


Abstract
The conformational dynamics of the polymorphous trigger loop (TL) in RNA polymerase (RNAP) underlie multiple steps in the nucleotide addition cycle and diverse regulatory mechanisms. These mechanisms include nascent RNA hairpin-stabilized pausing, which inhibits TL folding into the trigger helices (TH) required for rapid nucleotide addition. The nascent RNA pause hairpin forms in the RNA exit channel and promotes opening of the RNAP clamp domain, which in turn stabilizes a partially folded, paused TL conformation that disfavors TH formation. We report that inhibiting TH unfolding with a disulfide crosslink slowed multiround nucleotide addition only modestly but eliminated hairpin-stabilized pausing. Conversely, a substitution that disrupts the TH folding pathway and uncouples establishment of key TH–NTP contacts from complete TH formation and clamp movement allowed rapid catalysis and eliminated hairpin-stabilized pausing. We also report that the active-site distal arm of the TH aids TL folding, but that a 188-aa insertion in the Escherichia coli TL (sequence insertion 3; SI3) disfavors TH formation and stimulates pausing. The effect of SI3 depends on the jaw domain, but not on downstream duplex DNA. Our results support the view that both SI3 and the pause hairpin modulate TL folding in a constrained pathway of intermediate states.
INTRODUCTION
Structures of multisubunit RNA polymerases (RNAPs), structures of elongation complexes (ECs), and mechanistic studies reveal that the catalytic cycle of RNAP is highly regulated, for instance, at transcriptional pause sites. This regulation is mediated by mobile structural elements in the RNAP active site, among which the most important are a ∼37-aa bridge helix (BH) that spans the active-site cleft just downstream of the catalytic center and an adjacent ∼47-aa polymorphous loop called the trigger loop (TL) or trigger helices (TH), depending on its conformation.
Rapid, high-fidelity nucleotide addition in RNAP requires formation of the TH after cognate NTP binding. The helical hairpin TH form a three-helix bundle with the BH that aligns the NTP substrate through extensive contacts, dehydrates the active site, and shifts the positions of two catalytic Mg2+ ions, the RNA 3′ OH and the NTP α-phosphate to stabilize a trigonal bipyramidyl transition state (1–4). In most RNAP and EC crystal structures, however, both the BH and TH are deformed at Gly/Pro-containing ‘hinge’ regions that appear to destabilize the active, three-helix bundle conformation (5–9). The TH undergo the most dramatic change in which the loop-proximal portion of the helical hairpin completely unfolds when cognate NTP is absent. Thus, the TL/TH module can be functionally subdivided into seven discrete segments: the ‘base’ helices TH1b and TH2b that do not unfold; Hinge1 and Hinge2 that connect the base helices to the polymorphous region of the TL/TH; TH1a and TH2a, which transition between α-helix, partially folded (paused), and random-coil conformations; and a loop that connects TH1a and TH2a (Figure 1). TH1a contains all the side chains that contact NTP: β′M932, R933 and H936 in Escherichia coli RNAP (EcoRNAP; Figure 1). An additional TH1a-NTP contact to the 2′ and 3′ OH by a highly conserved Gln residue (β′Q929 in EcoRNAP) has been observed for Saccharomyes cerevisiae RNAPII (SceRNAPII) and Pyrococcus furiosis RNAP (PfuRNAP) (10,11), although this contact is not seen in a Thermus thermophilus RNAP (TthRNAP) NTP-EC structure (2). Thus, control of catalysis by RNAP can be viewed as the regulated establishment and dissociation of TH1a–NTP contacts that lower the activation energy for nucleotide addition. Inability to establish these contacts, for instance, in an RNAP containing two proline substitutions in TH1a (L930P, T931P in EcoRNAP; called LTPP) or truncation of the TL/TH at the hinge regions, causes a ≥104-fold decrease in RNAP kcat (2,4,12) and an ∼102-fold decrease in NTP selectivity (4,11,13,14). The F-loop, βDloopII, fork loop 2 and link modules together form a BH/TH cap, whose extensive contact network modulates TH formation (15–17).
Figure 1.
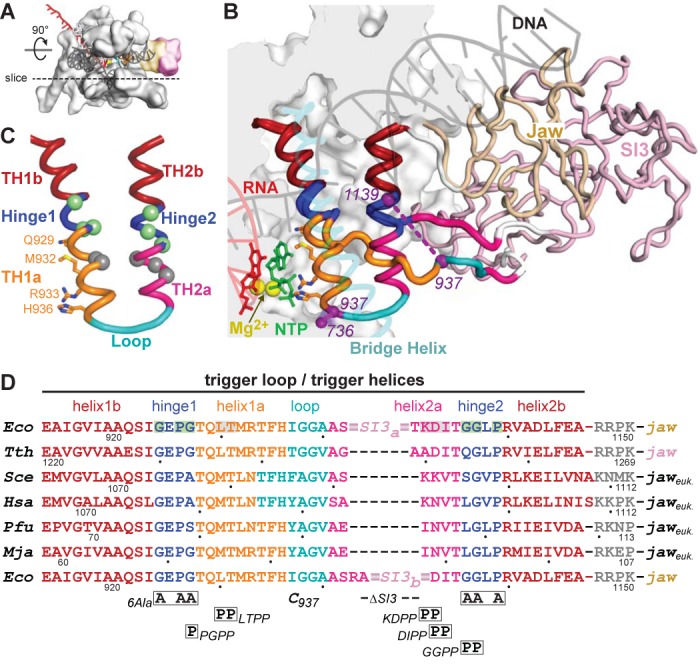
Structure and sequences of the TL/TH domain. (A) Model of the EcoRNAP EC in light gray space-fill showing the DNA (semi-transparent dark gray), RNA (red), active-site Mg2+ (yellow sphere) and the jaw (wheat) and SI3 (pink) domains. The dotted line depicts the approximate location of a slice through the EC that is rotated up ∼90° to give the main view in panel B. (B) Magnified view of TH and TL conformations in a model of the EcoRNAP EC based on the crystal structure of a TthRNAP EC bound to AMPCPP (PDB 2o5j (2) and a hydrid model of EcoRNAP (33)). The inset shows a space-filled rendering of EcoRNAP with some loops (BH, TL, lid, rudder, Zn-binding) shown as backbone worms for clarity. Nucleic acids, active-site Mg2+, jaw and SI3 domains are colored as in (A). The jaw (wheat) and SI3 (pink) domains are shown as worms, with only the connection of SI3 to the unfolded TL depicted for clarity (white segments are the connectors between the TL and SI3). The TH/TL is colored as follows: bases helices TH1b and TH2b (dark red), Hinges 1 and 2 (blue), TH1a (orange) and TH2a (magenta) and loop (cyan). The 188-aa SI3 is inserted between the loop and TH2a. Also shown are the incoming NTP (green), the BH (transparent cyan) and the Cys-pair reporters that detect the folded and unfolded conformation of the TH/TL (purple, F937–736 and U937–1139, respectively). Base helices TH1b and TH2b (dark red) do not change structure in the TL to TH transition. (C) Substructure of the TH and substitutions in EcoRNAP tested in this study. TH/TL is depicted in the same colors as in panel B also showing the positions of amino acids that make active site contacts to the NTP substrate to promote catalysis. The hinges (1 and 2; blue) contain Gly and Pro residues that destabilize the TH. Substitution locations depicted as spheres colored green (to stabilize) or gray (to destabilize) the TH, as highlighted in (D). (D) The sequences of the TL/TH in RNAPs from the three domains of life are shown colored as in panels B and C. Eco, Escherichia coli RNAP. Tth, Thermus thermophilus RNAP. Sce, Sacchaaromyces cerevisiae RNAPII. Hsa, Homo sapiens RNAPII. Pfu, Pyrococcus furiosis RNAP. Mja, Methanocaldococcus jannaschii RNAP. The locations of substitutions tested in this study are shown below the sequence alignment, with the two different SI3 deletions (SI3a and SI3b) shown in the top and bottom rows, respectively. The eukaryotic and archaeal jaw domains are not SBHM folds like the bacterial jaw domains.
In E. coli, several distinct types of pauses occur, with different effects on the active site. Elemental pauses occur about 10 times per kb on average and are thought to arise by a sequence-induced rearrangement of the RNAP active site that blocks loading of the template base. The elemental pauses help synchronize transcription with translation but also allow time for subsequent rearrangements and stabilization of the paused state by RNA structure formation, RNA backtracking or regulator interactions. Hairpin-stabilized pauses arise from elemental pauses when a nascent RNA structure forms in the exit channel and is thought to inhibit establishment of key TH1a-NTP interactions by forcing open the RNAP clamp domain and altering clamp–TH contacts to disfavor TH formation and favor a partially folded TL (17,18). Hairpin-stabilized pauses play key roles in gene regulation by coupling transcription to changes in nascent RNA structure caused by translation or ligand binding. At the well studied his leader pause, the hairpin-stabilized state generates a simple trigger that enables pause release when a translating ribosome melts the pause hairpin, resulting in precise coupling of transcription and translation in the his operon attenuation control region (12,17,18). Other hairpin-stabilized pauses guide formation of RNA structures into biologically active forms, for instance, for RNase P ribozyme function (19,20), to sense small molecule ligands in riboswitch control regions (21,22), and possibly to guide formation of intrinsic terminator hairpins (23).
In the context of these TL and pause models, key questions remain unanswered. First, is complete TH unfolding to the TL required for rapid nucleotide addition? TL unfolding is proposed to mediate translocation and allow PPi release (1,8,24–28). We recently reported that a TH-stabilizing disulfide between engineered Cys residues in the TH loop and the cap region (β′I736C-β′I937C in EcoRNAP; abbreviated F937–736; Figure 1B) slowed kcat only by ∼20-fold in a single-turnover assay, even though crosslinks stabilizing the partially or completely unfolded TL slowed kcat by ≥104 (17). This finding could indicate that modest TH1a unfolding is sufficient to admit NTP to the active site but it was unclear how the F937–736 crosslink would affect multiround transcription requiring PPi release and translocation.
Second, is formation of a complete TH1 and a complete TH2a necessary to form TH1a–NTP contacts? The need for TH2a folding is of particular interest in E. coli and related RNAPs where the TH2a-loop junction contains a large insertion of two or more sandwich-barrel hybrid motif domains (SBHMs) (29–32). In EcoRNAP, the 188-aa TL insertion, consisting of two SBHMs, is called sequence insertion 3 (SI3) (29) and interacts with a third SBHM domain called the jaw when the TL is unfolded (Figure 1B) (33). Alterations in SI3 or its binding to a mAb are known to affect nucleotide addition, hairpin-stabilized pausing, and intrinsic cleavage, in some cases with adaptive advantage for growth in minimal medium (29,34,35). Substitutions in Hinge2 or TH2a that alter nucleotide addition rates suggest TH2a folding affects TH1a folding (9,24,27,36), but whether TH2a is as important as TH1a and whether SI3 acts principally by modulating TL folding, or through other interactions within the EC or with transcription factors (37), remains unclear. Finally, the proposed linkage of clamp conformation to TH1a formation presumably is mediated by the hinge1-TH transition, but this linkage has not been tested to date.
To investigate the requirements for TH unfolding during multiround nucleotide addition, the interdependence of TH1a, TH2a and hinge folding in EcoRNAP, and the effects of SI3 on TL function, we constructed new TL variants in EcoRNAP and assessed their effects on nucleotide addition and pausing using in vitro transcription assays and their effects on TL folding using disulfide bond formation assays.
