

Abstract
Cells make decisions to differentiate, divide, or apoptose based on multiple signals of internal and external origin. These decisions are discrete outputs from dynamic networks comprised of signaling pathways. Yet the validity of this decomposition of regulatory proteins into distinct pathways is unclear because many regulatory proteins are pleiotropic and interact through cross-talk with components of other pathways. In addition to the deterministic complexity of interconnected networks, there is stochastic complexity arising from the fluctuations in concentrations of regulatory molecules. Even within a genetically identical population of cells grown in the same environment, cell-to-cell variations in mRNA and protein concentrations can be as high as 50% in yeast and even higher in mammalian cells. Thus, if everything is connected and stochastic, what hope could we have for a quantitative understanding of cellular decisions? Here we discuss the implications of recent advances in genomics, single-cell, and single-cell genomics technology for network modularity and cellular decisions. On the basis of these recent advances, we argue that most gene expression stochasticity and pathway interconnectivity is nonfunctional and that cellular decisions are likely much more predictable than previously expected.
INTRODUCTION
Modularity of a group of biological components is defined as the degree to which their collective function is insulated from other components and pathways (Hartwell et al., 1999). In the context of cellular decisions, a highly modular decision would be determined by a single pathway, whereas a nonmodular decision would be determined by many interconnected pathways. In general, genomics and high-throughput studies support the less modular view of cell signaling. Specifically, interaction maps based on protein–protein binding assays, phosphorylation interactions, or synthetic lethality of double mutants exhibit many interactions between components of different signaling pathways (Ficarro et al., 2002; Rual et al., 2005; Krogan et al., 2006; Boone et al., 2007). Accordingly, when plotted on a network graph, essentially all proteins in a cell are connected through few degrees of separation. If many of these proteins contributed equally to a biological decision or function, it would be very difficult, if not impossible, to understand how and why a certain input to a particular pathway affects a particular outcome.
Although understanding the computation underlying even the simplest cellular decisions seems such a formidable challenge in light of genomics studies, progress has been relentless in a large variety of networks. The past few decades of biological research has successfully used an increasingly automated forward-genetic framework to identify and classify signaling pathway components and their interactions. The success of forward-genetic approaches indicates that cellular pathways transmit information mostly unidirectionally and are relatively modular, that is, isolated from other pathways. If this were not the case, we would expect far fewer double mutants phenocopying the individual single mutants, and the ordering of genes into pathways would be much more difficult.
Moreover, it is likely that there are additional hierarchies of organization from network to pathway to even smaller groups of signaling molecules known as motifs (Figure 1). This hierarchical organization is in part due to the separate time scales of biological interactions. For instance, phosphorylation reactions are very fast, whereas protein synthesis and corresponding concentration changes are relatively slow. This allows analysis of phosphorylation kinetics of a few pathway components comprising a motif while treating protein concentrations as fixed. Thus the motif's dynamics can be studied in depth, and its function can be characterized. Indeed, motif analysis has made great progress in determining signaling principles (Alon, 2007), suggesting a view of biology as tractable, modular on several scales, and, it is hoped, ultimately predictable. According to this view, with sufficient understanding of networks and signaling principles, we should be able to understand how cells process information and predict what the output of a cellular decision will be from the measurements of regulatory proteins.
