

Abstract

Four 4-phthalimide derivatives of N-(3-chlorophenyl)-2-picolinamide were synthesized as potential ligands for the PET imaging of mGlu4 in the brain. Of these compounds, N-(3-chloro-4-(4-fluoro-1,3-dioxoisoindolin-2-yl)phenyl)-2-picolinamide (3, KALB001) exhibited improved binding affinity (IC50 = 5.1 nM) compared with ML128 (1) and was subsequently labeled with 18F. When finally formulated in 0.1 M citrate buffer (pH 4) with 10% ethanol, the specific activity of [18F]3 at the end of synthesis (EOS) was 233.5 ± 177.8 GBq/μmol (n = 4). The radiochemical yield of [18F]3 was 16.4 ± 4.8% (n = 4), and the purity was over 98%. In vivo imaging studies in a monkey showed that the radiotracer quickly penetrated the brain with the highest accumulation in the brain areas known to express mGlu4. Despite some unfavorable radiotracer properties like fast washout in rodent studies, [18F]3 is the first 18F-labeled mGlu4 radioligand, which can be further modified to improve pharmacokinetics and brain penetrability for future human studies.
Introduction
l-Glutamate is the most abundant excitatory neurotransmitter in the CNS of vertebrates.1,2 mGluRs and iGluRs are two major classes involved in glutamate signal transfer.2 While iGluRs are dedicated to quick synaptic responses to open ion channels, mGluRs create slow synaptic responses and their subsequent biochemical effects.2 The mGluRs belong to class C of the GPCR superfamily. mGluRs are found preferably as dimers and have a distinct large extracellular N-terminus,. This extracellular N-terminal domain contains an orthosteric binding site for the endogenous ligand, formed by two hinged globular domains, referred to as the Venus flytrap domain (VFD).3 The mGluRs can be further divided into three subgroups including eight known receptor subtypes (group I consisting of mGlu1 and mGlu5, group II consisting mGlu2 and mGlu3, and group III consisting of mGlu4, mGlu6, mGlu7, and mGlu8) based on their structural similarity, ligand specificity, and preferred coupling mechanisms.4 The mGluRs are involved in glutamate signal transfer in almost every excitatory synapse in CNS, and they have distinctive biodistribution in CNS depending on subtypes and subgroups.5 In recent years, mGlu4 has received a lot of attention because of the potential benefits of mGlu4 activation in several diseases such as Parkinson’s disease (PD).6,7 PD is caused by the degeneration of dopaminergic neurons in the basal ganglia and results in motor symptoms such as tremors and bradykinesia.8 As a group III mGluR, mGlu4 interacts with the Gαi/o subunit of the G-protein which negatively couples with adenylate cyclase to inhibit cAMP dependent signal pathways.9,10 The mGlu4 is expressed at multiple synapses throughout the basal ganglia, mainly localized presynaptically and expressed in the striatum, hippocampus, thalamus, and cerebellum.4,11−15 Its activation reduces neurotransmitter release, a mechanism implicated in the pathophysiology of PD. Since most orthosteric ligands of mGluRs lack clear subtype selectivity and BBB penetration, much effort has been done in the development of allosteric modulators, which target the seven-transmembrane spanning domain. In particular, mGlu4 has received much interest following the discovery that PHCCC (N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide), a partially selective mGlu4 positive allosteric modulator (PAM), demonstrated activity in models of neuroprotection and PD. Subsequent results with other PAMs of mGlu4 have further validated the antiparkinsonian activity in animal models of PD.14,15 This approach has opened a new avenue for developing nondopaminergic treatments for PD and for identifying novel disease modifying therapeutics. In 2009, Engers et al. (Vanderbilt University) reported a series of N-phenyl-2-picolinamide derivatives as mGlu4 PAMs, which were potent and centrally penetrating.16 Compounds 1 (N-(chloro-3-methoxyphenyl)-2-picolinamide, ML128) and 2 (N-(3-(difluoromethoxy)phenyl)-2-picolinamide) were the most potent mGlu4 PAMs in this series, in which 1 was the first mGlu4 PAM to show efficacy in a preclinical PD model upon systemic dosing.6
To better understand the role of mGlu4 under normal and disease conditions, we are interested in developing an mGlu4 selective radioligand for in vivo study. As a noninvasive medical imaging technique and powerful tool in neurological research, PET offers the possibility to visualize and analyze target receptor expression under physiological and pathophysiological conditions. Moreover, PET tracers serve as invaluable biomarkers during the development of potential therapeutic drugs. Recently, we have reported a carbon-11 labeled mGlu4 PAM [11C]1 (N-(chloro-3-[11C]methoxyphenyl)-2-picolinamide) as a PET radioligand.17 This compound demonstrated some promising features as a PET radioligand such as fast uptake into the brain and the specific accumulation of [11C]1 in mGlu4-rich regions of the brain. However, in comparison to one of the best mGlu5 PET tracers, [18F]FPEB (3-[18F]fluoro-5-(2-pyridinylethynyl)benzonitrile),18,19 [11C]1 showed decreased retention time in the brain, which may affect the quality of the imaging. As a result of our efforts to further develop mGlu4 PET ligands, we report here the synthesis and evaluation of [18F]3, the first fluorine-18 labeled PET radioligand for mGlu4 imaging.
Results and Discussion
Chemistry
Aimed at improving the potency of the previous PET ligand, we have looked for a new type of mGlu4 PAM with improved properties for developing a new PET tracer. In recent years, a series of phthalimide derivatives of N-phenyl-2-picolinamides have been developed as mGlu4 PAMs by the Merck Research Laboratory and Vanderbilt University.20,21 Four representative compounds (3–6) from this series are shown in Figure 1. ML182 (6) was a saturated version of the phthalimide, which proved to be orally active in the haloperidol induced catalepsy model, a well-established antiparkinsonian model. It was also shown that compounds 3 (hEC50 = 50.4 nM; IC50 = 36 nM), 4 (hEC50 = 43.6 nM; IC50 = 11 nM), and 5 (hEC50 = 101 nM; IC50 = 12 nM) had enhanced activity compared with 1 (hEC50 = 240 nM) and gave excellent binding affinity (depicted as the IC50 value) to mGlu4. Compound 4 was radiolabeled with tritium as a tool compound for in vitro studies.20 When the structures of 3–5 were checked, it was determined that the picolinamide (left side) was critical for the mGlu4 potency,16 and the phthalimide group at the 4-position on the phenyl ring enhanced potency compared to 1. In addition, although both the chloro- and bromo-substitutions at the 3-position of the phenyl ring (4 and 5) gave good binding affinities, the chloro-substitution was more favorable, since the bromo-substitution increased molecular weight as well as clogP value but not the potency at mGlu4. Because of their promising affinity to mGlu4, we have synthesized four phthalimide derivatives including 3 and 4 as well as their analogs 7 and 8 for further studies. 3, 4, 7, and 8 only differ in the halide and methyl groups on the phenyl ring of the phthalimide, which may offer an appropriate site for labeling the corresponding positron emitting radionuclides such as fluorine-18, carbon-11, bromine-76, and iodine-124, respectively.
Figure 1.

Representative mGlu4 PAMs from N-phenyl-2-picolinamides and the corresponding phthalimide derivatives.