MATERIALS AND METHODS
Reagents and materials
Plasmids and oligonucleotides are listed in Supplementary Table S1. RNA and DNA oligonucleotides were obtained from Integrated DNA Technologies (IDT) (Corvalville, IA, USA) and purified by denaturing polyacrylamide gel electrophoresis (PAGE) before use. The 3′deoxyG13 RNA was obtained from Thermo Fisher Scientific Biosciences Inc. [γ-32P]ATP, [α-32P]CTP and [α-32P]GTP were from PerkinElmer Life Sciences; rNTPs were from Promega (Madison, WI, USA).
Proteins
His-tagged wild-type and mutant core RNAPs were purified from E. coli strain RL2657 transformed with the appropriate plasmids (Supplementary Table S1). Cells were grown in 4 l Luria broth supplemented with kanamycin (25 μg/ml) on a platform shaker (250 revolutions per minute (rpm) at 37°C) to an apparent OD600 of 0.5. Protein expression was induced by addition of isopropyl-beta-D-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. The induced cells were then incubated at 37°C for 2.5 h or 25°C–30°C for 3–12 h at 200 rpm and harvested by centrifugation (3440 × g, 15 min, 4°C). The cell pellet from 1 l of cells was resuspended in 25 ml lysis buffer (50 mM Tris-Cl, pH 8.0, 2 mM ethylenediaminetetraacetic acid (EDTA), 5% v/v glycerol, 1 mM β-mercaptoethanol, 1 mM DTT, 300 mM NaCl, 0.25 ml of 10 mg phenylmethylsulfonyl fluoride (PMSF)/ml) in ethanol and 0.5 ml of a protease inhibitor cocktail containing 31.2 mg benzamide, 0.5 mg chymostatin, 0.5 mg leupeptin, 0.1 mg pepstatin, 1 mg aprotonin and 1mg antipain per ml in ethanol). The resuspended cells were lysed by sonication. Subsequent purification steps were carried out at 4°C unless otherwise indicated. Crude RNAPs were enriched by polyethylenimine (PEI) precipitation (38). PEI (ave MW 60K; Acros Organics #17857) was added to 0.6% final with gentle stirring and the precipitate was recovered by centrifugation (20 000 × g, 15 min, 4°C). The PEI pellets were washed by gentle resuspension in 25 ml TGEDZ buffer (10 mM Tris-Cl pH 8, 0.1 mM EDTA, 5 μM ZnCl2, 1 mM DTT, 5% glycerol) plus 500 mM NaCl followed by centrifugation (20 000 × g,15 min, 4°C). RNAPs were eluted by gentle resuspension of the pellets in 25 ml TGEDZ buffer +1M NaCl followed by centrifugation (20 000 x g,15 min, 4°C). RNAP was precipitated from the supernatant at 4°C by slow addition of solid ammonium sulfate with gentle stirring to 37% w/v final, allowed to stand at 4°C overnight and then recovered by centrifugation (20 000 × g, 15 min, 4°C). The precipitated RNAP was redissolved in 35 ml of binding buffer (20 mM Tris-Cl, pH 8, 500 mM NaCl, 5 mM βME; beta-mercaptoethanol) containing 5 mM imidazole, loaded onto a pre-charged nickel agarose (5 ml HisTrap) column, washed and then eluted with a gradient of 5–500 mM imidazole. Fractions containing RNAP were located by PAGE, pooled and diluted in TGEDZ buffer to 200 mM NaCl. The dialyzed RNAP was then loaded onto a heparin-sepharose column (5 ml HiTrap), washed with 25 ml of TGEDZ buffer plus 200 mM NaCl and eluted with TGEDZ buffer plus 500 mM NaCl. Purified RNAPs were dialyzed into storage buffer (20 mM Tris-Cl, pH 8, 250 mM NaCl, 20 μM ZnCl2, 1 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 25% glycerol) and stored in small aliquots at –80°C.
Cys pair reporter (CPR) crosslinking assays
CPR crosslinking assays (Figures 3, 6 and 7) were performed as described previously (17). Nucleic acid scaffolds were prepared by annealing 10 μM RNA, 12 μM template DNA (T-DNA) and 15 μM non-template DNA (NT-DNA) in reconstitution buffer (20 mM Tris-HCl pH 8, 20 mM NaCl and 1 mM EDTA). ECs (elongation complexes) and paused elongation complexes (PECs) were formed by incubating 1 μM RNAP and 2 μM scaffold in buffer A (50 mM Tris-HCl pH 8, 20 mM NaCl, 10 mM MgCl2, 1 mM EDTA and 2.5 ug/ml acetylated bovine serum albumin (BSA)) for 15 min at room temperature (RT). For crosslinking reactions with NTP, ECs were incubated for 15 min at RT with 10 mM GTP. Free RNAP, EC, or PEC (final RNAP 0.8 uM and scaffold 1.6 uM) were incubated for 60 min with 2.5 mM CSSC and 0.05–20 mM DTT (E = –0.140 to –0.414 V; 0.1 mM CSSC and 0.002–0.8 mM DTT was used for U937–1139 RNAP) and stopped with 50 mM iodoacetamide. Samples were separated by native PAGE to verify reconstitution efficiency and by sodium dodecyl sulphate-PAGE (SDS-PAGE) 7.5% GE Healthcare PhastGel (10–15% PhastGel for any U937–1139 RNAP) to quantify formation of crosslinks. Gels were silverstained or Coomassie brilliant blue R-250 stained, image digitized (FluorChem CCD; Protein Simple, Inc.) and quantified (ImageJ).
Figure 3.
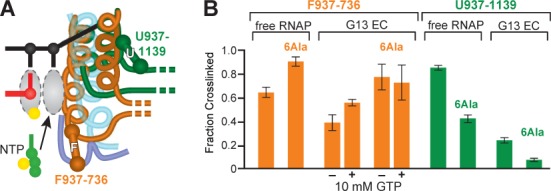
6Ala TH stabilization detected by CPR mapping of TL/TH conformation. (A) Schematic of F937–736 (orange) and U937–1139 (green) CPRs detecting the folded TH and unfolded TL conformations. (B) F937–736 and 6Ala F937–736 fraction crosslinked (orange) and U937–1139 and 6Ala U937–1139 (green; contains 5 of the 6Ala residues in the 6Ala RNAP, sixth residue is Cys in this variant). Bar graphs show the fraction crosslinked at saturating levels (EH –0.136 V) for free core RNAP or EC 3′deoxyG13 (ScaffoldGTP; Figure 2A; EC G13 containing 3′-dGMP) in the presence and absence of 10 mM GTP. Data are means ± SD of experimental triplicates.
Figure 6.
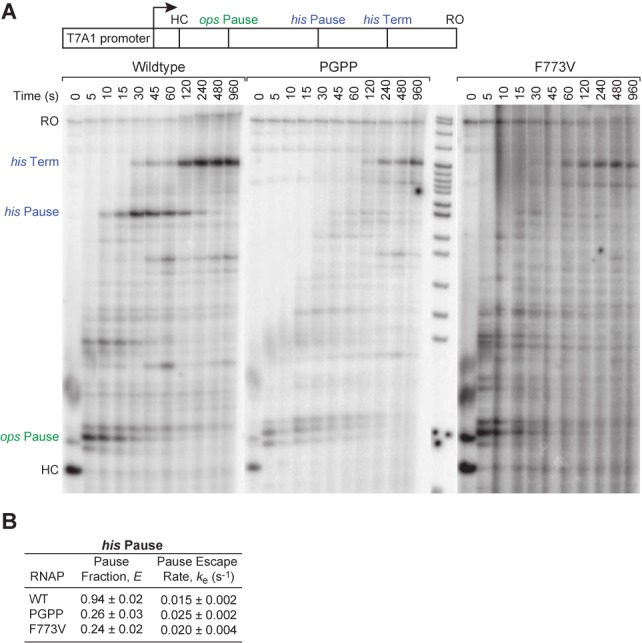
Uncoupling TH1a formation from TH1b promotes catalysis and prevents hairpin-stabilization of pausing. (A) Processivity and pausing of wild-type, PGPP and F773V RNAPs analyzed by promoter-initiated transcription on a long template containing the ops pause, his pause and his terminator (pIA349, Supplementary Table S1), reactions separated by 8% PAGE. Reactions proceed from halted complexes (HC) after addition of all four NTP at time 0. RO, run-off product. (B) The pause fraction and escape rate on the his pause for wild-type, PGPP and F773V RNAPs were quantified on reconstituted scaffoldPEC as in Figure 5 but starting reactions at U29 (pause) at time 0 and measuring pause escape. Data are means ± SD of experimental triplicates.
Figure 7.
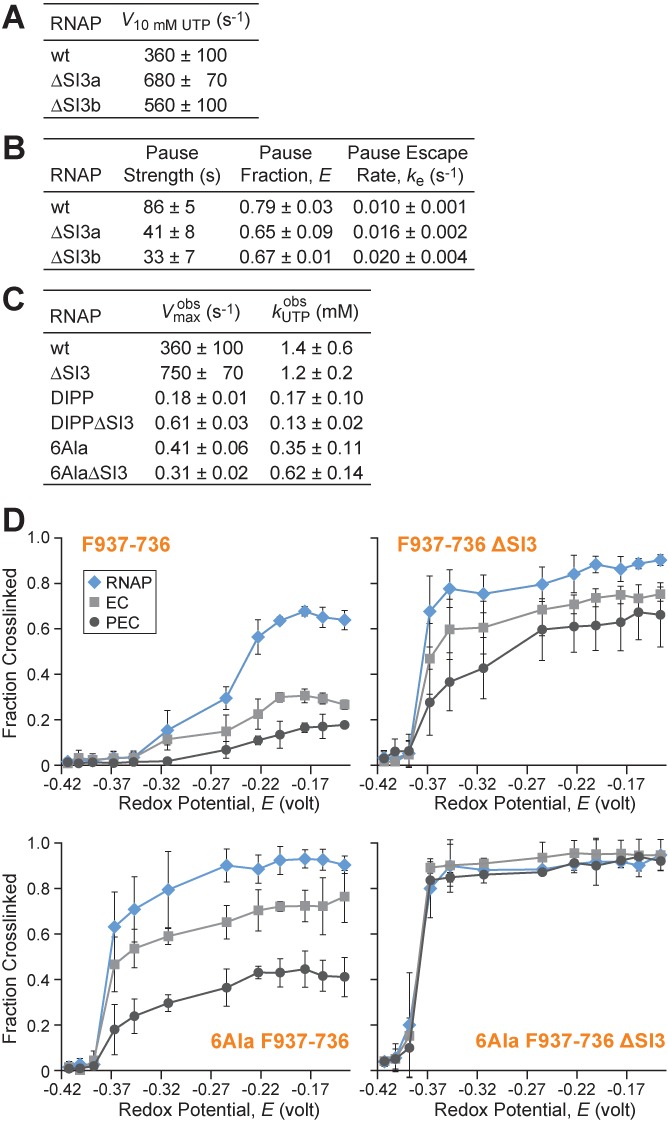
SI3 effects on elongation, pausing, and CPR mapping of TL/TH conformation. (A) Single-round NTP addition rates with ScaffoldUTP (Fgure 4) for wild-type and ΔSI3 RNAPs under predicted UTP saturation levels (10 mM). ΔSI3a corresponds to Δ1043–1130 and ΔSI3b corresponds to Δ1045–1132. (B) Comparison of his pause kinetics on scaffoldPEC (as in Figure 5) for wild-type, ΔSI3a and ΔSI3b RNAPs. (C) Comparison of UTP addition kinetics (as in Figure 4) for RNAP variants ± ΔSI3. (D) F937–736 ± ΔSI3 and 6Ala F937–736 ± ΔSI3 fraction crosslinked as a function of redox potential (2.5 mM CSSC, varying DTT) in free core RNAP, EC (ScaffoldGTP) and PEC (ScaffoldPEC at pause). Data are means ± SD of experimental triplicates.
