

Mammalian cell abscission was recently shown to be driven by the ESCRT machinery, but the mechanism has not been fully resolved. This work identifies the ESCRT components ESCRT-II and CHMP6 as essential components of ESCRT-mediated abscission and introduces a new approach for inhibition of abscission using the first 52 amino acids of CHMP6.
Abstract
Recently the ESCRT-III filamentous complex was designated as the driving force for mammalian cell abscission, that is, fission of the intercellular membrane bridge connecting daughter cells at the end of cytokinesis. However, how ESCRT-III is activated to set on abscission has not been resolved. Here we revisit the role of the upstream canonical ESCRT players ESCRT-II and CHMP6 in abscission. Using high-resolution imaging, we show that these proteins form highly ordered structures at the intercellular bridge during abscission progression. Furthermore, we demonstrate that a truncated version of CHMP6, composed of its first 52 amino acids (CHMP6-N), arrives at the intercellular bridge, blocks abscission, and subsequently leads to cell death. This phenotype is abolished in a mutated version of CHMP6-N designed to prevent CHMP6-N binding to its ESCRT-II partner. Of interest, deleting the first 10 amino acids from CHMP6-N does not interfere with its arrival at the intercellular bridge but almost completely abolishes the abscission failure phenotype. Taken together, these data suggest an active role for ESCRT-II and CHMP6 in ESCRT-mediated abscission. Our work advances the mechanistic understanding of ESCRT-mediated membrane fission in cells and introduces an easily applicable tool for upstream inhibition of the ESCRT pathway in live mammalian cells.
INTRODUCTION
Mammalian cell division ends with abscission—the cleavage of a thin intercellular membrane bridge connecting two daughter cells at the end of cytokinesis (Figure 1A). Recently the endosomal sorting complexes required for transport (ESCRT) membrane fission machinery (composed of five different subfamilies: ESCRT-0, -I, -II, and -III and VPS4) has been shown to mediate cytokinetic abscission (Carlton and Martin-Serrano, 2007; Morita et al., 2007; Elia et al., 2011, 2013; Guizetti et al., 2011). During cytokinesis, components of the ESCRT machinery assemble into cortical rings on the membrane of the midbody dark zone, an electron-dense structure at the center of the bridge (Figure 1A). Abscission is accompanied by rearrangement of ESCRT-III components to the abscission sites, located ∼1 μm away from either side of the center of the bridge (Figure 1A; Elia et al., 2011, 2012, 2013; Guizetti et al., 2011). Although these studies identified the ESCRT complex as the molecular machinery that drives abscission, many of the mechanistic steps that lead to ESCRT-mediated abscission and their regulation remained unresolved.
FIGURE 1:
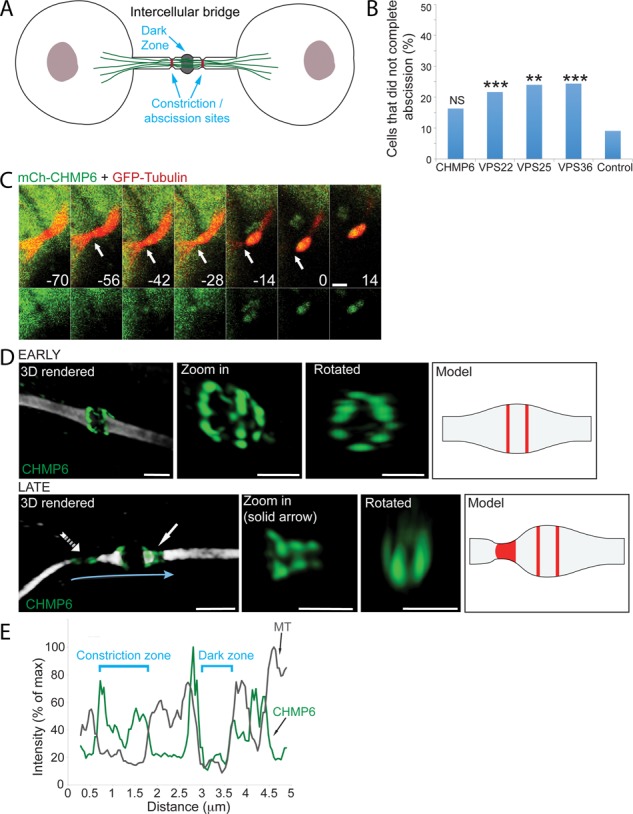
The ESCRT-III component CHMP6 localizes to the intercellular bridge during cytokinesis. (A) Schematic model of the late intercellular bridge. The dark zone is located at the center of the intercellular bridge. Bridge cleavage occurs at narrow constriction sites located peripherally on both sides of the dark zone. (B) Percentage of abscission failure in MDCK cells overexpressing ESCRT-II and CHMP6 components. Cells were cotransfected with GFP/mCherry-VPS25, GFP/mCherry-VPS36, GFP/mCherry-VPS22, or mCherry-CHMP6 together with mCherry/GFP-tubulin, respectively, and were imaged for 3–4 h using a confocal spinning disk. CHMP6 (n = 49), VPS22 (n = 60), VPS25 (n = 25), VPS36 (n = 82), and control cells (expressing either GFP/mCherry-tubulin or mCherry/GFP empty plasmids; n = 44). The rate of abscission failure in MDCK cells overexpressing CHMP6 was not significantly different from that in control cells (χ12 test, p = 0.068), whereas the rates of abscission failure in MDCK cells overexpressing VPS22, VPS25, or VPS36 were significantly higher than in control cells (**χ12 test, p < 0.01; ***χ12 test, p < 0.001). (C) Live-cell imaging of MDCK cells undergoing cytokinesis reveals acute recruitment of CHMP6 to the intercellular bridge. Cells expressing low levels of mCherry-CHMP6 together with GFP-tubulin were imaged using a spinning-disk confocal microscope at 7-min intervals. Shown are maximum-intensity projections of different time points during cytokinesis from a representative cell. Top, overlay of CHMP6 (green) and microtubules (red); bottom, CHMP6 signal alone. Time (indicated in minutes) is relative to abscission (Supplemental Video S1; n = 10; bar, 2 μm). Arrows indicate the position of the first abscission site. (D) Spatial organization of CHMP6, in early (top) and late (bottom) intercellular bridges. MDCK cells expressing Flag-CHMP6 were fixed, stained with anti–α-tubulin and anti-Flag antibodies, and imaged by SIM. Early and late bridges were categorized based on the diameter of the intercellular bridge at the constriction site (see Materials and Methods). Each panel shows (from left to right) a three-dimensional (3D) reconstruction of an overlay of CHMP6 (green) and tubulin (white; bar, 2 μm); a zoomed-in, 3D rendered image of the protein structure alone (bar, 1 μm); a zoomed-in, 3D rendered image of the protein structure rotated 90° (bar, 1 μm); and a schematic model for CHMP6 organization at the intercellular bridge based on SIM measurements. Zoomed-in and rotated images of late bridges are of the structure labeled by a solid arrow in the full 3D reconstructed image. The model for late bridges refers to the structure indicated by a dashed arrow in the full 3D reconstructed image, as this is a more advanced time point in the process. In early intercellular bridges, CHMP6 concentrates in two rings located on the rims of the dark zone. The rings are 0.47 ± 0.11 μm apart (n = 17). The dark zone is 0.69 ± 0.12 μm (n = 36) wide. The diameter of the rings is 1.11 ± 0.19 μm (n = 37). In late intercellular bridges, the CHMP6 structure elongates asymmetrically, peripherally to the center of the bridge (solid arrow), forming a series of cortical rings with decreasing diameters (ring diameters in zoomed-in image: 900, 500, and 450 nm). Finally, CHMP6 is located in two separate pools: one on the rims of the dark zone, and one that colocalizes with the site of microtubule constriction (dashed arrow; see also E). (E) The peripheral CHMP6 pool is located at the site of microtubule constriction. Plot shows tubulin (gray) and CHMP6 (green) line intensity profiles along the intercellular bridge (indicated by a blue arrow in D).