As shown in Scheme 1, compounds 3, 4, 7 and the nitro-precursor 17 that is used for radiolabeling were prepared in three steps from commercially available chemicals. Iodine derivative 8 was synthesized in five steps. 2-Picolinic acid (9) was converted into the corresponding acid chloride and then coupled with aniline 10 to give picolinamide derivative 11 in 41% yield. The nitro group in 11 was subsequently reduced by hydrogenation to afford aniline 12 in 91% yield. Aniline 12 was coupled with the phthalic anhydride derivatives 13–16 in acetic acid to give the corresponding phthalimides derivatives, 3, 4, 7, and 17 in 78%, 66%, 92%, and 80% yields, respectively. To prepare iodine derivative 8, bromo-derivative 7 was converted into trimethyltin derivative 18, which subsequently reacted with iodine to obtain 8 in 7% yield.
Scheme 1. Syntheses of the Phthalimide Derivatives.

Binding Affinity to mGlu4
Since we had studied [11C]1 as a PET ligand and there was no report on its affinity to mGlu4, we would like to directly compare the affinity of the phthalimide derivatives 3, 4, 7, and 8 with compound 1. Thus, we performed radiolabeling of compound 1 with tritium for a competitive binding assay (Scheme 2). The chromatography of [3H]1 radiosynthesis is shown in Supporting Information A.
Scheme 2. Radiosynthesis of [3H]1.

Table 1 presents the IC50 values of compounds 1–4, 7, and 8 obtained from in vitro competitive binding assay using [3H]1. The results show that the lead compounds from N-phenyl-2-picolinamide series 1 and 2 have IC50 values of 12.8 and 9.9 nM, respectively. Compound 2 has been used for in vivo mGlu4 blocking experiments for [11C]1.17 The phthalimide derivatives, 3 (5.1 nM) and 4 (4.2 nM) gave improved affinity compared to 1 while compounds 7 (11.8 nM) and 8 (26.3 nM) showed similar or less affinity to mGlu4 compared to 1. This result shows that 3 and 4 have similar affinity while Merck’s patent20 presents that 4 (11 nM) has better affinity than 3 (36 nM), which may be caused from using cells expressing different mGlu4 receptors. We used the rat mGlu4, while Merck employed the human mGlu4. The results verified that 3 and 4 are more potent compounds for developing an mGlu4 PET ligand. The results also indicate that these phthalimide compounds interact with the same allosteric site in mGlu4 where 1 binds because the quantitative specific binding curves of these compounds show complete replacement of the radioligand at a higher concentration (Figure 2). In addition, the selectivities of 3 to mGlu1, mGlu5, and mGlu8 were determined by the functional assays, which showed little activity against to these mGluRs (Supporting Information B).22,23
Table 1. Results from in Vitro Competitive Binding Assay.
| compd | mGlu4 IC50 (nM) | log(IC50) ± SD | RBI ± SD a (%) (10 μM) |
|---|---|---|---|
| 1 | 12.8 | –7.89 ± 0.09 | 100.1 ± 2.0 |
| 2 | 9.9 | –8.00 ± 0.09 | 99.1 ± 0.5 |
| 3 | 5.1 | –8.29 ± 0.08 | 99.4 ± 0.7 |
| 4 | 4.2 | –8.38 ± 0.07 | 99.4 ± 0.9 |
| 7 | 11.8 | –7.93 ± 0.09 | 96.6 ± 1.2 |
| 8 | 26.3 | –7.53 ± 0.09 | 96.0 ± 3.0 |
RBI = relative binding inhibition. SD = standard deviation.
Figure 2.
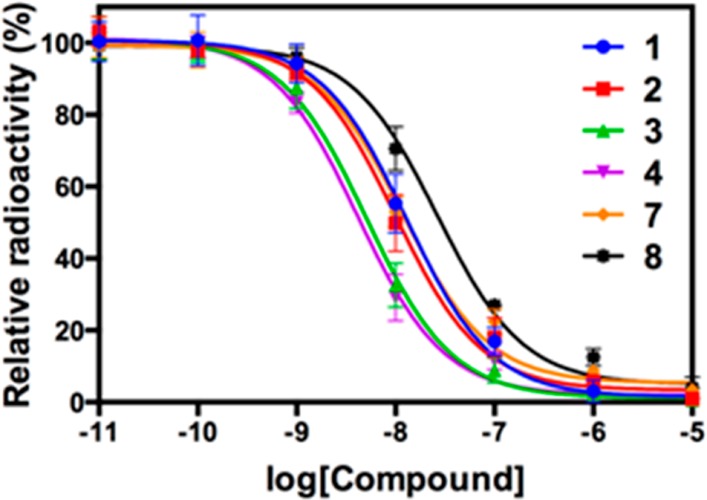
Binding curves from competitive binding assay (mGlu4).
On the basis of their affinity, both compounds 3 and 4 can be the ideal candidates for developing PET radioligands. Nonetheless, fluorine-18 is often the radionuclide of choice for both its physical and nuclear characteristics. Its half-life is long enough to carry out relatively extended imaging protocols when compared to what is possible with carbon-11, which facilitates kinetic studies and high-quality metabolic and plasma analyses. Therefore, we have selected compound 3 for radiolabeling and in vivo PET imaging.
Radiochemistry
As shown in Scheme 3, [18F]3 was prepared from the reaction of 17 in DMSO with non-carrier-added (nca) [18F]fluoride in the presence of Kryptofix 222 (K222) and potassium carbonate at 120 °C for 5 min. It was necessary to use an adequate amount of 17 (6 mg) for this reaction because our result showed that K222 activated carbonate anion and could cause the decomposition of substrate 17. The radioactive product was purified by semipreparative HPLC, eluting with a 0.1% formic acid solution of water and acetonitrile (52:48) at a flow rate of 4 mL/min. [18F]3 was eluted at 17.4–18.8 min and was well separated from starting material 17 (tR of 19.6–22.4 min) as well as other impurities (Supporting Information C). It was subsequently found that the radiochemical purity of [18F]3 was only 79% when the purified [18F]3 was first formulated in a neutral 10% ethanol–saline solution (Supporting Information D). This result indicates that the phthalimide derivative was not very stable and underwent hydrolysis in a neutral condition.24 To confirm this issue, we studied the stability of 3 in both neutral and acidic conditions by using a LC–MS method. When 3 was dissolved in a neutral HPLC solution (0.1 M ammonium formate water–acetonitrile solution) and analyzed by using the same neutral eluants, two side products were detected in which one was 12 and the other was the hydrolysis product 20a and/or 20b (Supporting Information E). Conversely, the stability test in acidic media gave minimal hydrolysis products. Thus, [18F]3 was purified and formulated in acidic media after radiosynthesis to minimize unwanted hydrolysis. When finally formulated in 0.1 M citrate buffer (pH 4) with 10% ethanol, the specific activity of [18F]3 at the end of synthesis (EOS) was 233.5 ± 177.8 GBq/μmol (n = 4). The average decay corrected radiochemical yield (RCY) of [18F]3 was 16.4 ± 4.8% (n = 4). The radiochemical purity of [18F]3 was over 98%.
Scheme 3. Radiosynthesis of [18F]3.

QC analysis using analytical HPLC and radio-TLC by co-injection with nonradioactive 3 has confirmed the isolated radioactive product as [18F]3 (Supporting Information F). Both methods validated that the radiochemical purity of [18F]3 was over 98%.