Nucleotide addition rate measurements
Wild-type and mutant ECs were assembled using EC reconstitution essentially as described elsewhere (17), but with 50 nM scaffold and 200 nM core RNAPs, or in vivo after initiation from a promoter. For UTP incorporation assays (Figure 4), 1 μM RNA (7718), 1.2 μM T-DNA (7713) and 2 μM NT-DNA (7712) were annealed in reconstitution buffer (above). Note that 200 nM scaffold was reconstituted with 250 nM RNAP (1 μM for LTPP and ΔTL mutants) in buffer B (20 mM Tris-HCl pH 8, 20 mM NaCl, 0.1 mM EDTA, 15 mM MgCl2, 5% glycerol, 50 μg/mL BSA and 2 mM βME) for 15 min at RT. Experiments that required sampling at less than 5 s reaction times were performed on a Kin-Tek quench-flow apparatus. Complexes were diluted 2-fold (for quench-flow processing) or 4-fold (for bench top processing) in buffer B and incorporation labeled with 2 μM [α-32P]CTP and ATP for 10 min (30 min for LTPP and ΔTL mutants) at 37°C. For quench-flow processing, diluted ECs (20 μl) were injected into one sample loop and NTPs (20 μl; varying [UTP]) in transcription buffer were injected into the other loop. Reactions were performed for predetermined times at 37°C. The samples were quenched with 2 M HCl (40 μl) and immediately neutralized to pH 8.0 by addition of 3 M Tris base (40 μl). The RNA was phenol extracted, ethanol precipitated, suspended in formamide loading dye and fractionated on a denaturing polyacrylamide (17.5%) gel and quantified using a Phosphorimager. Bench top-processed samples were quenched by adding equal volume of stop buffer (8 M urea, 50 mM EDTA, 90 mM Tris-borate buffer pH8, 0.02% bromophenol blue and 0.02% xylene cyanol) and resolved by 15% denaturing polyacrylamide gel. Gels were exposed to phosphor screens and quantified using a Typhoon PhosphorImager and ImageQuant software (GE Healthcare). Note that 16 nt RNA decay (C16/total RNA in the lane) was fitted to a single or double exponential equation (fast rate, with over 80% of complexes consistently at this rate, was taken as U17 addition rate (the small slow fraction was shown in other experiments to result from slow reconstitution of a small number of complexes) using KaleidaGraph (Synergy Software). Average rates were calculated ± standard deviation for 0–15 mM UTP.
Figure 4.
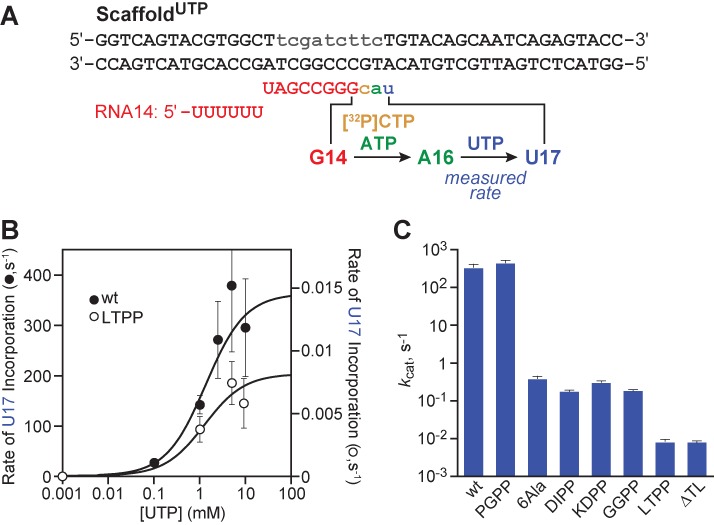
Kinetics of TH2a mutants for UTP addition. (A) ScaffoldUTP allows single-round NTP incorporation measurement upon EC RNAP reconstitution at G14, incorporation of 32P-CTP and ATP to A16, and extension of this EC with addition of UTP at time zero. (B) Rate of single round UTP incorporation for wild-type (filled circles, left y-axis) and LTPP RNAPs (open circles, right y-axis). (C) Comparison of kcat between wt and TL mutant RNAPs, derived from kinetic parameters derived from experiments including ones presented in (B). Data are means ± SD of experimental triplicates.
GTP incorporation assays (Figure 5) were performed as described previously (17). Briefly, 1 uM 5′-(32P)RNA (7636), 2 uM T-DNA (4903) and 3 uM NT-DNA (5069) were annealed, and reconstituted with RNAP (300 nM RNAP, 100 nM scaffold) in buffer A for 15 min at RT. Complexes were diluted 2-fold (for quench-flow processing) or 4-fold (for bench-top processing) in buffer A, supplemented with MgCl2 to 5 mM greater than the NTP concentration. Note that 20 mM oxidized DTT was incubated with ECs for 2 h at RT prior to the start of the experiment for oxidizing conditions. After addition of 0–15 mM GTP to 50 nM ECs, samples were taken at indicated times and processed as described above. Unless noted, 10 mM GTP was used (above saturating [GTP] levels for wild-type). The RNAs of length 13, 14 and ≥15 nt were quantified as a fraction of the total RNA in each lane. G14 formation rate was determined by using KaleidaGraph (Synergy Software) to fit 13 nt decay to a single or double exponential decay. To deconvolute crosslinked and uncrosslinked complexes in assays with F937–736 oxidized, we used a double exponential fit with the fast fraction set to the rate of uncrosslinked F937–736 (300 s−1; Nayak et al. and this study). To calculate the rate of G14 to G15, we used Runge–Kutta fourth-order numerical integration in Berkeley Madonna v8.3.22 (www.berkeleymadonna.com).
Figure 5.
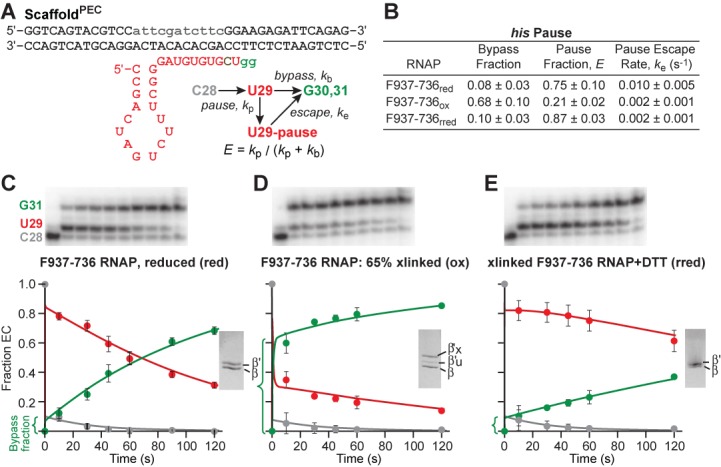
Blocking TH1a unfolding prevents hairpin-stabilization of pausing. (A) ScaffoldPEC allows his pause escape measurement by reconstituting ECs upstream of the pause, adding 32P-CTP to advance to C28 (pause-1), and then adding UTP (100 μM) and GTP (10 μM) at time zero to elongate through the pause site. (B) Kinetics of pause behavior for F937–736 RNAP under reduced, oxidized and re-reduced (oxidized and then reduced, rred) conditions, determined from kinetic fits of data included in C, D and E. (C) F937–736 RNAP (reduced) transcription products were resolved through denaturing PAGE. Fraction of C28, U29 and G30/G31 were fit and rates determined by numerical integration algorithm using the reaction scheme in (A) (Materials and Methods). The reduced RNAP state was confirmed through SDS-PAGE (inset). (D) As in (C), but using F937–736 RNAP under oxidized conditions. Transcription products show increased bypass fraction with crosslinking (65% confirmed through SDS-PAGE, inset). (E) As in (C), but using F937–736 RNAP that has been re-reduced (rred). The treatment reverses the pausing effects of crosslinking F937–736. Data are means ± SD of experimental triplicates.
For his pause assays (Figures 5, 6B, and 7B), EC27 were reconstituted 2 nt upstream from the pause site, incubated with 10 μM [α-32P]CTP (30 Ci/mmol) for 5 min at 37°C to form complexes halted 1 nt before the pause site (C28). If crosslinking was performed, complexes were then treated with 0.5 mM CSSC or oxidied DTT for 1 h at RT. Transcription was restarted by addition of 10 μM GTP and 100 μM M UTP. Samples were removed at different time points and quenched by the addition of an equal volume of 2× urea STOP buffer (7 M urea, 50 mM EDTA, 90 mM Tris-borate buffer [pH 8.0], 0.02% bromophenol blue and 0.02% xylene cyanol). Samples were heated for 2 min at 95°C and separated by electrophoresis though 16% denaturing polyacrylamide (19:1) gels in 0.5× TBE. Rates were determined by performing exponential fits of the fraction of RNA at the pause position, U29, relative to the total amount of RNA at this position and beyond (U29/≥U29) as a function of time.
To look at overall elongation behavior with RNAP variants (Figure 6A and Supplementary Figure S2), transcription was initiated with RNAP holoenzymes (40 nM) on a linear DNA template generated by PCR amplification (25 nM), combined with ApU (100 μM) and an NTP subset to allow formation of halted complexes (for elongation assays, 1 μM CTP, 5 μM ATP and GTP, 10 μCi [α32P]-CTP, 3000 Ci/mmol). For elongation assays, after incubation for 15 min at 37°C, transcription was restarted by addition of NTPs (10 μM GTP, 150 μM ATP, CTP and UTP) and rifampicin to 100 μg/ml at 37°C. Samples were removed at various times, quenched by addition of an equal volume 2× urea STOP buffer and separated on 8% denaturing polyacrylamide (19:1) gels in 0.5× TBE.
To look at nucleotide addition of more than two nucleotides by some mutant RNAPs, we assembled EC as described above on a longer scaffold, ScaffoldLong (Supplementary Figure S1 and Supplementary Table S1) that allowed formation of halted G15 complexes that could be extended with 1 mM NTP to transcribe a 78 nt RNA product.
G15 addition kinetic fitting
To determine NTP addition rates for nucleotides added after the first nucleotide in the time course (determining G15 addition rates), we used Runge–Kutta fourth-order numerical integration in Berkely Madonna v8.3.22 (www.berkeleymadonna.com) and fixed the first addition rate (G14; rates from KaleidaGraph) in the reaction scheme. For LTPP and DIPP experiments, the simplest reaction setup (Scheme 1, below) fit the data closely: k1 was fixed at the KaleidaGraph G13 exponential decay rate and the data (fraction of complexes at G13, G14 and G15 at each time point) were fit to determine k2. For wild-type ECs and reduced Cys mutant ECs, the Scheme 1 fitting had systematic error in the longer time points, likely due to a small fraction proceeding at a secondary slower rate. Scheme 2, with over 65% of complexes at the fast rate k1a, yielded a closer fit and G14 to G15 rate was taken as kcompilation. kcomp was calculated from x*k3+y*k4, where x and y represent the fraction of G14 complexes going at that rate: x = k3/(k3+k4) and y = k4/(k3+k4). (k3 from the Scheme 1 fit and kcomp from the Scheme 2 fit were similar for each individual data set.)
Scheme 1.

Scheme 2.