ESCRT-mediated membrane fission has been extensively studied in the context of other ESCRT-driven processes in cells, including the formation of multivesicular bodies (MVBs) during receptor degradation and budding of retroviruses from host cells (McCullough et al., 2013). All of these processes share a similar topology of budding away from the cytosol. On the basis of these studies and in vitro reconstitution studies, a mechanistic model for ESCRT-mediated membrane fission has been proposed. According to this model, activation of the ESCRT cascade begins with sequential recruitment of early ESCRT components (ESCRT-0, -I, and -II) to the designated membrane (Hurley and Hanson, 2010; McCullough et al., 2013). The mechanical act of membrane fission has been attributed to self-polymerization and remodeling of ESCRT-III components on the inner side of the membrane into helical filaments (Hanson et al., 2008; Hurley and Hanson, 2010; Lata et al., 2008; McCullough et al., 2013; Cashikar et al., 2014).
Self-polymerization and membrane recruitment of ESCRT-III were proposed to be induced by direct binding of the ESCRT-II component VPS25 to the N-terminal of the ESCRT-III protein CHMP6 (Im et al., 2009; Teis et al., 2010; Fyfe et al., 2011; Carlson and Hurley, 2012; Henne et al., 2012; McCullough et al., 2013), and both the ESCRT-II complex and CHMP6 were shown to be essential for ESCRT-mediated membrane fission in MVB biogenesis and in vitro reconstitution experiments (Babst et al., 2002; Malerod et al., 2007; Im et al., 2009; Wollert et al., 2009; Wollert and Hurley, 2010). That said, depletion (using small interfering RNA [siRNA]) or overexpression of CHMP6 or ESCRT-II components in cells did not affect HIV viral budding and did not lead to an increase in multinucleated cells (Langelier et al., 2006; Carlton and Martin-Serrano, 2007; Morita et al., 2007, 2010, 2011; Zhadina and Bieniasz, 2010). As a result, CHMP6 and the ESCRT-II proteins were considered dispensable for ESCRT-mediated cytokinetic abscission and HIV viral budding. Alix, which binds both ESCRT-I and ‑III, was suggested to substitute ESCRT-II in these cases (McCullough et al., 2013). However, Alix was unable to replace ESCRT-II and CHMP6 through the canonical ESCRT pathway in in vitro reconstitution of HIV budding (Carlson and Hurley, 2012). Therefore, how ESCRT-III is activated in ESCRT-mediated abscission and HIV viral budding remained unresolved.
Here we revisit the role of ESCRT-II and CHMP6 in cytokinetic abscission. We find, using structured illumination microscopy (SIM) and live-cell video recording, that both ESCRT-II and CHMP6 acutely recruit to the intercellular bridge, forming highly ordered assemblies at the bridge membrane, supporting a role for these proteins in abscission. Of note, whereas CHMP6 exhibits a similar localization pattern to that observed for other ESCRT-III components, the ESCRT-II component VPS36 appears to have a unique localization pattern that differs from that previously described for either ESCRT-I or ESCRT-III. We substantiated our findings by showing that a truncated version of CHMP6 composed of the first 52 amino acids (aa) of CHMP6 (CHMP6-N) arrives at the intercellular bridge, blocks abscission, and subsequently leads to cell death. A mutated version of CHMP6-N designed to prevent CHMP6-N binding to its ESCRT-II partner VPS25 (Im et al., 2009) does not arrive at the intercellular bridge and does not interfere with abscission. A CHMP6-N version deleted of the first 10 aa, a site of myristoylation, arrives at the intercellular bridge but has only a mild effect on abscission. Therefore, whereas VPS25 governs CHMP6 localization to the intercellular bridge, both the VPS25 interaction domain (aa 11–42) and the region comprising the first 10 aa are crucial for CHMP6-N– induced inhibition of ESCRT-driven abscission. Our work identifies the ESCRT-II complex and CHMP6 as essential players in ESCRT-mediated abscission and introduces a new tool for inhibition of the ESCRT pathway in live mammalian cells.
RESULTS
To revisit the role of ESCRT-II and of CHMP6 in cytokinetic abscission, we examined the spatiotemporal recruitment patterns of these proteins to the intercellular bridge in mammalian cells undergoing abscission. The ESCRT-II complex is composed of three subunits: VPS22, VPS25, and VPS36. We therefore began by exogenously expressing fluorescently tagged versions of each ESCRT-II protein and of CHMP6 together with mCherry/green fluorescent protein (GFP)–tagged tubulin in live Madin–Darby canine kidney (MDCK) cells. In these experiments, we selectively chose cells overexpressing low to medium levels of fluorescently tagged ESCRT components (Elia et al., 2011). Even under these selective conditions, a noticeable number of cells expressing ESCRT-II components failed to complete abscission during the time course of the experiment (Figure 1B), indicating that exogenous expression of ESCRT-II constructs is able to interfere with proper cytokinetic abscission. Owing to difficulties in generating specific antibodies for immunolabeling of ESCRT proteins in mammalian cells, our experiments in fixed cells often relied on exogenous expression of tagged versions of these proteins. To avoid reporting possible phenotypes resulting from the overexpression, we considered only results that were verified in live cells committing abscission with normal spatiotemporal kinetics (as previously described; Elia et al., 2011).