PET Imaging of [18F]3
The characterization of [18F]3 was first conducted with rat (male Sprague–Dawley) studies. These studies demonstrated that [18F]3 crossed BBB and occupied brain areas known to express mGlu4. Time−activity curves (TACs) showed fast uptake and washout in different brain regions within 20 min (Figure 3a). These results are similar to our previous PET studies with [11C]1.17
Figure 3.

(a) TACs derived from the rat brain PET images [18F]3 (n = 7). (b) mGlu4 blocking studies demonstrated reduced uptake in the brain regions by 10–20%, while mGlu5 blocking agents displayed minimal blocking effect indicating good selectivity to mGlu4. Tissue uptake was calculated as area under TAC expressed as (% dose/cc) × min: Str = striatum, Hipp = hippocampus, Thal = thalamus, Cort = cortex, Cereb = cerebellum, WB = whole brain. Shown are results from baseline studies (n = 7), mGlu4 (2) blocking studies (n = 4), and mGlu5 (MTEP) blocking studies (n = 3). (c) Percentage of [18F]3 in the plasma over time. (n = 2).
Pretreatment with 10 mg/kg 2·HCl as an mGlu4 blocking agent16,17 and 10 mg/kg 3-((2-methyl-1,3-thiazole-4-yl)ethynyl)pyridine (MTEP) as a selective mGlu5 blocking agent25,26 resulted in modest blocking effects in rat brains of 10–20% and 0–5% reduced uptake, respectively (Figure 3b).
The low blocking effects induced by 2 can be attributed to insufficient dosage and fast pharmacokinetics behavior of 2 in brain.16 10 mg/kg of 2·HCl may not be enough to achieve noticeable blocking effects in the brain. The low blocking effects can also be due to fast pharmacokinetic properties of 2 in the brain.16 The time difference between the injection of the blocking agent and radiotracer was 5 min, and some of blocking agent 2 might have washed off before [18F]3 injection. Therefore, the current results might be improved to obtain better specificity and selectivity with further adjusted blocking.
In addition, the arterial plasma analysis (Figure 3c) showed that after the intravenous bolus injection, about 90% of [18F]3 was quickly metabolized within 5 min. It is important to improve its metabolic stability.
After the characterization of [18F]3 in rats studies, combined PET–MRI studies showed similar results in the monkey brain with high accumulation of [18F]3 in the regions such as the striatum, thalamus, hippocampus, and cerebellum where previous literature has reported high expression of mGlu4 (Figure 4a).4,15 T1-weighed MRI study (Figure 4c) and fused PET–MRI studies (Figure 4b) illustrated anatomical details and the regional distribution of [18F]3.
Figure 4.
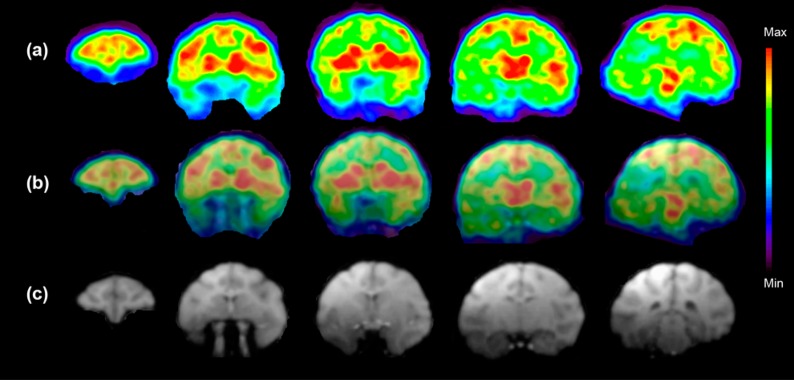
(a) Color coded PET images show accumulation of [18F]3 in the selected five coronal slices of the monkey brain at the time point 5–15 min after the injection of radioactivity. These images show the highest accumulation of [18F]3 in the caudate, putamen, thalamus, and several cortical areas. (b) Fused PET and MR images from (a) and (c). (c) T1-weighted coronal MR images show the anatomical structures of selected brain areas. (n = 1) 189 MBq [18F]3 (37 ng/kg) was injected.
TACs derived from the different monkey brain areas showed fast accumulation of [18F]3 with the highest accumulation about 3 min after the administration of the radioligand. About 50% of the activity stayed for 20 min followed by slower washout until 40 min, allowing suitable retention time for binding. The binding potential determined by the metabolite corrected plasma input function and volume distribution model was highest in the thalamus (2.35) followed by the putamen (2.13), the caudate (1.91), the prefrontal cortex (1.47), and the cerebellum (1.19). These studies confirm that the cerebellum cannot be used as a reference tissue in quantitative modeling.
The fast pharmacokinetics of [18F]3 in the brain can also be related to metabolic instability of the radiotracer. The percent of parent [18F]3 in plasma was quickly dropped within 5 min in both rats and the monkey (Figure 3c and Figure 5). However, [18F]3 showed better permeability in the monkey brain than in the rat brain and was retained in the brain until 40 min (Figure 3a and Figure 5).
Figure 5.

TACs derived from the different areas of the monkey brain show fast accumulation of [18F]3 and reversible binding.
Conclusion
Four 4-phthalimide derivatives of N-(3-chlorophenyl)-2-picolinamide 3, 4, 7, and 8 and precursor 17 were synthesized by three or five steps of syntheses from commercially available chemicals. Compounds 3, 4, 7, and 8 were studied for their binding affinity using rat mGlu4 transfected Chinese hamster ovary (CHO) cells. Among these compounds, 3 and 4 exhibited the highest binding affinity, which demonstrated improved affinity compared to the previously reported mGlu4 PET ligand, [11C]1. 3 was selected and labeled with fluorine-18, since fluorine-18 has been the choice of radionuclide for PET.
One-step labeling reaction with fluorine-18 was successfully performed after careful optimization of reaction conditions with 16.4 ± 4.8% of RCY (n = 4) and 233.5 ± 177.8 GBq/μmol of specific activity (n = 4). [18F]3 was formulated with 10% ethanol in pH 4 citrate buffer to achieve over 98% radiochemical purity. In vivo imaging studies in the monkey showed that the radiotracer quickly penetrated the brain with the highest accumulation in the brain areas known to express mGlu4. In the rat studies, the fast accumulation was followed with fast washout from the brain within 20 min while it was 40 min in the monkey study. Even though [18F]3 did not improve the problems of fast washout in rodent studies which we reported with [11C]1, it might be a more promising radiotracer for further development for use in monkey and possibly human studies because it displayed higher peak uptake in the brain and it was retained in the brain for a longer period. In vitro tests also indicated that [18F]3 would give better results with mGlu4 human cell lines than [11C]1.6,16,20 Despite of some unfavorable properties as a radiotracer such as fast washout in rodent studies, [18F]3 is the first 18F-labeled radioligand and can be further modified to improve pharmacokinetic properties and brain penetrability as a candidate for future human studies.
Experimental Section
Animal Procedures
The animal studies were approved and done under the strict supervision of the Subcommittee on Research Animals of the Massachusetts General Hospital and Harvard Medical School and performed in accordance with the Guide of NIH for the Care and Use of Laboratory Animals.
