imidazole (40 mg, 0.60 mmol) in dry CH2Cl2 (0.8 mL) at 0 °C was added dropwise tert-butyldiphenylchlorosilane (143 mg, 0.52 mmol). After stirring at 0 °C for 30 min, the reaction was continued at rt for 16 h. The mixture was washed with saturated NaHCO3 solution and water (2 mL) and dried over anhydrous Na2SO4, and all volatiles were evaporated in vacuo. Purification of the residue via silica gel column chromatography using 0–20% ethyl acetate/hexane as eluent gave 26 (140 mg, 80%) as a clear oil. TLC: Rf ≈ 0.3 (hexanes). 1H NMR (400 MHz, CDCl3) δ 7.70–7.64 (m, 4H), 7.44–7.33 (m, 6H), 6.26 (dd, J = 15.0, 10.9 Hz, 1H), 6.08 (d, J = 10.8 Hz, 1H), 5.65 (dt, J = 14.6, 7.0 Hz, 1H), 4.08 (s, 2H), 2.10 (q, J = 7.2 Hz, 2H), 1.69 (s, 3H), 1.46–1.34 (m, 2H), 1.34–1.20 (m, 8H), 1.05 (s, 9H), 0.87 (t, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 135.5(4), 134.3, 134.2, 133.7, 129.5(2), 127.6(5), 125.9, 123.8, 68.6, 33.0, 31.8, 29.5, 29.21, 29.19, 26.8(3), 22.7, 19.3, 14.1, 13.9. Molecular ion could not be found by HRMS.
(E)-tert-Butyl((3-(3-heptyloxiran-2-yl)-2-methylallyl)oxy)diphenylsilane (27)/(E)-tert-Butyl((2-methyl-3-(non-1-en-1-yl)oxiran-2-yl)methoxy)diphenylsilane (28)
Following the general epoxidation procedure, 26 (66 mg, 0.17 mmol), MTO (2 mg, 5 mol %), pyridine (2 μL, 12 mol %), and 30% H2O2 (39 μL, 0.34 mmol) were stirred in dry CH2Cl2 (2.5 mL) at −5 °C for 20 h. Chromatographic purification of the crude product afforded the title products as a colorless oil (46 mg, 66%, ∼1:1 mixture of regioisomers). TLC: Rf ≈ 0.75 and 0.72 for 27 and 28, respectively (4% EtOAc/hexanes). 1H NMR of 27 (400 MHz, CDCl3) δ 7.72–7.62 (m, 4H), 7.48–7.34 (m, 6H), 5.24 (dq, J = 8.8, 1.6 Hz, 1H), 4.05 (d, J = 1.5 Hz, 2H), 3.34 (dd, J = 8.9, 2.3 Hz, 1H), 2.84 (td, J = 5.6, 2.3 Hz, 1H), 1.74 (d, J = 1.3 Hz, 3H), 1.64–1.54 (m, 3H), 1.54–1.40 (m, 1H), 1.40–1.22 (m, 8H), 1.06 (s, 9H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 141.1, 135.50(2), 135.49, 133.5, 133.4, 129.6(2), 127.6(5), 121.2, 67.9, 60.4, 55.1, 32.2, 31.8, 29.4, 29.2, 26.8(3), 26.0, 22.6, 19.3, 14.1, 13.8. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C29H42O2Si, 473.2846; found, 473.2859.
1H NMR of 28 (400 MHz, CDCl3) δ 7.72–7.64 (m, 4H), 7.46–7.34 (m, 6H), 5.87 (dt, J = 15.5, 6.8 Hz, 1H), 5.40–5.22 (m, 1H), 3.75–3.56 (m, 2H), 3.28 (d, J = 7.9 Hz, 1H), 2.17–1.98 (m, 2H), 1.46–1.20 (m, 13H), 1.06 (s, 9H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 138.1, 135.7(2), 135.6(2), 133.34, 133.30, 129.7(2), 127.67(2), 127.65(2), 124.3, 67.9, 62.6, 61.0, 32.6, 31.8, 29.11, 29.08, 29.0, 26.8(3), 22.6, 19.3, 14.5, 14.1. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C29H42O2Si, 473.2846; found, 473.2846.