Live-cell recording of MDCK cells expressing mCherry-CHMP6 and GFP-tubulin revealed that CHMP6 is recruited to the intercellular bridge shortly before abscission, with spatiotemporal localization correlating with acute constriction and fission of the intercellular bridge (Figure 1C and Supplemental Video S1). To determine the spatial organization of CHMP6 at different time points of cytokinesis, we used SIM to image MDCK cells expressing Flag-CHMP6 and stained with anti-tubulin and anti-Flag antibodies. In these experiments, the diameter of the microtubule stalk within the intercellular bridge was used as an indication for abscission progression, as previously described (Elia et al., 2011, 2012). This analysis revealed that CHMP6 is initially organized in two cortical rings at the rims of the dark zone (average diameter, 1.1 μm). It then redistributes peripherally toward the abscission sites until it forms two distinct structures—one on the rim of the dark zone and another at the abscission site (Figure 1, D and E). These data are consistent with a previous study showing localization of CHMP6 at the intercellular bridge (Guizetti et al., 2011). Careful examination of the spatial organization of CHMP6 at intermediate stages revealed that CHMP6 forms an array of cortical ring structures as it grows away from the initial pools (Figure 1D, late; solid arrow). We were able to identify up to three rings with decreasing diameters located adjacently along one side of the intercellular bridge (see zoomed-in image in Figure 1D, late). The smallest inner diameter we measured in SIM images was 150 nm. Overall the spatiotemporal organization of CHMP6 described here highly resembles the spatiotemporal organization previously described for other essential ESCRT-III components in abscission (Elia et al., 2011; Guizetti et al., 2011), suggesting that CHMP6 is part of the canonical ESCRT-mediated abscission pathway.
We next examined the spatiotemporal organization of the ESCRT-II complex during cytokinesis. Live-cell imaging revealed that the ESCRT-II components VPS36 and VPS22 localize to the intercellular bridge during late cytokinesis (Figure 2, A and B, Supplemental Figure S1A, and Supplemental Videos S2–S4, respectively). Quantitative analysis of the recruitment pattern of VPS36 to the intercellular bridge revealed that VPS36 begins accumulating at the bridge only ∼20 min before final scission and reaches its peak levels at the time of scission (Figure 2A and Supplemental Video S2). The increase in VPS36 levels is temporally correlated with acute constriction of the microtubules on either side of the intercellular bridge (Figure 2B and Supplemental Video S3). VPS36 was also clearly observed in postabscission remnants (Figure 2B and Supplemental Video S3).
FIGURE 2:
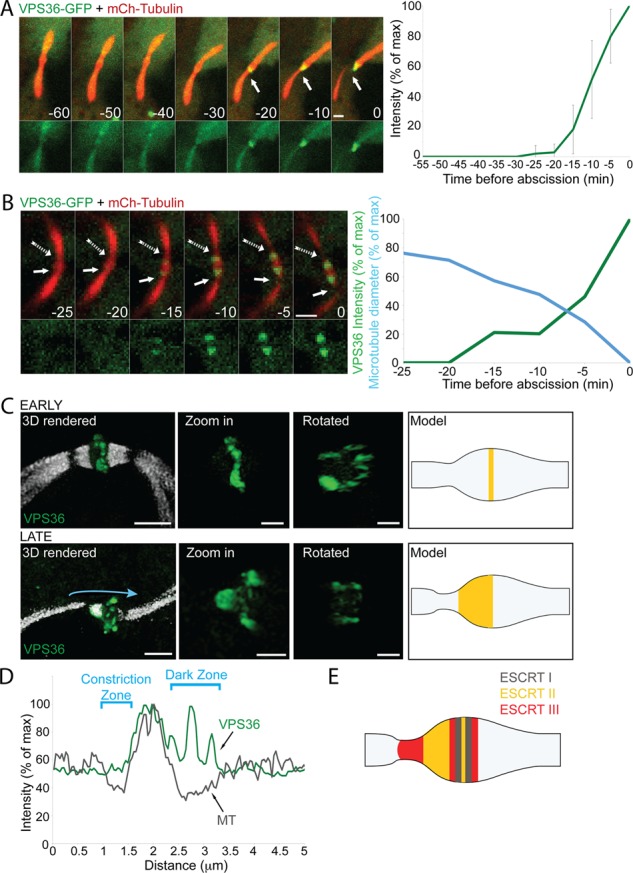
Spatiotemporal recruitment of the ESCRT-II protein VPS36 to the intercellular bridge during cytokinesis. (A and B) Live-cell imaging of MDCK cells undergoing cytokinesis reveals acute recruitment of VPS36 to the intercellular bridge. Cells expressing low levels of GFP-VPS36 together with mCherry-tubulin were imaged using a spinning-disk confocal microscope at 5-min intervals. Shown are maximum-intensity projections of different time points throughout cytokinesis (A) or during abscission (B) from representative cells. Top, overlay of VPS36 (green) and microtubules (red). Bottom, VPS36 alone. Recruitment of VPS36 to the bridge was observed in 30 cells. Solid arrows indicate the position of the first abscission site. Dashed arrows (in B) indicate the second abscission site. Time (indicated in minutes) is relative to abscission of the first site. Plot in A, changes in VPS36 intensity at the intercellular bridge relative to abscission, as quantified from seven independent experiments. Plot in B, changes in microtubule diameter (blue) and VPS36 fluorescence intensity (green), quantified from the first abscission site (Supplemental Videos S2 and S3; bar, 2 μm). (C) Spatial organization of VPS36 in early (top) and late (bottom) intercellular bridges. MDCK cells expressing GFP-VPS36 were fixed, stained with anti–α-tubulin antibodies, and imaged by SIM. Early and late bridges were categorized based on the diameter of the intercellular bridge at the constriction site (see Materials and Methods). Each panel shows (from left to right) a 3D reconstruction of an overlay of VPS36 (green) and tubulin (white; bar, 2 μm); a zoomed-in, 3D rendered image of the protein structure alone (bar, 1 μm); a zoomed-in, 3D rendered image of the protein structure rotated 90° (bar, 1 μm); and a schematic model for VPS36 organization at the intercellular bridge based on SIM measurements. In early intercellular bridges, VPS36 concentrates in a ring located at the center of the dark zone. Ring diameter, 1.7 ± 0.25 μm; ring thickness, 0.61± 0.3 μm (n = 22). In late intercellular bridges, VPS36 distributes peripherally to the central ring, forming a structure that stretches 1.01 ± 0.23 μm (n = 6) away from the center. (D) VPS36 organizes in the area between the dark zone and the constriction sites in late intercellular bridges but does not reach the constriction site. Plot shows tubulin (gray) and VPS36 (green) line intensity profiles along the intercellular bridge (indicated by the blue arrow in C). Unlike CHMP6 and other ESCRT-III components, VPS36 signal is low at the constriction site. (E) Model for ESCRT organization at the intercellular bridge, integrating the SIM measurement obtained for ESCRT-II and CHMP6. ESCRT-I, gray; ESCRT-II, yellow; ESCRT-III, red.