Materials and General Methods
Unless otherwise specified, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO). The reactions were monitored by TLC using a UV lamp monitored at 254 nm. If necessary, the reactions were also checked by LC–MS using the Agilent 1200 series HPLC system coupled with a multiwavelength UV detector and a model 6310 ion trap mass spectrometer (Santa Clara, CA) equipped with an Agilent Eclipse C8 analytical column (150 mm × 4.6 mm, 5 μm). Elution was with a 0.1% formic acid solution of water (A) and acetonitrile (B). Flash chromatography was performed with a CombiFlash Rf Purification System (Teledyne Isco) using a Silica ReadySep Rf column. Hydrogenation reaction was performed using a Parr shaker hydrogenation apparatus (Moline, IL). The products were identified by LC–MS and 1H NMR and 13C NMR using a Varian 500 MHz spectrometer. 19F NMR was used to identify the fluorine compound. All NMR samples were dissolved in chloroform-d (CDCl3) containing tetramethylsilane as a reference standard. Chemical shifts were expressed as ppm and calculated downfield or upfield from the NMR signal of reference standard. J was expressed as Hz, and its splitting patterns were reported as s, d, t, q, or m. HRMS was obtained from the High Resolution Mass Spectrometry Facility at the University California, Riverside, using electrospray ionization (ESI)/atmospheric pressure chemical ionization (APCI) technique (Agilent Time of Flight (TOF) LC–MS). Melting points were measured using a Mettler MP50 melting point system. Unless otherwise specified, the purities of all new compounds were over 95% determined by HPLC.
The mGlu4 blocking agent 2 and its hydrochloride salt were prepared by a published method.17 [3H]Iodomethane (37 GBq/mL in DMF) was purchased from American Radiolabeled Chemicals Inc. and used without further purification.
The National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract HHSN-271-2008-00025-C (NIMH PDSP), generously performed selectivity tests for mGlu1 and mGlu5 using intracellular inositol phosphates (IPs) accumulation assay with CHO cells that stably expressed mGlu1a and mGlu5a. Detailed procedures of selectivity tests were described in the previous literature.17,22,23 For the selectivity test for mGlu8, split luciferase biosensor cAMP assay was performed using mGlu8 transfected CHO cells.22,23
Syntheses of Phthalimide Derivatives 3, 4, 7, and 17
A solution of 12 and phthalic anhydride derivatives (13, 14, 15, and 16) in acetic acid was heated to 100 °C for 5 h. After acetic acid was removed at reduced pressure, residue was recrystallized in a solution of ethyl acetate and hexane to give the corresponding phthalimide derivatives 3, 4, 7, and 17 as brown powders, respectively.
N-(3-Chloro-4-(4-fluoro-1,3-dioxoisoindolin-2-yl)phenyl)-2-picolinamide (3)
12 (0.20 mmol, 50 mg), 3-fluorophthalic anhydride (13, 0.24 mmol, 40 mg), and acetic acid (4.0 mL) were used to afford 3 (62 mg, 78% yield); mp 218–220 °C. 1H NMR (500 MHz, CDCl3): δ 10.21 (s, 1H), 8.64 (d, J = 4.5 Hz, 1H), 8.31 (d, J = 8.0 Hz, 1H), 8.18 (d, J = 2.0 Hz, 1H), 7.95 (t, J = 8.0 Hz, 1H), 7.80–7.83 (m, 3H), 7.53 (dd, J = 7.5 Hz, 5.0 Hz, 1H), 7.48 (t, J = 8.0 Hz, 1H), 7.35 (d, J = 8.5 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 165.97 (d, J = 2.6 Hz), 163.71, 162.57, 158.30 (d, J = 265.6 Hz), 149.46, 148.43, 140.23, 138.23, 137.38 (d, J = 6.8 Hz), 134.39, 134.17, 131.28, 127.27, 124.92, 123.18 (d, J = 20.5 Hz), 122.99, 121.31, 120.52 (d, J = 3.9 Hz), 118.88, 118.16 (d, J = 1.3 Hz). 19F NMR (470 MHz, CDCl3): δ −111.6 (t, J = 5.9 Hz). HRMS m/z calcd for C20H12ClFN3O3 (M + H)+, 396.0546; found 396.0554.
N-(3-Chloro-4-(4-methyl-1,3-dioxoisoindolin-2-yl)phenyl)-2-picolinamide (4)
12 (0.40 mmol, 100 mg), 3-methylphthalic anhydride (14, 0.42 mmol, 39 mg), and acetic acid (8.0 mL) were used to afford 4 (66 mg, 66% yield); mp 184–185 °C. 1H NMR (500 MHz, CDCl3): δ 10.19 (s, 1H), 8.63 (d, J = 5.0 Hz, 1H), 8.31 (d, J = 8.0 Hz, 1H), 8.17 (d, J = 2.0 Hz, 1H), 7.94 (td, J = 8.0 Hz, 1.0 Hz, 1H), 7.81 (m, 2H), 7.66 (t, J = 8.0 Hz, 1H), 7.52–7.57 (m, 2H), 7.35 (d, J = 8.5 Hz, 1H), 2.76 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 167.91, 167.19, 162.52, 149.51, 148.41, 139.95, 139.07, 138.19, 137.15, 134.29, 134.23, 132.71, 131.35, 128.99, 127.21, 125.54, 122.93, 121.93, 121.24, 118.78, 18.13. HRMS m/z calcd for C21H15ClN3O3 (M + H)+, 392.0796; found 392.0805.
N-(3-Chloro-4-(4-bromo-1,3-dioxoisoindolin-2-yl)phenyl)-2-picolinamide (7)
12 (2.02 mmol, 0.500 g), 3-bromophthalic anhydride (15, 2.22 mmol, 0.504 g), and acetic acid (10 mL) were used to afford 7 (844 mg, 92%); mp 187–189 °C. 1H NMR (500 MHz, CDCl3): δ 10.21 (s, 1H), 8.62 (d, J = 4.5 Hz, 1H), 8.30 (d, J = 7.5 Hz, 1H), 8.17 (d, J = 1.0 Hz, 1H), 7.92–7.95 (m, 3H), 7.79 (dd, J = 8.5 Hz, 1.5 Hz, 1H), 7.65 (t, J = 7.5 Hz, 1H), 7.51–7.54 (m, 1H), 7.35 (d, J = 8.5 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 165.47, 165.08, 162.55, 149.42, 148.41, 140.17, 139.71, 138.21, 135.63, 134.43, 134.10, 131.21, 129.81, 127.25, 125.02, 123.33, 122.96, 121.24, 119.55, 118.82. HRMS m/z calcd for C20H12ClBrN3O3 (M + H)+, 455.9745; found 455.9742.