FIGURE 1:
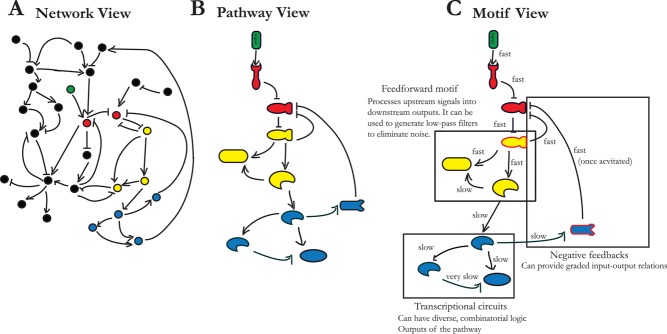
Hierarchies of organization from network to pathway to motifs. (A) In the network view, pathways are usually not readily discernible because of many apparent interactions between pathways from genomic studies (the pathway in question similarly colored in all panels). We note that in many network analyses, there is no information about the functional significance of interactions. (B) Even though the identity of components (receptors, kinases, transcription factors, downstream targets) is usually indicated in the pathway view, it is a static description of a dynamic system. (C) The separation of time scales may allow the analysis of groups of network components as functionally distinct motifs. The example shown here is idealized because for most pathways; not all components can be so easily broken down into motifs. In this example, “fast” indicates the phosphorylation time scale (∼1 min), and “slow” signifies transcription time scale (∼15 min to 1 h) typical of yeast.
STOCHASTICITY IN CELLULAR DECISIONS
The protein concentrations of key regulatory molecules exhibit cell-to-cell variation even within genetically identical cells in highly similar conditions. A study of all budding yeast genes fused to green fluorescent protein revealed significant cell-to-cell variability ranging from as low as 10% for some species to nearly 50% for others (Newman et al., 2006). Such noise is often even larger in mammalian cells (Raj et al., 2006). These studies have increasingly emphasized the view in which each cell is unique and its behavior is variable due to fluctuations in the expression of individual genes arising from either differences in RNA polymerase and ribosome number between cells (extrinsic noise) or inherent randomness in the synthesis of small numbers of molecules within a cell (intrinsic noise) (Elowitz et al., 2002; Blake et al., 2003; Brown et al., 2013).
Perhaps predictably, the less-than-predictable stochastic gene expression has been associated with cell-to-cell variability in cellular decisions in a wide variety of organisms (Raj and van Oudenaarden, 2008). Even though these high noise levels are mostly viewed as a problem that cells need to overcome to reliably respond to environmental inputs, they might also perform useful functions. Noise can allow genetically identical cells to have remarkably different gene expression levels when combined with positive feedback–driven switches (Losick and Desplan, 2008). Gene expression heterogeneity might allow isogenic cells to be better adapted to different or fluctuating environments (Kussell and Leibler, 2005; Acar et al., 2008; Salathé et al., 2009). Indeed, this phenomenon has been observed in the competence behavior of Bacillus subtilis, as well as in the latent phase of human immunodeficiency virus infection, and is also thought to underlie the persistence of subpopulations of Escherichia coli (Maamar et al., 2007; Süel et al., 2007; Weinberger et al., 2008; Maisonneuve et al., 2013). In eukaryotes, noise determines the specification of blue-sensitive versus yellow-sensitive photoreceptors in Drosophila eyes, blood-cell differentiation in mammalian cells, and incomplete penetrance of mutants in Caenorhabditis elegans (Wernet et al., 2006; Chang et al., 2008; Raj et al., 2010). Moreover, cell-to-cell differences in gene expression have been suggested to be further enhanced in tumors due to microenvironment and genetic differences, which may be related to highly variable therapeutic response (Navin et al., 2011; Burrell et al., 2013; Patel et al., 2014). All in all, the stochasticity of cellular decisions has been increasingly recognized, emphasized, and perhaps even celebrated in the past 10–15 years.
For both modular and nonmodular networks, stochastic cell-to-cell variation in regulatory proteins will make cellular computation less predictable. However, the effect is expected to be much worse in nonmodular networks. In a nonmodular network, by definition, many components significantly contribute to a cellular decision. Fluctuations in each of these components would combine to affect the outcome in a manner not unlike how measurement errors in different parts of a computation propagate to the final result. Of course, the time scales at which these components fluctuate will determine the extent to which fluctuations affect the decision. If fluctuations were fast relative to system response, we would only expect the mean level to affect the decision. However, in general, the more components involved, the more we expect noise to decrease the predictability of a cellular decision.
