

Abstract
DNA mismatch repair functions to correct replication errors in newly synthesized DNA and to prevent recombination between related, but not identical (homeologous), DNA sequences. The mechanism of mismatch repair is best understood in Escherichia coli and is the main focus of this review. The early genetic studies of mismatch repair are described as a basis for the subsequent biochemical characterization of the system. The effects of mismatch repair on homologous and homeologous recombination are described. The relationship of mismatch repair to cell toxicity induced by various drugs is included. The VSP (Very Short Patch) repair system is described in detail.
Keywords: MutS, MutL, MutH, UvrD, exodeoxyribonucleases, DNA polymerase, DNA damage, DNA repair, DNA replication, Escherichia coli, alkylating agents, cisplatin, DNA methylation, recombination, chromosome, bacterial genetics, mutation, drug effects
INTRODUCTION
There are several DNA mismatch repair systems in E. coli. The methyl-directed (or dam-directed or MutHLS) mismatch repair system (referred to as mismatch repair hereafter) occupies most of the space in this article. A smaller amount is devoted to Very-Short-Patch (VSP) mismatch repair. No further space is devoted to U-G mispairs, which arise from spontaneous DNA cytosine deaminations, and are substrates for base excision repair involving uracil N-glycosylase, DNA polymerase I and DNA ligase, is not considered further. Spontaneous oxidation of guanine residues in DNA yields 8-oxo-guanine (among others) that is a substrate for base excision repair initiated by MutM. During replication 8-oxoG-A mispairs can be formed that are substrates for MutY glycosylases. Mismatch repair competes with VSP, MutY and Ung for substrate but usually loses the race to the competing system which has a higher affinity for the specific mispairs.
Mismatch repair is a highly conserved repair mechanism found in all kingdoms of life with only a few exceptions (e.g., Mycobacterium tuberculosis)(190). In general, the role of mismatch repair is to conserve DNA sequence by removing base mispairs created by replication or homologous recombination. In higher organisms, it is also required for successful meiosis and mitosis and immunoglobulin diversity. In humans defective mismatch repair is associated with sporadic and hereditary cancers especially non-polyposis colon cancer. Almost all recent reviews on mismatch repair concentrate on eukaryotic systems, and a few are referenced below.
Mismatch repair was part of the chapter on DNA Repair Mechanisms in the second edition of EcoSal. It’s elevation to chapter status is a reflection of the importance that it has gained since then. In this review, the genetic foundations of mismatch repair will be covered first, as this subject has been eclipsed by the biochemistry of the system in many reviews. There are other recent reviews of mismatch repair that include details not covered here on biochemistry, structural information, and mismatch repair in other organisms (68,72,76,85,206).
EARLY GENETIC STUDIES
Although the mismatch repair system of E. coli, a Gram negative bacterium, is known in more detail than that of any other organism at both the genetic and biochemical levels, the early genetic work which laid the foundations of our understanding of mismatch repair was actually done in fungi and Streptococcus pneumoniae, a Gram-positive bacterium.
Gene Conversion in Fungi
In many fungi, it is possible to examine directly the products of meiosis. For a given heterozygous marker, the meiotic products typically occur with the expected Mendelian ratios. However, occasionally, aberrant segregation was observed with products in a 5:3 or 6:2 configuration, and this phenomenon was referred to as gene conversion (Fig. 1). Genetic analysis in fungi could distinguish gene conversion from localized multiple reciprocal exchanges which would give the same result. Holliday (64) proposed a model to account for gene conversion by invoking a breakage-reunion model according to which DNA strands from homologous duplexes could be exchanged (Fig. 2). The result of such an exchange would be a region of heteroduplex DNA if two different alleles of a heterozygous marker were paired. The formation of such heteroduplex DNA could occur with or without reciprocal exchange of flanking markers. Repair of the mismatches in heteroduplex regions either toward the wildtype or mutant configurations would explain the aberrant segregation in Fig. 1. That is, no recombination leads to normal segregation (Fig. 1A), but a reciprocal exchange and heteroduplex formation leads to aberrant 4:4 segregation (Fig. 1B). A reciprocal exchange followed by mismatch repair in favor of the wildtype in one (Fig. 1C) or both (Fig. 1D) strands of the heteroduplex respectively yields 5:3 and 6:2 segregation, while mismatch repair in favor of the mutant allele yields respectively 3:5 and 2:6 segregation (Fig. 1E and F). It is important to note that although the concept explained much experimental data there existed, at that time, no physical evidence for either heteroduplex DNA or mismatch repair.
FIGURE 1.

Certain fungi, such as Sordaria finicola, have spore color mutant alleles allowing the direct visualization of meiotic products shown here as black and red ovals. Each chromatid is shown as two lines with each line representing one DNA strand at the pachytene stage of meiosis. The formation of heteroduplex DNA could occur with or without reciprocal exchange of flanking markers as shown by the mixed black and red lines. mismatch repair of the heterozygosity (a/+) in heteroduplex regions either toward the wildtype or mutant configurations would explain the aberrant segregation. That is, no recombination leads to normal segregation (Fig. 1A) but a reciprocal exchange and heteroduplex formation leads to aberrant 4:4 segregation (Fig. 1B). A reciprocal exchange followed by mismatch repair in favor of the wildtype in one (Fig. 1C) or both (Fig. 1D) strands of the heteroduplex yields 5:3 and 6:2 segregation, while mismatch repair in favor of the mutant allele yields 3:5 and 2:6 segregation (Fig. 1E and F).
FIGURE 2.
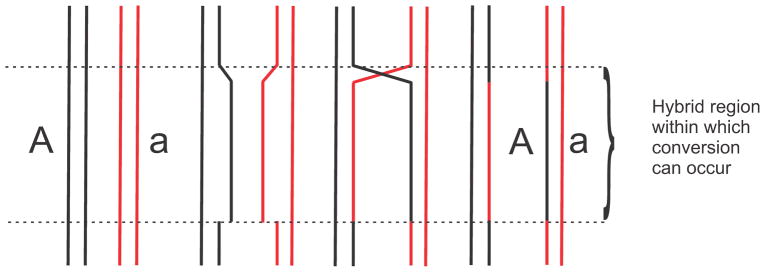
Break and rejoining of DNA chains to form heteroduplex (“hybrid” DNA. Figure modified from (64).
The Holliday model made two important points with regard to mismatch repair. First, it was proposed that mismatch repair is intimately involved with the recombination process and second, that the existence of a mismatch repair process could explain gene conversion.
Streptococcus pneumoniae Transformation
The detailed history of mismatch repair in S. pneumoniae up to 1986 is covered in the superb review by Claverys and Lacks (25). Purification and identification of the “transforming principle” as DNA by Avery et al (5) explained genetic transformation of S. pneumonia. It was subsequently shown that double-stranded DNA is taken up by S. pneumoniae and then, at random, one of the strands is degraded. The surviving strand can pair with its homologous region on the chromosome and form a mismatched region if there is any heterozygosity. It was found that genetic markers could be placed into two groups: low efficiency of transformation and high efficiency. This difference extended to mutations in the same gene. To explain the difference in transformation frequency it was postulated that a mismatch correction mechanism acted on low-efficiency markers to eliminate the donor marker (Fig. 3) (38).
FIGURE 3.
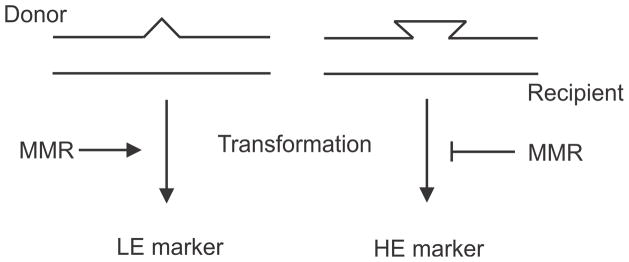
High (HE) and low (LE) transformation efficiency of markers in S. pneumoniae. Transformation leads to the formation of heteroduplex DNA susceptible to mismatch repair (left) thereby reducing transformation frequency. Heteroduplexes not susceptible to mismatch repair yield high transformation frequencies.
This hypothesis received support through the isolation and characterization of hex (E. coli equivalent = mutSL) mutant strains (87) (hex = high efficiency, unknown (x) or heteroduplex repair deficiency). The hex bacteria are deficient in mismatch repair and have two important characteristics. First, all markers transform hex strains with high efficiency; that is, low efficiency markers in wildtype strains transform hex strains with high efficiency. Second, hex strains have a mutator phenotype with spontaneous mutation frequencies 4- to 30-fold higher than wildtype strains. In hex bacteria, radioactive label in donor DNA could be detected in specific restriction fragments from the recipient chromosome, whereas in wildtype recipients the label was drastically reduced (131). By the mid-1970s, it was clear that the Hex mismatch repair system played a role in correcting replication errors using a long-patch repair tract (25). This concept later helped formulate the same proposed role for the mismatch repair mut genes. The hexA and hexB genes correspond to the E. coli mutS and mutL genes respectively based on amino acid sequence conservation (161,164). The types of mismatches corrected by the Hex and Mut systems are very similar (see “Specificity of Mismatch Correction,” below).
The low efficiency markers indicate that the Hex system discriminates against the donor marker, and the problem arose of how does the Hex system discriminate between donor and recipient (or parental and daughter) DNA strands. This important question has not been settled for any organism other than E. coli and its relatives that use Dam methylation for strand discrimination (see below)(Module 4.4.5.DNA Methylation). The explanation currently used for strand discrimination, in both prokaryotic and eukaryotic mismatch repair, is based on the research results using S. pneumoniae. It has been postulated that integration of the donor strand into the genome would create transient single-strand breaks at the ends of the fragment (57). Such nicks would serve as entry sites for a mismatch repair-specific helicase(s) acting in conjunction with HexA and HexB. Note that single-strand breaks are not present in the recipient DNA strand. The above model for transformation can be extended to explain correction of base mismatches in replicating DNA. Transient nicks in the lagging or leading strand due to Okazaki fragments or to excision of uracil residues could serve as entry points for the mismatch repair-specific helicase (25).
Mutator Strains of E. coli
An E. coli strain with high spontaneous mutation frequency was first described by Treffers (196) but until the 1990s the defect in this and other mutator strains remained largely unknown (28,66,136). When E. coli genetic nomenclature was standardized, this mutant strain was referred to as mutT and since then most mutator genes have taken on the name of the person who first isolated them. For example, mutS, mutL and mutH bacteria were first described by E. C. Siegel, R. M. Lieberfarb and R. F. Hill, respectively. A mutator strain sensitive to ultra-violet radiation was originally named mutU by Siegel (182) (thereby making an exception in the naming process), but the mutU4 allele was subsequently found to map in uvrD. The mutator strains were isolated either spontaneously (e.g., mutT) or by selecting for mutant bacteria showing increased resistance to an antibiotic, a phenotype which is easy to screen for, or by a papillation assay (see below). Another mutator strain, dam, isolated as deficient in DNA adenine methylation at GATC sequences and was subsequently found to have a mutator phenotype (125) (Module 4.4.5.DNA Methylation).
5-Bromouracil (5-BU) is normally incorporated into DNA in place of thymine but it can occasionally pair with guanine giving rise to GC to AT transition mutations. Given the double-stranded structure of DNA and its semi-conservative mode of replication, it would be predicted that 5-BU-induced mutations should occur only in mixed clones. To test this prediction, Witkin and Sicurella (213) studied the frequency of 5-BU-induced E. coli Lac− mutant bacteria by plating treated cells on indicator medium. The number of sectored Lac− colonies was about the same as the pure Lac− colonies. This discordance from the predicted result led the authors to conclude that “intracellular repair mechanisms functioning to restore normal hydrogen bonding in damaged DNA” might be responsible.