In SIM images, VPS36 is found in a large-diameter (1.7 μm on average) cortical ring located at the center of the dark zone of both early and late bridges (Figure 2C). This measurement is consistent with ring diameters previously measured for the ESCRT-I component TSG101 (Elia et al., 2011). GFP-VPS22 was also occasionally observed in a large-diameter cortical ring located at the center of the bridge (1.5 μm on average; Supplemental Figure S1B). However, VPS22 ring formation was five times more frequent in cells that failed to complete abscission within the time course of the experiment (3–4 h), suggesting that this localization pattern is associated with the abscission delay observed in these cells and could be a result of the overexpression (Supplemental Figure S1C). Cells that successfully complete abscission exhibit a diffused pattern of VPS22 at the center of the intercellular bridge (Supplemental Figure S1B).
In late intercellular bridges (indicated by reduced diameter of the microtubule stalk), an additional VPS36 pool, extending from the central ring toward the constriction site, is observed (Figure 2C, late). In contrast to the localization pattern observed for CHMP6 (Figure 1, D and E) and other ESCRT-III components (Elia et al., 2011), we could not detect VPS36 at the abscission sites even when microtubule diameter was minimal (compare line scans in Figures 1E and 2D). VPS36 therefore has a distinct localization pattern from those previously observed for either early or late ESCRT components (Elia et al., 2011); it is positioned in the area between the dark zone, where early ESCRTs are found, and the constriction site, where late ESCRTs function. This localization pattern supports a role for ESCRT-II in bridging the spatial gap between early ESCRT components and late ESCRT-III components. A revised model for ESCRT organization at the intercellular bridge, integrating previous and present data, is depicted in Figure 2E.
The spatiotemporal organization of ESCRT-II and CHMP6 in cytokinetic abscission and its agreement with the spatiotemporal mapping previously described for other ESCRT components in cytokinesis points to the involvement of these proteins in ESCRT-mediated abscission. To substantiate a role for ESCRT-II and CHMP6 in cytokinetic abscission, we designed a tool for specific inhibition of ESCRT-II–CHMP6 interaction in live cells using exogenous expression of the first 52 aa of CHMP6 in cells (CHMP6-N). siRNA depletion of these components was reported to have no effect on cytokinetic abscission (Morita et al., 2007) and was therefore avoided. According to in vitro studies and work in yeast, binding of CHMP6 to the ESCRT-II component VPS25 facilitates ESCRT-III activation and polymerization (Peel et al., 2011; McCullough et al., 2013). CHMP6 binding to ESCRT-II is mediated by direct interactions between the N-terminus of CHMP6 (aa 11–42) and the ESCRT-II component VPS25 (Im et al., 2009), whereas induction of ESCRT-III polymerization is attributed to the C- terminal domains of CHMP6 (Teis et al., 2010; Peel et al., 2011; McCullough et al., 2013; Figure 3A). Therefore, if CHMP6 arrives at the intercellular bridge via interactions with VPS25, then exogenous CHMP6-N is expected to localize to the intercellular bridge, compete with endogenous CHMP6 over binding to VPS25, and inhibit ESCRT-mediated abscission. Indeed, a CHMP6-N-GFP construct arrives to the intercellular bridge in MDCK cells undergoing cytokinesis (Figure 3B, bottom, arrow). Of note, >80% of cells expressing CHMP6-N-GFP failed to complete abscission within the 3–4 h of the experiment (Figure 3, B and D, and Supplemental Video S5). Late ESCRT components arrived to the intercellular bridges of these cells but failed to form the characteristic ring-like structures or to localize to the constriction sites (Supplemental Figure S2). In 40% of the cells, the inability to complete abscission was accompanied by premature death of the daughter cells, characterized by acute cell contraction and membrane blebbing (asterisks in Figure 3, B and D, and Supplemental Video S5). Such morphology was observed in <10% of the daughter cells upon expression of GFP/mCherry-tubulin plasmids alone or with an mCherry/GFP control plasmid (Figure 3D). Similar results were obtained with CHMP6-N-mCherry.
FIGURE 3:
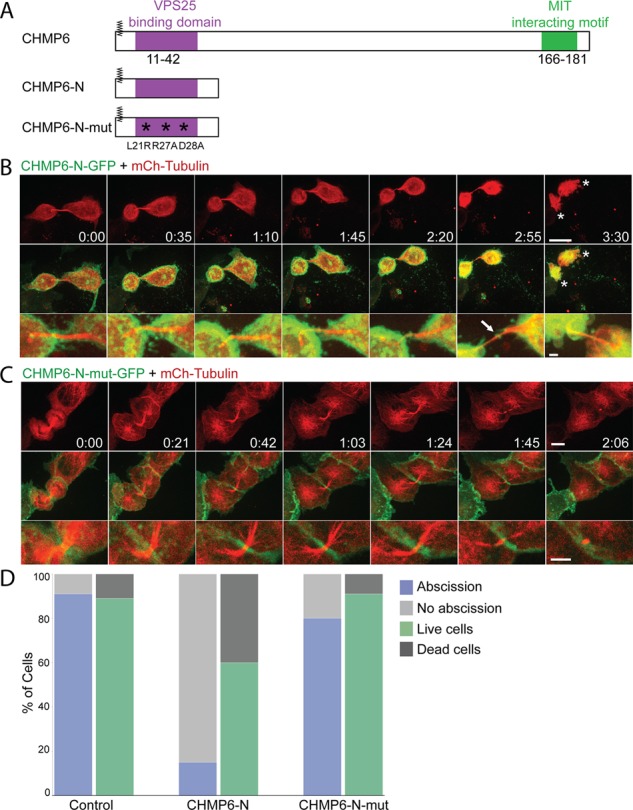
Exogenous expression of CHMP6-N-GFP inhibits abscission through specific interaction with the ESCRT-II protein VPS25 and leads to cell death. (A) Schematic view of the CHMP6 constructs used in the study. Highlighted are CHMP6 myristoylation site (zigzag line), VPS25-binding domain (purple), and CHMP6–ESCRT-III interaction motif (green). CHMP6-N is composed of the first 52 aa of CHMP6. Asterisks indicate three point mutations that were introduced in CHMP6-N-mut in order to interfere with its binding to VPS25. All constructs were fused to GFP/mCherry fluorescent tags. (B and C) Dividing MDCK cells expressing CHMP6-N-GFP (A) or CHMP6-N-mut-GFP (C) together with mCherry-tubulin were imaged using a spinning-disk confocal microscope at 7-min intervals. Shown are maximum-intensity projections of different time points during cytokinesis from representative cells. Top, tubulin signal alone (bar, 10 μm); middle, overlay of CHMP6-N-GFP/CHMP6-N-mut-GFP (green) and tubulin (red); bottom, enlargement of the intercellular bridge taken from the overlay images (bar, 5 μm). Of note, CHMP6-N-GFP arrived at the center of the bridge (arrows). Asterisks in B indicate dying cells exhibiting acute cell contraction and membrane blebbing. While exogenous expression of CHMP6-N-GFP delays abscission and increases cell death, exogenous expression of CHMP6-N-mut-GFP (L21R, R27A and D28A) does not affect abscission or cell viability (Supplemental Videos S5 and S6; bars, 5 μm). (D) Percentage of cells that completed abscission successfully and percentage of cell death observed in daughter cells within the time course of the experiments (3–4 h) under the different conditions. Cell death was determined based on changes in cell morphology as observed in the movie sequences (see asterisks in B). CHMP6-N–GFP (n = 40); CHMP6-N-mut-GFP (n = 44); and control cells expressing GFP (n = 44).