SINGLE-CELL GENOMICS AND THE DEFINITION OF CELL STATE
Terms such as cell state or type have a long history of usage. However, their definition was based either on morphological features or the expression of single marker genes. Therefore it was not known to what extent cell types corresponded to distinct categories of cell states. Although earlier single-cell studies examining expression of single genes emphasized stochasticity, more recent studies measuring transcription genome-wide in single cells emphasize the coherence of distinct cell states or types. For instance, in a recent study, the clustering of RNA profiles of cells from splenic tissue was determined to be sufficient to classify thousands of cells into a few, well-defined cell types (Jaitin et al., 2014). In a similar study, another complex tissue—distal lung epithelium—was shown to consist of five distinct types of cells associated with previously known marker genes (Treutlein et al., 2014). Given the inherent stochasticity of gene expression, it is surprising that these studies found only a few distinct, but not intermediate, cell types, and different cells could unequivocally be categorized simply by their expression of a few markers. Thus transitions between states must be rapid relative to time spent within states because if the time scale of the transition were slow, then states would not appear well defined in single-cell transcriptome studies. Taken together, these results argue that the decision-making networks determining cell types have only a few output states and that the idea of cell state is well defined on the level of gene expression. The clustering of cells extracted from complex tissues into discrete types represents an extraordinary simplification in the description of a cell from a vector of thousands of protein concentrations to a single integer number corresponding to a particular cell type.
In developmental contexts, stochasticity does not seem desirable, because patterns must be formed reliably. Boundaries between groups of different cell types must appear nearly in the same place from organism to organism within a species. For instance, in Drosophila embryos, for neighboring cells to have distinct fates as dictated by developmental patterning, neighboring nuclei need to distinguish a 10% difference in a Bicoid gradient that sets up the anterior–posterior axis. This is very difficult to achieve, as it is close to the physical limits of noise imposed by the absolute number of Bicoid molecules. Nevertheless, it was shown that this level of precision is achieved (Gregor et al., 2007). Moreover, information transferred from Bicoid to the downstream Hunchback gradient is nearly 90% of the theoretical maximum (Tkacik et al., 2008). These results indicate that patterns of morphogens are reliably read out by signaling pathways, and decision-making networks perform accurate computations based on these signals to specify the correct cell type.
Finally, while stochasticity was initially thought to be very important for embryonic stem cells, recent reports indicate that previously documented heterogeneity might be an artifact of experiment conditions. Earlier research found that Nanog, a key determinant of pluripotency, alternates stochastically between low and high states. However, it was later shown that these fluctuations in Nanog primarily arise from conflicting stimuli present in serum and are unlikely to be relevant to development (Smith, 2013).
HISTORY DEPENDENCE AND MEMORY
Cells in different states respond differently to similar inputs. Thus the history of various input signals specifies the current discrete cell state to indirectly determine the current cellular response. For example, positive feedback between the cell division cycle protein kinase Cdk1 and Erk mitogen-activated kinase transforms a transient induction stimulus to irreversible oocyte maturation in Xenopus and thereby separates two distinct cell states (Xiong and Ferrell, 2003). The uninduced state can dramatically respond to hormone signals to trigger development, whereas the induced state continues development independent of the hormone signal. Similarly, a positive feedback loop of G1 cyclin expression separates a G1 phase from a post-G1 cycling cell state in budding yeast. Whereas the G1 cell responds to pheromone signaling, the cycling cell has largely dismantled this pathway and does not respond (Strickfaden et al., 2007; Garrenton et al., 2009).