N-(3-Chloro-4-(4-nitro-1,3-dioxoisoindolin-2-yl)phenyl)-2-picolinamide (17)
12 (0.606 mmol, 0.150 g), 3-nitrophthalic anhydride (16, 0.666 mmol, 0.129 g), and acetic acid (6 mL) were used to afford 17 (0.204 g, 80% yield); mp 199–201 °C. 1H NMR (500 MHz, CDCl3): δ 10.23 (s, 1H), 8.65 (d, J = 5.0 Hz, 1H), 8.32 (d, J = 8.0 Hz, 1H), 8.25 (d, J = 7.0 Hz, 1H), 8.20–8.22 (m, 2H), 8.01 (t, J = 7.8 Hz, 1H), 7.96 (td, J = 8.0 Hz, 1.5 Hz, 1H), 7.82 (dd, J = 8.8 Hz, 2.3 Hz, 1H), 7.54 (dd, J = 7.0 Hz, 5.0 Hz, 1H), 7.37 (d, J = 8.5 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 164.62, 162.61, 161.68, 149.41, 148.45, 145.87, 140.50, 138.26, 136.14, 134.25, 134.05, 131.17, 129.41, 128.03, 127.31, 124.50, 124.00, 123.02, 121.31, 118.91. HRMS m/z calcd for C20H12ClN4O5 (M + H)+, 423.0491; found 423.0487.
N-(3-Chloro-4-(4-iodo-1,3-dioxoisoindolin-2-yl)phenyl)-2-picolinamide (8)
Iodine (0.116 mmol, 29.4 mg) was added to a solution of 18 (72.5 μmol, 39.2 mg) in DCM (1.4 mL), and the mixture was stirred at room temperature for 2 h. The solution was quenched with 5% sodium metabisulfite solution until its color disappeared. The solution was then extracted with ethyl acetate three times. Organic layers were collected, dried over anhydrous sodium sulfate (Na2SO4), filtered, and evaporated. Residue was separated with 25% ethyl acetate in hexane by flash column chromatography to give 8 as a yellowish solid (2.5 mg, 7% yield); mp 140–142 °C. 1H NMR (500 MHz, CD3CN): δ 10.37 (s, 1H), 8.72 (d, J = 4.5 Hz, 1H), 8.30 (d, J = 8.0 Hz, 1H), 8.26–8.27 (m, 2H), 8.05 (t, J = 7.5 Hz, 1H), 7.98 (d, J = 7.5 Hz, 1H), 7.92 (dd, J = 8.5 Hz, 2.0 Hz, 1H), 7.64 (dd, J = 8.0 Hz, 4.0 Hz, 1H), 7.59 (t, J = 7.8 Hz, 1H), 7.48 (d, J = 9.0 Hz, 1H). HRMS m/z calcd for C20H12ClN3O3I (M + H)+, 503.9606; found 503.9417.
N-(3-Chloro-4-nitrophenyl)-2-picolinamide (11)
Thionyl chloride (163 mmol, 11.8 mL) was added to 2-picolinic acid (9, 40.6 mmol, 4.00 g) in benzene (60 mL), and the mixture was refluxed for 2 h. Then the solvent and extra thionyl chloride were removed by vacuum distillation. The resulting 2-picolinic acid chloride was isolated as a gray salt powder. A solution of 3-chloro-4-nitroaniline (6, 10.2 mmol, 1.75 g) (Alfa Aesar) and triethylamine (122 mmol, 17.0 mL) in 30 mL of THF was then added to the gray salt powder in 30 mL of THF. The mixture was refluxed for 2 h and was then quenched with saturated ammonium chloride solution. The reaction mixture solution was extracted with ethyl acetate three times and dried over anhydrous Na2SO4, filtered, and evaporated. The residue was separated by flash chromatography with eluants of ethyl acetate (20%) and hexane (80%) to give the crude product as a solid, which was recrystallized in a solution of ethyl acetate and hexane to give 11 as a white crystal (1.153 g, 41% yield); mp 179–180 °C. 1H NMR (500 MHz, CDCl3): δ 10.35 (s, 1H), 8.65 (d, J = 5.0 Hz, 1H), 8.31 (d, J = 8.0 Hz, 1H), 8.15 (d, J = 2.0 Hz, 1H), 8.05 (d, J = 9.0 Hz, 1H), 7.97 (td, J = 8.0 Hz, 1.0 Hz, 1H), 7.57 (dd, J = 7.0 Hz, 5.0 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 162.77, 148.90, 148.53, 143.16, 142.61, 138.39, 129.45, 127.69, 127.66, 123.12, 122.04, 117.87. HRMS m/z calcd for C12H9ClN3O3 (M + H)+, 278.0327; found 278.0329.
N-(3-Chloro-4-aminophenyl)-2-picolinamide (12)
Compound 11 (3.96 mmol, 1.10 g) and 10% palladium/charcoal (0.220 g) were added to a solution of methanol (70 mL) and ethyl acetate (70 mL). The mixture was shaken under 3 atm of hydrogen for 20 h at room temperature. The solution was filtered and evaporated. The residue was separated by flash chromatography with eluants of 30% ethyl acetate and 70% hexane to give a crude product, which was recrystallized in a solution of ethyl acetate and hexane to afford 12 as a yellowish solid (0.889 g, 91% yield); mp 144–145 °C. 1H NMR (500 MHz, CDCl3): δ 9.85 (s, 1H), 8.60 (d, J = 5.0 Hz, 1H), 8.29 (d, J = 8.0 Hz, 1H), 7.91 (td, J = 8.0 Hz, 1.0 Hz, 1H), 7.84 (d, J = 2.0 Hz, 1H), 7.48 (dd, J = 7.5 Hz, 5.0 Hz, 1H), 7.44 (dd, J = 9.0 Hz, 2.0 Hz, 1H), 6.80 (d, J = 8.5 Hz, 1H), 4.00 (s, 2H). 13C NMR (125 MHz, CDCl3): δ 161.98, 150.12, 148.27, 140.13, 137.98, 129.87, 126.67, 122.60, 121.56, 120.11, 119.69, 116.28.
N-(3-Chloro-4-(4-trimethylstannyl-1,3-dioxoisoindolin-2-yl)phenyl)-2-picolinamide (18)
Bis(trimethyltin) (0.40 mmol, 84 μL) was added to a mixed solution of 7 (0.18 mmol, 80 mg) and tetrakis(triphenylphosphine)palladium(0) (1.8 μmol, 2.0 mg) in degassed toluene (2 mL) at room temperature, and then the mixture was refluxed for 8 h. The resulting mixture was diluted with saturated ammonium chloride solution and extracted three times with ethyl acetate. The organic layers were combined, dried over anhydrous Na2SO4, filtered, and evaporated. The residue was separated by flash chromatography with 10% ethyl acetate in hexane to give 18 as a white solid (44 mg, 46% yield); mp 213-214 °C. 1H NMR (500 MHz, CDCl3): δ 10.19 (s, 1H), 8.63 (d, J = 4.0 Hz, 1H), 8.30 (d, J = 8.0 Hz, 1H), 8.18 (s, 1H), 7.90–7.96 (m, 3H), 7.80 (d, J = 8.0 Hz), 7.72 (t, J = 5.5 Hz, 1H), 7.38 (d, J = 8.5 Hz, 1H), 0.41 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 169.16, 167.78, 162.48, 149.46, 148.39, 142.39, 142.09, 139.92, 138.17, 134.08, 133.28, 131.75, 131.23, 127.20, 125.49, 124.02, 122.92, 121.27, 118.78, −8.37. HRMS m/z calcd for C23H21ClN3O3120Sn (M + H)+, 542.0288; found 542.0294.