4-Methylhepta-2(E),4(E)-dien-1-yl Acetate (29)
To a 0 °C solution of ethyl 4-methylhepta-2(E),4(E)-dienoate (41) (2.0 g, 11.9 mmol) in CH2Cl2 (50 mL) was slowly added DIBAL (28.7 mL, 1 M solution in toluene). After 1 h, the reaction was quenched using MeOH (10 mL), diluted with CH2Cl2 (100 mL), washed with water (2 × 50 mL) and brine (50 mL), and dried over Na2SO4, and all volatiles were evaporated. The residue was purified by flash chromatography to give 4-methylhepta-2(E),4(E)-dien-1-ol (1.13 g, 75%) as a colorless oil whose spectral data were in accord with literature values.11a
Following the acylation procedure above, 4-methylhepta-2(E),4(E)-dien-1-ol (200 mg, 1.6 mmol) was treated with acetic anhydride (0.20 mL, 1.90 mmol) and triethylamine (0.25 mL, 1.90 mmol) in CH2Cl2 (10 mL) at rt for 3 h. Chromatographic purification of the crude product using 5–10% EtOAc/hexanes as eluent afforded 29 (240 mg, 91%) as a clear oil. TLC: Rf ≈ 0.5 (7% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 6.29 (d, J = 15.6 Hz, 1H), 5.62 (dt, J = 6.8, 14.4 Hz, 1H), 5.52 (t, J = 7.2 Hz, 1H), 4.59 (d, J = 6.8 Hz, 2H), 2.17–2.08 (m, 2H), 2.06 (s, 3H), 1.73 (s, 3H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 171.0, 139.9, 136.4, 132.3, 119.9, 65.7, 21.2, 21.1, 14.1, 12.3; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C10H16O2, 191.1050; found, 191.1044.
(E)-3-(3-Ethyl-2-methyloxiran-2-yl)allyl Acetate (30)
Following the general epoxidation procedure, 29 (50 mg, 0.29 mmol), MTO (4 mg, 5 mol %), pyridine (4 μL, 12 mol %), and 30% H2O2 (60 μL, 0.43 mmol) were stirred in dry CH2Cl2 (5 mL) at −10 °C for 14 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (40 mg, 73%). TLC: Rf ≈ 0.5 (20% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.85 (dt, J = 6.0, 15.5 Hz, 1H), 5.62 (dd, J = 1.0, 15.5 Hz, 1H), 4.58 (d, J = 6.5 Hz, 1H), 2.78 (t, J = 6.5 Hz, 1H), 2.08 (s, 3H), 1.69–1.53 (m, 2H), 1.40 (s, 3H), 1.04 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 171.0, 137.6, 125.5, 66.8, 64.5, 59.0, 22.2, 21.2, 15.4, 10.7. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C10H17O3, 185.1178; found, 185.1184.
(E)-5-Methylhexa-2,4-dien-1-yl Acetate (31)
Following the acylation procedure above, (E)-5-methylhexa-2,4-dien-1-ol49 (180 mg, 1.6 mmol) was treated with acetic anhydride (0.18 mL, 1.9 mmol) and pyridine (0.22 mL, 2.4 mmol) in CH2Cl2 (10 mL) at rt for 12 h. Chromatographic purification of the crude product using 10–20% EtOAc/hexanes as eluent afforded 31(50) (210 mg, 85%) as a clear oil. TLC: Rf ≈ 0.6 (10% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 6.49 (dd, J = 10.8, 14.8 Hz, 1H), 5.82 (d, J = 10.8 Hz, 1H), 5.60 (dt, J = 7.6, 15.2 Hz, 1H), 4.58 (d, J = 6.8 Hz, 2H), 2.04 (s, 3H), 1.77 (s, 3H), 1.75 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 170.9, 137.6, 131.5, 123.9, 123.3, 65.3, 26.0, 21.0, 18.3.