Although the current cell state determines much of the cell-to-cell variability in signaling response, cells of the same type may respond differently to similar input signals. For example, in budding yeast, the cell-cycle inhibitor Far1, whose level determines the length of mating arrest, accumulates more rapidly at high pheromone concentrations. Therefore current Far1 levels reflect a history of exposure to pheromone. As a result, cells in the same G1 state that experienced different histories of pheromone signal will respond differently to future pheromone input signals (Doncic and Skotheim, 2013). However, this history dependence would not be readily observable in single-cell RNA-sequencing studies because mRNAs quickly reach steady-state values due to their rapid turnover. In contrast to rapidly degraded mRNAs, proteins are mostly long-lived, so their accumulation will mark how long ago a particular cell adopted its current fate. In fact, a protein that was produced even in a previous state can still be present and affect decisions in subsequent cell cycles (Doncic, Atay, Grande, Bush, Valk, Loog, Colman-Lerner, and Skotheim, unpublished data). Thus the proteome will contain memories of previous states and input signals, which can be used to inform subsequent decisions. This may at least partially explain the cell-to-cell variability in the response to input signals without having to refer to noise in gene expression.
Of importance, novel mass cytometry tools that allow quantification of >30 protein markers in single cells have started to be used to describe the gene expression trajectory of state transitions, including multistep differentiation processes (Bendall et al., 2011, 2014). By combining these tools with more focused approaches such as fluorescence tagging and tracking of important cell type markers across transitions, we can begin to understand not only the mechanisms of cellular decisions that specify distinct cell types, but also the history dependence of these decisions. We anticipate that history dependence of cell signaling will be an emerging theme in this decade, as even the classical vertebrate morphogen TGF-β signal, previously thought to statically read out a gradient, likely depends on the dynamics of ligand stimulation (Warmflash et al., 2012; Sorre et al., 2014). It is hoped that this line of research will reveal why development is so robustly reproducible.
HIGHLY PREDICTABLE CELLULAR DECISIONS
Perhaps one of the most fundamental state transitions in yeast is the commitment to cell division. After commitment, yeast is insensitive to even high levels of mating pheromone (Hartwell et al., 1974). When this commitment point was investigated quantitatively, it was found that the measurement of cell size or time after the previous cell division is a poor predictor of whether a cell has committed to cell division, that is, whether it would not arrest upon pheromone exposure. However, the measurement of the nuclear localization of a single protein, Whi5, could predict the subsequent divide-or-arrest decision in >97% of cells (Doncic et al., 2011; Figure 2). That is, almost all cells that have inactivated more than a threshold level of Whi5 do not arrest upon pheromone exposure, whereas cells that have inactivated less Whi5 arrest immediately. Of interest, the corresponding cell division commitment decision in mammalian cells could be similarly predicted by the measurement of only Cdk2 activity (Johnson, 2014). Note that in both cases, the prediction relies only on information gathered before the step change in inputs. We note that the use of measurements after a step change to “predict” previous cell state is not really a prediction, as is commonly done (e.g., Spencer et al., 2013), but rather a postdiction.
FIGURE 2:
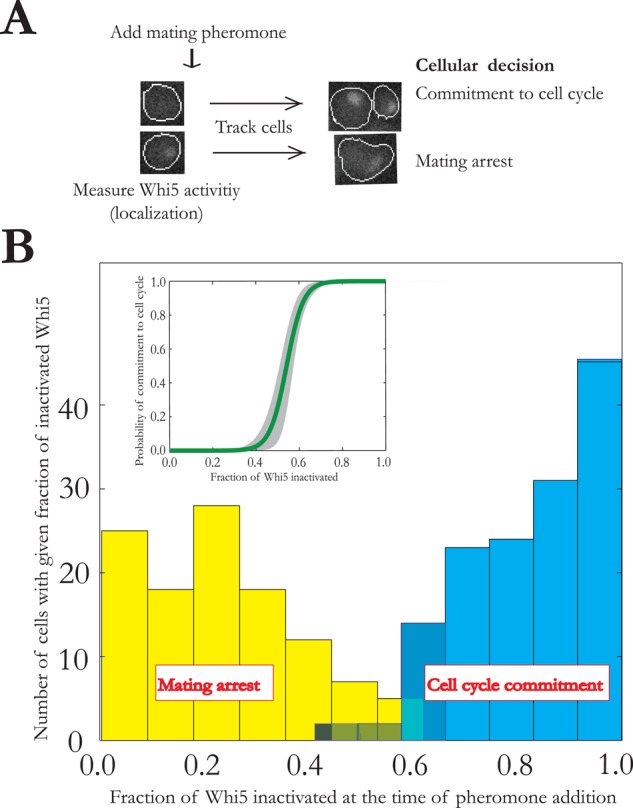
Predictability of the proliferation decision in budding yeast. (A) Cells that are committed to the cell cycle do not arrest upon exposure to mating pheromone. Whi5 activity was measured up until pheromone addition, and this information was used to predict whether a cell would arrest or divide when exposed to pheromone. (B) Histograms and logistic regression curve showing the ability of a single Whi5 measurement at the time of pheromone exposure to predict cell state. The shaded region in the logistic regression indicates 95% confidence interval (by bootstrapping). Data from Doncic et al. (2011).