Radiosynthesis of [3H]1
[3H]Iodomethane (1 Ci/ml in DMF) was added to a DMF solution of N-(4-chloro-3-hydroxyphenyl)picolinamide (19, 1.5 mg, 6.0 μmol) and 5 M potassium hydroxide solution (3.0 μL, 15 μmol). The resulting mixture was heated at 90 °C for 10 min and diluted with 1.0 mL of HPLC solvents. Then the aliquot was injected into HPLC equipped with a Gemini-NX C18 semipreparative column (250 mm × 10 mm, 5 μm, Phenomenex Inc.), a flow scintillation detector, and an internal UV detector, eluting with a solution of 55% acetonitrile and 45% 0.1 M ammonium formate at a flow rate of 4 mL/min. An eluant containing [3H]1 was collected between 11 and 13 min. The radioactive product was diluted with 30 mL of water and passed through a C18 Sap-Pak Plus followed by additional wash with 5 mL of sterile water. [3H]1 was then eluted from cartridge with ethanol. The final product was diluted with 9 volumes of sterile saline to make a 10% ethanol solution in saline for cell studies.
Preparation of mGlu4 Transfected CHO Cells
The vector of mGlu4 from rat was obtained as a gift from Drs. Tanabe and Nakanishi’s laboratory (Osaka Bioscience Institute, Osaka, Japan), and its structure was described in their previous literature.27 To extract an mGlu4 cDNA insert out of its backbone, pBluescript II KS(+), EcoR1 (New England Biolabs) was treated and incubated for 4 h at 37 °C. Then electrophoresis was performed and the mGlu4 insert (3704 bp) was isolated from its backbone (2954 bp) using a gel purification kit (Qiagen Inc.) following the procedure provided by the kit. The target backbone, pcDNA, was also digested by the same procedure. After their concentrations were measured using a NanoDrop (Thermo Scientific), pcDNA (30 fmol) and the mGlu4 insert (90 fmol) were mixed and incubated at room temperature for 2 h mediated by T4 ligase (Invitrogen Life Technology). After the ligase was deactivated by heating, the combined vectors were incorporated into DH5α-E competent cells (Invitrogen Life Technology) by electroporation. The aliquot was transferred to a LB agar dish containing ampicillin and grown at 37 °C overnight. Some clones were selected and amplified in LB media containing 0.1% ampicillin. Successful clones were screened by electrophoresis and DNA sequencing. They continued to culture in LB broth (1 L) containing 0.1% ampicillin stock solution. The resultant vectors containing mGlu4 cDNA was obtained using a Maxi-Prep column kit (Qiagen Inc.) and its standard procedure.
The mGlu4 DNA and Plus reagent (Invitrogen Life Technology) were mixed in reduced serum medium (Opti-MEM Life Technology) and added to the reduced serum medium containing Lipofectamine (Invitrogen Life Technology). The resulting aliquot was incubated for 15 min at room temperature. Meanwhile, CHO cells were loaded to a six-wall plate and cultured overnight until confluence at 37 °C in the culture medium. After the culture medium in the six-wall plate was replaced with reduced serum medium, the mGlu4 aliquot containing Lipofectamine and Plus reagent was loaded to the six-wall plate. Transfection was performed at 37 °C for 3 h. The medium was replaced with culture medium containing G418 to select and grow only transfected cells. The cells in each wall continued to grow until nontransfected cells died. The selection medium was replaced every 3 days during culturing. The successful colonies were cropped and moved to bigger culture bottles. These cells continued to grow until confluence by changing medium containing G418 every 3 days. Each colony was further selected to identify successful colonies by Western blot.
In Vitro mGlu4 Binding Assay
CHO cells expressing mGlu4 were used for all binding assay protocols. For competitive binding, 10 nM [3H]1 was used together with increased concentrations of test compounds ranging from 0.01 nM to 10 μM. Each test tube contained 50 000 freshly harvested CHO cells expressing mGlu4 cultured in HAMs cell culture medium, penicillin–streptomycin (100 units), and 1 mM G418. [3H]1 (10 nM) was added to the cell extract with and without test compounds on ice and then incubated for 30 min at room temperature. Samples were centrifuged at 1200 rpm for 10 min at 4 °C and washed three times with a cold cell culture medium as a washing buffer. Samples were lysed by adding 100 μL of 0.5% NaOH and heated using a heating block (56 °C, 30 min). Samples were cooled with an ice bath and transferred to Solvent-Saver scintillation vials (VWR International LLC.). To obtain binding parameters, the scintillation liquid (PerkinElmer, Optima Gold) was added prior to counting with a scintillation counter (Packard TriCarb Model, 1 min/vial). Nonspecific binding was determined using 10 μM nonradioactive 1, and specific binding was determined by extracting the nonspecific binding from total binding. All measurements were done in triplicate and analyzed with GraphPad Prism software (GraphPad Software Inc.).
Radiosynthesis of [18F]3
[18F]Fluoride was generated by a Siemens Eclipse HP 11 MeV cyclotron (Malvern, PA) using 18O-enriched water (Isoflex Isotope, San Francisco, CA) with proton bombardment. Fluorine-18 labeling chemistry was carried out in a Siemens Explora GN synthetic module. A [18F]fluoride reagent was passed through a QMA Sep-Pak Cartridge (Waters, Milford, MA) to trap [18F]fluoride ions. A solution of potassium carbonate (1 mg) and K222 (3 mg) in acetonitrile and water (1:1, 0.4 mL) was passed through the cartridge to elute [18F]fluoride ions. The solvents were evaporated at 115 °C in a stream of nitrogen. To remove water completely, 1 mL of acetonitrile was added and evaporated twice. 17 (6 mg) in 0.6 mL of DMSO was added to the residue, and the mixture was heated to 120 °C for 5 min. The resulting mixture was cooled to 100 °C, and 1.5 mL of the HPLC solvent was added. The mixture was then purified by HPLC (Agilent 1100 series equipped with UV detector and radioactivity detector) using an Eclipse XDB-phenyl semipreparative column (250 mm × 10 mm, 5 μm) and eluting with a 0.1% formic acid solution of water and acetonitrile (52:48) at a flow rate of 4 mL/min. The fraction containing [18F]3 was diluted with a 0.1 M citrate buffer solution (pH 4) to 20 mL and loaded on a C18 Sep-Pak column. The final product was obtained after eluting 1 mL of ethanol through the C18 Sep-Pak column, and the collected product was diluted with a sterile 0.1 M citrate buffer solution (pH 4) to make a 0.1 M citrate buffer solution with 10% ethanol.
QC Analysis of [18F]3
A purified radioactive aliquot (10 μL) and nonradioactive standard (3) were injected into the HPLC (Agilent 1100 series equipped with a UV detector monitored at 254 nm and a Ramsey model 105S single channel radiation detector (Berkeley, CA)) using an Eclipse XDB-phenyl analytical column (150 mm × 4.6 mm, 5 μm, Agilent) and eluting with a solution of acetonitrile and 0.1% formic acid (55:45) at a flow rate of 1 mL/min.
An AR-2000 radio-TLC imaging scanner (Bioscan Inc.) was also used to check radio-TLC for further confirmation of radioactive product. The radioactive aliquot was cospotted with the nonradioactive 3 (in acetonitrile) on TLC and developed with a solution of ethyl acetate and hexane (1:1). The same Rf values of the radioactive spot and UV active spot were confirmed by the radio-TLC scanner and UV lamp at 254 nm, respectively.