For F937–736oxidized ECs, Scheme 3 yielded the closest fit to the data. Consistent with percentage of crosslinked to uncrosslinked RNAP determined experimentally, a double exponential fit the F937–736ox G13 decay data well: with a large slow fraction (crosslinked ECs), a smaller fast fraction (∼300 s−1) and a third fraction (∼0 s−1). The ‘unreactive’ third fraction was <14% of complexes proceeding at a rate too slow to see in the timescale of the experiment. To account for the uncrosslinked fraction of ECs (∼40%) proceeding at a fast rate, we added a completely separate species to the scheme (complex b line in Scheme 3) with the G14 addition rate of this ‘species b’ (the uncrosslinked fraction of F937–736oxidized) fixed to the F937–736reduced G14 addition rate (300 s−1). Supporting the addition of this secondary species reaction line to Scheme 1 to approximate the uncrosslinked fraction, the rate of G15 appearance obtained through the fit for this ‘species b’ was fast (65 ± 6 s−1). Each replicate data set was fitted independently, and averages of k2 ± standard deviation were reported.
Scheme 3.

Exonuclease III footprinting
Exonuclease III footprinting was performed as described previously (12). Briefly, ECs were reconstituted as described above from DNA strands including a template DNA strand that was 5′-32P-labeled using [γ-32P]ATP and T4 polynucleotide kinase. ExoIII (2.5 U/μl) was added to 50 nM ECs and incubated for 0.25–2 min at 37°C. Reactions were stopped by the addition of an equal volume of 2×STOP buffer (8 M urea, 50 mM EDTA, 90 mM Tris-borate, pH 8.3), separated by 20% PAGE alongside products of a G+A nucleotide-sequencing reaction of the same DNA fragment, and visualized by Phosphorimager. We averaged the ratio of −15/−14 bands for the 0.25–2 min time points and present that averaged ratio.
RESULTS
Blocking TH1a unfolding is less inhibitory than blocking TH1a formation
The surprisingly small effect of the F937–736 disulfide crosslink on single-round nucleotide addition (17) could be explained if TL unfolding were only required for the PPi and translocation steps necessary for multiple rounds of nucleotide addition. To test whether the F937–736 crosslink caused greater effects on a second round of nucleotide addition, we formed ECs on a scaffold that allowed two rounds of GTP addition to convert EC G13 (bearing a 5′-32P-labeled, 13mer RNA) to EC G15 (Figure 2A; ScaffoldGTP; Supplementary Table S1). We measured rates of G14 and G15 formation at saturating GTP (10 mM; 17) to eliminate effects of NTP binding and used a kinetic fitting algorithm to deconvolute the G15 addition rate from the G14 addition rate (see Materials and Methods). Under these conditions, wild-type RNAP added G14 at ∼500 s−1 and subsequently added G15 at ∼150 s−1 (Figure 2B–D and Supplementary Table S2). The slower addition of G15 is consistent with a rate-limiting translocation event at ∼150 s−1 at 37°C. This rate of translocation is within range of the maximal processive elongation rate of EcoRNAP (∼100 s−1; (39)), the measured 3′-G translocation rate at 30°C (60 s−1; (26)) and evidence that both translocation and catalysis can contribute to multiround transcription rates (23,40). PPi release is thought to occur concomitantly with and slightly faster than translocation (26,41). F937–736 RNAP under reducing conditions and wild-type RNAP under oxidizing conditions exhibited similar rates of G14 and G15 addition (Figure 2D and Supplementary Table S2). Treatment of F937–736 G13 ECs with 20 mM oxidized DTT converted ∼60% to crosslinked form (Figure 2C, inset). We deconvoluted the rates G14 and G15 addition by crosslinked and uncrosslinked F937–736 ECs by assuming that ∼40% of the ECs exhibited rates comparable to the reduced F937–736 ECs (see Materials and Methods). This yielded close fits of the data and estimates of 12 s−1 and 15 s−1 for G14 and G15 addition, respectively, by crosslinked F937–736 ECs (Figure 2D). Thus, the second round of GTP addition by crosslinked F937–736 RNAP was not slower than the first, indicating that unfolding of the TH into the TL conformation in wild-type RNAP cannot facilitate translocation by more than a factor of ∼10 (the ratio of G15 addition in crosslinked F937–736 ECs to that in wild-type ECs).
Figure 2.
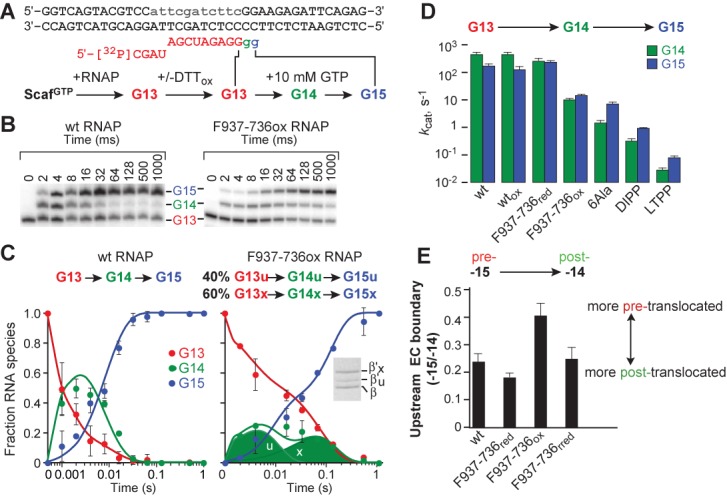
Effect of crosslinking TH in folded state and of 6Ala substitutions on multiround transcription. (A) ScaffoldGTP allows two rounds of GTP addition to reconstituted G13 ECs. The rates of GTP addition saturate below 10 mM GTP, allowing measurements of kcat for each GTP addition step at 1–10 mM GTP (17). Lowercase gray portion of NT strand denotes non-complementary bases to promote reconstitution. (B) Transcription reactions were performed using 5′-32P-labeled RNA on a rapid quench-flow device. Products were separated by polyacrylamide gel electrophoresis and quantified using a phosphorimager. A representative gel is shown for wild-type and F937–736 (ox) RNAPs. (C) Deconvolution of the rates of G14 and G15 addition for wild-type RNAP (left) and crosslinked F937–736 RNAP (right). Kinetic fitting to two sequential steps of GTP addition for one population (wild-type) or two populations (F937–736) of RNAP by a numerical integration algorithm (see Materials and Methods) allowed separation of the distinct species. The faster uncrosslinked (∼40%, u) and slower crosslinked (∼60%, x) fractions are clearly evident in the reaction progress curves (circles) and total RNAs predicted by the kinetic fitting algorithm (solid lines). The predicted levels of the individual uncrosslinked (u) and slower crosslinked (x) fractions for G14 are shown as semi-transparent areas. Percent crosslinked (β’X/total β’) was detected by SDS-PAGE (inset). (D) Calculated rates for G14 and G15 addition by wild-type, F937–736, 6Ala, DIPP and LTPP RNAPs at 1 mM GTP (wt) and 10 mM GTP for slowed RNAPs. (E) Exonuclease mapping of upstream edge of ECs. ECs were assembled on scaffold shown in (A) containing a 5′-32P-labeled template strand. Exonuclease digestion was performed on the complexes of wild-type or F937–736 RNAP to map the upstream edge of the transcription bubble. Data are presented as fraction of complexes that map to –15 relative to –14 and is derived from quantitation of the gel shown in Supplementary Figure S2. Data are means ± SD of experimental triplicates.
To test whether a similar rate of nucleotide addition persisted during synthesis of a longer transcription, we next tested the crosslinked F937–736 ECs formed on a variant of the same scaffold with a fully complementary NT strand and 50 additional bp downstream (Supplementary Figure S1; ScaffoldLong; see Materials and Methods). ScaffoldLong allowed G15 complexes formed by reaction of G13 complexes with [α-32P]GTP to proceed through 63 rounds of nucleotide addition requiring all 4 NTPs to form a 78 nt run-off transcript. Wild-type ECs and reduced F937–736 ECs synthesized the 78-nt RNA in 2–6 s at 37°C, 1 mM each NTP, suggesting an average transcription rate of 7–20 s−1. This slower overall rate relative to the single- or double-turnover assays (Figure 2; 17) is comparable to prior observations for processive transcription under similar conditions (42,43) and reflects template positions at which translocation or catalysis may be slower than GTP addition on ScaffoldGTP or at which pausing occurs. In reactions containing crosslinked F937–736 ECs, a distinct, slower elongating species is evident (red boxes, Supplementary Figure S1) with an average elongation rate of ∼1 s−1. Thus, the F937–736 crosslink slowed processive transcription on average on ScaffoldLong by about the same factor of ∼10–20 that it slowed the single- or double-turnover GTP reactions at both 1 and 10 mM GTP (Figure 2; 17). We conclude that EcoRNAP containing a crosslink that blocks complete TH unfolding into the TL conformation allows rates of processive nucleotide addition 200–400 times faster than the rates observed for an RNAP in which TH formation is blocked (e.g. LTPP; Figure 2D and Supplementary Table S2).
Although it is possible that reduced NTP binding or PPi release rates contributed to the effect of the F937–736 crosslink on processive translocation, we propose the major effect arises from slower translocation, consistent with prior results showing that the folded TH stabilize the pre-translocated register of transcription complexes (1,8,24–27). To determine directly whether the F937–736 crosslink stabilizes the pre-translocated register, we used exonuclease III footprinting to detect translocation states on ScaffoldGTP and found that stabilizing the TH with the F937–736 crosslink shifted translocation bias toward the pre-translocated register (Figure 2E and Supplementary Figure S2).
TH stabilization by hinge substitutions partially inhibits nucleotide addition
As an additional test of the requirement for TL formation during processive transcription, we constructed a variant of the TL in which all Gly and Pro residues in the two hinge regions were converted to Ala (6Ala RNAP; Figure 1D). We reasoned that these substitutions should greatly stabilize the folded TH conformation by creating strong, continuous TH1 and TH2 α-helices lacking the destabilizing effects of the hinge regions. To test whether the 6Ala substitutions indeed stabilized the TH conformation, we also constructed 6Ala variants containing the F937–736 and U937–1139 CPRs that detect folded and unfolded conformations of the TL/TH (Figures 1B and 3A; 17). We measured the relative extent of CPR crosslinking for isolated core RNAPs and for G13 ECs containing a 3′dG in the presence and absence of 10 mM GTP (Figure 3; see Materials and Methods). Consistent with stabilization of the TH, the 6Ala F937–736 crosslink formed to greater equilibrium extent at high Eh (∼0.9 versus ∼0.6 fraction crosslinking in free RNAP and ∼0.7 versus ∼0.35 fraction crosslinked in EC G13, respectively, at Eh = 0.136 V; Figure 3B). The 6Ala substitution also eliminated the ability of GTP binding to stimulate the F937–736 crosslink seen previously in wild-type RNAP (Figure 3B; 17), suggesting 6Ala stabilized the TH in the absence of NTP, greatly lessened formation of a crosslink specific to the unfolded conformation in free RNAP (U937–1139; Figure 3B), and essentially eliminated the U937–1139 crosslink in EC G14. Taken together, these results suggest that the 6Ala substitutions significantly increase the stability of the TH conformation relative to the TL conformation.
To test the effect of TH stabilization by the 6Ala substitutions, we examined the rates of G14 and G15 addition on ScaffoldGTP and processive transcription on ScaffoldLong, as described above for the F937–736 crosslink RNAP (Figure 2D and Supplementary Figure S1). G14 addition was significantly slower for the 6Ala RNAP than for wild-type RNAP (∼300-fold slower), but was less reduced for G15 addition (∼25-fold; Figure 2D). Processive elongation on ScaffoldLong appeared to be approximately equivalent to the crosslinked F937–736 EC, although a fraction of ECs were unable to elongate past positions 16–24 (Supplementary Figure S1).