Rydberg (172) found that at low levels of 5-BU incorporation, there was a non-linear response between dose and induced mutagenesis in E. coli (Fig. 4A). Such a response was compatible with a model whereby 5-BU-G mismatches were corrected at low level of incorporation of the base analog, but at higher concentrations the correction system was saturated leading to increased mispairs and increased mutation frequency (172). In order to identify the correction system, Rydberg (173) devised a papillation assay using MacConkey-galactose plates containing 5-BU at a level provoking few papillae (Gal+ microcolonies in Gal− colonies) in the wildtype strain (Fig. 4B). The wildtype strain was mutagenized with UV light and survivors showing increased papillation were isolated (Fig. 4B). It was found that the increased papillation mutant bacteria showed a mutator phenotype even in the absence of 5-BU (Fig. 4A). The alleles showing increased papillation mapped to the mutH, mutL, mutS and uvrD genes. This experiment identified these mutator genes as part of the correction system active on 5-BU-G induced mispairs, as well as endogenously formed mismatches.
FIGURE 4.
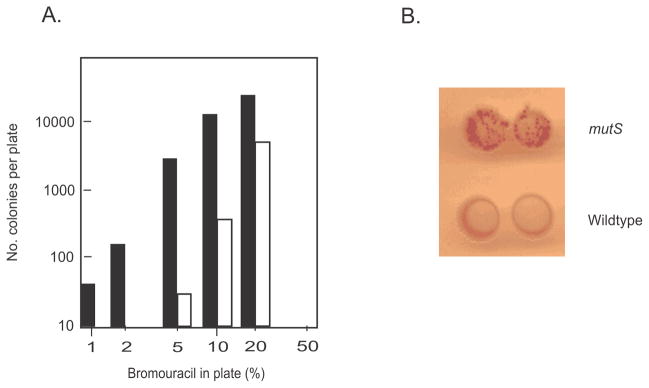
Bromouracil-induced mutagenesis. A. Mutation frequency of induced mutant strains as a function of bromouracil concentration for the wildtype strain (open circles) and a uvrD mutant strain (filled circles). The wildtype strain shows no mutant induction at low (2–4%) bromouracil concentrations, consistent with a repair mechanism that becomes saturated at higher concentrations, yielding a concentration-dependent dose-response. A mutant strain defective in repair (uvrD) yields induced mutant cells at low (2–4%) bromouracil concentrations but also shows a mutator phenotype in the absence of mutagen. The Figure has been modified from reference (173). B. Spontaneous Gal+ papillation on MacConkey agar of a mutS strain (top) and the wildtype strain (bottom) (image supplied by the author).
As mentioned above, dam mutant strains are deficient in methylation of adenines in DNA at -GATC- sequences and show a mutator phenotype (124,125). Furthermore, synthetic lethality occurs when mutations in the dam and recA genes are combined (125). Second-site suppressor mutations were isolated which allowed growth and these mapped to the mutS and mutL genes (130). Similarly, dam mutant cells have reduced survival when exposed to the base analog 2-aminopurine (see below) and selection for resistance yielded second-site suppressors in mutH, mutL and mutS (51). These studies suggested that there was a link between DNA methylation and mutator genes that functioned in mismatch repair.
Transfection with Phage Lambda Heteroduplexes
Various elements of mismatch repair suggested by mutator studies, Dam methylation, heteroduplexes and mismatch correction, were brought together in the classic paper by Pukkila et al (166). Although there was the suggestion that methylation could impart directionality in the mismatch repair process (205) and some preliminary experiments supported this idea (51), it was the results of the Pukkila et al paper, that formed the basic model of mismatch correction in E. coli and served as the foundation for further experimentation. A wonderful review by Meselson (135) explains the historical experiments leading up to and including those in the Pukkila et al paper as well as a review of mismatch repair up to that time.
Before discussing the implications of the results obtained by Pukkila et al., a short synopsis of Dam methylation in E. coli follows. In the wildtype strain, almost all Dam recognition sites (-GATC-) are methylated except for the region just behind the replication fork which has hemi-methylated DNA (the parental strand methylated and the 3′-end of the newly synthesized strand is unmethylated). This occurs because the Dam concentration is limiting and methylation lags behind replication causing transient hemi-methylation (Module 4.4.5. DNA Methylation).
When phage lambda DNA is subjected to density-gradient centrifugation in CsCl, two bands, designated light (l) and heavy (h) in Table 1, are formed corresponding to each strand of the DNA. This technique allows for the creation of heteroduplex molecules using wildtype and mutant DNA by mixing the complementary strands of mutant and wildtype lambda phage. Wildtype lambda produces turbid plaques, while cI mutant phage produce clear plaques on lawns of E. coli indicator bacteria. The strands of each of these can be separated and combined to form the two possible heteroduplex configurations designated I and II in Table 1 (166). Since Dam methylation is inefficient during propagation of phage lambda DNA on a wildtype host, in vitro methylation with Dam was used to ensure complete methylation of both lambda DNA strands. Lambda DNA strands were also prepared after passage through dam bacteria yielding completely unmethylated chains. These procedures allowed for the construction of four types of heteroduplexes for each configuration (I and II) differing in methylation state (me+ and me−) and genotype (+ or c) (Table 1). Phage with the +/+ genotype formed turbid plaques and those with c/c genotype formed clear plaques. Phage with the +/c genotype produced both types of progeny phage in the plaque leading to a mottled appearance. These are referred to as “mixed”: (from mixed burst) in Table 1.
Table 1.
Mismatch repair of heteroduplexes
| Plaques | |||||||
|---|---|---|---|---|---|---|---|
| Heteroduplex | Wild-type host | mutL host | |||||
| Strand methylation | c | Mixed | + | c | Mixed | + | |

|
+/+ | 33 | 32 | 35 | 42 | 38 | 20 |
| −/− | 64 | 3 | 33 | 52 | 21 | 27 | |
| +/− | 95 | 3 | 2 | 66 | 18 | 16 | |
| −/+ | 4 | 2 | 94 | 22 | 28 | 50 | |
| +/+ | 29 | 26 | 45 | 34 | 35 | 30 | |
| −/− | 40 | 16 | 44 | 33 | 27 | 40 | |
| +/− | 38 | 29 | 32 | 34 | 37 | 29 | |
| −/+ | 33 | 30 | 37 | 30 | 30 | 40 | |
This Table has been modified from reference 166.
Transfection of the bacterial hosts (wildtype, dam or mutL) with heteroduplex I will be considered first (Table 1). When the wildtype strain was transfected with the me+/me− heteroduplex (c/+), 95% of the progeny formed clear plaques, while the me−/me+ heteroduplex of the same genotype resulted in 94% turbid plaques. This result shows a clear-cut bias of correction to the marker on the methylated strand. The unmethylated heteroduplex shows some bias toward clear plaques (64% clear, 33% turbid) but there is clearly less directional bias than with the hemi-methylated (me−/me+) duplexes. Surprisingly, the fully methylated duplex shows about one third clear, one third turbid and one-third mixed (mottled) plaques. Clearly, the proportion of mixed plaques is far greater than that for any other heteroduplex indicating that the fully methylated heteroduplex was not corrected. If there was no correction of the fully methylated +/c heteroduplex, then why aren’t all plaques mottled? The answer is not known and is referred to as “strand loss”. It appears from the data in Table 1 that in most cells (about 60–70%) the uncorrected heteroduplex replicates and either one or the other genotype predominates (compare c and + for the fully methylated duplex) to yield either clear or turbid plaques, as if one of the strands is “lost.”
Transfection of configuration I heteroduplexes into the dam mutant gives essentially the same result as transfection into the wild type (not shown in Table 1). This indicates that the Dam methylation status of the host cell after correction of the heteroduplex DNA is not important. It shows, however, that dam cells are fully proficient at mismatch correction.
It is expected that transfection of the heteroduplexes into a mutL strain should result in little, if any, correction. The results in Table 1 support this view, especially when the proportion of mottled (mixed) plaques is compared with the other two hosts.
With heteroduplexes in configuration II, no correction is detected in any of the heteroduplexes in any strain, as indicated by the proportion of mixed plaques arising after transfection.
The implications of the Pukkila et al results are as follows. First, correction of mismatches does not occur on fully methylated DNA duplexes. Second, mismatch repair occurs preferentially in hemi-methylated DNA with the parental methylated strand as template and removal of the mismatched base in the unmethylated strand. Since hemi-methylated DNA occurs only transiently behind the replication fork in wild-type cells, mismatch repair must occur here with removal of the base in the newly synthesized strand and is terminated by the action of Dam methyltransferase.
A primary function of mismatch repair is, therefore, the correction of replication errors. Third, the failure to repair heteroduplexes in configuration II indicates that the correction machinery has specificity. The mismatch in configuration I is different to that in configuration II. For example, a G-T mismatch in configuration I would be an A-C mismatch in configuration II. The actual mutational identity of the cI mutation in DNA used by Pukkila et al is not known, but this does not matter for the interpretation of the results. The lack of correction in configuration II indicates specificity in the system. Fourth, the mutL strain is deficient in mismatch correction. Fifth, unmethylated heteroduplexes are subject to correction but not in a directed manner.
The basic results of Pukkila et al. remain the conceptual cornerstone of our understanding of mismatch correction in E. coli and will be expanded in the next section.
It is interesting to recall the earlier work with S. pneumoniae and how it fits-in nicely with the results of Pukkila et al. There remains the question as to the mechanism of strand discrimination at the replication fork. When the GATC methylation for strand preference is removed from E. coli, is the correction mechanism operative in S. pneumoniae still active? This question was addressed by measuring the reversion rate in a cell with plasmids bearing the cat (chloramphenicol acetyltransferase) gene with an amber mutation (26). The 3.9-kb plasmids contained 0, 1 or 2 GATC sites. In wild-type cells, there was a 10-fold increase in mutation rate when the number of GATCs was reduced from two to zero. Similarly, a two- to threefold increase was found in mutS and mutL but not mutH or wildtype strains. In other words, 50% to 70% of the potential mutations are removed by MutS,L in the absence of GATCs on the plasmid. The lack of mutation increase in the mutH strain was explained by an alternate mechanism by which nicks or gaps in DNA are generated to allow UvrD access during mismatch repair. This is consistent with both in vivo and in vitro data showing that if a nick is already present near the mismatch on the correct strand, then the need for MutH is obviated (90,92). It was proposed that, like S. pneumoniae, single-strand breaks in the newly synthesized strand allow for strand discrimination. Another argument for an additional system is that the mutation rate in dam bacteria is about 10-fold less than in mut cells, but still about 10- to 20-fold higher than in wild-type cells. The occurrence of double-strand breaks in dam cells and the possible loss of mutant bacteria complicates this explanation (146,208).
At any rate, it is still possible that the dam-directed system overlays the primordial mismatch repair system present in other organisms, but further experimental data are needed.