To examine the specificity of the observed phenotype to binding of CHMP6 to VPS25, we introduced three point mutations (L21R, R27A, and D28A) in the VPS25-binding domain of CHMP6-N-GFP (CHMP6-N-mut-GFP). These three mutations were shown to abolish CHMP6 binding to VPS25 in vitro (Im et al., 2009). Exogenous CHMP6-N-mut-GFP does not localize to the intercellular bridge, interfere with cytokinetic progression or abscission, or with the organization of late ESCRT components at the intercellular bridge and does not affect cell mortality (Figure 3, C and D, Supplemental Figure S2, and Supplemental Video S6). This strongly indicates that binding of CHMP6-N to VPS25 is essential for its ability to inhibit abscission and that CHMP6 localization to the intercellular bridge is mediated through VPS25 binding.
To test whether binding of CHMP6-N to VPS25 is sufficient for inhibiting abscission, we generated a series of CHMP6-N truncations without perturbing the VPS25-CHMP6 interaction domain (aa 11–42; Im et al., 2009). Three additional CHMP6-N truncations were conjugated to GFP: CHMP6-N-11-52, CHMP6-N-1-42, and CHMP6-N-11-42 (Figure 4A). All three constructs efficiently localize to the intercellular bridge of dividing MDCK cells (Figure 4, B–D, bottom, arrows, and Supplemental Videos S7–S9), confirming that CHMP6-N localization to the intercellular bridge is mediated through binding to VPS25. Of interest, constructs lacking the first 10 aa of CHMP6 show only mild inhibition of abscission and no cell death (Figure 4, B, C, and E, and Supplemental Videos S7 and S8), and the only construct that is able to recapitulate the inhibition observed for CHMP6-N–GFP is CHMP6-N-1-42–GFP (Figure 4, D and E, and Supplemental Video S9). This indicates that although binding to VPS25 is sufficient for CHMP6-N localization to the intercellular bridge, it is not sufficient for CHMP6-N–induced inhibition of abscission. The first 10 aa of CHMP6 include a myristoylation moiety and a basic patch of amino acids that presumably assigns CHMP6 affinity to the plasma membrane (Saksena et al., 2007). Perhaps membrane binding of CHMP6-N is essential for its ability to inhibit abscission.
FIGURE 4:
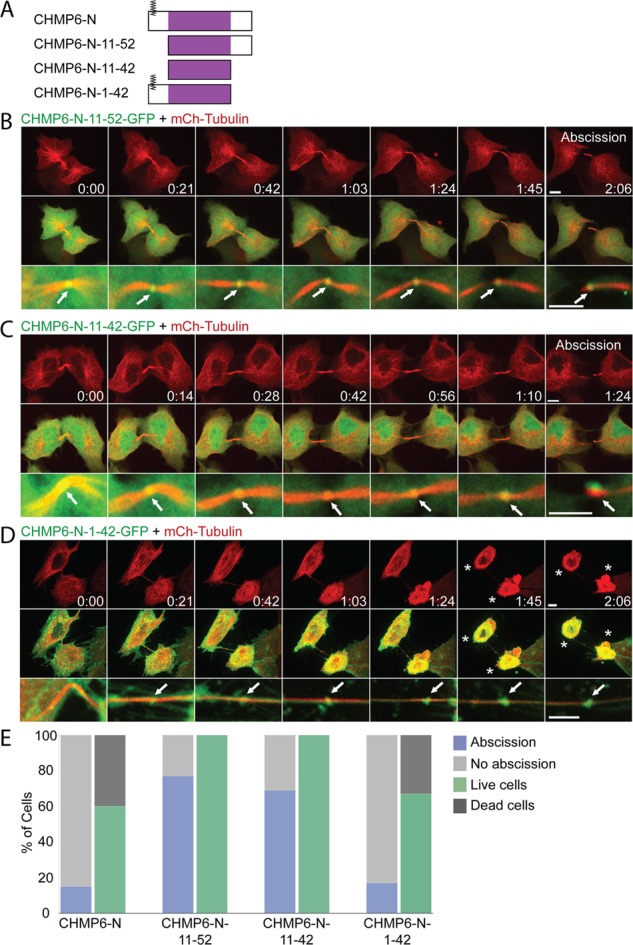
Deletions in CHMP6-N suggest a role for the first 10 aa in CHMP6-N–induced inhibition of ESCRT-driven abscission. (A) Schematic view of the CHMP6-N deletion constructs used in the study. VPS25-binding domain is depicted in purple. All constructs were fused to GFP/mCherry fluorescent tags on their C-terminal. (B–D) MDCK cells expressing CHMP6-N-11-52–GFP (B), CHMP6-N-11-42–GFP (C), or CHMP6-N-1-42–GFP (D) together with mCherry-tubulin were imaged during cytokinesis using a spinning-disk confocal microscope at 7-min intervals. Each panel shows a maximum-intensity projection of different time points taken from a representative movie sequence (Supplemental Videos S7–S9 respectively). Top, tubulin signal alone (bars, 10 μm); middle, overlay of the CHMP6-N construct (green) and tubulin (red); bottom, enlargement of the intercellular bridge from the overlay images (bars, 5 μm). All constructs localize to the center of the intercellular bridge during abscission (arrows). Of note, whereas exogenous expression of either CHMP6-N-11-52–GFP or CHMP6-N-11-42–GFP has only a mild effect on abscission and has no effect on cell viability, expression of CHMP6-N-1-42–GFP delays abscission and increases cell death. Asterisks indicate dying cells exhibiting acute cell contraction and membrane blebbing. (E) Percentage of cells that completed abscission successfully and percentage of daughter cell death observed within the time course of the experiments (3–4 h) upon expression of the different constructs. Cell death was determined based on cell morphology (see asterisks in D). CHMP6-N-11-52–GFP (n = 43); CHMP6-N-11-42–GFP (n = 49); and CHMP6-N-1-42–GFP (n = 40).