It seems unlikely that Whi5 and Cdk2 are the only special markers that can predict commitment. Whi5, in particular, is neither essential nor a hub in interaction networks. However, both Whi5 and Cdk2 activities correlate with the abrupt activation of positive feedback loops driving commitment (Skotheim et al., 2008; Yao et al., 2008). Because the transition is very rapid relative to the time spent in states, there will be relatively few cells near the separatrix—the hypothetical boundary dividing different states of distinct expression patterns. This likely makes it relatively easy to predict with high accuracy what the current cell state is and the final state will be in response to a given input. Indeed, any protein that significantly changes during a transition is a good candidate for a predictor of the current cell state. Because transitions from one cell state to another are often extensive and likely somewhat similar from cell to cell, we expect that the changes in concentration or localization of many proteins will be sufficient to predict cell state. Of course, as previously discussed, two cells of the same type might have different histories of proteins from their previous states. For these two cells, if the protein that we choose for prediction is sensitive to concentrations of these historic proteins, its threshold value that determines the cell state would not be the same. In this case, the measurement of such a protein would be expected to be less predictive of cell state. Therefore, for the measurement of a single protein to be predictive of cell state, either that protein must be insensitive to other protein concentrations or the cells must have a shared history of initial states. Nevertheless, the fact that two proteins that were chosen for predicting commitment to the cell cycle in two different organisms were both similarly accurate suggests that these conditions are not rare. Thus it might be possible to predict the final state of a cell with only a few measurements, even when cellular decisions involve multiple pathways.
POTENTIAL RECONCILIATION OF NOISE, INTERCONNECTED NETWORKS, AND PREDICTABILITY
The success of forward genetics, the distinct clustering of cell types, and the reproducibility of development all favor the tractable view of biology. However, it is unclear why this might be, given the high degree of interconnectivity between pathways and the stochasticity associated with each network node. In fact, naively, one would expect that the measurement of levels of tens of proteins would be required to be able to predict and understand any nontrivial cellular decision. However, the examples of cell cycle commitment in yeast and mammalian cells suggest that the measurement of even a single protein can be enough. However, how can we reconcile this high degree of predictability with the high degree of interconnectivity in protein and genetic networks and the known stochastic nature of gene expression?
Although most protein–protein interactions or phosphorylation interactions are unlikely to be experimental artifacts, many of them may not be functionally significant. In particular, studies that compare transcription factor–target gene interactions across species find evidence of high levels of rewiring even at short evolutionary time scales, likely through nonadaptive neutral evolution (Lynch, 2007; Tuch et al., 2008). Similarly, protein–protein interactions rapidly evolve, as even interactions between homologues are usually lost across species (Lewis et al., 2012). Anecdotally, many unpublished studies were unable to find phenotypes by mutating phosphorylation sites. Of course, it is possible that some of these phosphorylation sites may be important in conditions not yet tested or in ways that are not assayed. However, consistent with the notion that most phosphorylation sites are nonfunctional and evolve neutrally, they tend not to be conserved even among closely related yeast species (Holt et al., 2009).