MISMATCH CORRECTION
Specificity of Mismatch Correction
The specificity of mismatch correction has been determined both in vivo and in vitro. In vivo methods include determining the types of mutations produced in dam, mutS, mutL and mutH strains, as well as measuring the frequency with which artificially-constructed heteroduplexes are corrected after transfection or transformation. Early studies with mutHLS strains using both forward and back mutation assays showed a specificity for frameshift and transition mutations ((25,28) and references therein). These data were similar to those obtained with S. pneumoniae hexA and hexB mutant bacteria (25) and subsequently in S. eneterica serovar Typhimurium mutHLS strains (179). In some of these studies it was possible to show a specificity mainly for GC to AT and AT to GC transitions implying A-C and G-T mismatches were the major repair substrates (e.g., (94)). In other studies, deletion of a T residue in a run of six was observed (e.g., (183). U-G mispairs were also subject to mismatch repair (181) as were O6-methylguanine-adenine pairs (220). The advent of DNA sequencing allowed further refinement to confirm this specificity (23,175,186). The dam mutant strain showed the same mutation spectrum as the mutHLS strains, confirming the involvement of Dam methylation in mismatch repair (24,50).
Transfection of wild-type E. coli with M13 heteroduplexes showed a hierarchy of correction of T-G, C-A, G-G>A-A>G-A, A-G, T-T, C-T, T-C and C-C base mismatches (84). The hierarchy is more-or-less the same in S. pneumoniae with C-A and T-G pairs corrected most efficiently, C-C not at all, and the others in between (25). It should be noted that the frequency of correction can be influenced greatly by flanking sequence and the presence of other mismatches. Plasmid heteroduplexes with one, two, or three base insertions/deletions (IDL) were corrected with the same high efficiency as T-G after transformation into wildtype E. coli. A four base IDL was repaired marginally, and a five base IDL was not detectably repaired (152). The affinity of MutS for these IDLs using an electrophoresis mobility shift assay in vitro showed the ranking for the IDLs was one > two > three = G-T > four > five (152). The affinity of MutS using a band-shift assay for base mismatches was determined to be T-G, C-A>A-A, G-G, T-T>A-G, C-T, C-C, which is quite similar to the in vivo results (194).
Reconstitution of mismatch repair in vitro
The genetic studies on mismatch correction in E. coli indicated a central role for the products of the mutH, mutL, mutS and uvrD genes, as well as defining Dam methylation as the mechanism for strand discrimination during replication error correction. The mechanism of action of the biochemical products of these genes in E. coli was determined in a tour de force by P. Modrich and colleagues. Creating a reliable assay for mismatch repair in vitro was the starting point of the investigation (110). This consisted of a covalently-closed f1 phage supercoiled molecule with a single Dam recognition site (-GATC-) and a G-T mismatch. Correction of the mismatch on one strand generated a unique restriction site for HindIII, while correction on the other strand generated a restriction site for XhoI (Fig. 5). Crude extracts from the wildtype supported mismatch correction with this substrate, while those from mutS, mutL, mutH and uvrD mutant strains did not. Using a complementation assay, purified fractions from extracts of wildtype bacteria were used to complement extracts of the mutant bacteria to restore in vitro repair. In short order, this led to the purification of the MutS, MutL, MutH and UvrD proteins (see reviews by Modrich et al (54,89,139–141) and references therein for complete details).
FIGURE 5.
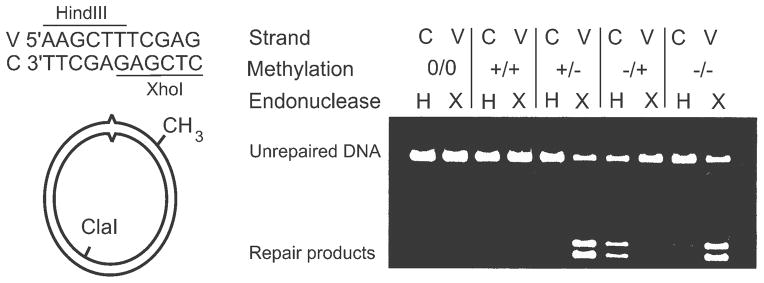
In vitro assay for mismatch correction. The substrate supercoiled molecule contains a single GATC sequence which can be methylated on either the viral (V) or complementary (C) strand (+/− or −/+) or both (+/+) or neither (−/−). A control molecule (0/0) contains a GATT sequence in place of GATC. The substrate also contains a T-G mismatch (carats) in the indicated sequence which is part of both a HindIII and an XhoI recognition site. Upon completion of the in vitro reaction in extracts or with purified components, the substrate DNA is digested with ClaI, HindIII and XhoI. Correction of the mismatch will lead to resistance to one of the restriction enzymes. The substrate without the GATC (0/0) or a fully methylated GATC (+/+) shows no repair products while the hemi-methylated (+/− or −/+) or unmethylated substrates (−/−) show repair products derived preferentially from one strand. The Figure has been modified from reference (88).
The culmination of this biochemical approach was the complete reconstitution of E. coli mismatch correction in vitro (Fig. 6) (88). In addition to MutS, MutL, MutH and UvrD, Exonuclease I, single-strand binding (SSB) protein, DNA ligase and DNA polymerase III holoenzyme (11 proteins) were necessary, as well as MgCl2, ATP and dNTPs. Subsequently, it was found that other exonucleases could also participate in the reaction (see below). The DNA substrate requirements for the in vitro mismatch repair reaction reflected those required in vivo. The f1 supercoiled molecule required a base mismatch and a GATC site which could not be substituted for by GATT. Strand targeting could be controlled by GATC hemi-methylation and a fully methylated GATC was refractory to correction, but an unmethylated GATC was not. In the latter case, some molecules were repaired on one strand, and some on the other. The product of the reaction was a covalently-closed f1 molecule without the mismatch. The specificity of repair of base mismatches was similar to that observed in vivo (see above): the rank order was T-G, G-G, T-T, A-A, C-A, C-T, A-G and little, if any, repair of C-C. It is quite amazing that an ensemble of 20 or so proteins, and the excision and replacement of up to a thousand nucleotides, is required for the correction of a single base in a mispair.
FUGURE 6.

Bidirectional mechanism of mismatch repair. The mismatch (carets) is bound by MutS (pierced circle) and MutL (red circles) leading to activation of the MutH endonuclease which can cleave either side of the mismatch. The mechanism leading to MutH endonuclease activation is not known. After cleavage, MutH is displaced by UvrD helicase which unwinds the DNA towards the mismatch. Unwinding in the 5′ to 3′ direction results in digestion of the single-stranded end by ExoVII or RecJ. Unwinding in the opposite direction requires ExoI, ExoVII or ExoX nucleases. The gap produced by excision is filled by DNA polymerase III holoenzyme. The resultant nick is sealed by DNA ligase, and Dam methylates the GATCs on the unmethylated strand, preventing further repair. The Figure has been modified from reference (53).
If the f1 heteroduplex molecule was first nicked in the umethylated strand by MutH at the hemi-methylated -GATC- and the protein removed, this substrate was processed by the in vitro system (lacking MutH) as efficiently as the optimal substrate with the complete in vitro system (29,90,209). That is, a nick in the unmethylated strand could bypass the need for MutH. This is of interest because adenine methylation is not present in organisms other than E. coli and some related enteric bacteria, and if a nick is present in the appropriate strand, there is no need for methylation and MutH. Recall that studies with S. pneumoniae transformation also suggested that a transient nick was sufficient for the Hex system.
The basic reactions of mismatch repair are diagrammed in Fig. 6. A hemi-methylated molecule with two -GATC- sites (CH3) and a mismatch (carats) is shown. Binding of MutS to the mismatch and to MutL and their interaction with MutH activates the latter’s latent endonuclease activity that cleaves 5′ to the G of the GATC sequence. UvrD (helicase II) is recruited to the nick which can be either 5′ or 3′ to the mismatch whichever is closest. UvrD unwinding is accompanied by exonuclease digestion: in the 5′-direction by ExoVII or RecJ, while in the 3′-direction by ExoI, ExoVII or ExoX. The ssDNA gap (which may be up to 2 kb) produced by nuclease excision is filled by DNA polymerase III holoenzyme, and then DNA ligase seals the nick. Finally, Dam methylation occurs on the unmethylated strand thereby rendering the DNA inert for further mismatch repair.
It is useful to contrast the mismatch repair reaction with other DNA repair pathways, such as base excision repair (BER) and nucleotide excision repair (NER). First, the mismatch repair system uses many more proteins (about 20 versus a few). Second, both NER and BER use DNA polymerase I, while DNA polymerase III holoenzyme is used in mismatch repair. Third, repair tracts are short for BER (1–2 nt) and NER (about 15 nt), but are long (up to 2 kb) for mismatch repair.
MutS
The Gene
The mutS gene was first sequenced from S. enterica (58) as was the hexA gene in S. pneumoniae (161), and there was conservation of the predicted amino acid sequences. The hexA gene, however, did not complement E. coli mutS bacteria for mutator phenotype, but it did produce a dominant-negative phenotype in wildtype E. coli, probably through competition for mismatch binding (165). Many mutations have been described in this gene including those that are dominant-negative (215) and temperature-sensitive (65).
Regulation
Since MutS initiates mismatch repair, this gene would be a good target for regulating the process. In exponentially growing cells, the level of MutS was found, by western blotting, to be two to threefold higher in an hfq mutant strain compared to wildtype strain (197). The mutS transcript encoded only this gene and was initiated about 75 bp upstream of the AUG codon. RNA polymerase is a multisubunit protein which can bind to specific promoter regions using alternative “sigma” factors (169). There were no obvious RNA polymerase RpoD (sigma-70) recognition −10 and −35 consensus hexamers upstream of the translation initiation codon. The level of mutS mRNA was increased about threefold in the hfq mutant strain but there was no difference in half-life, thereby correlating mRNA amount with the increase in protein level. Hfq is an RNA chaperone protein that promotes binding of complementary regions of RNA molecules. The amount of beta-galactosidase in hfq mutant and wildtype cells containing a mutS::lacZYA transcriptional fusion was the same but a translational fusion showed a threefold increase in the hfq mutant bacterium. These results were consistent with a posttranscriptional mechanism whereby Hfq destabilizes mutS transcripts (197). This could occur directly by Hfq binding to transcript or indirectly through the chaperone action of Hfq with mutS mRNA and a small regulatory RNA. The small non-coding regulatory RNA molecules have been implicated in the translational regulation of many E. coli genes but their involvement with mutS regulation has not yet been tested (193).