(E)-3-(3,3-Dimethyloxiran-2-yl)allyl Acetate (32)
Following the general epoxidation procedure, 31 (50 mg, 0.32 mmol), MTO (4 mg, 5 mol %), pyridine (3.1 μL, 12 mol %), and 30% H2O2 (55 μL, 0.48 mmol) were stirred in dry CH2Cl2 (4 mL) at −10 °C for 14 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (43 mg, 78%). TLC: Rf ≈ 0.4 (10% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.98 (dt, J = 6.0, 13.5 Hz, 1H), 5.66 (dd, J = 7.0, 15.5 Hz, 1H), 4.60 (d, J = 6.5 Hz, 2H), 3.22 (d, J = 7.5 Hz, 1H), 2.08 (s, 3H), 1.37 (s, 3H), 1.28 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 170.7, 129.84, 129.75, 64.1, 63.2, 60.5, 24.7, 21.0, 18.8. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C9H15O3, 171.1022; found, 171.1018.
(E)-3-(Cyclohex-1-en-1-yl)allyl Acetate (33)
Following the acylation procedure above, (E)-3-(cyclohex-1-en-1-yl)prop-2-en-1-ol51 (880 mg, 1.6 mmol) was treated with acetic anhydride (0.78 mL, 8.30 mmol) and pyridine (1.3 mL, 8.83 mmol) in CH2Cl2 (10 mL) at rt for 12 h. Chromatographic purification of the crude product using 10–20% EtOAc/hexanes as eluent afforded the title compound 33 (1.1 g, 91%) as a clear oil. TLC: Rf ≈ 0.6 (10% ethyl acetate/hexane). 1H NMR (400 MHz, CDCl3) δ 6.27 (d, J = 15.6 Hz, 1H), 5.79 (br s, 1H), 5.61 (dt, J = 6.4, 14.4 Hz, 1H), 4.59 (d, J = 6.8 Hz, 2H), 2.16–1.99 (m, 4H), 2.06 (s, 3H), 1.69–1.56 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 170.9, 138.5, 135.0, 131.3, 119.0, 65.7, 26.0, 24.5, 22.6, 22.5, 21.1. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C11H16O2Na, 203.1050; found, 203.1044.
(E)-3-(7-Oxabicyclo[4.1.0]heptan-1-yl)allyl Acetate (34)
Following the general epoxidation procedure, 33 (30 mg, 0.16 mmol), MTO (2 mg, 5 mol %), pyridine (1.6 μL, 12 mol %), and 30% H2O2 (29 μL, 0.24 mmol) were stirred in dry CH2Cl2 (5 mL) at −10 °C for 14 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (26 mg, 79%). TLC: Rf ≈ 0.6 (20% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.85 (dt, J = 6.0, 15.5 Hz, 1H), 5.65 (dd, J = 1.5, 15.5 Hz, 1H), 4.57–4.56 (m, 2H), 3.03 (t, J = 1.5 Hz, 1H), 2.08 (s, 3H), 1.98–1.90 (m, 4H), 1.50–1.37 (m, 2H), 1.35–1.20 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 171.0, 136.9, 125.4, 64.5, 61.2, 58.3, 26.7, 24.7, 21.2, 19.9, 19.8. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C11H17O3, 197.1178; found, 197.1172.
(E)-4-(2,6,6-Trimethylcyclohex-1-en-1-yl)but-3-en-2-yl Acetate (35)
Following the acylation procedure above, (E)-4-(2,6,6-trimethylcyclohex-1-en-1-yl)but-3-en-2-ol (500 mg, 2.6 mmol) was treated with acetic anhydride (0.53 mL, 5.2 mmol) and pyridine (0.32 mL, 3.9 mmol) in CH2Cl2 (20 mL) at rt for 12 h. Chromatographic purification of the crude product using 5–10% EtOAc/hexanes as eluent afforded 35 (430 mg, 71%) as a clear oil whose spectral values were consistent with literature data.52 TLC: Rf ≈ 0.6 (20% EtOAc/hexanes).