Similarly, cell-to-cell concentration variations of each network component are unlikely to be functional. Outputs of cellular decisions may be robust to fluctuations in all but a few components of a network (Kitano, 2004; Masel and Siegal, 2009). It seems that single copies of >95% of all genes in diploid S. cerevisiae and Drosophila are sufficient for near-wild-type fitness and normal development (Lindsley et al., 1972; Deutschbauer et al., 2005). Moreover, this lack of fitness cost does not stem from any dosage compensation mechanisms that adjust the level of proteins in yeast deletion strains. In the vast majority of tested genes, single-copy deletions in diploid yeast resulted in 50% decrease in protein expression without a corresponding fitness cost (Springer et al., 2010). This result is in stark contrast to double-knockout screens, which typically find all biological processes connected with each other. However, double knockouts are severe perturbations that completely remove nodes from a network. They alter network architecture and completely eliminate information flow from dozens of other nodes. Therefore, whereas severe perturbations such as double knockouts may result in genetic interactions, stochastic fluctuations normally experienced by a cell will be much smaller and often be nonfunctional.
Moreover, even functionally significant components shared between two pathways do not necessarily indicate cross-talk between pathways. This is because these functional components may be in excess, so that use by one pathway does not limit the other. For example, in budding yeast, pheromone and hyperosmotic stress pathways share upstream components that are essential for signal transduction. Nevertheless, the amplitude of pheromone-induced transcription is nearly unaffected by the presence of osmotic stress. That is, pheromone signals and osmotic stress signals are insulated (Patterson et al., 2010). Moreover, even when the signal to one pathway affects the output of another pathway, it does not necessarily mean that these two pathways cross-talk. For instance, although addition of mating pheromone can trigger osmotic stress response in cells adapted to high osmolarity, this signal does not reflect a loss of insulation. Instead, the pheromone signal induces morphological changes as an output, which leads to activation of the cell wall integrity pathway, which in turn activates the osmotic stress kinase Hog1 (Baltanás et al., 2013). This analysis shows that even when two pathways appear to cross-talk molecularly, a careful temporal analysis may order their activities to reveal that the output of one pathway serves as an input to activate the second pathway.
Finally, whereas most protein–protein interactions, phosphorylation events, and stochasticity do not seem to significantly affect many aspects of cellular decisions, some are likely to influence when a cell switches from one cell state to another. This is because the timing of transitions is sensitive to where the separatrix between two states is located. Small changes in protein activity or abundance due to mutation can affect separatrix location and thereby alter transition timing. Similarly, close to a separatrix, dynamical systems are sensitive to stochastic fluctuations in protein concentrations, which can be amplified by positive feedback loops to abruptly trigger a transition. Thus, whereas the precise timing of future transitions may be variable and difficult to predict, we emphasize that we can likely determine a cell's current state and predict many aspects of a cellular decision from the measurement of a few, judiciously chosen proteins.
CONCLUDING REMARK
At first glance, apparent interconnectivity of pathways and inherent stochasticity of gene expression paint a bleak picture for the understanding of quantitative principles underlying cellular decisions. However, recent advances in single-cell studies indicate that such interconnectivity and stochasticity may be largely nonfunctional. Indeed, cellular decisions are likely much more predictable than previously expected or even imagined.
Acknowledgments
We thank Daniel S. Fisher, Arjun Raj, and an anonymous reviewer for careful reading of the manuscript and insightful comments and suggestions. This work was supported by a Stanford Graduate Fellowship (O.A.), the National Institutes of Health (R01 GM092925 to J.M.S.), and a Career Award at the Scientific Interface from the Burroughs Wellcome Fund (J.M.S.).