In stationary-phase wildtype cells, the amount of MutS decreases fourfold relative to exponentially growing cells but in an rpoS mutant no decrease is observed (41,197). RpoS, the sigma-38 subunit of RNA polymerase, is important in regulating the transition from logarithmic growth into stationary phase cells (62). In addition, in growing cells, it is important in response to stresses such as starvation for glucose, phosphate or amino acids and osmotic, acid or heat shock. Although Hfq is required for translation of rpoS mRNA, the level of MutS was increased almost six fold in hfq and hfq rpoS cells. The actual mechanism(s) that regulate the amount of MutS in stationary or stressed cells remains unknown but does not involve RNAse E, or the proteases Clp, Lon or HflAB (197). The lower MutS level in stationary phase may reflect the decreased rate of DNA synthesis, and hence the reduced need for error correction, in stressed or non-growing cells. Another possible rationale for low MutS levels in stationary phase cells is that too much would interfere with VSP repair through competition with MutL.(see VSP Repair)
The MutS Protein
The MutS protein is composed of 853 amino acids. The amino (N)-terminal end contains the DNA binding domain, and the conserved connector domain (211–212) for interaction with MutL (133); the middle domain harbors ATPase activity (568–765) and the (C)-terminal end (766–853) contains information for oligomerization. The 95 kDa protein exists in solution as a mixture of dimers and tetramers and initiates mismatch repair by binding to a mismatch (194). To obtain co-crystals with DNA containing a heteroduplex, the last 53 amino acids were deleted from the protein (MutSdelta800) to obtain a predominantly dimer population (91,145,150). MutSdelta800 in the crystals is a dimer and has two large channels but only one of these binds oligonucleotide. The complex can be visualized as a pair of hands held together as if in prayer with the DNA passing through the gap formed between the finger tips and the thumbs. Of particular interest is that the MutS homodimer behaves as a heterodimer in that only one of the subunits (the tip of one thumb) contacts the mismatch. The binding of MutS distorts the DNA conformation to induce a 60° bend that is stabilized by the insertion of the highly conserved Phe-39 residue into the DNA helix. ATP binding and hydrolysis are important to MutS function (the protein has weak ATPase activity) but the role of this cofactor is controversial. In the MutSdelta800 crystal structure, however, only one molecule of ADP, which stabilizes DNA binding, was present. Asymmetric binding of nucleotide to MutS is induced by binding to DNA with base mismatches (143). The presence of nucleotide in only one of the monomers may explain the heterodimeric asymmetry of the protein. Although a gene encoding the MutSdelta800 protein, when present in multiple copies, complements the mutator phenotype of a mutS deletion strain, in single copy it does not (22). MutSdelta800 has reduced affinity for certain base mismatches (14,21) and has reduced ability, when complexed with MutL, to activate the endonuclease activity of MutH (14). These results indicate that the C-terminal 53 amino acids of MutS are essential for full MutS activity (21). The role of these amino acids is to form multimers (117) although whether a tetramer (14,69,73) or a stable dimer (134) constitutes the active form of the protein is still in dispute.
Deficiency of mismatch repair in E. coli creates a mutator phenotype and increased recombination in interspecies crosses (Antirecombination, see below). In at least two laboratories, surveys of chromosomal mutS mutations that uncouple these functions were unsuccessful. The MutSdelta800 protein expressed from a multicopy plasmid is able to complement a mutS mutant for mutator phenotype but not for antirecombination (21). This observation suggests that higher levels of active MutS are required to prevent antirecombination than mutation avoidance. The need for a higher MutS level is probably because many more mismatches are expected to be formed during interspecies crosses than arise as replication errors. This explanation may also apply to the mutS60 temperature-sensitive strain which has no mutator activity at 37°C but has a modest increase in antirecombination relative to wildtype (65).
In addition to interaction with MutL and MutH, the MutS protein has two binding motifs for the beta clamp, a component of DNA polymerase III holoenzyme (108) and this interaction is described in more detail below (Beta Clamp).
MutL
The Gene
The mutL gene was first cloned from S. enterica (151) and S. pneumoniae (hexB) (164), and a comparison of the predicted amino acid sequences showed substantial sequence conservation, especially at the N-terminal end. As with MutS, a screen for dominant-negative E. coli mutL mutant strains showed that many of the mutations altered amino acids that are highly conserved (3). A temperature-sensitive allele has also been characterized (65). Overproduction of MutL is detrimental, leads to plasmid instability and is mutagenic (1,3). Like MutS (21), altering the level of MutL allows separation of antirecombination and mutation frequency functions with the former requiring a higher level than the latter (37).
Regulation
The level of MutL as determined by western blotting is unchanged in stationary and growing cells and is unchanged in hfq or rpoS mutant strains, and there is no posttranscriptional regulation (197). Multiple promoters produce mutL transcripts using RpoD (sigma 70) and RpoS (sigma 38)-containing RNA polymerases but if their synthesis is regulated is unknown.
The MutL Protein
MutL interacts with MutS, MutH, UvrD, as well as the, alpha, beta clamp, and gamma subunits of polymerase III holoenzyme and, hence, has a central role in mismatch repair (72,85). This 615 amino acid DNA binding protein has an N-terminal region that is highly conserved in the MutL superfamily and which contains ATP and a beta clamp binding motif. The C-terminal region is important for dimerization, interaction with other mismatch repair proteins and the beta clamp. Mutational inactivation of the N- or C-terminal beta clamp motifs of MutL reduces mismatch repair (see Beta Clamp).
Separate molecular structures are available for the N-terminal 349 amino acid domain and for a C-terminal dimerization domain of MutL (residues 432–615) (7,8,56). In the presence of the non-hydrolyzable ATP analog, AMPPNP, the N-terminal domain dimerizes to form two active ATPase sites which are essential for enzyme activity. The ATPase activity is low indicating that the role of nucleotide is to allow conformational changes for protein-protein interactions. The C-terminal domain crystallizes as a dimer, and by molecular modeling with the N-terminal domain, a channel is proposed where DNA binding depends on ATP hydrolysis (56).
The N-terminal domain dimer bound with AMPPNP, has surfaces which, using molecular modeling, could interact with MutS and MutH (7). On the other hand, both N- and C-terminal domains appear to interact with UvrD (56).
MutL Endonuclease Activity
In organisms that do not have Dam methylation, there is no equivalent to the MutH protein to nick the newly-synthesized strand. At present, how the initial nick is introduced either 3′ or 5′ to the mismatch in such organisms is not known. In such organisms, there is usually only a single exonuclease that has 5′ to 3′ directionality and this raises the problem of how excision occurs when the initial nick is on the 3′ side of the mismatch. In human cells, it was discovered that MutL contained a latent endonuclease activity (78) that introduced a nick on the distal side of the mismatch. That is, if the initial nick is on the 3′ side of the mismatch, MutL introduces a second nick on the 5′ side, thereby allowing the exonuclease to remove nucleotides in the 5′ to 3′ direction. Latent MutL endonuclease activity has since been found in several other organisms including B. subtilis, where the crystal structure of MutL was solved (155). In this structure, the endonuclease sub-domain has many negatively charged residues, which led the authors to propose that electrostatic repulsion prevents binding of the site to DNA. Binding would require a conformational change that may be induced by nucleotide binding, as well as interaction with other proteins, such as MutS and the beta clamp (see beta clamp).
MutH
The Gene and Regulation
The E. coli mutH gene was cloned and sequenced by Grafstrom and Hoess (52), and it encodes a 229 amino acid protein. The RNA polymerase RpoD (sigma 70) subunit recognition −10 and −35 hexamers are present upstream of the gene and their deletion abolishes MutH production. The hexamers differ significantly from consensus and the spacing between them is 18 bp, instead of the consensus 17, suggesting low constitutive activity or recognition for another sigma factor. Further investigation showed a two-fold decrease in the level of MutH in stationary-phase cells compared to logarithmic phase cells and this decrease did not occur in hfq or rpoS or hfq rpoS double mutant strain (197). This regulation, however, may simply reflect the decreased need for mismatch repair in stationary phase cells where the amount of DNA replication is less than that in logarithmic phase cells. These data support the idea that the promoter region in front of mutH is also bound by RNA polymerase containing RpoS (sigma 38). The involvement of Hfq is puzzling and might reflect the action of a small regulatory RNA to decrease the level of mRNA or its translation.
The MutH Protein
The MutH protein is a monomer in solution, and its structure (free, not bound) has been determined at the atomic level. It resembles a large clamp with a cleft in which the DNA substrate is thought to bind. The movement of the clamp arms appears to be modulated by the position of the N-terminal helix which can be likened to a lever. Although both MutS and MutL are required to activate MutH for cleavage in extracts, MutL alone, in the presence of ATP, can activate the latent endonuclease activity of MutH. An attractive feature of the structural model is that MutL binding allows movement of the lever to close the clamp holding the bound DNA. This forces the DNA into proximity of the catalytic residues at the bottom of the cleft and on one of the arms (9,107).
The next step in this crystallography saga will be the atomic structure of the MutSLH complex bound to DNA with a mismatch. The use of MutH variants defective in incision may help to realize this goal. Such a structure should help to identify the binding faces of the proteins and the conformational changes that probably occur in the ternary complex.
UvrD
The uvrD locus, encoding a DNA helicase, has many alleles which differ in their mutator phenotype, sensitivity to ultra-violet light, uvrABC nucleotide excision repair, SOS-induction, postreplication repair, transposition, homologous recombination, replication and inviability of a polA uvrD mutant strain. This myriad of traits has caused this gene to have several designations - uvrD, uvrE, recL, mutU, and pdeB. These features imply that the UvrD helicase has many roles in DNA metabolism, including mismatch repair (217)
A transcriptional terminator (“attenuator”) is located between two promoter regions upstream of the uvrD gene. The major promoter, P1, is LexA-regulated and lies upstream of the attenuator. The constitutive level of transcription was unchanged following mutational inactivation of the attenuator but was increased substantially relative to the unmutagenized control following induction of the SOS response (4,34). The role of the attenuator, therefore, is to limit transcription after LexA cleavage since too much UvrD reduces viability of cells. There is no evidence for any genetic regulation of mismatch repair through the expression of UvrD or vice versa.
After MutHLS binding to mismatched DNA, MutH nicking of the unmethylated strand can occur at a GATC sequence either 5′ or 3′ to the mismatch (Fig. 6). How this choice is made is not known. After the endonucleolytic incision, MutH must be released from the MutLS-DNA complex and UvrD recruited. Since UvrD unwinds with a 3′ to 5′ polarity, MutS-MutL must load UvrD on different strands depending on the polarity and again, the mechanism by which this is achieved is not known. There must be interaction between DNA-bound MutS-MutL and UvrD, but the focus has been on MutL as it can load UvrD at a nick on homoduplex DNA (129). Molecular modeling suggests that both N- and C-terminal domains of MutL interact with UvrD (56), but biochemical and yeast two-hybrid data have not clearly defined the interacting regions between MutLand UvrD (129).
Exonucleases
After unwinding of the mismatched DNA by UvrD, single-strands with either 3′ or 5′ termini are produced, and these are acted upon by exonucleases of the corresponding polarity (Fig. 6). Although the initial reconstitution of mismatch repair in vitro included only ExoI (88), subsequent work showed that other nucleases can participate in mismatch repair (72,142). The 3′-specific exonucleases are ExoI, ExoVII or ExoX and the 5′-specific ones are ExoVII and RecJ (ExoVII is non-specific). Elimination of ExoVII and RecJ abolishes 5′-directed mismatch repair, while inactivation of ExoI, ExoVII and ExoX prevents 3′-directed mismatch repair in E. coli cell extracts (203). The single-stranded DNA exposed by exonuclease action is covered with single-strand binding protein (SSB). Omission of SSB severely decreases repair efficiency (88).
The redundancy of exonucleases predicts that single mutant strains lacking each exonuclease should not have a mutator phenotype, and this is, indeed, the case, as only the triple and quadruple mutant strains show phenotypic alterations compared to the wildtype strain (17,203). Only the quadruple mutant strain has a mutator phenotype and the mutation rate is at least 10-fold lower than expected compared to mutHLS bacteria. The elevated mutation rate in the quadruple mutant bacteria, however, correlates with a substantial decrease in mismatch repair activity in assays in vitro. The lower-than-expected mutation rate of the quadruple mutant strain was attributed to lethal events triggered by mismatch repair. Support for this hypothesis was the finding that the quadruple mutant strain is cold-sensitive for growth in rich (but not minimal) media at 30° C, undergoes filamentation, and is very sensitive to the toxic effect of 2-aminopurine (2-AP). 2-AP is a base analogue that when incorporated into DNA by growing cells promotes mispairs that are substrates for mismatch repair (see 2-Aminopurine). These phenotypic properties were abolished in the quadruple mutant strain upon inactivating mismatch repair by mutations in mutS, mutL, mutH and uvrD. The RecJ− ExoVII− ExoI− triple mutant strain was also sensitive to 2-AP suggesting that these three exonucleases remove most, if not all, 2-AP mismatches. The molecular basis for the lethality associated with the quadruple mutant strain upon activation of mismatch repair is not known. The accumulation of mismatch repair intermediates and single-stranded DNA, could present serious problems during chromosome replication, leading to double-strand breaks or other DNA structures that are difficult to repair. As with the multifunctional UvrD helicase, there is no evidence that exonuclease gene expression regulates mismatch repair or are regulated by mismatch repair components.