We conclude that stabilization of the TH conformation by hinge substitutions that eliminate helix-destabilizing Gly and Pro residues decreases nucleotide addition and elongation rates, but is much less inhibitory than alterations that block TL folding into the TH. Together with the similar effect of the F937–736 disulfide crosslink that should block complete TL unfolding, these results suggest that complete TL unfolding is not necessary to achieve multiple rounds of nucleotide addition, but that effects on translocation may slow processive transcription when TL unfolded is inhibited.
Rapid nucleotide addition by EcoRNAP requires formation of TH2a
We next sought to determine whether TH2a formation is required for TL function in EcoRNAP despite the presence of the large SI3 insertion at the loop/TH2a junction. For this purpose, we constructed three sets of double-Pro substitutions, KDPP, DIPP and GGPP that should preclude α-helix formation at different points in TH2a or the TH2a-hinge2 junction (Figure 1D). To test the effects of these substitutions, we chose a new scaffold design that allowed measurement of UTP addition rates in a single-turnover assay (ScaffoldUTP; Figure 4A). We reasoned that UTP incorporation might exhibit a greater TH requirement than GTP addition because the weak rUTP/dA bp would contribute less to proper alignment of the α phosphate with the RNA 3′ OH than an rGTP/dC bp. We formed A16 ECs on ScaffoldUTP by reaction of G14 ECs with [α-32P]CTP and ATP and found the A16 ECs added UTP with simple hyperbolic kinetics (Figure 4B). LTPP RNAP, blocked in formation of TH1a, exhibited a 4.5 × 104-fold defect in kcat, with no defect in UTP binding (Figure 4B and C, KUTP ≈ 1.5 mM; Supplementary Table S2). Thus, blocking TH1a formation causes a 3–4 times greater defect in UTP addition than in GTP or CTP addition (Figures 2D and 4B, and Supplementary Table S2; (2)). In comparison, we found that the three TL mutants blocked in TH2a formation gave defects in kcat for UTP of 1–2 × 103 (Figure 4C and Supplementary Table S2). Relative to the LTPP substitution blocking TH1a formation, the TH2a substitutions are 25–50 times faster at UTP addition. These data indicate that rapid nucleotide addition by EcoRNAP requires formation of TH2a despite the presence of the SI3 insertion. However, the 25–50× faster catalysis when formation of TH2a is blocked relative to when TH1a formation is blocked suggests that TH1a may still form to some extent even when formation of TH2a is not possible.
To confirm this interpretation, we tested the TH2a-blocking DIPP substitution on ScaffoldGTP and found a 1.5 × 103-fold defect in kcat, a rate about 10 times faster than that of the TH1a-blocking LTPP mutant (Figure 2D). For both the DIPP and LTPP substitutions, the kcat defect was less for the second round of GTP addition (∼2 × 102 and ∼2 × 103, respectively, versus ∼2 × 103 and 2 × 104 for the first round of GTP addition; Supplementary Table S2). These data confirm the overall importance of TH2a formation in EcoRNAP for rapid nucleotide addition, but show a ∼10-fold lower contribution than for TH1a. The 10× lesser effect for the second round of nucleotide addition for both TH1a- and TH2a-blocking substitutions could suggest that blocking TL folding increases rates of translocation, which contribute to the rates of complete rounds of nucleotide addition. Taken together, these kinetic measurements strongly suggest that TH2a formation aids catalysis by RNAP almost as much as the direct TH1a–NTP contacts, likely indirectly by stabilizing folding of the TH/BH module.
TH stabilization interferes with hairpin-stabilization of pausing
We next sought to test whether stabilizing the folded TH using the F937–736 crosslink would affect hairpin-stabilized pausing. Our current understanding is that formation of a nascent hairpin in the RNAP exit channel stabilizes an open-clamp conformation of the paused EC in which TH formation is inhibited by loss of clamp/BH and clamp/TH contacts (12,17,18,44). The exit-channel hairpin also appears to slow translocation (12,18). The F937–736 crosslink afforded a way to distinguish these effects, since it should reduce the effect of pause-hairpin inhibition of TL folding on catalysis while increasing effects on translocation per se by favoring the pre-translocated register. Thus, we compared pausing in wild-type ECs to pausing in F937–736 crosslinked ECs using a scaffold that forms the hairpin-stabilized, paused state (Figure 5A; 17,45). In the presence of DTT at levels that inhibit the crosslink, F937–736, ECs pause essentially like wild-type ECs with ∼75% entering the long-lived, hairpin stabilized state (Figure 5B and C). Upon crosslinking at ∼65% efficiency with oxidized DTT, only ∼20% of the ECs entered the long-lived pause state and ∼60% were observed to elongate past the pause site rapidly (Figure 5B and D). Treatment of the crosslinked ECs with DTT restored pausing to about the same levels observed without crosslinking (Figure 5B and E). The slower escape of the 20% of ECs that pause is likely attributable to effects of oxidation on non-crosslinked RNAP (Figure 5E). However, the dramatic reduction in pausing in the 60% F937–736 crosslinked fraction suggests that stabilizing the TH eliminates the ability of the pause hairpin to inhibit nucleotide addition. This finding is readily explained by the model that the pause hairpin stabilizes an open-clamp conformation of RNAP that favors a partially folded (or paused) TL conformation unable to stimulate catalysis (17).
Uncoupling TH1a formation from TH1b promotes catalysis and prevents hairpin-stabilization of pausing
We next investigated the hairpin/clamp/TL model by investigating the effects of the G927P substitution, which creates a tandem PP in hinge1 and should prevent formation of a continuous TH1 (Figure 1), and the F773V substitution in the BH that is predicted to inhibit pausing by stabilizing TL folding (29,46,47). The so-called PGPP RNAP was surprisingly active, completing synthesis of a 668-nt run-off transcript almost as fast as wild-type RNAP (Figure 6A). This minimal effect of the PGPP mutant strikingly contrasts to the dramatic inhibition of nucleotide addition caused by disruption of TH1a or TH2a by the LTPP or DIPP substitutions (Figure 2D), and suggests TH1a must be able to form independently of TH1b and contact NTP in the active site. However, despite the near normal elongation rate and single round UTP addition rate (Figure 4C), the PGPP mutant essentially eliminated the hairpin-stabilized his pause even though it allowed near normal pausing at the elemental/backtrack ops pause earlier on the template (Figure 6A and B; we see no evidence for misincorporation by this mutant RNAP, Supplementary Figure S4). F773V RNAP behaved similarly, eliminating hairpin-stabilized his pausing while retaining ops pausing. To quantitate the effect on hairpin-stabilized pausing, we compared pausing of wild-type RNAP to pausing by PGPP or the F773V RNAP on the his pause scaffold (Figures 5A and 6B; 17,45). Both the PGPP and F773V RNAPs exhibited a marked decrease in the fraction of complexes entering the stabilized pause state. We conclude that TH1a can fold independently of TH2a, but that loss of structural linkage between TH1a and TH2a in PGPP RNAP or stabilization of TH formation in F773V RNAP prevents hairpin-stabilized clamp opening from trapping the TL in a paused state. In contrast, elemental and backtrack pausing at the ops site is largely unaffected by changes in TL folding. The PGPP effect is particularly interesting because it suggests that the effect of clamp opening on TH formation is disrupted when TH1b is disconnected from TH1a, whereas the F773V effect likely reflects stabilization of TH formation similar to the F937–736 crosslink. That neither substitution has an equivalently dramatic effect on the ops pause agrees with recent evidence that elemental/backtrack pauses occur by a mechanism that differs from hairpin-stabilized pausing (48). In particular, our current results suggest that elemental pausing may not involve inhibition of TL folding as found for hairpin-stabilized pausing.
SI3 inhibits TH formation and nucleotide addition, but promotes hairpin-stabilization of pausing
Given that formation of both arms of the EcoRNAP TH appeared necessary for efficient catalysis, we next wished to test how SI3, located at the loop/TH2a junction, affects the TL-TH equilibrium. Our previous results suggested that SI3 contributed to slow TH formation during escape from the hairpin-stabilized his pause and also slowed the overall rate of elongation on a long DNA template (29,34). However, the effects of SI3 on single-turnover reactions and on TH formation have not been studied directly. To test the effects of SI3, we first compared two different deletions, ΔSI3a and ΔSI3b, that removed the 188-aa SI3 insertion in two different ways to create TLs equal in length to that found in bacterial RNAPs lacking SI3 (e.g. TthRNAP). ΔSI3a (Figure 1D, Supplementary Table S1) was identical to the SI3 deletion we designed and tested previously based on alignments of diverse bacterial RNAP sequences (29). Miropolskaya et al. (49) subsequently argued that the deletion point in ΔSI3a was two amino acids more N-terminal than the SI3 insertion point predicted by their sequence alignments. Therefore, we constructed the second deletion, ΔSI3b, corresponding to this alternative SI3 insertion point (Figure 1D). We compared ΔSI3a and ΔSI3b EcoRNAPs for single-turnover UTP addition, for pausing at the his pause site, and for overall elongation rate on a long DNA template. We observed no significant differences in their behaviors. Both SI3 deletions increased the overall elongation rate up to 2-fold (the 60 s time point of ΔSI3a and ΔSI3b RNAPs resembled the 120 s timepoint of wild-type RNAP; Supplementary Figure S3). Similarly, both deletions increased single-turnover kcat for UTP (Figure 7A; 10 mM UTP). We also observed no major differences in the 2–3× reduction in pause strength measured here with the scaffold-based assay (Figures 5A and 7B), roughly comparable to the ∼3× effect seen previously on a promoter-initiated template (29). It is possible the slightly reduced effect of SI3 on hairpin-stabilized pausing on the scaffold reflects the absence of some downstream DNA–clamp contacts (45). We conclude that there is no precise sequence requirement for TH2a in ΔSI3 EcoRNAP. ΔSI3a and ΔSI3b create TH2a arms with two different sequences (ASTKDIT and ASRADIT, respectively), with comparable properties for catalysis and pausing. Thus, TH2a appears to exhibit a lesser requirement for specific sequence than TH1a where substitutions have drastic effects (2,4,9,11,13,14), consistent with the role of TH1a in direct NTP contacts. As a consequence, we cannot deduce the evolutionarily relevant insertion point of SI3 from our biochemical results.
We next asked if SI3 could affect nucleotide addition when folding of TH2a was blocked by the DIPP substitution or when the TH were stabilized by the 6Ala substitutions. Using single-turnover UTP addition, we found that the presence of SI3 inhibited nucleotide addition for the DIPP mutant, indicating that removal of SI3 promotes formation of TH1a even when TH2a folding is blocked (Figure 7C). This result is consistent with the location of SI3 at the loop–TH2a junction, since removal of SI3 may allow the loop to more readily accommodate TH1a folding. However, SI3 had no detectable effect on UTP addition in the 6Ala mutant RNAP (Figure 7C). This result suggests that increasing the propensity for TH formation by removing SI3 does not alter the rate-limiting step in the 6Ala nucleotide addition cycle, which we reasoned above may involve a step other than TH formation.