(E)-4-(2,2,6-Trimethyl-7-oxabicyclo[4.1.0]heptan-1-yl)but-3-en-2-yl Acetate (36)
Following the general epoxidation procedure, 35 (50 mg, 0.20 mmol), MTO (2.2 mg, 5 mol %), pyridine (2 μL, 12 mol %), and 30% H2O2 (34 μL, 0.30 mmol) were stirred in dry CH2Cl2 (3 mL) at −5 °C for 24 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (43 mg, 82%, ∼1:1 mixture of diastereomers).53 TLC: Rf ≈ 0.4 (20% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 5.90 (d, J = 15.4 Hz, 1H), 5.67–5.61 (m, 1H), 5.38–5.36 (m, 1H), 2.05 (s, 3H), 1.87 (dt, J = 15.3, 7.7 Hz, 1H), 1.75–1.70 (m, 1H), 1.50–1.34 (m, 2H), 1.32–1.31 (m, 2H), 1.14 (s, 3H), 1.07 (s, 3H), 1.05 (s, 3H), 0.91 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 170.57, 170.56, 133.23, 133.17, 127.9, 71.0, 70.8, 70.5, 65.4, 65.2, 35.82, 35.81, 33.5, 30.2, 30.1, 26.0, 25.95, 25.94, 25.92, 21.63, 21.59, 21.12, 21.07, 20.9, 20.7, 17.3.
(E)-3-(3-((E)-Prop-1-en-1-yl)oxiran-2-yl)allyl Acetate (38)
Following the general epoxidation procedure, 37(54) (100 mg, 0.60 mmol), MTO (7.5 mg, 5 mol %), pyridine (6 μL, 12 mol %), and 30% H2O2 (103 μL, 0.90 mmol) were stirred in dry CH2Cl2 (5 mL) at −5 °C for 12 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (71 mg, 65%) and ∼10% bis-epoxide that was not further characterized. TLC: Rf ≈ 0.4 (20% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3) δ 6.02–5.86 (m, 1H), 5.54 (dd, J = 15.6, 7.5 Hz, 1H), 5.25–5.13 (m, 1H), 4.57 (d, J = 5.9 Hz, 2H), 3.29–3.16 (m, 2H), 2.07 (s, 2H), 1.74 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 170.8, 132.2, 131.4, 128.8, 127.9, 63.9, 60.6, 59.2, 21.0, 18.1. HRMS (ESI-TOF) m/z [M + 1]+ calcd for C10H14O3, 183.1016; found, 183.1011.
Ethyl (E)-3-(3-Heptyloxiran-2-yl)acrylate (40)
Following the general epoxidation procedure, 39(47) (100 mg, 0.45 mmol), MTO (11 mg, 5 mol %), pyridine (8 μL, 12 mol %), and 30% H2O2 (100 μL, 0.90 mmol) were stirred in dry CH2Cl2 (5 mL) at −10 °C for 60 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (58 mg, 64%) whose spectral data were consistent with published values.55
Ethyl (E)-3-(3-Ethyl-2-methyloxiran-2-yl)acrylate (42)
Following the general epoxidation procedure, 41(11a) (200 mg, 1.20 mmol), MTO (15 mg, 5 mol %), pyridine (11 μL, 12 mol %), and 30% H2O2 (272 μL, 2.4 mmol) were stirred in dry CH2Cl2 (10 mL) at −10 °C for 24 h. Chromatographic purification of the crude product afforded the title product as a colorless oil (210 mg, 94%) whose spectral values were consistent with literature data.11a TLC: Rf ≈ 0.4 (20% EtOAc/hexanes).