An obvious problem with our understanding of the excision step of mismatch repair, is that the signals that make the helicase/exonuclease machinery to stop are not known. In an in vitro system with purified ExoI or ExoVII excision begins at the GATC and terminates at a number of sites within 100 bp after the mismatch (53). The mechanism of this termination is not known but must rely on some component(s) of the in vitro system.
Genetic studies with phage lambda have shown that markers separated by three thousand base pairs are subject to co-ordinate mismatch repair-dependent correction (25,135). In these cases, the nearest GATC is expected to be bypassed since the predicted average distribution of Dam-recognition sites in the E. coli chromosome is 256 bp. This observation suggests that use of the nearest GATC may not be absolute in vivo.
DNA polymerase III holoenzyme
The excision step of mismatch repair produces repair tracts of up to 3 kb long, and in vitro reconstitution of the mismatch repair system shows that DNA polymerase III holoenzyme is used for re-synthesis (88). From models for DNA replication, the beta clamp is loaded onto the 3′-end of the gap followed by loading of the other holoenzyme proteins. Polymerization ensues until gap-filling is completed, leading to dissociation of the holoenzyme from the DNA and recruitment (to the beta clamp?) of DNA ligase to restore the intact DNA chain (Fig. 6).
There is no genetic information about the mismatch repair-dependent DNA synthesis step. Part of the problem is that strains mutant for the genes encoding DNA polymerase III holoenzyme subunits, such as dnaB (helicase), dnaE (catalytic alpha sub-unit) and dnaN (beta sliding clamp) are constitutively activated for the SOS response, resulting in increased mutability, that obscures any mismatch repair mutator effects. Other experimental approaches, such as in vivo heteroduplex correction in temperature-sensitive strains, encoding DNA polymerase III holoenzyme subunits, have not been reported.
Extracts of dnaX (Ts) cells do not support mismatch repair-dependent repair synthesis at the restrictive temperature (88) (dnaX is referred to as dnaZ in this reference). The dnaX gene encodes two proteins, one a truncated form of the other, and both are part of the clamp-loader complex which loads the beta clamp onto the DNA (83,149). It is the beta clamp that is expected to be the key requirement for initiating repair synthesis, so anything that prevents clamp loading will prevent synthesis. In vitro assays of mismatch repair in other DNA polymerase III holoenzyme subunit mutant strains may be worthwhile.
The MutS and MutL proteins interact with the alpha catalytic subunit of DNA polymerase III holoenzyme (159). Since, in E. coli, these interactions are of limited importance during the initiation and excision steps of mismatch repair, Pluciennik et al (159) have suggested that the mismatch repair proteins could provide a regulatory mechanism to modulate replication fork activity in response to various types of DNA damage. Such modulation could integrate the repair and replication events during error removal. This suggestion could also explain the interaction of the MutL and MutS proteins with the gamma complex (clamp loader) of DNA polymerase III holoenzyme (95,159).
Further evidence for an intimate connection between mismatch repair proteins and the replication machinery has been obtained in B. subtilis by Klocko et al (81). Using fluorescently labeled proteins in living cells, it was shown that the essential lagging-strand maturation polymerase (DnaE) foci decreased following the introduction of mismatches by 2-aminopurine. Furthermore, the decrease was dependent on BsMutS and BsMutL which can directly bind DnaE. Surprisingly, the leading- and lagging-strand polymerase, PolC, was not affected. However, since DnaE is essential and required for replication, it could be a target for modulation of replication fork activity by BsMutS as suggested for E. coli (159). Klocko et al (81) speculate that the encounter of DnaE with a mismatch could lead to binding of the beta clamp by BsMutS, thereby decreasing the amount of beta clamp for use by the polymerases.
Beta Clamp
Although it is part of the DNA polymerase III holoenzyme, because of its importance, the beta clamp deserves a separate heading. The clamp is encoded by the dnaN gene, which is located between dnaA and recF genes in an operon; transcription of the dnaN gene is controlled by six promoters. Two of these also control transcription of dnaA and are cell cycle regulated. The major promoter, dnaAP1, has a Dam-recognition site, GATC, that overlaps the −35 hexamer, and methylation of this sequence is required for maximal transcription initiation (15). The dnaA, dnaN and recF genes lie close to the origin of replication, oriC, and after initiation these genes are “sequestered” by binding of SeqA. The dnaA promoter is also auto-regulated via the ATP-bound form of DnaA (15). This regulatory scheme ensures that transcription of the dnaA and dnaN genes is maximal at the beginning of the cell cycle.
The other four promoters are located in front of dnaN and transcribe it as well as recF. The transcripts from these promoters are at a low level in logarithmic phase cells but increase substantially in stationary phase cells (202). This increase is dependent on the alternate RpoS (sigma 38) promoter recognition sequence. Since it is expected that transcription from the dnaA promoter is down-regulated in stationary phase, induction of dnaN transcripts by RpoS-containing RNA polymerase suggests a need to maintain the level of clamp proteins in stationary phase or stressed cells. This requirement may be for stationary or starved cell specific DNA replication or repair processes (including mismatch repair), some of which are discussed below (see Adaptive Mutation).
The beta clamp is a dimer which is loaded onto the 3′-end of duplex DNA at a gap or on the 3′-end of primer RNA-DNA hybrid during chromosome replication (149). The clamp tethers the polymerase III alpha catalytic subunit by binding it directly and sliding along with it during chain elongation, thereby endowing it with high processivity. The clamp is able to bind many cellular proteins including all other DNA polymerases, DNA ligase, MutS and MutL (108). The dimeric structure and the ability to bind more than one ligand at the same time might endow it with the ability to serve as a mobile scaffold for multiple enzymes that perform sequential actions on DNA. This feature is particularly attractive to model events occurring during mismatch repair and deserves to be tested experimentally.
The MutS protein has two binding motifs for the beta clamp, one at the N-terminus (QQYLRL) and the other at the C-terminus ((QMSLL) (108). In vitro experiments using a competition binding assay between MutS peptides and the beta-clamp indicate that the MutS C-terminal motif (residues 812–816) binds the beta-clamp much more strongly than does the N-terminal motif (Met13-Arg19). Although the N-terminal beta-binding motif is close to the crucial Phe-39 required for DNA intercalation at the mismatch, the MutS protein with the altered motif displays normal kinetics of binding to heteroduplex DNA. Although inactivation, by alanine substitution, of the N-terminal motif imparted a mutator phenotype in E. coli (108), it was subsequently shown that this mutant protein was unstable (159). Surprisingly, a mutS mutation deleting the C-terminal motif imparts a wildtype phenotype on E. coli (108)
A similar competition binding assay with MutL peptides and the beta-clamp identified a region in the N-terminus loop 2 that specifically bound the clamp (108). However, specific binding could only occur in the presence of single-stranded DNA. Mutation of both Leu150 and Phe151 residues to alanines produced a protein incapable of supporting mismatch repair in vivo. The mutant Leu150 Phe151 protein has wildtype ATPase activity and promotes UvrD unwinding at the same rate as the wildtype protein. Subsequently, a second beta binding site was identified in the C-terminus (482-QPLLIP) and inactivation of it reduced mismatch repair activity in vivo (156).
In contrast to the results in E. coli, interaction of the beta clamp with the mismatch repair proteins in B. subtilis is essential for successful mismatch repair. BsMutS has the beta recognition motif at the C-terminal end of the protein, like that of E. coli MutS. Inactivation of this motif by mutation results in a mutator phenotype and inability of BsMutS to localize properly at sites of mismatched bases in the chromosome (184). Dupes et al (33) have isolated mutations in the beta clamp that allow for normal DNA replication but are defective for mismatch repair.
It is not known at present why the requirement for the beta clamp interaction with mismatch repair proteins is essential in B. subtilis but not in E. coli. Perhaps the basis for this observation lies in the possession of Dam methylation in E. coli but not in B. subtilis. The interaction between MutS and the beta clamp has been suggested as a basis for strand discrimination during mismatch repair (see (74) for review). The interaction of the clamp with the C-terminal motif in BsMutLhas been proposed to activate the latent endonuclease activity of the protein (156). Assuming that the interaction of the beta clamp with MutS and MutL is important for successful mismatch repair, it is not clear how these interactions fit into the current model of mismatch repair (Fig. 6). It should be noted that the in vitro reconstituted mismatch repair system contained the beta clamp as part of the DNA polymerase III holoenzyme (88). In wildtype E. coli, the trailing hemimethylated DNA is close to the replication fork and it would not be surprising if the replication machinery and mismatch repair proteins work in close proximity. One way to link them would be through the beta clamp, although this may require that some clamps remain on the DNA after passage of the fork. Clamps used sequentially by the primase, DNA polymerase and ligase might be bound by MutS and/or MutL in cases where a mismatch has been detected or the binding might facilitate MutS recognition of the mismatched bases. Perhaps a complex of MutS-Clamp or MutS-MutL-Clamp binds the mismatch and might facilitate GATC location and interaction with MutH and its replacement with UvrD. After cleavage by MutH, the clamp would be positioned to bind the alpha-subunit of DNA polymerase III again for polymerizing across the gap produced by UvrD and exonuclease action. Binding of the alpha subunit to the clamp would still allow the second site to be bound by MutS or MutL, perhaps by the latter, since single-stranded DNA would be present at this stage. Finally, the alpha subunit or MutL would be displaced by DNA ligase to seal the nick as in normal DNA replication.
Models for the Initiation of Mismatch Repair
Although MutS binding to mismatched DNA would seem straightforward, this and the subsequent steps involving MutL and MutH are the subject of controversy. MutS binding to mismatched DNA is not observed in the presence of ATP, but the protein can be trapped on end-blocked DNA, suggesting that it moves away from the mismatch (72). There are at least three possible models currently under discussion (72). In the first model, MutS or MutS-MutL move from the mismatch to the nearest GATC by a sliding mechanism. In support of this model, addition of DNA binding sites and the corresponding DNA binding proteins can block MutS progression (160). Each monomer of MutS can bind nucleotide, and ATP hydrolysis is thought to stabilize mismatch binding, resulting in the ADP bound form found in the crystal structure. The ADP-MutS can bind a second ATP molecule, which is not hydrolyzed, but probably induces a conformational change allowing for sliding. Single or multiple MutS molecules with bound nucleotide could be involved (1).
In the second model, ATP-induced conformational changes in MutS provoke MutL nucleation along the DNA to the nearest GATC sequence, but MutS remains at the mismatch (138).
A third model also has the MutS-MutL complex remaining at the site of the mismatch but interaction with MutH bound at the GATC sequence occurs through either bending (2) or looping (67) of the intervening DNA. In all these models, interaction of DNA bound MutS-MutL with MutH activates endonuclease nicking of the unmethylated strand.
At present, there is not overwhelming support for any of these models. One problem may be that the models are based on results from in vitro biochemical experiments. What is needed is in vivo experiments that would lend support to one or more models.