Finally, to verify directly that SI3 affects TH formation, we measured effects of SI3 on the ease of forming the F936–736 disulfide crosslink, which reports the folded TH conformation. We found that removing SI3 increases the F937–736 crosslink in free RNAP, ECs and his paused ECs (Figure 7D). Further, these effects were additive with the TH-promoting effect of the 6Ala substitutions such that the combination of ΔSI3 and 6Ala gave highly efficient F937–736 crosslinking in all forms of RNAP (Figure 7D). Interestingly, both the 6Ala substitutions and ΔSI3 allowed the F937–736 crosslink to form at lower Eh than the wild-type F937–736 crosslink (∼−0.38 V versus ∼−0.24 V half-maximal Eh for wild-type RNAP). Although the disulfide assay is subject to multiple complications that lessen its quantitative reliability (17), assuming the chemical environments for disulfide formation are largely unaffected by these changes would suggest that either 6Ala or ΔSI3 in principle stabilize the TH by about a factor of 10 (Figure 7). These results thus suggest that the significant increase in pausing at the his pause site caused by SI3 (relative to a ΔSI3 RNAP) can be explained by SI3-inhibition of TH formation.
The SI3 and jaw domains cooperatively modulate hairpin-stabilized pausing through effects on TH formation independent of downstream DNA contact
Although SI3 appears to affect pausing and nucleotide addition through its effects on TH formation, it is possible that this effect is dependent on interactions of the unfolded TL or of the SI3/jaw submodule with downstream DNA since both the unfolded TL and the jaw may contact the downstream DNA (30,50–52). Further the jaw, like SI3, is known to increase pausing at the his pause site (34,50).
To test whether SI3 and the jaw affect the his pause through contacts to the downstream DNA, we first tested whether the effects of SI3 and the jaw were distinct or interdependent by measuring his pause half-lives in pause assays on a linear DNA template (Figure 8A). Consistent with previous work, deletion of SI3 or the jaw reduced the his pause lifetime by factors of ∼3 and ∼5, respectively (29,42,50). Combining the two deletions in ΔSI3Δjaw RNAP yielded the same reduction by a factor of ∼5 in his pause lifetime as observed for Δjaw RNAP (Figure 8B). Thus, the effects of ΔSI3 and Δjaw were not additive. We conclude that the presence of either SI3 or the jaw alone has minimal or no effect on his pause lifetime (relative to ΔSI3ΔjawRNAP), but that the presence of both elements together increases his pause lifetime by a factor of ∼5, suggesting that the effects of the jaw and SI3 are interdependent.
Figure 8.
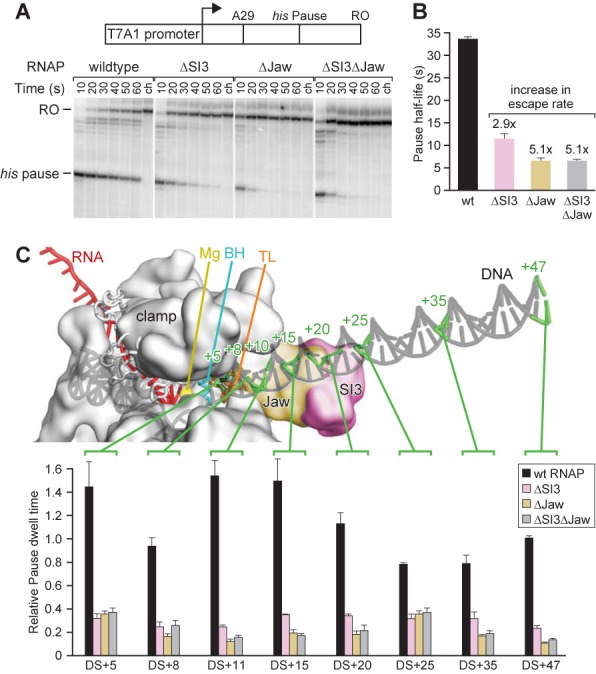
SI3 and the jaw do not modulate the active site through their putative downstream DNA contacts. (A) His pause escape on a promoter-driven his pause template (29) for wild-type, ΔSI3, Δjaw, and ΔSI3Δjaw RNAPs. Preformed [α-32P]-CTP-labeled A29 complexes were incubated with 20 μM GTP, 150 μM ATP, CTP and UTP, together with 50 μg heparin/ml. Position of pause and run-off (RO) transcripts are indicated. Samples were taken at the times indicated (ch, chase). (B) Bar graph showing the average pause half-life values of wild-type, ΔSI3, Δjaw, and ΔSI3Δjaw RNAPs at his pause site derived from the experiment shown in (A) and two additional experiments shown as means ± SD. Numbers above the bars indicate the fold-increase in pause escape rate. (C) Pausing for wild-type, ΔSI3, Δjaw, and ΔSI3Δjaw RNAPs on templates with different amounts of downstream sequence. Top: schematic of paused elongation complex illustrating the progressive downstream-DNA truncations. The RNAP (gray spacefill), DNA (gray) and RNA (red) are shown. Positions of duplex DNA truncations are indicated on the schematic drawing by green lines and numbers indicating the number of base pairs downstream of the pause site, where the pause site with RNAP in pre-translocated register is denoted as the +1 position. Bottom: normalized pause half-lives of wild-type, ΔSI3, Δjaw, and ΔSI3Δjaw RNAPs on truncation templates. The half-life of wild-type RNAP on DS+47 (47 bp of downstream DNA duplex) was normalized to 1 and all other pause half-life values were normalized accordingly. Data are means ± SD of experimental triplicates.
To ask if the interdependent effect of SI3 and the jaw depended on interactions with downstream DNA, we constructed and tested a series of mutant transcription templates bearing progressive truncations of the downstream DNA duplex (from DS+5 to DS+47; DS+5 means there are 5 bp of DNA downstream of the pause site; Figure 8C). Crosslinking and structural studies of ECs position the jaw and SI3 close to the downstream DNA around +10 or +15 (30,53). We found that shortening the downstream DNA to less than 20–25 bp prolonged pause lifetime (Figure 8C, black bars), consistent with results obtained at the HIV-1 pause site with mammalian RNAPII (51) and previously with his pause scaffolds (45). However, regardless of the length of downstream DNA present, the effects of the SI3 and jaw deletions on pausing and their lack of additivity persisted with about the same magnitude, including on the DS+5 and DS+8 templates (Figure 8C). These results show unambiguously that the major synergistic effects of the jaw and SI3 on his pause lifetime do not involve downstream DNA contacts and thus are likely to reflect a jaw–SI3 interaction that allows SI3 to inhibit TL folding.
DISCUSSION
We sought to define the roles of distinct parts of the E. coli RNAP TL (hinge1, TH2a and the large sequence insertion SI3) in nucleotide addition and pausing. In particular, we wished to test the model that hairpin-stabilization of pausing depends on inhibition of TL folding mediated by altered clamp domain contacts to the TL through switch 1 and switch 2. Using a disulfide bond that stabilized the folded form of the TL, we found that inhibition of TL unfolding had surprisingly little effect on multiround transcription but largely eliminated hairpin-stabilization of pausing. We also found that SI3 inhibited TL folding and aided hairpin-stabilized pausing independently of downstream DNA contacts. Finally, we found that disrupting the folding pathway of TH1 by creating a double-Pro sequence in hinge1 uncoupled rapid catalysis from the effects of the pause hairpin proposed to be mediated by clamp–TH1b contacts. These results, coupled with tests of other TL-altered RNAP mutants, provide important new insights into TL function generally and validate key aspects of a clamp–TL model of hairpin-stabilized pausing.
Roles of hinge1, TH1b, TH2a and TL unfolding in TL function
The requirement for the TL-to-TH transition and TH-NTP contacts for rapid catalysis of RNA extension by RNAP is supported by numerous results of both structural (2,3) and biochemical studies (4,5,8,9,11,12,14,17,27,49,54), and is now generally accepted. However, most results document the importance of TH1a-NTP contacts but do not necessarily establish the need to form the complete helical hairpin TH in the BH/TH 3-helix bundle. Establishment of the NTP contacts and formation of the TH have been generally assumed to be tightly linked, and the inhibitory effects of some alterations outside TH1a are best explained by this assumption (9,24,27,36).
Our findings suggest a refinement to this picture as follows. The folding of different parts of the TL (TH1a, TH2a and the hinges) to form the TH are ordinarily relatively tightly linked in a narrow pathway of folding intermediates that also involves the BH, anchor and cap modules. At least one intermediate can become trapped in a paused state. The proline substitutions in TH2a unambiguously establish that an inability to form TH2a is nearly as deleterious for rapid catalysis as an inability to form TH1a, in the context of otherwise intact TL (Figures 2 and 4). Thus in wild-type RNAP, TH–NTP contacts appear to be obligatorily established by concerted folding of the BH, TH, cap and anchor to form the BH–TL 3-helix bundle. This concerted folding explains the increased rates of nucleotide addition, involving effects on both translocation and catalysis, observed when substitutions in or near TH2a appear to favor TH2a folding (9,24,27,36). However, the presence of tandem prolines in hinge1 (PGPP), just one α-helical turn away from the TH1a LTPP substitutions that slow catalysis by ≥104, had surprisingly little effect on the overall rate of nucleotide addition (PGPP; Figure 4 and Supplementary Table S2). Thus, the hinge1 PGPP substitution appears to uncouple folding of TH1a from the normal pathway of TL folding intermediates and allow TH1a to fold relatively efficiently even when a continuous active-site proximal trigger helix cannot form (Figure 9).
Figure 9.
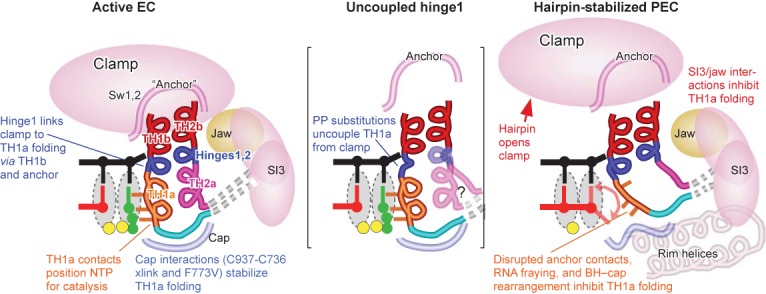
Folding of the TL into the TH is coupled with movements of other modules of RNAP. (Left) The BH, anchor and cap modules link clamp position with folding of the TL (TH1a, TH2a and the hinges) to form the TH. The TH1a contacts to the NTP allow correct positioning for catalysis in the active EC. (Center) The PGPP mutant uncouples TH1a from the clamp position. (Right) In the hairpin-stabilized PEC, the paused TL conformation is stabilized by clamp opening resulting from formation of the pause RNA hairpin and also involves rearrangements of the BH and the BH/TL cap. These BH and cap rearrangements inhibit the cap's interaction with the loop of the TH.
A second aspect of TL function that remains uncertain is the extent to which the TL must unfold during successive rounds of nucleotide addition. The folded TH conformation appears to favor the pre-translocated register of ECs, possibly through TH contacts to the 3′ RNA nucleotide (26). As a result, alterations that favor TL folding appear to bias the EC toward the pre-translocated register (27). We find that neither crosslinking the TH with a disulfide that should prevent unfolding nor Ala substitutions that should favor the TH conformation inhibit transcription as significantly as blocking TL folding. The disulfide result is most striking, the Ala substitutions could alter important TL contacts. Given that multiple rounds of nucleotide addition were inhibited no more than 20× by the crosslink, either the block to translocation, PPi release, and NTP binding by the folded TH must be incomplete or the crosslink must allow sufficient unfolding to release the pre-translocated state, release PPi, and enable NTP binding. We think the latter interpretation is most attractive, since the altered contacts and increased negative charge upon PPi generation likely destabilize the TH enough to allow thermal fluctuation to a more open state. However, this opening must involve only a partial unfolding of the TH since the disulfide essentially traps the loop of the TH within the BH/TL cap.