Specific Activity of [18F]3
Specific activity was measured by the published method.17 The radioactivity of the aliquot was measured and corrected to the time point of the EOS. The corresponding mass was measured by a calibration curve that showed the linear relationship between the molar amount of 3 and UV integration. The calibration curve was previously established with five different standard concentrations of 3. The specific activity was calculated and expressed as GBq/μmol.
PET Imaging of [18F]3 in Rats
PET imaging studies were conducted using seven normal Sprague–Dawley male rats. The rats were anesthetized with isoflurane/nitrous oxide (1.0–1.5% isoflurane, with oxygen flow of 1–1.5 L/min), and a catheter was installed in their tail veins for the administration of [18F]3. Dynamic volumetric PET data were acquired with a PET-CT scanner for 60 min (Triumph-II preclinical imaging system, Tri-Foil imaging, Northridge, CA). Vital signals such as heart and respiration rates were monitored during the scanning period. PET data acquisition started immediately after the administration of [18F]3 (37 MBq) followed by CT imaging to obtain anatomical information and data for attenuation correction of PET data. PET data were processed using a maximum-likelihood expectation–maximization (MLEM) algorithm with 30 iterations for dynamic volumetric images and were corrected for uniformity, scatter, and attenuation. The CT data were processed by a modified Feldkamp algorithm using matrix volumes of 512 × 512 × 512 with a pixel size of 170 μm. Co-registration of CT and PET images and analysis of PET images were performed using PMOD3.2 software (PMOD Technology, Zurich, Switzerland).
For blocking studies, the same imaging protocol was used for baseline studies. For mGlu4 blocking, 2·HCl salt (10 mg/kg) dissolved in a saline solution with 10% ethanol and 10% Tween20 was injected 5 min before iv administration of [18F]3 in four normal male rats. For mGlu5 blocking, 10 mg/kg of MTEP·HCl (Abcam Inc.) in 0.9% saline was intravenously administered to three normal male rats 5 min before [18F]3 injection.
Blood Metabolite Studies
Blood metabolite studies were performed by the procedure reported in previous literature.17 Blood samples were taken from rats (n = 2) at 10, 30, and 60 min after [18F]3 injection. Each blood sample was centrifuged for 3 min to separate plasma and blood coagulants. The plasma (0.3 mL) was retrieved, and 0.3 mL of acetonitrile was added to the plasma. The suspended solution was vortexed and centrifuged for 3 min to precipitate protein and to obtain protein-free plasma as supernatant. The resulting supernatant was divided into two parts. One part was used for measuring the total radioactivity, and another part (0.3 mL) was loaded to a Strata-X C18 cartridge (500 mg, Phenomenex Inc.) that had been purged with water. A series of elutions were performed using using 0.1% TFA/water and 0.1% TFA/acetonitrile in quantities 95:5, 90:10, 85:15, 80:20, 70:30, 60:40, 30:70, 100% of 0.1% TFA/acetonitrile at a volume of 4 mL. The eluted fractions were collected in eight test tubes. A control experiment by injection of a small amount of [18F]3 onto a test cartridge elution described above indicated that the parent [18F]3 was collected in the seventh test tube exclusively. The radioactivities were measured using a 2480 Wizard2 automatic gamma counter (PerkinElmer Inc., Waltham, MA).
PET Imaging of [18F]3 in a Rhesus Monkey
A female rhesus monkey was restricted from food for 12 h before the PET-MR imaging. At first, 10 mg/kg ketamine was administered intramuscularly. Anesthesia was maintained at 1–4% isoflurane (Forane) in a mixture of oxygen and nitrogen. The monkey was intubated, and catheters were introduced for radiotracer injection and a radial arterial line for blood drawing needed for the input function and metabolite analysis. MR-PET images were obtained using an integrated 3T MR-BrainPET camera (Siemens, Munich, Germany) with a resolution of 2.5 mm and an axial field of view (FOV) of 19.25 cm and a transaxial FOV of 30 cm. A multiecho magnetization-prepared rapid gradient-echo (MEMPRAGE) sequence was initiated to acquire MRI data for attenuation correction and anatomical coregistration. After the injection of 189 MBq [18F]3 (37 ng/kg) in a 10% ethanol solution in isotonic saline, dynamic PET data were acquired for 90 min. For quantitative analyses, blood samples were collected every 10 s for the first 2 min after the administration of [18F]3 and subsequently obtained at the following time points during the scan: 3, 5, 10, 15, 20, 30, 45, 60, and 90 min.
The images were reconstructed using an ordinary Poisson ordered-subset expectation maximization (OP-OSEM) 3D algorithm from prompt and random coincidences, normalization, attenuation, and scatter coincidences sinograms using 16 subsets and 6 iterations. The reconstructed volume consisted of 153 slices with 256 × 256 pixels (1.25 × 1.25 × 1.25 mm3). Regions of interest (ROIs) were drawn on all coronal levels, where structures were observed anatomically (fused data with MRI) and activity per unit volume, percent activity of injected dosage, and ligand concentration were calculated. Plasma data were corrected for counting efficiency, calibration factors, and measured metabolites of injected ligand. Percent activity of injected dose and ligand concentration were calculated. Plasma data were used as an input function in kinetic modeling. The binding potential values of [18F]3 in the ROI were determined by PMOD3.2.
Acknowledgments
The authors thank the technical staff of the PET/MRI and radiochemistry facilities at the Athinoula A. Martinos Center for Biomedical Imaging at Massachusetts General Hospital for the operation of cyclotron and synthesis modules. We thank Drs. Tanabe and Nakanishi’s laboratory (Osaka Bioscience Institute, Osaka, Japan) for providing the vector of mGlu4 from the rat as a gift. The authors acknowledge the following supporting instrument grants: Grants 1S10RR029495-01, 1S10RR026666-01, and 1S10RR023452-01. Financial support for PP from The Orion Farmos Research Foundation and Kuopio University Foundation is gratefully acknowledged. Finally, we also appreciate the generous help from the NIMH PDSP program (Contract HHSN-271-2008-00025-C) led by Dr. Bryan L. Roth, M.D., Ph.D., at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, U.S. Grants NIBIB-R01EB012864 and NIMH-R01MH91684 to A.-L.B. supported this work.