Abbreviations used:
- Cdk
cyclin-dependent kinase
- Erk
extracellular signal-regulated kinase
- G1
growth/gap 1
- TGF-β
transforming growth factor beta
Footnotes
REFERENCES
- Acar M, Mettetal JT, Van Oudenaarden A. Stochastic switching as a survival strategy in fluctuating environments. Nat Genet. 2008;40:471–475. doi: 10.1038/ng.110. [DOI] [PubMed] [Google Scholar]
- Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- Baltanás R, Bush A, Couto A, Durrieu L, Hohmann S, Colman-Lerner A. Pheromone-induced morphogenesis improves osmoadaptation capacity by activating the HOG MAPK pathway. Sci Signal. 2013;6:ra26. doi: 10.1126/scisignal.2003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir E-D, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, Pe'Er D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157:714–725. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir E-D, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake WJ, Kærn M, Cantor CR, Collins JJ. Noise in eukaryotic gene expression. Nature. 2003;422:633–637. doi: 10.1038/nature01546. [DOI] [PubMed] [Google Scholar]
- Boone C, Bussey H, Andrews BJ. Exploring genetic interactions and networks with yeast. Nat Rev Genet. 2007;8:437–449. doi: 10.1038/nrg2085. [DOI] [PubMed] [Google Scholar]
- Brown CR, Mao C, Falkovskaia E, Jurica MS, Boeger H. Linking stochastic fluctuations in chromatin structure and gene expression. PLoS Biol. 2013;11:e1001621. doi: 10.1371/journal.pbio.1001621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–547. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW, Nislow C, Giaever G. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169:1915–1925. doi: 10.1534/genetics.104.036871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncic A, Falleur-Fettig M, Skotheim J. Distinct interactions select and maintain a specific cell fate. Mol Cell. 2011;43:528–539. doi: 10.1016/j.molcel.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncic A, Skotheim JM. Feedforward regulation ensures stability and rapid reversibility of a cellular state. Mol Cell. 2013;50:856–868. doi: 10.1016/j.molcel.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20:301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- Garrenton LS, Braunwarth A, Irniger S, Hurt E, Kunzler M, Thorner J. Nucleus-specific and cell cycle-regulated degradation of mitogen-activated protein kinase scaffold protein Ste5 contributes to the control of signaling competence. Mol Cell Biol. 2009;29:582–601. doi: 10.1128/MCB.01019-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor T, Tank DW, Wieschaus EF, Bialek W. Probing the limits to positional information. Cell. 2007;130:153–164. doi: 10.1016/j.cell.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Culotti J, Pringle JR, Reid BJ. Genetic control of the cell division cycle in yeast. Science. 1974;183:46–51. doi: 10.1126/science.183.4120.46. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insiqhts into evolution. Science. 2009;325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A. Onset of CDK2 Activity Determines Passage Through the Restriction Point in Primary Cells. Thesis. Stanford University, Stanford, CA. 2014 [Google Scholar]
- Kitano H. Biological robustness. Nat Rev Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Kussell E, Leibler S. Ecology: phenotypic diversity, population growth, and information in fluctuating environments. Science. 2005;309:2075–2078. doi: 10.1126/science.1114383. [DOI] [PubMed] [Google Scholar]
- Lewis ACF, Jones NS, Porter MA, Deane CM. What evidence is there for the homology of protein-protein interactions. PLoS Comput Biol. 2012;8:e1002645. doi: 10.1371/journal.pcbi.1002645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley DL, Sandler L, Baker BS, Carpenter AT, Denell RE, Hall JC, Jacobs PA, Miklos GL, Davis BK, Gethmann RC, et al. Segmental aneuploidy and the genetic gross structure of the Drosophila genome. Genetics. 1972;71:157–184. doi: 10.1093/genetics/71.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick R, Desplan C. Stochasticity and cell fate. Science. 2008;320:65–68. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. The evolution of genetic networks by non-adaptive processes. Nat Rev Genet. 2007;8:803–813. doi: 10.1038/nrg2192. [DOI] [PubMed] [Google Scholar]