Acknowledgments
Financial support was provided by the Robert A. Welch Foundation (I-0011) and USPHS NIH (GM31278, DK38226, HL034300). Prof. Mats Hamberg is thanked for sharing information regarding standards. Analyses was kindly provided by the Shimadzu Center for Advanced Analytical Chemistry at U.T.-Arlington.
Supporting Information Available
1H and 13C NMR of all new compounds; HPLC chromatograms of epoxide standards, epoxides 5a/6a; and cochromatographic comparisons. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Oxenoids:; a Minko Y.; Marek I. Org. Biomol. Chem. 2014, 12, 1535–1546. [DOI] [PubMed] [Google Scholar]; b Bergmeier S. C.; Lapinsky D. J. Prog. Heterocycl. Chem. 2012, 24, 89–113. [Google Scholar]; Enzymatic:; c Larsen A. T.; May E. M.; Auclair K. J. Am. Chem. Soc. 2011, 133, 7853–7858. [DOI] [PubMed] [Google Scholar]; Dioxiranes:; d Adam W.; Saha-Möller C. R.; Zhao C.-G. Org. React. 2004, 61, 219–516. [Google Scholar]; Biomimetic:; e Aouf C.; Durand E.; Lecomte J.; Figueroa-Espinoza M.-C.; Dubreucq E.; Fulcrand H.; Villeneuve P. Green Chem. 2014, 16, 1740–1754. [Google Scholar]; Organocatalytic:; f Zhu Y.; Wang Q.; Cornwall R. G.; Shi Y. Chem. Rev. 2014, 114, 8199–8256. [DOI] [PubMed] [Google Scholar]
- a Aziridines and Epoxides in Organic Synthesis; Yudin A. K., Ed.; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]; b Das B.; Damodar K. In Heterocycles in Natural Product Synthesis; Majumdar K. C., Chattopadhyay S. K, Eds.; Wiley-VCH: Weinheim, Germany, 2011; Chapter 3, pp 63–95. [Google Scholar]; c Muzart J. Eur. J. Org. Chem. 2011, 4717–4741. [Google Scholar]; d Taylor S. K. Tetrahedron 2000, 56, 1149–1163. [Google Scholar]
- Marco-Contelles J.; Molina M. T.; Anjum S. Chem. Rev. 2004, 104, 2857–2900. [DOI] [PubMed] [Google Scholar]
- a Joergensen K. A. Chem. Rev. 1989, 89, 431–458. [Google Scholar]; b Mizuno N.; Yamaguchi K.; Kamata K. Coord. Chem. Rev. 2005, 249, 1944–1956. [Google Scholar]
- He J.; Ling J.; Chiu P. Chem. Rev. 2014, 114, 8037–8128. [DOI] [PubMed] [Google Scholar]
- Sheng M. N.; Zajacek J. G. J. Org. Chem. 1970, 35, 1839–1843. [Google Scholar]
- Ledon H. J.; Varescon F. Inorg. Chem. 1984, 23, 2735–2737. [Google Scholar]
- a Thomsen D. S.; Schiott B.; Joergensen K. A. J. Chem. Soc., Chem. Commun. 1992, 1072–1074. [Google Scholar]; b Rasmussen K. G.; Thomsen D. S.; Joergensen K. A. J. Chem. Soc., Perkin Trans. 1 1995, 2009–2017. [Google Scholar]
- a Lee N. L.; Jacobsen E. N. Tetrahedron Lett. 1991, 32, 6533–6536. [Google Scholar]; b Chang S.; Lee N. H.; Jacobsen E. N. J. Org. Chem. 1993, 58, 6939–6941. [Google Scholar]