DISCUSSION
It has been suggested, based on studies in HIV viral budding and cytokinesis, that ESCRT-II and CHMP6 are in some cases dispensable for ESCRT-mediated membrane fission (McCullough et al., 2013). In the context of cytokinesis, this suggestion relied on the observations that no increase in multinucleated cells occurs upon depletion or overexpression of ESCRT-II or CHMP6 (Carlton and Martin-Serrano, 2007; Morita et al., 2007). However, this assay, which is widely used for monitoring cytokinesis failure, is not specific for detection of abscission failure. For example, depletion of TSG101, an established player in ESCRT-mediated abscission, does not cause an increase in multinucleated cells (Carlton and Martin-Serrano, 2007; Morita et al., 2007). By directly monitoring abscission in live cells, our work provides evidence to suggest that, at least in the context of cytokinesis, ESCRT-II and CHMP6 are actively involved in the ESCRT-mediated membrane fission cascade. First, both ESCRT-II and CHMP6 localized to the intercellular bridge during abscission with a unique spatiotemporal pattern that correlates with abscission onset. Second, arrival of an exogenous CHMP6 construct composed of the first 52 aa of CHMP6 at the intercellular bridge was solely dependent on binding to VPS25. Third, the ability of the CHMP6-N construct to inhibit abscission was dependent on its interaction with VPS25. Recently ESCRT-II and CHMP6 were found to be essential for ESCRT-III recruitment in an in vitro reconstitution system of viral budding (Carlson and Hurley, 2012), suggesting that these proteins are also crucial for ESCRT-mediated viral budding. Future work will be needed to analyze the role of ESCRT-II and CHMP6 in viral budding under cellular conditions.
Analysis of the localization pattern of CHMP6 at the intercellular bridge revealed that in late intercellular bridges, CHMP6 is found in a series of adjacent cortical rings with decreasing diameters beginning from the initial ring and extending peripherally along the bridge. In previous work, we showed that during abscission, a pool of ESCRT-III extends from the initial ring, separates, and slides along the bridge until it stabilizes at the constriction sites where abscission occurs (Elia et al., 2012). Here we show for the first time that the intermediate pool, which extends from the initial pool, is composed of a series of cortical rings with reduced diameters, supporting a role for ESCRT-III filaments in bridge constriction. Cortical filaments have been previously documented in intercellular bridges using cryo–electron tomography; however, it could not be determined whether these filaments are ESCRT based (Guizetti et al., 2011). Our findings suggest that this is indeed the case.
Using SIM, we found that the ESCRT-II component VPS36 is initially localized in a cortical ring located at the center of the intercellular bridge. This localization is consistent with the localization previously documented for the ESCRT-I component TSG101. However, whereas TSG101 is found in two adjacent large-diameter rings, we could only resolve a single ring for VPS36. In late intercellular bridges, VPS36 redistributes along the bridge and is located in both the central ring and the area between the dark zone and constriction site. In contrast to what we observed for CHMP6 or other ESCRT-III components, this is not an intermediate localization pattern that ultimately leads to the formation of two structures, as a similar pattern is observed in bridges with obvious constriction sites and in postabscission remnants (Figure 2). This unique localization pattern positions ESCRT-II in the spatial gap between early and late ESCRTs, as described in the revised model for ESCRT localization during abscission presented in Figure 2E. The ESCRT-II complex is believed to provide the mechanistic cue for ESCRT-III polymerization and activation by binding to both ESCRT-I and ESCRT-III components. Our results suggest that it may also physically link between early and late ESCRT components along the constricting intercellular bridge.
Our analysis of different truncations of the N-terminal of CHMP6 shows that binding to VPS25 is sufficient for CHMP6-N localization to the intercellular bridge, whereas both the VPS25 interaction domain (aa 11–42) and the membrane-binding region (aa 1–10) of CHMP6 are essential for mediating the ability of CHMP6-N to delay abscission. The latter suggests that the first 10 aa of CHMP6 (which contain a site of myristoylation) contribute to the interaction between CHMP6-N and VPS25, enabling it to efficiently compete with endogenous CHMP6 over binding to VPS25. Perhaps myristoylation ensures proper orientation of CHMP6-N toward VPS25 to increase binding efficiency. This can be through either membrane interactions or binding to a third, currently unknown, partner. Importantly, these findings highlight a mechanistic role for the first 10 aa of CHMP6 in ESCRT activation. Future experiments are needed in order to determine the specific role of this region in activation of the ESCRT-mediated membrane fission pathway.
Our data show that inhibition of abscission using exogenous expression of a truncated version of CHMP6 leads to acute cell death. Induction of cell death as a result of abscission failure was previously reported upon inhibition of abscission using the dynamin inhibitor dynole 34-2 (Chircop et al., 2011). However, this is not always the case. In most reported cases, abscission failure led to cytokinetic regression and consequently to the formation of multinucleated cells (Green et al., 2012). In other cases, abscission failure does not affect cell survival or cell cycle progression, giving rise to the formation of multiple cells interconnected by bridges (Dambournet et al., 2011; Florindo et al., 2012). It therefore appears that different inhibition strategies of abscission can lead to different cell fates. Identifying the molecular switch that governs this critical decision will no doubt shed light on our understanding of abscission and how it is regulated in the cellular, cell cycle context.
The ESCRT machinery is involved in various processes in cells, including MVB biogenesis and viral budding. Up to now, however, inhibition of the pathway in cells was limited. Our work potentially provides a tool for specific inhibition of ESCRT in live mammalian cells using a synthetic peptide composed of the first 42 aa of CHMP6. Several new cellular functions have been recently identified for the late ESCRT proteins ESCRT-III and VPS4, including roles in wound healing and in membrane shedding from primary cilia (Jimenez et al., 2014; Soetedjo and Jin, 2014). However, the involvement of early ESCRT components in these pathways is still unclear. The CHMP6-N inhibition approach will provide an elegant, easily applicable tool for dissecting the role of early ESCRT components in these processes, as well as in any ESCRT-mediated cellular processes.
MATERIALS AND METHODS
Cell culture and transfection
MDCK II cells were grown in MEM supplemented with 5% fetal bovine serum (FBS), 2 mM glutamine, 10,000 U/ml penicillin, and 10 mg/ml streptomycin. Transfection was carried out using Lipofectamine 2000 (Life Technologies, Carlbad, CA) according to manufacturer’s guidelines.
Plasmid constructs
GFP-VPS22; mCherry-VPS22.
Full-length human vacuolar protein sorting 22 (VPS22) in pEGFP-C1 vector (Clontech, Mountain View, CA) and full-length human vacuolar protein sorting 22 (VPS22) in pmCherry-C1 vector (Clontech) were kind gifts of James H. Hurley (University of California, Berkeley, CA).
GFP-VPS25.
Full-length human vacuolar protein sorting 25 (VPS25) was amplified by PCR from pIRES-VPS25 vector, kindly provided by James H. Hurley, and cloned to pEGFP-C1 vector (Clontech).
GFP-VPS36; mCherry-VPS36.