RECOMBINATION
Homologous Recombination and MutS2 Proteins
As mentioned at the beginning of this article, mismatch repair was found to be involved with gene conversion in fungi and to affect transformation efficiency in S. pneumoniae. Prior to the advent of DNA sequencing, genetic mapping was used to locate genetic markers within a gene (see (135) for review). The basis of this technique was that the recombination frequency was directly related to the distance between alleles. Over short distances, however, this relationship often did not hold and this failure was termed interference. In hindsight, it is easy to see how mismatch repair in heteroduplex DNA could correct a mismatch one way or the other, thereby increasing or decreasing a particular recombinant class leading to errors in measuring distances between genes.
Recombination has been studied extensively in bacteriophage lambda. Unlike the E. coli host, however, there is under-methylation at GATC sites in the lambda genome (166) allowing for the formation of hemi-methylated GATC sites which are substrates for MutH action. As a consequence of under-methylation and its effect on mismatch repair, as well as VSP repair (see VSP Repair), there is variability in the yield of phage recombinant classes due to gene conversion. In E. coli, however, almost all GATC sites are methylated and if mismatch repair is to occur in heteroduplex DNA, how can MutH act on fully methylated DNA? Recall that in mismatch repair, MutH action can be by-passed if a nick is already present near the mismatch. It is probable that during the recombination process nicks or gaps are introduced into the DNA chains; if heterologies are present, these nicks or gaps are utilized as entry sites for the UvrD helicase. If so, then mismatch repair effects on E. coli recombination should be largely MutH-independent. The only experimental evidence on this point (40) is described below and is not in agreement with this prediction. Therefore, it is necessary at some point in the recombination process that there is new DNA synthesis to allow for the formation of hemi-methylated GATC sites.
In crosses between lambda phages, the mutHLS status of the host did not affect intergenic (between cI and R genes) recombination, but it did affect intragenic (Pam3 and Pam80) recombination (51). The results with mutH and uvrD mutant hosts are somewhat confusing because it was not recognized at the time that the Pam80 mutation is subject to VSP repair. Since efficient VSP repair requires MutS and MutL, the results obtained in hosts lacking either of these proteins reflect complete absence of both mismatch repair and VSP repair. Nevertheless, the reduction in intragenic recombination is consistent with the idea that mismatch correction could be responsible by its action on heteroduplex DNA. Although not stated in the paper, the reduction in P+ recombinants could be due to co-repair of both markers.
Six factor crosses with lambda phages indicated that only about 1–2% of primary recombination products plated on wildtype cells contained evidence of heteroduplex DNA (70). In a mutL host the percentage increased to 20%, thereby implicating the mismatch repair system in the frequency of heterozygous phage emerging from the cross. The six factor cross also allowed the estimation that the average heteroduplex region was about 4 kb, but could be as long as 10 kb.
Feinstein and Low (40) screened for hyper-recombination E. coli mutant bacteria using a pair of closely linked intragenic markers in both leu and lac genes and by searching for increased number of Leu+ Lac+ recombinants after conjugal crosses. The search yielded six hyper-recombination mutant strains, all of which also had a mutator phenotype, and five mapped to mutS and one to mutL. Known mutator alleles were then also used in the crosses, and mutS, mutL, mutH and uvrD alleles all showed the hyper-recombination phenotype. The mutU4 and uvrE502 alleles of uvrD gave a six- to ten-fold greater effect than other uvrD and mut alleles, which by themselves increased recombination by two- to seven-fold. Note that for each pair of intragenic leu or lac markers, and in all mut strains, there was an increase in recombination. This was in contrast to results in phage lambda crosses, where the selected recombinant class decreased dramatically in mut bacteria (e.g., (51)).
The question of how E. coli strains defective in mismatch correction of heteroduplex DNA consistently produce excess numbers of recombinants between closely-linked markers remains unanswered. Some possible explanations were considered by Feinstein and Low (40). First, that an increase in the number of uncorrected mismatches in heteroduplex DNA could lead to more recombination initiation events in a given DNA interval. This possibility seemed unlikely because mismatch-proficient mutator mutant strains also have an increased number of mismatches in DNA, but do not impart a hyper-recombination phenotype. Second, that mismatch correction of heteroduplexes in wildtype strains normally functions to decrease recombinant production seemed problematic. Third, that rapid co-correction of the two close markers in the heteroduplex region in wildtype bacteria would decrease recombinational potential while persistence of the heteroduplex region in mutHLS strains could increase it. Fourth, that the increase in recombination in mutHLS strains is not due directly to failure of mismatch correction but to some other alteration of MutHLS function.
The MutHLS system could prevent formation of heteroduplex DNA during the RecA strand transfer reaction (as in antirecombination, see below). Alternatively, or in addition, the heteroduplex intermediate is disassembled by UvrD (helicase II). The connection between UvrD and RecA actions is based on the observation that recA mutations increase the viability of uvrD lexA (Ind−)(a lexA allele preventing SOS induction) strains by preventing inappropriate recombination (104). UvrD reverses the effect of RecA-mediated strand exchange with homologous substrates in vitro (144) and removes RecA from single-stranded regions of DNA such as at the replication fork (201). This model would explain the reduction of heteroduplex DNA in phage lambda crosses in wildtype versus mutL hosts (70) and the hyper-recombination phenotype of mut and uvrD strains. In mut strains a greater amount of heteroduplex DNA is made leading to increased recombination potential. In uvrD strains, RecA is not removed as frequently from single-stranded DNA leading to increased RecA-mediated strand transfer (201).
Parenthetically, dam mutant strains also show a hyper-recombination phenotype (123) and were isolated in a screen for hyper-recombination mutant bacteria using an intrachromosomal recombination assay employing two widely separated inactive lac operons (82). The basis for the hyper-recombination phenotype in dam mutant cells, however, is most probably the single-and double-strand breaks introduced by the MutHLS system as a consequence of misdirected mismatch repair.
The MutS superfamily is divided into groups I and II. MutS-II family members differ from MutS-I by lacking the highly conserved N-terminal domain, although they do share the conserved ATPase domain. The E. coli K-12 MutS protein belongs to the MutS-I family of homologs while other bacteria contain either MutS-I and MutS-II or only MutS-II (35). Recent studies with Helicobacter pylori, which has only MutS-II, have begun to clarify the role of this homolog (79,157). Mutant bacteria deleted for the mutS2 gene did not have a mutator phenotype, but showed elevated transformation frequencies for homologous and homeologous (similar but not identical) DNA. The purified protein binds equally well to homoduplex and heteroduplex DNA, but has a preference for four-way-junction and open fork structures which stimulate MutS2 ATP hydrolysis (157). The protein also inhibits RecA-mediated strand exchange with homologous substrates in vitro. The H. pylori MutS2, therefore, is not part of a mismatch repair system for mutation avoidance, but appears to modulate homologous (and homeologous) recombination by a mechanism distinct from that of E. coli MutS.
Homeologous Recombination (Antirecombination)
Conjugational crosses between E. coli and S. enterica, which are about 85% homologous at the DNA level, yield very few recombinants, as expected from organisms that are separate species (Table 3). If the S. enterica recipient in this conjugal cross is defective in MutS or MutL, however, recombinants are obtained at about 1000-fold higher frequency. The recombinant frequency is about 66-fold less with mutH bacteria compared to mutS cells and 55-fold less with uvrD recipients (168). These increases in recombination frequency are RecA-dependent (Table 3). These results suggest that mismatch repair acts to prevent recombination between slightly diverged sequences that are expected to occur in genomes, and this process will be referred to here as antirecombination. For example, in E. coli the 3.7 kb rhsA (recombination hotspot) and rhsB sequences are 1% diverged at the DNA sequence level and 140 kb apart. The generation of chromosomal duplications between them is elevated about 10-fold in mutL and mutS bacteria (154).
Table 3.
Conjugal crosses between E. coli and S. entericaa
| E. coli Hfr donor | S. enterica recipient | E. coli recipient | Recombinant frequency |
|---|---|---|---|
| mut+ | mut+ | 9 × 10−7 | |
| mut+ | mutS | 2 × 10−3 | |
| mut+ | mutL | 3 × 10−3 | |
| mut+ | mutH | 6 × 10−5 | |
| mut+ | uvrD | 5 × 10−5 | |
| mut+ | mutS recA | 2 × 10−8 | |
| mut+ | mut+ | 3 × 10−1 |
This Table has been modified from reference 168.
Interspecies crosses activate the SOS response which, by inducing RecA and RuvAB proteins, has the effect of promoting recombination and antagonizing antirecombination (128)(Module 5.4.3. The SOS Regulatory Network and Module 7.2.8. The SOS Response). For example, in a mutS background the lexA1 (Ind−)(a lexA allele preventing SOS induction) allele reduces the yield of interspecies recombinants by 170-fold. The SOS inducing signal has not been identified but it is likely to be either the single-stranded DNA transferred from the donor during conjugation or some persistent recombination intermediate formed during the inter- but not intra-species matings.
The antagonizing effect of increased RecA and Ruv protein concentration on antirecombination suggests that antirecombination does not destroy recombination intermediates but either prevents their formation or returns them to the substrates from which they were formed (“heteroduplex rejection”). Support for this hypothesis comes from both in vivo and in vitro studies. Transformation of highly mismatched plasmid DNA into mutS or mutL hosts resulted in a greater frequency of colonies with two plasmids arising from both strands of the heteroduplex compared to wildtype (210). This result would not be expected from a heteroduplex destruction model. Since the plasmid molecules do not seem to have recombined in any way, the result is an example of the separation of repair from recombination.
Further support for a heteroduplex rejection model came from biochemical studies using RecA-mediated strand exchange which can be monitored by the conversion of double-stranded linear DNA and single-stranded circular DNA to form nicked circular DNA (Fig. 7). Neither MutS nor MutL, either separately or in combination, has any significant effect on RecA-mediated strand exchange with completely homologous DNA (214). With homeologous DNA, where there is about 3% sequence divergence, MutS alone is able to significantly reduce heteroduplex product, while MutL alone is without effect. The combination of MutS and MutL, however, is substantially greater effect than either protein alone and can completely prevent RecA-mediated strand exchange (Fig. 7). This result indicates that MutS and MutL can interfere with homeologous recombination by suppressing RecA-mediated strand transfer which is an early step in the process.
FIGURE 7.
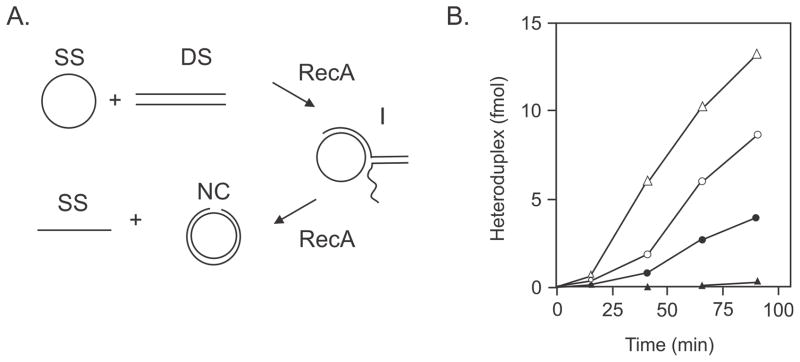
RecA strand transfer reaction. A. Single-stranded circular (SS) DNA anneals to its complement from double-stranded linear (DS) DNA to form a three-stranded intermediate (I) structure which forms a nicked circle (NC) and SS DNA as the products of the reaction. B. The RecA strand transfer reaction using homologous M13-M13 DNA substrates (open triangles) is more rapid than the homeologous M13-fd reaction (open circles). Inclusion of MutS and/or MutL has no effect on the reaction with homologous DNA (data not shown). Inclusion of MutS in the reaction with homeologous DNA depresses the rate of the reaction (filled circles) and inclusion of both MutS and MutL (filled triangles) almost completely prevents formation of product. Figure modified with permission from (214) Copyright (1994) National Academy of Sciences, U.S.A.