- Baunstark A.; Vasquez P. C.; Michelena-Baez E.; Chen H.-H. Heterocycl. Commun. 2012, 18, 75–78. [Google Scholar]
- a Frohn M.; Dalkiewicz M.; Tu Y.; Wang Z.-X.; Shi Y. J. Org. Chem. 1998, 63, 2948–2953. [Google Scholar]; b Burke C. P.; Shi Y. Angew. Chem., Int. Ed. 2006, 45, 4475–4478. [DOI] [PubMed] [Google Scholar]
- The formation of allylic E- or Z-epoxyols via rearrangement of fatty acid hydroperoxides is an important source of autacoids.; a Mammals: Lederer M. O.; Schuler A.; Ohmenhauser M. J. Agric. Food Chem. 1999, 47, 4611–4620. [DOI] [PubMed] [Google Scholar]; b Plants: Kato T.; Yamaguchi Y.; Ohnuma S.; Uyehara T.; Namai T.; Kodama M.; Shiobara Y. J. Chem. Soc., Chem. Commun. 1986, 743–744. [Google Scholar]; c Marine organisms: Piomelli D.; Shapiro E.; Zipkin R.; Schwartz J. H.; Feinmark S. J. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 1721–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Stagonolide D.; Vadhadiya P. M.; Puranik V. G.; Ramana C. V. J. Org. Chem. 2012, 77, 2169–2175. [DOI] [PubMed] [Google Scholar]; b Fostriecin: Reddy Y. K.; Falck J. R. Org. Lett. 2002, 4, 969–971. [DOI] [PubMed] [Google Scholar]; c Aigilomycin B.; Xu L.; He Z.; Xue J.; Chen X.; Wei X. J. Nat. Prod. 2010, 73, 885–889. [DOI] [PubMed] [Google Scholar]; d Mueggelone: Motoyoshi H.; Ishigami K.; Kitahara T. Tetrahedron 2001, 57, 3899–3908. [Google Scholar]; e Palmerolide A.; Lisboa M. P.; Dudley G. B. Chem.—Eur. J. 2013, 19, 16146–16168. [DOI] [PubMed] [Google Scholar]; f Hepoxilin A3: Yu Z.; Schneider C.; Boeglin W. E.; Marnett L. J.; Brash A. R. Proc. Nat. Acad. Sci. U.S.A. 2003, 100, 9162–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuki T.; Martin V. S. Org. React. 1996, 48, 1–300. [Google Scholar]
- Zhang W.; Basak A.; Kosugi Y.; Hoshino Y.; Hisashi Yamamoto H. Angew. Chem., Int. Ed. 2005, 44, 4389–4391. [DOI] [PubMed] [Google Scholar]
- a Falck J. R.; Manna S.; Siddhanta A. K.; Capdevila J.; Buynak J. D. Tetrahedron Lett. 1983, 24, 5715–5718. [Google Scholar]; b Falck J. R.; Manna S.; Capdevila J.; Buynak J. D. Tetrahedron Lett. 1983, 24, 5719–5720. [Google Scholar]
- For an alternative approach to this class, see:; Tortosa M. Angew. Chem., Int. Ed. 2011, 50, 3950–3953. [DOI] [PubMed] [Google Scholar]; Related study:; Kurashina Y.; Kuwahara S. Biosci. Biotechnol. Biochem. 2012, 76, 605–607. [DOI] [PubMed] [Google Scholar]
- Williams D. E.; Sturgeon C. M.; Roberge M.; Andersen R. J. J. Am. Chem. Soc. 2007, 129, 5822–5823. [DOI] [PubMed] [Google Scholar]
- Baldwin J. E.; Davies D. I.; Hughes L.; Gutteridge N. J. J. Chem. Soc., Perkin Trans. 1 1979, 115–121. [Google Scholar]