Full-length human vacuolar protein sorting 36 (VPS36) was amplified by PCR from pIRES-VPS25-VPS36 vector, kindly provided by James H. Hurley, and cloned to pEGFP-C1 and to pmCherry-C1 vector (Clontech).
Flag-CHMP6.
Full-length human charged multivesicular body protein 6 (CHMP6) in flag vector was kindly provided by Wesley Sundquist (University of Utah School of Medicine, Salt Lake City, UT).
mCherry-CHMP6.
The CHMP6 sequence was amplified by PCR from Flag-CHMP6 plasmid and cloned to mCherry-C1 vector (Clontech).
CHMP6-N-mCherry; CHMP6-N-GFP.
The first 52 aa from the N-terminal of CHMP6 were amplified by PCR and cloned to mCherry-N1 vector and to pEGFP-N1 vector (Clontech).
CHMP6-N-mut-GFP.
Three point mutations were introduced into the CHMP6-N-GFP sequence by overlapping PCR as follows: L21R codon CTG was replaced by CGG; R27A codon CGG was replaced by GCG; and D28A codon GAC was replaced by GCC.
CHMP6-N-11-52–GFP; CHMP6-N-1-42–GFP; CHMP6-N-11-42–GFP.
Amino acids 11–52, 11–42, and 1–42, respectively, from the N-terminal of CHMP6 were amplified by PCR and cloned to mCherry-N1 vector and to pEGFP-N1 vector (Clontech).
GFP–α-tubulin; mCherry–α-tubulin.
Full-length human α-tubulin was cloned into pEGFP-C1 and to pmCherry-C1 as previously described (Elia et al., 2011).
All constructs were confirmed by sequencing.
Live-cell recording and image processing
MDCK cells were plated in low density on a four-well chamber slide (Nunc, Rochester, NY, or ibidi, Martinsried, Germany), transfected 24 h later with the plasmids indicated in the figure legends, and imaged 24–40 h later. Z-stacks of selected low-expressing cells undergoing cytokinesis were collected at the specified intervals using a fully incubated confocal spinning-disk microscope (Marianas; Intelligent Imaging, Denver, CO) with a 63× oil objective (numerical aperture, 1.4) and were video recorded on an electron-multiplying charge-coupled device camera (pixel size, 0.079 μm; Evolve; Photometrics, Tucson, AZ). Only cells that successfully completed cytokinesis within the time course of the experiment (3–4 h) were analyzed. Image processing and analysis were done using SlideBook version 5 or 6 (Intelligent Imaging). Intensity values of ESCRT proteins at the intercellular bridge were calculated by measuring the total intensity fluorescence of a mask object applied to the sum Z-projection of the movie series. Intensity levels at the intercellular bridge before ESCRT recruitment were set as zero and subtracted from all time points.
Microtubule diameter was determined based on the microtubule fluorescence intensity profile of a line positioned perpendicular to the intracellular bridge at the most constricted region, ∼1 μm from the center of the bridge.
SIM imaging
MDCK cells were plated at 10% density on #1.5 coverslips (Marienfeld, Lauda-Konigshfen, Germany) and transfected 24 h later with the designated proteins (as described). Cells were fixed 24 h later using 4% paraformaldehyde for 15 min at room temperature. All samples were subjected to immunostaining as described later. Thin z-sections (0.11–0.15 μm) of high-resolution images were collected in five rotations for each channel using an ELYRA PS.1 microscope (Carl Zeiss MicroImaging). Images were reconstructed using ZEN software (Carl Zeiss MicroImaging, Jena, Germany) based on the structured illumination algorithm developed by Heintzmann and Cremer (1999). All measurements were performed on reconstructed superresolution images in ZEN. Geometric measurements of protein structures (Figures 1D and 2C and Supplemental Figure S1B) were obtained by measuring the distance between the lowest-intensity pixel located at the beginning of the structure and the lowest-intensity pixel located at the end of the structure from a line intensity of the protein signal in ZEN. Line intensity profile measurements (Figures 1E and 2D) were obtained by stretching a curved line along the intercellular bridge. Three-dimensional rendering was done using Volocity 6 (PerkinElmer, Waltham, MA). Microtubule diameter was measured as described and used to distinguish between early and late bridges as previously described (Elia et al., 2012). Early bridges correspond to ∼1 μm in diameter, late bridges to <0.7 μm in diameter.
Immunostaining
For SIM, cells were permeabilized with 0.5% Triton X-100 for 10 min and blocked with 10% FBS for 15 min. All cells were stained with monoclonal anti α-tubulin antibodies (DM1A; Sigma-Aldrich). Whenever indicated, cells were also stained with rabbit polyclonal anti-Flag antibodies (kindly provided by Ramanujan Hedge, MRC Laboratory of Molecular Biology, Cambridge, United Kingdom). Cells were then subjected to a secondary antibody staining using Alexa Fluor 488 or Alexa Fluor 594 anti-mouse or anti-rabbit secondary antibodies, respectively (Life Technologies). Finally, cells were mounted with Fluoromount-G (SouthernBiotech, Birmingham, AL).
Supplementary Material
Acknowledgments
We thank James H. Hurley for advice and support and for his generosity in sharing plasmids. We thank Mehtap Abu-Qarn for feedback on the manuscript. The Elia lab is funded by Israeli Science Foundation Grant 455/13, United States-Israel Binational Science Foundation Grant 2011309, a Marie Curie Integration grant, and the National Institute for Biotechnology in the Negev.
Abbreviations used:
- aa
amino acids
- 3D
three-dimensional
- ESCRT
endosomal sorting complexes required for transport
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- MDCK
Madin–Darby canine kidney
- MVBs
multivesicular bodies
- SIM
structured illumination microscopy
- siRNA
small interfering RNA.
Footnotes
I.G. conducted all the experiments, including the data analysis, presented in the manuscript and helped with writing the manuscript; D.N. provided technical help and support and helped with experimental design; O.G. was responsible for statistical analysis and helped with writing the manuscript; N.E. designed the experiments, was involved in data analysis, and wrote the manuscript.
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-08-1317) on September 17, 2014.