Hairpin enhancement of pausing rearranges the TL, BH and cap to resist TH formation
Currently available data suggests that the hairpin-stabilized state arises by formation of a nascent RNA hairpin 11–12 nt upstream from the RNA 3′ end in an initially formed elemental paused complex caused by consensus sequences in the RNA:DNA hybrid, active site and downstream DNA (48,55,56). Although the elemental pause mechanism remains uncertain, the consensus sequences may loosen contacts that stabilize the closed clamp and may increase the barrier to complete translocation to load the template base into the RNAP active site (57). Hairpin formation then stabilizes the open clamp conformation (18,44,57), causing prolonged pausing principally because the open clamp disrupts contacts of the BH and TL to switch 1 and switch 2 in the anchor that connect the clamp to the body of RNAP (Figure 9). This loss of contacts appears to favor a partially folded conformation of the TL in which the loop region is near the so-called rim helices and that cannot be shifted to the folded TH even after NTP binding (17). Indeed, the stimulatory effect of clamp opening on pausing can be observed by stabilizing the open-clamp conformation with a disulfide crosslink even when no nascent RNA hairpin is present (18). An additional, but lesser, contribution of the pause hairpin appears to be to inhibit translocation.
Our results suggest two important refinements to the model for hairpin-stabilized pausing. First, the partially folded TL state, detected by preferential disulfide formation between Cys residues placed in the TL loop and the rim helix in a hairpin-stabilized PEC (17), is not just a consequence of the paused conformation but directly contributes to the hairpin-induced increase in pause dwell time. Second, our results implicate BH–TL–cap rearrangements in creating the paused TL state.
The causal nature of the paused TL state is supported by two separate and interesting results. First, stabilizing the folded TH conformation with a disulfide crosslink between the TH loop and the BH/TL cap, which prevents formation of the partially folded state, robustly inhibits formation of the hairpin-stabilized pause (Figure 5). Thus, preventing the TH loop from moving to the position at which it is observed in the hairpin-stabilized PEC (near the rim helices) actually blocks the hairpin effect on pausing. Since the folded TH state stabilized by the disulfide should increase inhibition of translocation and thus be synergistic with an effect of the hairpin on translocation, this result is particularly striking. Apparently, the partially folded TL conformation detected previously (17) is unable to promote catalysis and can therefore be described mechanistically as a paused TL conformation.
Second, the disruption of hairpin-stabilized pausing by tandem proline residues in hinge1 suggests that the pause hairpin's effect depends on structural communication between TH1b and TH1a through the extended network of α-helical H-bonds. In this PGPP mutant RNAP, nucleotide addition at the elemental pause remains inhibited, as detected at the ops pause site (Figure 6). However, the hairpin-stabilized state is not generated in PGPP RNAP at the his pause site. The contacts between switch 1 and TH1b should not be directly affected by the PGPP substitution, since G927 is not involved in these interactions. Thus, the simplest interpretation is that disrupting structural communication between TH1b and TH1a eliminates the consequence of clamp opening on TH1a formation, with the result that the paused TL conformation does not become a stable intermediate (Figure 9). Consistent with this view, a crystal structure that captures the TL in the conformation closest to that postulated for the paused conformation (with the loop near the rim helices) reveals partial helical character at positions equivalent to P926 and G927 (yeast RNAPII ECs 2nvq and 3gtg; 3,58).
Technically, we have not excluded the possibility that the PGPP variant prevents pause hairpin formation, but we consider this extremely unlikely because even blocking clamp opening with a disulfide does not prevent hairpin formation (18). We also cannot exclude the possibilities that BH interactions with the anchor, cap, and TH contribute to the paused TL stabilization or that BH-TH1a or TH1a-TH2a interactions aid NTP contacts when the PGPP substitution is present, but both of these possibilities are consistent with the model that clamp opening inhibits TH formation. Rather, we think the BH and TH2a are likely involved in both situations.
Indeed, our results strongly implicate BH–TL–cap rearrangements in creating the paused TL state. We propose that the paused TL conformation that is stabilized by clamp opening also involves rearrangements of the BH and the BH/TL cap (consisting of βDloopII, link, fork loop 2, and the F-loop; 17) and that these BH and cap rearrangements alter the cap to inhibit its interaction with the loop of the TH (Figure 9). The exact nature of the BH–cap rearrangement is unclear, but multiple findings implicate its involvement: (i) the BH takes on an altered conformation when the clamp is open (57,59); (ii) the F773V substitution appears to alter BH–cap interactions in ways that disrupt the hairpin-stabilized pause state (Figure 6; 29,46,47) and (iii) the RNAP inhibitor CBR703, which binds to the BH and may alter its interaction with the cap possibly by repositioning His777, reverses pause suppression by F773V and generally enhances hairpin-stabilized pausing (46,60). In wild-type RNAP, the restricted folding pathway for the TH (17) may be incompatible with this BH–cap state, inhibiting the transition to the fully folded TH. By disconnecting TH1a folding from this restricted folding space, the PGPP substitution may free TH1a from this inhibition and allow TH1a to form and catalyze nucleotide addition with less influence of the surrounding RNAP conformation.
Role of SI3 in hairpin-stabilized pausing
Our results also define a mechanistic contribution of SI3 to the hairpin-stabilized pausing model. SI3 appears to buttress the hairpin-stabilized pause state, increasing pause dwell time ∼3× (Figure 8) and generating much larger increases or decreases in pausing when mutated or bound by antibodies (34,35). Our disulfide crosslinking assay directly establishes that SI3 inhibits TH formation, consistent with a role of SI3 in stabilizing the partially folded, paused TL conformation (Figure 7). Additionally, we showed that this effect of SI3 (and of its interacting partner, the jaw domain) in increasing the dwell time of the hairpin-stabilized state does not depend on interaction with the downstream DNA (Figure 8). Thus, even though SI3 and the jaw are located near the downstream DNA, the effects of alterations in the jaw and SI3 that decrease pausing (34) are likely to be mediated by direct effects on folding of the TL.
Although it is possible that SI3 plays additional roles in controlling access to the secondary channel (37) or through interactions with as yet unidentified regulators, we suggest that the primary effect of the sequence insertion is in modulating TL folding. The centrality of TL folding to RNAP function may explain why similar insertions into the TL have arisen in clearly independent bacterial lineages, including cyanobacteria (32). The ease with which adaptive mutations arise within SI3 raises the possibility that a principal selective advantage to its presence could be to increase the evolvability (61) of RNAP. By providing a larger target for substitutions that can alter catalytic function, TL insertions like SI3 could make it easier for organisms to acquire adaptive mutations that confer advantage in new conditions where greater or lesser pause strengths might compensate for changes in the efficiency of translation or ease of RNA folding.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank members of the Landick laboratory for helpful discussions and for comments on the manuscript.
FUNDING
U.S. National Institutes of Health (NIH) [GM038660 to R.L.]. Funding for open access charge: NIH [R.L.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Seibold S.A., Singh B.N., Zhang C., Kireeva M., Domecq C., Bouchard A., Nazione A.M., Feig M., Cukier R.I., Coulombe B., et al. Conformational coupling, bridge helix dynamics and active site dehydration in catalysis by RNA polymerase. Biochim. Biophys. Acta. 2010;1799:575–587. doi: 10.1016/j.bbagrm.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vassylyev D., Vassylyeva M., Zhang J., Palangat M., Artsimovitch I., Landick R. Structural basis for substrate loading in bacterial RNA polymerase. Nature. 2007;448:163–168. doi: 10.1038/nature05931. [DOI] [PubMed] [Google Scholar]
- 3.Wang D., Bushnell D., Westover K., Kaplan C., Kornberg R. Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis. Cell. 2006;127:941–954. doi: 10.1016/j.cell.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J., Palangat M., Landick R. Role of the RNA polymerase trigger loop in catalysis and pausing. Nat. Struct. Mol. Biol. 2010;17:99–104. doi: 10.1038/nsmb.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan L., Wiesler S., Trzaska D., Carney H.C., Weinzierl R.O. Bridge helix and trigger loop perturbations generate superactive RNA polymerases. J. Biol. 2008;7:40.1–40.15. doi: 10.1186/jbiol98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinzierl R.O. The nucleotide addition cycle of RNA polymerase is controlled by two molecular hinges in the Bridge Helix domain. BMC Biol. 2010;8:134.1–134.15. doi: 10.1186/1741-7007-8-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jovanovic M., Burrows P.C., Bose D., Camara B., Wiesler S., Zhang X., Wigneshweraraj S., Weinzierl R.O., Buck M. Activity map of the Escherichia coli RNA polymerase bridge helix. J. Biol. Chem. 2011;286:14469–14479. doi: 10.1074/jbc.M110.212902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kireeva M.L., Opron K., Seibold S.A., Domecq C., Cukier R.I., Coulombe B., Kashlev M., Burton Z.F. Molecular dynamics and mutational analysis of the catalytic and translocation cycle of RNA polymerase. BMC Biophys. 2012;5:1–18. doi: 10.1186/2046-1682-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaplan C.D., Jin H., Zhang I.L., Belyanin A. Dissection of Pol II trigger loop function and Pol II activity-dependent control of start site selection in vivo. PLoS Genet. 2012;8:e1002627. doi: 10.1371/journal.pgen.1002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheung A.C., Sainsbury S., Cramer P. Structural basis of initial RNA polymerase II transcription. EMBO J. 2011;30:4755–4763. doi: 10.1038/emboj.2011.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fouqueau T., Zeller M.E., Cheung A.C., Cramer P., Thomm M. The RNA polymerase trigger loop functions in all three phases of the transcription cycle. Nucleic Acids Res. 2013;41:7048–7059. doi: 10.1093/nar/gkt433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toulokhonov I., Zhang J., Palangat M., Landick R. A central role of the RNA polymerase trigger loop in active-site rearrangement during transcriptional pausing. Mol. Cell. 2007;27:406–419. doi: 10.1016/j.molcel.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Yuzenkova Y., Bochkareva A., Tadigotla V.R., Roghanian M., Zorov S., Severinov K., Zenkin N. Stepwise mechanism for transcription fidelity. BMC Biol. 2010;8:1–15. doi: 10.1186/1741-7007-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaplan C.D., Larsson K.M., Kornberg R.D. The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by alpha-amanitin. Mol. Cell. 2008;30:547–556. doi: 10.1016/j.molcel.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miropolskaya N., Artsimovitch I., Klimasauskas S., Nikiforov V., Kulbachinskiy A. Allosteric control of catalysis by the F loop of RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18942–18947. doi: 10.1073/pnas.0905402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miropolskaya N., Esyunina D., Klimasauskas S., Nikiforov V., Artsimovitch I., Kulbachinskiy A. Interplay between the trigger loop and the F loop during RNA polymerase catalysis. Nucleic Acids Res. 2014;42:544–552. doi: 10.1093/nar/gkt877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nayak D., Voss M., Windgassen T., Mooney R.A., Landick R. Cys-pair reporters detect a constrained trigger loop in a paused RNA polymerase. Mol. Cell. 2013;50:882–893. doi: 10.1016/j.molcel.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hein P.P., Kolb K.E., Windgassen T., Bellecourt M.J., Darst S.A., Mooney R.A., Landick R. RNA polymerase pausing and nascent RNA structure formation are energetically linked through clamp domain movement. Nat. Struct. Mol. Biol. 2014;21:794–802. doi: 10.1038/nsmb.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan T., Artsimovitch I., Fang X.W., Landick R., Sosnick T.R. Folding of a large ribozyme during transcription and the effect of the elongation factor NusA. Proc. Natl. Acad. Sci. U.S.A. 1999;96:9545–9550. doi: 10.1073/pnas.96.17.9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong T.N., Sosnick T.R., Pan T. Folding of noncoding RNAs during transcription facilitated by pausing-induced nonnative structures. Proc. Natl. Acad. Sci. U.S.A. 2007;104:17995–18000. doi: 10.1073/pnas.0705038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollands K., Sevostiyanova A., Groisman E.A. Unusually long-lived pause required for regulation of a Rho-dependent transcription terminator. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E1999–E2007. doi: 10.1073/pnas.1319193111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong T.N., Pan T. RNA folding during transcription: protocols and studies. Methods Enzymol. 2009;468:167–193. doi: 10.1016/S0076-6879(09)68009-5. [DOI] [PubMed] [Google Scholar]