Glossary
Abbreviations Used
- mGlu4
metabotropic glutamate receptor subtype 4
- CHO
Chinese hamster ovary
- RCY
radiochemical yield
- EOS
end of synthesis
- K222
Kryptofix 222
- TAC
time–activity curve
- ROI
region of interest
- MTEP
3-((2-methyl-1,3-thiazole-4-yl)ethynyl)pyridine
- FOV
field of view
- PAM
positive allosteric modulator
Supporting Information Available
Chromatograms for the separation and analyses of [3H]1 and [18F]3 and the stability study and in vitro selectivity studies of 3. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† K.J.: Department of Neurobiology, A. I. Virtanen Institute, University of Eastern Finland, Kuopio, Finland.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Conn P. J. Physiological roles and therapeutic potential of metabotropic glutamate receptors. Ann. N.Y. Acad. Sci. 2003, 1003, 12–21. [DOI] [PubMed] [Google Scholar]
- Watkins J. C.; Jane D. E. The glutamate story. Br. J. Pharmacol. 2006, 147S1S100–S108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson D. R.; Rose E. M.; Antflick J. E.. The Structure of Metabotropic Glutamate Receptors. In The Glutamate Receptors; Gereau R. W., Swanson G. T., Eds.; Human Press: Totowa, NJ, 2008; pp 363–386. [Google Scholar]
- Conn P. J.; Pin J. P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 205–237. [DOI] [PubMed] [Google Scholar]
- Riedel G.; Platt B.; Micheau J. Glutamate receptor function in learning and memory. Behav. Brain Res. 2003, 140, 1–47. [DOI] [PubMed] [Google Scholar]
- Robichaud A. J.; Engers D. W.; Lindsley C. W.; Hopkins C. R. Recent progress on the identification of metabotropic glutamate 4 receptor ligands and their potential utility as CNS therapeutics. ACS Chem. Neurosci. 2011, 17, 433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amalric M.; Lopez S.; Goudet C.; Fisone G.; Battaglia G.; Nicoletti F.; Pin J. P.; Acher F. C. Group III and subtype 4 metabotropic glutamate receptor agonists: discovery and pathophysiological applications in Parkinson’s disease. Neuropharmacology 2013, 66, 53–64. [DOI] [PubMed] [Google Scholar]
- Schapira A. H. V.; Bezard E.; Brotchie J.; Calon F.; Collingridge G. L.; Ferger B.; Hengerer B.; Hirsch E.; Jenner P.; Le Novere N.; Obeso J. A.; Schwarzschild M. A.; Spampinato U.; Davidai G. Novel pharmacological targets for the treatment of Parkinson’s disease. Nat. Rev. Drug Discovery 2006, 5, 845–854. [DOI] [PubMed] [Google Scholar]
- Fettagutti F.; Balani-Guerra B.; Corsi M.; Nakanishi S.; Corti C. Activation of the extracellular signal regulated kinase 2 by metabotropic glutamate receptors. Eur. J. Neurosci. 1999, 11, 2073–2082. [DOI] [PubMed] [Google Scholar]
- Niswender C. M.; Conn P. J. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti C.; Aldegheri L.; Somogyi P.; Ferraguti F. Distribution and synaptic localization of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience 2002, 110, 403–420. [DOI] [PubMed] [Google Scholar]
- Kinoshita A.; Ohishi H.; Nomura S.; Shigemoto R.; Nakanishi S.; Mizuno N. Presynaptic localization of a metabotropic glutamate receptor, mGluR4a, in the cerebellar cortex: a light and electron microscope study in the rat. Neurosci. Lett. 1996, 207, 199–202. [DOI] [PubMed] [Google Scholar]
- Bradley S. R.; Standaert D. G.; Rhodes K. J.; Rees H. D.; Testa C. M.; Levey A. I.; Conn P. J. Immunohistochemical localization of subtype 4a metabotropic glutamate receptors in the rat and mouse basal ganglia. J. Comp. Neurol. 1999, 407, 33–46. [PubMed] [Google Scholar]
- Valenti O.; Marino M. J.; Wittmann M.; Lis E.; DiLella A. G.; Kinney G. G.; Conn P. J. Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J. Neurosci. 2003, 23, 7218–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O.; Mannaioni G.; Seabrook G. R.; Conn P. J.; Marino M. J. Group III metabotropic glutamate-receptor-mediated modulation of excitatory transmission in rodent substantia nigra pars compacta dopamine neurons. J. Pharmcol. Exp. Ther. 2005, 313, 1296–1304. [DOI] [PubMed] [Google Scholar]
- Engers D. W.; Niswender C. M.; Weaver C. D.; Jadhav S.; Menon U. N.; Zamorano R.; Conn P. J.; Lindsley C. W.; Hopkins C. R. Synthesis and evaluation of a series of heterobiaryl amides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs). J. Med. Chem. 2009, 52, 4115–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kil K.-E.; Zhang Z.; Jokivarsi K.; Gong C.; Choi J.-K.; Kura S.; Brownell A.-L. Radiosynthesis of N-(4-chloro-3-[11C]methoxyphenyl)-2-picolinamide ([11C]ML128) as a PET radiotracer for metabotropic glutamate receptor subtype 4 (mGlu4). Bioorg. Med. Chem. 2013, 21, 5955–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S.; Hamill T. G.; Connolly B.; Jagoda E.; Li W.; Gibson R. E. Species differences in mGluR5 binding sites in mammalian central nervous system determined using in vitro binding with [18F]F-PEB. Nucl. Med. Biol. 2007, 34, 1009–1017. [DOI] [PubMed] [Google Scholar]
- Wang J.; Tueckmantel W.; Zhu A.; Pellegrino D.; Brownell A.-L. Synthesis and preliminary biological evaluation of 3-[18F]fluoro-5-(2-pyridinylethynyl)benzonitrile as a PET radiotracer for imaging metabotropic glutamate receptor subtype 5. Synapse 2007, 61, 951–961. [DOI] [PubMed] [Google Scholar]
- McCauley J. A.; Hess J. W.; Liverton N. J.; McIntyre C. J.; Romano J. J.; Rudd M. T.. Phthalimide derivative metabotropic glutamate R4 ligands. WO2010033349A1, 2010; Merck Research Lab.
- Jones C. K.; Engers D. W.; Thompson A. D.; Field J. R.; Blobaum A. L.; Lindsley S. R.; Zhou Y.; Gogliotti R. D.; Jadhav S.; Zamorano R.; Bogenpohl J.; Smith Y.; Morrison R.; Daniels J. S.; Weaver C. D.; Conn P. J.; Lindsley C. W.; Niswender C. M.; Hopkins C. R. Discovery, synthesis, and structure–activity relationship development of a series of N-4-(2,5-dioxopyrrolidin-1-yl)phenylpicolinamides (VU0400195, ML182): characterization of a novel positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu4) with oral efficacy in an antiparkinsonian animal model. J. Med. Chem. 2011, 54, 7639–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q.; Savage J. E.; Hufeisen S. J.; Rauser L.; Grajkowska E.; Ernsberger P.; Wroblewski J. T.; Nadeau J. H.; Roth B. L. l-Homocysteine sulfinic acid and other acidic homocysteine derivatives are potent and selective metabotropic glutamate receptor agonists. J. Pharmacol. Exp. Ther. 2003, 305, 131–142. [DOI] [PubMed] [Google Scholar]
- Roth B. L.: National Institute of Mental Health Psychoactive Drug Screening Program. Assay Protocol Book. http://pdsp.med.unc.edu/PDSP%20Protocols%20II%202013-03-28.pdf (accessed October 20, 2014).
- Ernst M. L.; Schmir G. L. Isoimides. A kinetic study of the reaction of neucleophiles with N-phenylphthalisoimides. J. Am. Chem. Soc. 1966, 88, 5001–5009. [Google Scholar]
- Cosford N. D. P.; Tehrani L.; Roppe J.; Schweiger E.; Smith N. D.; Andersen J.; Bristow L.; Brodkin J.; Jiang X.; McDonald I.; Rao S.; Washburn M.; Varney M. A. 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J. Med. Chem. 2003, 46, 204–206. [DOI] [PubMed] [Google Scholar]
- Lea P. M. IV.; Faden A. I. Metabotropic glutamate receptor subtype 5 antagonists MPEP and MTEP. CNS Drug Rev. 2006, 12, 149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y.; Ishii T.; Shigemoto R.; Nakanishi S. A family of metabotropic glutamate receptors. Neuron 1992, 8, 169–179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.