- a Rudolph J.; Reddy L. K.; Chiang J. P.; Sharpless K. B. J. Am. Chem. Soc. 1997, 119, 6189–6190. [Google Scholar]; b Epoxidation of homoallylic alcohols: Yamazaki S. J. Org. Chem. 2012, 77, 9884–9888. [DOI] [PubMed] [Google Scholar]
- Lane B. S.; Burgess K. J. Am. Chem. Soc. 2001, 123, 2933–2934. [DOI] [PubMed] [Google Scholar]
- Katsuki T.; Sharpless K. B. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar]
- Barlan A. U.; Basak A.; Yamamoto H. Angew. Chem., Int. Ed. 2006, 45, 5849–5852. [DOI] [PubMed] [Google Scholar]
- Gelalcha F. G.; Bitterlich B.; Anilkumar G.; Tse M. N.; Beller M. Angew. Chem., Int. Ed. 2007, 46, 7293–7296. [DOI] [PubMed] [Google Scholar]
- a Wu M.; Wang B.; Wang S.; Xia C.; Sun W. Org. Lett. 2009, 11, 3622–3625. [DOI] [PubMed] [Google Scholar]; b Maity N. C.; Kumar Bera P.; Ghosh D.; Abdi S. H. R.; Kureshy R. I.; Khan N.-u. H.; Bajaj H. C.; Suresh E. Catal.: Sci. Technol. 2014, 4, 208–217. [Google Scholar]
- Dubois G.; Murphy A.; Stack T. D. Org. Lett. 2003, 5, 2469–2472. [DOI] [PubMed] [Google Scholar]
- a Catalytic system: White M. C.; Doyle A. G.; Jacobsen E. N. J. Am. Chem. Soc. 2001, 123, 7194–7195. [DOI] [PubMed] [Google Scholar]; b BPBP ligand: Suzuki K.; Oldenburg P. D.; Que L. Jr. Angew. Chem., Int. Ed. 2008, 47, 1887–1889. [DOI] [PubMed] [Google Scholar]
- A 4:1 mixture of methyl 9(R),10(S)- and methyl 9(S),10(R)-epoxy-13(S)-hydroxy-11(E)-octadecenoate was prepared enzymatically and acetylated to give 2 and 3, respectively.Hamberg M.; Hamberg G. Arch. Biochem. Biophys. 1990, 283, 409–416. [DOI] [PubMed] [Google Scholar]; Additionally, an authentic sample of methyl 9(S),10(R)-epoxy-13(S)-hydroxy-11(E)-octadecenoate was purchased from Larodan Fine Chemicals AB, Malmö, Sweden. The mixture of 2 and 3 was deacetylated using K2CO3 in MeOH, and the structures of the C(13)-alcohols were confirmed via HPLC: Ascentis Express (Sigma-Aldrich) (15 cm × 4.6 mm, 2.7 μ), hexanes/IPA (100:1), 2 mL/min, 205 nm. Commercial methyl 9(S),10(R)-epoxy-13(S)-hydroxy-11(E)-octadecenoate and the alcohol derived from 3 had tR ∼ 20.5, and the alcohol derived from 2 had tR ∼ 18.4 min. Structure assignments of other epoxides were made by analogy or using standards prepared as noted in ref (29).
- Authentic, individual diastereomeric standards were also prepared by catalytic asymmetric distal epoxidation of the corresponding 1-acyl(silyl)oxypenta-2,4-diene using a chiral Ti(salan) complex and H2O2:Jat J. L.; De S. R.; Kumar G.; Adebesin A. M.; Gandham S. K.; Falck J. R., submitted for publication.