Further biochemical studies using the RecA-mediated transfer assay, employed the intermediates formed during the reaction (“I” in Fig. 7). These branched structures are those expected to be acted on by RuvAB subsequent to RecA action and, when RuvAB was added, a two- to threefold increase in product formation was detected with homeologous, but not homologous, substrates (39). This RuvAB stimulation was abrogated in the presence of MutS and MutL. Overall, the experiments with the strand-transfer assay show that MutS and MutL are capable of destabilizing RecA-DNA and RuvAB-DNA structures on heteroduplex DNA. There are some unanswered questions about these results. The primary one is how do MutS and MutL accomplish the reversal reaction? How do MutS and MutL enter the RecA nucleoprotein filament in which the mismatch is imbedded? How is RecA action stopped by MutS and MutL when it has already gone past the mismatch? Does the action of the Mut proteins occur at the mismatch? Similar question pertain to the RuvAB reaction. It would seem that there are still many biochemical experiments that can be done with the strand transfer assay to try to answer these questions.
The experiments described above show that MutS and MutL can act at the early stages of the recombination process and this probably accounts for the large effects on recombination in mutS or mutL recipient bacteria in either conjugational or transductional crosses. The smaller effect of MutH on antirecombination has been suggested to occur after D-loop formation and extension of the 3′-end of the invading strand when it copies a GATC sequence in the template strand (191). It was proposed that methylation of the newly-synthesized strand is delayed thereby allowing MutH to cleave and facilitate removal of mismatches by mismatch repair. More experimental evidence is needed to validate this mechanism.
Even though relief of antirecombination in crosses between S. enterica and E. coli occurs with mutS and mutL recipients, the frequency of recombinants is still at least 100-fold lower than with homologous crosses. In transductional crosses with S. enterica and S. typhi, which have only 1–2% sequence divergence, it was shown that the frequency of recombinants using a mutS recD mutant strain was almost as high as that for the homologous crosses (218). The increase in recombination frequency in both homologous and homeologous crosses with the double mutant strain was greater than in each of the single mutant strains. This result suggested two distinct mechanisms; one dependent on mismatch repair and the other mediated by RecBCD. It was suggested that heteroduplex reversal by MutSL of recombination intermediates would regenerate the double-stranded end of the donor DNA thereby making it susceptible to RecBCD nuclease action. Interestingly, there is little difference in recombinant yield in E. coli - S. enterica crosses between mutS or mutS recD strains (128). It was suggested that the larger amount of DNA transfer during conjugation allows for more attempts to initiate recombination to occur before DNA degradation (218). The high efficiency of homeologous recombination in mutS recD bacteria makes them useful in constructing genetic hybrids (116).
MISMATCH REPAIR AND DNA DAMAGE
E. coli dam mutant strains are much more sensitive than wildtype strains to the killing action of certain base analogues and chemicals while mismatch repair-deficient derivatives are resistant (46,50,80) (Fig. 8). The same increase in sensitivity occurs in mammalian cells, and mismatch repair-deficient derivatives are resistant to these agents (13). The phenomenon is, therefore, a conserved feature of mismatch repair, and is detrimental to mismatch repair-proficient cells. Mismatch repair can be likened to a Dr. Jekyll and Mr. Hyde duality: on the one hand it is beneficial to the cell because it prevents mutations and genome rearrangements, but on the other hand it kills when the cell is exposed to certain cytotoxic agents (121). It is not known why mismatch repair provokes this response, nor is the molecular mechanism by which it kills the cell. Part of the reason for this lack of understanding, is that there appears to be a different mechanism for each agent, although mismatch repair-induced DNA double-strand breaks are found in all cases and probably contribute substantially to cell death. Unlike the wildtype where mismatch repair is confined to the region just behind the replication fork, in E. coli dam mutant cells mismatch repair can be operative anywhere on the chromosome. This raises the possibility that gaps in the DNA created by mismatch repair at these sites ahead of the replication fork can lead to fork collapse (86). Collapse would generate a double-strand end and initiate homologous recombination by the RecBCD pathway. Other models are described below.
FIGURE 8.

Sensitivity of dam and dam mut mutant strains to MNNG and cisplatin. Cells in the exponential phase of growth were exposed to cisplatin or MNNG at the indicated doses, and survival was determined by plating cells on nutrient agar plates.
2-Aminopurine
As described above, E. coli dam mutant bacteria were found to have a decreased survival after exposure to the base analogue 2-aminopurine (2-AP) and this sensitivity was suppressed by second site mutations in the mutHLS genes (50). The molecular basis for 2-AP killing is not known, but may involve futile cycling (see below). Subsequent to the initiating event, extensive mismatch repair-induced degradation leads to dam cells without any DNA (126). Clearly DNA replication is necessary to incorporate the analog, and it is assumed that overlapping mismatch repair excision tracts lead to double-strand breaks (DSBs) and subsequent DNA degradation by the RecBCD nuclease.
MNNG and Cisplatin
Methylating agents are found in the environment and are used as anticancer agents (177). Exposure of cells to these agents results in the formation of methylated bases in DNA and RNA at available oxygen and nitrogen atoms. Methylated bases in DNA are subject to removal by base excision repair (BER), which produces abasic sites after the action of glycosylases. Subsequent action by AP-endonucleases, DNA polymerase I and DNA ligase restores the correct nucleotide sequence (177). Alternatively, the methyl group of O6-methylguanine (O6meG) can be removed directly from DNA by the Ada or Ogt methyltransferases. (Module 7.2.4. DNA Damage Reversal and Excision Repair). Recombination-deficient (recBCD, recFOR, ruvABC) strains of E. coli are more sensitive to methylating agents than the wildtype strain indicating that recombination proteins are required to repair DSBs and gaps produced by DNA replication at blocking lesions or at single-strand nicks resulting from AP-endonuclease action (148). Cells deficient in both BER and recombination show greater sensitivity to methylating agents than cells lacking one of these repair mechanisms (148).
E. coli dam mutant cells are also more sensitive than the wildtype strain to methylating agents such as N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) while dam mutHLS strains are resistant (Fig. 8) (75,80). Sensitivity was found only to MNNG and not other simple methylating agents. MNNG exposure results in the formation of larger amounts of O6-methylguanine (O6meG) in DNA than with other methylating agents and it was proposed that this base was the basis for cell killing (80). To explain the MNNG sensitivity, it was hypothesized that O6meG could pair with either C or T during replication, but that both base pairs were substrates for mismatch repair, and therefore a futile cycle of repair ensued (Fig. 9A)(80). Futile cycling could lead to several undesirable outcomes, such as DSB formation at a GATC sequence (Fig. 9A), or replication fork collapse at sites of futile cycling when a second replication fork comes through (Fig. 9B). DSBs were detected, using pulse field gel electrophoresis (PFGE), in dam recBC mutant cells exposed to MNNG. The DSB frequency was severely reduced in a dam recBC mutS mutant derivative, a result that correlated with increased survival (147). DSB formation was also dramatically reduced in MNNG-exposed dam recBC mutant cells overproducing the Ada protein, which converts O6meG back to guanine. This result confirms that O6meG is the key methylated base recognized by mismatch repair in vivo which correlates with the ability of MutS to bind to O6meG base pairs in vitro (167). Unexpectedly, DSB formation in dam recBC mutant bacteria was independent of DNA replication. It was suggested that DSBs arose as a consequence of overlapping mismatch and base excision repair, since a large number of heat-labile (abasic) sites (112) were detected at the low MNNG doses used in the experiment (147). Since survival of dam cells after MNNG exposure was found to be related to growth rate (fast growing cells are killed more efficiently than slow growing cells), it is possible that replication-dependent DSBs are formed, but are masked by the greater number of replication-independent DSBs. Alternatively, replication-dependent DSBs present a greater challenge to repair than replication-independent DSBs.
FIGURE 9.

Double-strand break formation in dam mutant strains exposed to MNNG. A. Replication of an O6meG base in the parental strand can result in the insertion of a T or G in the complementary strand. The mismatch repair system treats both types of base pairs as substrates, and a futile cycle of repair ensues. Repair can occur on either strand, and if it occurs on both strands simultaneously, a DSB may result due to overlapping repair tracts or by MutH (arrowheads) nicking at the same or closely spaced GATCs (black rectangles). B. During futile cycling a second replication fork encounters the gap present during mismatch repair excision, which results in replication fork collapse.
Cisplatin produces both inter- and intra-strand crosslinks in DNA of cells exposed to it (207). However, it is the intra-strand diguanyl-crosslinks that are recognized by MutS (45) and initiate mismatch repair-induced cell killing. The intra-strand crosslinks are removed from DNA by nucleotide excision repair utilizing the uvrABCD gene products, DNA polymerase I and DNA ligase. (Module 7.2.4. DNA Damage Reversal and Excision Repair). Consequently, there must be competition between NER and mismatch repair for substrate. Homologous recombination is also required to prevent cell death after cisplatin exposure, since recombination-deficient mutant strains are as sensitive to cisplatin damage as uvrABC mutant cells (219). Cells deficient in both NER and recombination are more sensitive to cisplatin than either mutant cells alone indicating that NER and recombination act independently from each other (219).
E. coli dam mutant cells are more sensitive to the killing action of cisplatin, but dam mutHLS mutant cells are resistant (Fig. 8)(46). A dose-dependent accumulation of DSBs was detected in dam recBC mutant cells exposed to cisplatin, but not in dam recBC mutHLS mutant derivatives (146). These DSBs are found only in cells undergoing chromosome replication (147) suggesting that replication forks encounter gaps produced during the excision phase of mismatch repair. Such an encounter leads to replication fork collapse, and recombination proteins are required to restore the integrity of the fork (86). An alternative model in which DSBs occur immediately behind the replication fork is difficult to build because chromosome replication results in gaps at the site of lesions in a similar manner to that described for ultra-violet irradiation (171). These gaps are repaired by the RecFOR recombination pathway and so it is difficult to imagine how a mismatched base is incorporated opposite the lesion.
E. coli and other organisms have translesion polymerases that are able to polymerize across DNA damage. There are three of these in E. coli: PolII, PolIV and PolV encoded by the polB, dinB and umuDC genes. To determine the contribution of these polymerases to mismatch repair-dependent killing by MNNG and cisplatin, a dam mutant strain deleted for the genes encoding them was constructed. The survival of this strain after exposure to MNNG and cisplatin was not different from that of the parental dam strain which had all three translesion polymerases (42). This result shows that translesion polymerases do not contribute to mismatch repair-dependent killing.
Methylating agents are used in the laboratory as mutagens. If methylating agents produce DNA damage recognized and processed by mismatch repair, then in the absence of repair, mutability should be increased. This is indeed the case. Shanabruch et al (180) found that mutS, mutL, mutH and uvrD mutant strains were hypermutable by methyl methane sulfonate, ethyl methane sulfonate, MNNG and N-methyl-N-nitrosourea. No increased mutability was seen with 4-nitroquinoline oxide or ultra-violet irradiation. It is a general phenomenon that agents producing substrates for mismatch repair are hyper-mutable. In addition to the above agents, these include 5-bromouracil (172,213), 2-aminopurine (158) and 9-aminoacridine (186).