REFERENCES
- Babst M, Katzmann DJ, Snyder WB, Wendland B, Emr SD. Endosome-associated complex, ESCRT-II, recruits transport machinery for protein sorting at the multivesicular body. Dev Cell. 2002;3:283–289. doi: 10.1016/s1534-5807(02)00219-8. [DOI] [PubMed] [Google Scholar]
- Carlson LA, Hurley JH. In vitro reconstitution of the ordered assembly of the endosomal sorting complex required for transport at membrane-bound HIV-1 Gag clusters. Proc Natl Acad Sci USA. 2012;109:16928–16933. doi: 10.1073/pnas.1211759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Sci. 2007;316:1908–1912. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- Cashikar AG, Shim S, Roth R, Maldazys MR, Heuser JE, Hanson PI. Structure of cellular ESCRT-III spirals and their relationship to HIV budding. eLife. 2014:e02184. doi: 10.7554/eLife.02184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chircop M, Perera S, Mariana A, Lau H, Ma MP, Gilbert J, Jones NC, Gordon CP, Young KA, Morokoff A, et al. Inhibition of dynamin by dynole 34-2 induces cell death following cytokinesis failure in cancer cells. Mol Cancer Ther. 2011;10:1553–1562. doi: 10.1158/1535-7163.MCT-11-0067. [DOI] [PubMed] [Google Scholar]
- Dambournet D, Machicoane M, Chesneau L, Sachse M, Rocancourt M, El Marjou A, Formstecher E, Salomon R, Goud B, Echard A. Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat Cell Biol. 2011;13:981–988. doi: 10.1038/ncb2279. [DOI] [PubMed] [Google Scholar]
- Elia N, Fabrikant G, Kozlov M, Lippincott-Schwartz J. Computational model of cytokinetic abscission driven by ESCRT-III polymerization and remodeling. Biophys J. 2012;102:2309–2320. doi: 10.1016/j.bpj.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia N, Ott C, Lippincott-Schwartz J. Incisive imaging and computation for cellular mysteries: lessons from abscission. Cell. 2013;155:1220–1231. doi: 10.1016/j.cell.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia N, Sougrat R, Spurlin TA, Hurley JH, Lippincott-Schwartz J. Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc Natl Acad Sci USA. 2011;108:4846–4851. doi: 10.1073/pnas.1102714108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florindo C, Perdigao J, Fesquet D, Schiebel E, Pines J, Tavares AA. Human Mob1 proteins are required for cytokinesis by controlling microtubule stability. J Cell Sci. 2012;125:3085–3090. doi: 10.1242/jcs.097147. [DOI] [PubMed] [Google Scholar]
- Fyfe I, Schuh AL, Edwardson JM, Audhya A. Association of the endosomal sorting complex ESCRT-II with the Vps20 subunit of ESCRT-III generates a curvature-sensitive complex capable of nucleating ESCRT-III filaments. J Biol Chem. 2011;286:34262–34270. doi: 10.1074/jbc.M111.266411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RA, Paluch E, Oegema K. Cytokinesis in animal cells. Annu Rev Cell Dev Biol. 2012;28:29–58. doi: 10.1146/annurev-cellbio-101011-155718. [DOI] [PubMed] [Google Scholar]
- Guizetti J, Schermelleh L, Mantler J, Maar S, Poser I, Leonhardt H, Muller-Reichert T, Gerlich DW. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science. 2011;331:1616–1620. doi: 10.1126/science.1201847. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Roth R, Lin Y, Heuser JE. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J Cell Biol. 2008;180:389–402. doi: 10.1083/jcb.200707031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzmann R, Cremer CG. Laterally modulated excitation microscopy: improvement of resolution by using a diffraction grating. Proc SPIE. 1999;3568:185–196. [Google Scholar]
- Henne WM, Buchkovich NJ, Zhao Y, Emr SD. The endosomal sorting complex ESCRT-II mediates the assembly and architecture of ESCRT-III helices. Cell. 2012;151:356–371. doi: 10.1016/j.cell.2012.08.039. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT machinery: it’s all in the neck. Nat Rev Mol Cell Biol. 2010;11:556–566. doi: 10.1038/nrm2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im YJ, Wollert T, Boura E, Hurley JH. Structure and function of the ESCRT-II-III interface in multivesicular body biogenesis. Dev Cell. 2009;17:234–243. doi: 10.1016/j.devcel.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. ESCRT machinery is required for plasma membrane repair. Science. 2014;343:1247136. doi: 10.1126/science.1247136. [DOI] [PubMed] [Google Scholar]
- Langelier C, von Schwedler UK, Fisher RD, De Domenico I, White PL, Hill CP, Kaplan J, Ward D, Sundquist WI. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J Virol. 2006;80:9465–9480. doi: 10.1128/JVI.01049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S, Schoehn G, Jain A, Pires R, Piehler J, Gottlinger HG, Weissenhorn W. Helical structures of ESCRT-III are disassembled by VPS4. Science. 2008;321:1354–1357. doi: 10.1126/science.1161070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malerod L, Stuffers S, Brech A, Stenmark H. Vps22/EAP30 in ESCRT-II mediates endosomal sorting of growth factor and chemokine receptors destined for lysosomal degradation. Traffic. 2007;8:1617–1629. doi: 10.1111/j.1600-0854.2007.00630.x. [DOI] [PubMed] [Google Scholar]
- McCullough J, Colf LA, Sundquist WI. Membrane fission reactions of the mammalian ESCRT pathway. Annu Rev Biochem. 2013;82:663–692. doi: 10.1146/annurev-biochem-072909-101058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Colf LA, Karren MA, Sandrin V, Rodesch CK, Sundquist WI. Human ESCRT-III and VPS4 proteins are required for centrosome and spindle maintenance. Proc Natl Acad Sci USA. 2010;107:12889–12894. doi: 10.1073/pnas.1005938107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sandrin V, Chung HY, Morham SG, Gygi SP, Rodesch CK, Sundquist WI. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007;26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sandrin V, McCullough J, Katsuyama A, Baci Hamilton I, Sundquist WI. ESCRT-III protein requirements for HIV-1 budding. Cell Host Microbe. 2011;9:235–242. doi: 10.1016/j.chom.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel S, Macheboeuf P, Martinelli N, Weissenhorn W. Divergent pathways lead to ESCRT-III-catalyzed membrane fission. Trends Biochem Sci. 2011;36:199–210. doi: 10.1016/j.tibs.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Saksena S, Sun J, Chu T, Emr SD. ESCRTing proteins in the endocytic pathway. Trends Biochem Sci. 2007;32:561–573. doi: 10.1016/j.tibs.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Soetedjo L, Jin H. Agonist-induced GPCR shedding from the ciliary surface is dependent on ESCRT-III and VPS4. Curr Biol. 2014;24:509–518. doi: 10.1016/j.cub.2014.01.010. [DOI] [PubMed] [Google Scholar]
- Teis D, Saksena S, Judson BL, Emr SD. ESCRT-II coordinates the assembly of ESCRT-III filaments for cargo sorting and multivesicular body vesicle formation. EMBO J. 2010;29:871–883. doi: 10.1038/emboj.2009.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464:864–869. doi: 10.1038/nature08849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollert T, Wunder C, Lippincott-Schwartz J, Hurley JH. Membrane scission by the ESCRT-III complex. Nature. 2009;458:172–177. doi: 10.1038/nature07836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhadina M, Bieniasz PD. Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathog. 2010;6:e1001153. doi: 10.1371/journal.ppat.1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.