- 23.Larson M.H., Greenleaf W.J., Landick R., Block S.M. Applied force reveals mechanistic and energetic details of transcription termination. Cell. 2008;132:971–982. doi: 10.1016/j.cell.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bar-Nahum G., Epshtein V., Ruckenstein A., Rafikov R., Mustaev A., Nudler E. A ratchet mechanism of transcription elongation and its control. Cell. 2005;120:183–193. doi: 10.1016/j.cell.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 25.Kireeva M., Kashlev M., Burton Z.F. Translocation by multi-subunit RNA polymerases. Biochim. Biophys. Acta. 2010;1799:389–401. doi: 10.1016/j.bbagrm.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Malinen A.M., Turtola M., Parthiban M., Vainonen L., Johnson M.S., Belogurov G.A. Active site opening and closure control translocation of multisubunit RNA polymerase. Nucleic Acids Res. 2012;40:7442–7451. doi: 10.1093/nar/gks383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larson M.H., Zhou J., Kaplan C.D., Palangat M., Kornberg R.D., Landick R., Block S.M. Trigger loop dynamics mediate the balance between the transcriptional fidelity and speed of RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 2012;109:6555–6560. doi: 10.1073/pnas.1200939109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Da L.T., Wang D., Huang X. Dynamics of pyrophosphate ion release and its coupled trigger loop motion from closed to open state in RNA polymerase II. J. Am. Chem. Soc. 2012;134:2399–2406. doi: 10.1021/ja210656k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Artsimovitch I., Svetlov V., Murakami K., Landick R. Co-overexpression of E. coli RNA polymerase subunits allows isolation and analysis of mutant enzymes lacking lineage-specific sequence insertions. J. Biol. Chem. 2003;278:12344–12355. doi: 10.1074/jbc.M211214200. [DOI] [PubMed] [Google Scholar]
- 30.Chlenov M., Masuda S., Murakami K., Nikiforov V., Darst S., Mustaev A. Structure and function of lineage-specific sequence insertions in the bacteria RNA polymerase ß′ subunit. J. Mol. Biol. 2005;353:38–54. doi: 10.1016/j.jmb.2005.07.073. [DOI] [PubMed] [Google Scholar]
- 31.Iyer L.M., Koonin E.V., Aravind L. Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct. Biol. 2003;3:1–23. doi: 10.1186/1472-6807-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lane W.J., Darst S.A. Molecular evolution of multisubunit RNA polymerases: sequence analysis. J. Mol. Biol. 2010;395:671–685. doi: 10.1016/j.jmb.2009.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Opalka N., Brown J., Lane W.J., Twist K.A., Landick R., Asturias F.J., Darst S.A. Complete structural model of Escherichia coli RNA polymerase from a hybrid approach. PLoS Biol. 2010;8:e1000483. doi: 10.1371/journal.pbio.1000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conrad T., Frazier M., Joyce A., Cho B., Knight E., Lewis N., Landick R., Palsson B. RNA polymerase mutants found through adaptive evolution re-program Escherichia coli for optimal growth in minimal media. Proc. Natl. Acad. Sci. U.S.A. 2010;107:20500–20505. doi: 10.1073/pnas.0911253107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zakharova N., Bass I., Arsenieva E., Nikiforov V., Severinov K. Mutations in and monoclonal antibody binding to evolutionary hypervariable region of Escherichia coli RNA polymerase beta’ subunit inhibit transcript cleavage and transcript elongation. J. Biol. Chem. 1998;273:24912–24920. doi: 10.1074/jbc.273.38.24912. [DOI] [PubMed] [Google Scholar]
- 36.Kireeva M.L., Nedialkov Y.A., Cremona G.H., Purtov Y.A., Lubkowska L., Malagon F., Burton Z.F., Strathern J.N., Kashlev M. Transient reversal of RNA polymerase II active site closing controls fidelity of transcription elongation. Mol. Cell. 2008;30:557–566. doi: 10.1016/j.molcel.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furman R., Tsodikov O.V., Wolf Y.I., Artsimovitch I. An insertion in the catalytic trigger loop gates the secondary channel of RNA polymerase. J. Mol. Biol. 2013;425:82–93. doi: 10.1016/j.jmb.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jendrisak J.J., Burgess R.R. A new method for the large-scale purification of wheat germ DNA-dependent RNA polymerase II. Biochemistry. 1975;14:4639–4645. doi: 10.1021/bi00692a012. [DOI] [PubMed] [Google Scholar]
- 39.Vogel U., Jensen K.F. The RNA chain elongation rate in Escherichia coli depends on the growth rate. J. Bacteriol. 1994;176:2807–2813. doi: 10.1128/jb.176.10.2807-2813.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dangkulwanich M., Ishibashi T., Liu S., Kireeva M.L., Lubkowska L., Kashlev M., Bustamante C.J. Complete dissection of transcription elongation reveals slow translocation of RNA polymerase II in a linear ratchet mechanism. eLife. 2013;2:e00971. doi: 10.7554/eLife.00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hein P.P., Palangat M., Landick R. RNA transcript 3′-proximal sequence affects translocation bias of RNA polymerase. Biochemistry. 2011;50:7002–7014. doi: 10.1021/bi200437q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ederth J., Mooney R., Isaksson L., Landick R. Functional interplay between the downstream DNA-Jaw domain of bacterial RNA polymerase and allele-specific residues in the product RNA-binding pocket. J. Mol. Biol. 2006;356:1163–1179. doi: 10.1016/j.jmb.2005.11.080. [DOI] [PubMed] [Google Scholar]
- 43.Svetlov V., Belogurov G.A., Shabrova E., Vassylyev D.G., Artsimovitch I. Allosteric control of the RNA polymerase by the elongation factor RfaH. Nucleic Acids Res. 2007;35:5694–5705. doi: 10.1093/nar/gkm600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kolb K.E., Hein P.P., Landick R. Antisense oligonucleotide-stimulated transcriptional pausing reveals RNA exit channel specificity of RNA polymerase and mechanistic contributions of NusA and RfaH. J. Biol. Chem. 2014;289:1151–1163. doi: 10.1074/jbc.M113.521393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kyzer S., Ha K.S., Landick R., Palangat M. Direct versus limited-step reconstitution reveals key features of an RNA hairpin-stabilized paused transcription complex. J. Biol. Chem. 2007;282:19020–19028. doi: 10.1074/jbc.M701483200. [DOI] [PubMed] [Google Scholar]
- 46.Malinen A.M., Nandymazumdar M., Turtola M., Malmi H., Grocholski T., Artsimovitch I., Belogurov G.A. CBR antimicrobials alter coupling between the bridge helix and the beta subunit in RNA polymerase. Nat. Commun. 2014;5:3408.1–3408.9. doi: 10.1038/ncomms4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nedialkov Y.A., Opron K., Assaf F., Artsimovitch I., Kireeva M.L., Kashlev M., Cukier R.I., Nudler E., Burton Z.F. The RNA polymerase bridge helix YFI motif in catalysis, fidelity and translocation. Biochim. Biophys. Acta. 2013;1829:187–198. doi: 10.1016/j.bbagrm.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larson M.H., Mooney R.A., Peters J.M., Windgassen T., Nayak D., Gross C.A., Block S.M., Greenleaf W.J., Landick R., Weissman J.S. A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science. 2014;344:1042–1047. doi: 10.1126/science.1251871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miropolskaya N., Nikiforov V., Klimasauskas S., Artsimovitch I., Kulbachinskiy A. Modulation of RNA polymerase activity through the trigger loop folding. Transcription. 2010;1:89–94. doi: 10.4161/trns.1.2.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ederth J., Artsimovitch I., Isaksson L., Landick R. The downstream DNA jaw of bacterial RNA polymerase facilitates both transcriptional initiation and pausing. J. Biol. Chem. 2002;277:37456–37463. doi: 10.1074/jbc.M207038200. [DOI] [PubMed] [Google Scholar]
- 51.Palangat M., Hittinger C.T., Landick R. Downstream DNA selectively affects a paused conformation of human RNA polymerase II. J. Mol. Biol. 2004;341:429–442. doi: 10.1016/j.jmb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 52.James E., Liu M., Sheppard C., Mekler V., Camara B., Liu B., Simpson P., Cota E., Severinov K., Matthews S., et al. Structural and mechanistic basis for the inhibition of Escherichia coli RNA polymerase by T7 Gp2. Mol. Cell. 2012;47:755–766. doi: 10.1016/j.molcel.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Korzheva N., Mustaev A., Kozlov M., Malhotra A., Nikiforov V., Goldfarb A., Darst S.A. A structural model of transcription elongation. Science. 2000;289:619–625. doi: 10.1126/science.289.5479.619. [DOI] [PubMed] [Google Scholar]
- 54.Xu L., Butler K.V., Chong J., Wengel J., Kool E.T., Wang D. Dissecting the chemical interactions and substrate structural signatures governing RNA polymerase II trigger loop closure by synthetic nucleic acid analogues. Nucleic Acids Res. 2014;42:5863–5870. doi: 10.1093/nar/gku238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan C.L., Wang D., Landick R. Multiple interactions stabilize a single paused transcription intermediate in which hairpin to 3′ end spacing distinguishes pause and termination pathways. J. Mol. Biol. 1997;268:54–68. doi: 10.1006/jmbi.1997.0935. [DOI] [PubMed] [Google Scholar]
- 56.Wang D., Meier T.I., Chan C.L., Feng G., Lee D.N., Landick R. Discontinuous movements of DNA and RNA in RNA polymerase accompany formation of a paused transcription complex. Cell. 1995;81:341–350. doi: 10.1016/0092-8674(95)90387-9. [DOI] [PubMed] [Google Scholar]
- 57.Weixlbaumer A., Leon K., Landick R., Darst S.A. Structural basis of transcriptional pausing in bacteria. Cell. 2013;152:431–441. doi: 10.1016/j.cell.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang D., Bushnell D.A., Huang X., Westover K.D., Levitt M., Kornberg R.D. Structural basis of transcription: backtracked RNA polymerase II at 3.4 angstrom resolution. Science. 2009;324:1203–1206. doi: 10.1126/science.1168729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tagami S., Sekine S.I., Kumarevel T., Hino N., Murayama Y., Kamegamori S., Yamamoto M., Sakamoto K., Yokoyama S. Crystal structure of bacterial RNA polymerase bound with a transcription inhibitor protein. Nature. 2010;468:978–982. doi: 10.1038/nature09573. [DOI] [PubMed] [Google Scholar]
- 60.Artsimovitch I., Chu C., Lynch A.S., Landick R. A new class of bacterial RNA polymerase inhibitor affects nucleotide addition. Science. 2003;302:650–654. doi: 10.1126/science.1087526. [DOI] [PubMed] [Google Scholar]
- 61.Kirschner M., Gerhart J. Evolvability. Proc. Natl. Acad. Sci. U.S.A. 1998;95:8420–8427. doi: 10.1073/pnas.95.15.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.