- Maamar H, Raj A, Dubnau D. Noise in gene expression determines cell fate in Bacillus subtilis. Science. 2007;317:526–529. doi: 10.1126/science.1140818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonneuve E, Castro-Camargo M, Gerdes K. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell. 2013;154:1140–1150. doi: 10.1016/j.cell.2013.07.048. [DOI] [PubMed] [Google Scholar]
- Masel J, Siegal ML. Robustness: mechanisms and consequences. Trends Genet. 2009;25:395–403. doi: 10.1016/j.tig.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–95. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JRS, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, Weissman JS. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson JC, Klimenko ES, Thorner J. Single-cell analysis reveals that insulation maintains signaling specificity between two yeast MAPK pathways with common components. Sci Signal. 2010;3:ra75. doi: 10.1126/scisignal.2001275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Rifkin SA, Andersen E, Van Oudenaarden A. Variability in gene expression underlies incomplete penetrance. Nature. 2010;463:913–918. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Salathé M, Van Cleve J, Feldman MW. Evolution of stochastic switching rates in asymmetric fitness landscapes. Genetics. 2009;182:1159–1164. doi: 10.1534/genetics.109.103333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotheim JM, Di Talia S, Siggia ED, Cross FR. Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature. 2008;454:291–296. doi: 10.1038/nature07118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. Nanog heterogeneity: tilting at windmills. Cell Stem Cell. 2013;13:6–7. doi: 10.1016/j.stem.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorre B, Warmflash A, Brivanlou AH, Siggia E. Encoding of temporal signals by the TGF-ß pathway and implications for embryonic patterning. Dev Cell. 2014;30:334–342. doi: 10.1016/j.devcel.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, Meyer T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell. 2013;155:369–383. doi: 10.1016/j.cell.2013.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer M, Weissman JS, Kirschner MW. A general lack of compensation for gene dosage in yeast. Mol Syst Biol. 2010;6:368. doi: 10.1038/msb.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickfaden SC, Winters MJ, Ben-Ari G, Lamson RE, Tyers M, Pryciak P. A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway. Cell. 2007;128:519–531. doi: 10.1016/j.cell.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Süel GM, Kulkarni RP, Dworkin J, Garcia-Ojalvo J, Elowitz MB. Tunability and noise dependence in differentiation dynamics. Science. 2007;315:1716–1719. doi: 10.1126/science.1137455. [DOI] [PubMed] [Google Scholar]
- Tkacik G, Callan CG, Bialek W. Information flow and optimization in transcriptional regulation. Proc Natl Acad Sci USA. 2008;105:12265–12270. doi: 10.1073/pnas.0806077105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuch BB, Li H, Johnson AD. Evolution of eukaryotic transcription circuits. Science. 2008;319:1797–1799. doi: 10.1126/science.1152398. [DOI] [PubMed] [Google Scholar]
- Warmflash A, Zhang Q, Sorre B, Vonic A, Siggia ED, Brivanlou AH. Dynamics of TGF-ß signaling reveal adaptive and pulsatile behaviors reflected in the nuclear localization of transcription factor Smad4. Proc Natl Acad Sci USA. 2012;109:E1947–E1956. doi: 10.1073/pnas.1207607109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger LS, Dar RD, Simpson ML. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat Genet. 2008;40:466–470. doi: 10.1038/ng.116. [DOI] [PubMed] [Google Scholar]
- Wernet MF, Mazzoni EO, Çelik A, Duncan DM, Duncan I, Desplan C. Stochastic spineless expression creates the retinal mosaic for colour vision. Nature. 2006;440:174–180. doi: 10.1038/nature04615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Ferrell JE. A positive-feedback-based bistable “memory module” that governs a cell fate decision. Nature. 2003;426:460–465. doi: 10.1038/nature02089. [DOI] [PubMed] [Google Scholar]
- Yao G, Lee TJ, Mori S, Nevins JR, You L. A bistable Rb-E2F switch underlies the restriction point. Nat Cell Biol. 2008;10:476–482. doi: 10.1038/ncb1711. [DOI] [PubMed] [Google Scholar]