- Effect of ligands upon Re catalyzed epoxidations:van Vliet M. C. A.; Arends I. W. C. E.; Sheldon R. A. J. Chem. Soc., Perkin Trans. 1 2000, 377–380. [Google Scholar]
- Kühn F. E.; Zhao J.; Herrmann W. A. Tetrahedron: Asymmetry 2005, 16, 3469–3479. [Google Scholar]
- Review of MTO reactions:Kühn F. E.; Scherbaum A.; Herrmann W. A. J. Organomet. Chem. 2004, 689, 4149–4164. [Google Scholar]
- a Adam W.; Mitchell C. M. Angew. Chem., Int. Ed. 1996, 35, 533–535. [Google Scholar]; b Boehlow T. R.; Spilling C. D. Tetrahedron Lett. 1996, 37, 2717–2720. [Google Scholar]
- Seminal, but brief, reports of diene epoxidation using MTO:; a Jie M. S. F. L. K.; Lam C. N. W.; Ho J. C. M.; Lau M. M. L. Eur. J. Lipid Sci. Technol. 2003, 105, 391–396. [Google Scholar]; b Musumeci D.; Sica D. Steroids 2002, 67, 661–668. [DOI] [PubMed] [Google Scholar]
- Gardner H. W.; Weisleder D. Lipids 1972, 7, 191–193. [Google Scholar]
- Kato T.; Nakai T.; Ishikawa R.; Karasawa A.; Namaib T. Tetrahedron: Asymmetry 2001, 12, 2695–2701. [Google Scholar]
- Denis C.; Gerard L. Synthesis 1993, 4, 377–379. [Google Scholar]
- Zamora R.; Gallardo E.; Hidalgo F. J. J. Agric. Food Chem. 2008, 56, 7970–7975. [DOI] [PubMed] [Google Scholar]
- Tranchepain I.; Berre F. L.; DurBault A.; Merrer Y. L.; Depezay J. C. Tetrahedron 1989, 45, 2057–2065. [Google Scholar]
- Johnson D. V.; Griengl H. Tetrahedron. 1997, 53, 617–624. [Google Scholar]
- Kato T.; Watanabe T.; Hirukawa T.; Tomita N.; Namai T. Bull. Chem. Soc. Jpn. 1996, 69, 1663–1666. [Google Scholar]
- Piazza G. J.; Nuñez A.; Foglia T. A. Lipids 2003, 38, 255–261. [DOI] [PubMed] [Google Scholar]
- Aggarwal V. K.; Alonso E.; Bae I.; Hynd G.; Lydon K. K.; Palmer M. J.; Patel M.; Porcelloni M.; Richardson J.; Stenson R. A.; Studley J. R.; Vasse J.-L.; Winn C. L. J. Am. Chem. Soc. 2003, 125, 10926–10940. [DOI] [PubMed] [Google Scholar]
- Ma E.; Kim H.; Kim E. Steroids 2005, 70, 245–250. [DOI] [PubMed] [Google Scholar]
- Ma E.; Kim E. Steroids 2007, 72, 360–367. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Ma J.; Cheon H. S.; Kishi Y. Angew. Chem., Int. Ed. 2007, 46, 1333–1336. [DOI] [PubMed] [Google Scholar]
- Falck J. R.; He A.; Fukui H.; Tsutsui H.; Radha A. Angew. Chem., Int. Ed. 2007, 46, 4527–4529. [DOI] [PubMed] [Google Scholar]
- DeBoef B.; Counts W. R.; Gilbertson S. R. J. Org. Chem. 2007, 72, 799–804. [DOI] [PubMed] [Google Scholar]
- Takacs J. M.; Clement F.; Zhu J.; Chandramouli S. V.; Gong X. J. Am. Chem. Soc. 1997, 119, 5804–5817. [Google Scholar]
- Liu B.; Li K.-N.; Luo S.-W.; Huang J.-Z.; Pang H.; Gong L.-Z. J. Am. Chem. Soc. 2013, 135, 3323–3326. [DOI] [PubMed] [Google Scholar]
- Azzari E.; Faggi C.; Gelsomini N.; Taddei M. J. Org. Chem. 1990, 55, 1106–1108. [Google Scholar]
- Aleu J.; Brenna E.; Fuganti C.; Serra S. J. Chem. Soc., Perkin Trans. 1 1999, 271–278. [Google Scholar]
- Sun H.; Kong R.; Zhu D.; Lu M.; Ji Q.; Liew C. W.; Lescar J.; Zhong G.; Liang Z.-X. Chem. Commun. 2009, 47, 7399–7401. [DOI] [PubMed] [Google Scholar]
- Sabitha G.; Reddy C. S.; Srihari P.; Yadav J. S. Synthesis 2003, 17, 2699–2704. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.