Bile
S. enterica is a pathogen that causes gastrointestinal disorders and systemic infection in humans and livestock animals. As a defense against pathogenic bacteria, higher eukaryotes produce bile that has strong antibacterial activity. Both E. coli and S. enterica are relatively resistant to bile; although the mechanisms are not completely understood, they involve envelope structure, porins and efflux pumps. Bile induces the SOS response and is mutagenic. Mutant xthA nfo (AP endonuclease), dinB (polymerase IV), lexA(Ind−), recA, and recBCD strains are more sensitive to the bile killing action than wildtype strains (162,163). These data suggest that BER and RecBCD- initiated recombination are necessary to ensure survival after DNA damage by bile. In addition to the mutant strains above, dam mutant cells are also sensitive to bile, and this sensitivity is suppressed by mutations in mutS, mutL and mutH. In vivo studies indicate that S. enterica dam mut mutant bacteria cause partial relief of virulence attenuation seen with dam mutant cells by the oral route, but less so with intraperitoneal administration.
The bile-induced DNA lesion(s) is probably an oxidation product, consistent with the observation in E. coli that dam mutant bacteria are sensitive to peroxide, and that mutHLS derivatives are more resistant (216). Overall, the data are consistent with the hypothesis that mismatch repair is also an important mechanism for bile resistance. It is probable that a bile-induced oxidative lesion in DNA is recognized by the mismatch repair system leading to the formation of DSBs requiring RecBCD recombinational repair.
Antirecombination and Drug Sensitivity
In the previous section, it was shown that homologous recombination is required for dam mutant cells to resist the cytotoxic action of cisplatin and MNNG. What role might antirecombination play, if any, in response to DNA damage in these cells? To test this possibility, the RecA strand transfer assay (Fig. 7), described above for antirecombination, was utilized. DNA substrate molecules were prepared with unmodified, or methylated, or platinated adducts and assayed in the absence or presence of MutS and MutL (19,20). With unmodified DNA, MutS and MutL had no effect on RecA-mediated strand transfer (Fig. 10). If the single-stranded circular DNA was modified and reacted with unmodified linear double-stranded DNA in the presence of RecA, again there was no difference in the strand transfer rate compared to the unmodified substrate. If MutS was included in this reaction, however, there was a sharp decrease in rate and yield of product (Fig. 10). Inclusion of MutL in the reaction augmented the inhibition produced by MutS. Switching the modification to the other substrate molecule did not alter the result. Surprisingly, one OMG or platinum crosslink residue per 1000 bp was sufficient to depress the RecA reaction (19,20). In these reactions, the modified base is paired with its normal partner. However, the affinity of MutS for oligonucleotides containing platinated GG-CC crosslinks and OMG-C base pairs is higher than for unmodified DNA but 10- to 40-fold lower than for oligonucleotides with G-T mismatches (21,45,167). The data above suggest that MutS and MutL can block the RecA strand-transfer reaction if one of the substrate molecules contains platinated or methylated adducts. At present there is only in vitro evidence for such an antirecombination effect by mismatch repair on drug-modified DNA and in vivo confirmation is needed.
FIGURE 10.
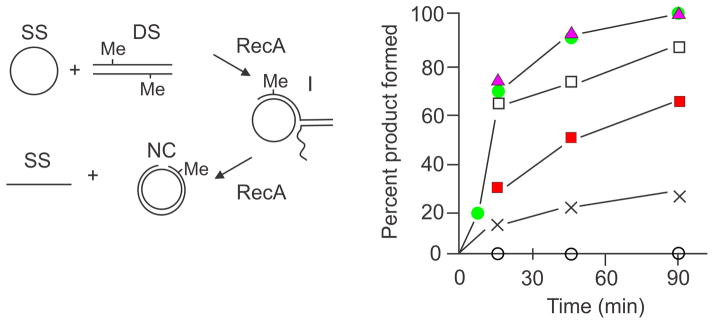
Antirecombination with modified homologous DNA. The RecA strand transfer reaction is as described in the legend to Fig. 7. The rate of reaction with homologous DNA substrate with or without MutS and MutL addition is shown by the green circles. Modified substrate DNA with 5–10 O6meG residues per molecule reacted with homologous DNA is shown by the filled triangles. Modified substrate DNA with 5–10 O6meG residues per molecule reacted with homologous DNA and 25 μM MutS (open squares), 100 μM MutS (red squares), 25 μM MutS plus 50 μM MutL (crosses), 100 μM MutS plus 100 μM MutL (open circles). The Figure has been modified from reference (19) published by the Oxford University Press.
When dam bacteria are exposed to the various agents discussed above, mismatch repair-induced DSBs, are formed. If antirecombination is also active in vivo, the repair of DSBs will be compromised leading to unrepaired DSBs and subsequent cell death. The mechanism of cell death would, therefore, result not only from DSB formation but also to the inability to repair them.
Mismatch Repair and Transcription-Coupled Nucleotide Excision Repair
Nucleotide excision repair (NER) recognizes and removes bulky adducts, such as ultraviolet (UV) light-induced pyrimidine dimers, from DNA using the UvrABCD proteins plus DNA polymerase I and DNA ligase (47). These enzymes efficiently remove pyrimidine dimers from DNA that is not being transcribed. In transcribed genes, removal of photoproducts additionally requires the Mfd (mutation frequency decline) protein and occurs at a faster rate than in non-transcribed DNA (Fig. 11)(132). Mutation frequency decline (MFD) was first described by Witkin (211) as a decrease in the yield of UV-light induced mutations in cells in which protein synthesis was transiently inhibited immediately after radiation. MFD is dependent on functional uvr and mfd genes (212) and in the latter mutant strain, excision of pyrimidine dimers from DNA is much slower suggesting that MFD might be a specialized kind of NER. It was shown subsequently in the lac operon that pyrimidine dimers were more rapidly removed if the operon was being transcribed than when it was not (Fig. 11)(132). Biochemical studies show that Mfd dissociates transcription complexes stalled at UV-induced lesions and recruits the Uvr proteins to remove the lesions (178).
FIGURE 11.

Mismatch repair and transcription-coupled NER. A. E. coli cultures of the indicated strains were irradiated with ultraviolet light, and samples removed at the indicated times to extract DNA. The DNA was then digested with T4 endonuclease V which nicks the DNA 5′ to the photoproducts. After electrophoresis in alkaline agarose the DNA was transferred to a membrane and hybridized with probes specific for the transcribed (T) or non-transcribed (NT) strands of the lac operon. The mfd mutant strain is deficient in transcription-coupled NER. B. Graphical representation of the results in A. Closed symbols represent the data for the transcribed strand and open symbols for the non-transcribed strand. The Figure has been modified from reference (132) Copyright (1996) National Academy of Sciences, U.S.A.
In mutS and mutL, but not mutH, bacteria transcription-coupled NER is abolished and excision of photoproducts is slowed (Fig. 11)(132). Clearly, there must be some feature of the damaged DNA-RNA transcript-RNA polymerase-Mfd complex that is recognized and bound by MutS and MutL to enhance transcription-coupled NER. In spite of the number of years since its discovery in E. coli, there has been no further progress in this area, nor has there been confirmation of the observations from other laboratories.
OTHER ASPECTS OF MISMATCH REPAIR
Enhancement of Tn10 Transposition
Transposon Tn10 is able to excise itself from the bacterial chromosome either partially or completely. To determine which host functions may play a role in the excision process, Lundblad and Kleckner (111) used a lacZ::Tn10 strain to screen mutagenized (by nitrosoguanidine) derivatives that had a greater frequency of Lac+ papillae in single colonies. The majority of these mutant bacteria had mutations in the mutHLS, uvrD or dam genes. The uvrD mutations had the greatest effects on precise excision not only of Tn10, but of Tn5 and Tn9 as well.
How mismatch repair reduces the frequency of precise events is not known. Each transposon has inverted repeats (IS elements) at its ends which are not identical. If pairing of these inverted repeats is involved in the excision process, base mismatches will be formed thereby providing substrates for mismatch repair (111). The mismatch repair complex might then interfere with the transposition machinery.
Pathogenic Bacteria and Mismatch Repair
Among pathogenic E. coli and S. enterica strains, LeClerc et al (93) found that the frequency of mutator bacteria was about one per cent, which was higher than expected from previous studies of the K-12 laboratory strain (55,77) and that almost all of these mutator strains were defective in mismatch repair. A subsequent study found that the frequency of mismatch repair-deficient mutator strains was the same in pathogenic and commensal E. coli strains (127). The increased mutation rate and homeologous recombination of mismatch repair-deficient bacteria led to the suggestion that such a mutator sub-population could enhance the acquisition and spread of virulence and antibiotic resistance genes in pathogens (93). In the laboratory E. coli K-12 strain, selection for Lac+ bacteria, using reversion of a lac frameshift mutation, increases the percentage of mutHLS mutators in the population from less than 1 per 100,000 to 1 per 200 cells (119). Addition of a single selection step to a mutagenized population can increase this value to 50%, showing that a mutator strain can rapidly dominate a bacterial population under selection (119). Since pathogens must adapt to different conditions during invasion of the host, or adapt to different hosts, as well as tolerating the external environment, the mutator sub-population could be advantageous in producing variants better suited to each niche. Bacterial populations of both pathogens and commensals would, therefore, be constantly changing depending on external conditions. It would also require that as soon as a new variant becomes dominant, it should lose its mutator phenotype (for the majority of the population), since high mutation rates are deleterious (48,137). At present, although there is some experimental support for these ideas, basic mechanisms are still lacking (see reviews (59,195)).
Retrons and Saturation of Mismatch Repair
E. coli and other bacteria can contain retrons which encode multicopy single-stranded DNA linked to an RNA molecule (71). The single-stranded DNA has characteristic stem-loop structures containing mismatched base pairs. When expressed in high copy in E. coli, they impart a mutator phenotype and increase homeologous recombination (113). These effects can be suppressed by increasing the concentration of MutS suggesting that the retron-encoded DNA titrates out the MutS protein to convert the cell to a mutS phenocopy.
Saturation of the mismatch repair system can also be achieved by increasing the number of replication errors by the use of a dnaQ (=mutD, epsilon proof-reading subunit of DNA polymerase III holoenzyme) (176) strain or by using a base analog such as 5-bromouracil (Fig. 4A) (172).
Adaptive Mutation
Adaptive mutation refers to a process that produces advantageous as well as non-selected mutations during selection (18,43) (Module 7.2.3. Stress-induced Mutagenesis). For example, reversion of a +1 frameshift in the lac operon of E. coli K-12 strain FC40 occurs in two defined phases after plating cultures on minimal lactose plates (reviewed in (43)). First, Lac+ mutant bacteria pre-existing in the culture before plating form colonies within 2–3 days with a Luria-Delbruck fluctuation frequency. Second, the number of Lac+ mutant colonies continues to increase for several days with a Poisson distribution indicating that Lac+ cells arose after plating (adaptive mutations). Strain FC40 has the lac operon on an F-plasmid and the chromosomal lac operon deleted, and adaptive mutations are almost exclusively −1 bp frameshifts in runs of iterated bases. The rate of adaptive mutations is decreased about 90% in rpoS (stress-induced RNA polymerase sigma factor) mutant bacteria and the bulk of this decrease is due to the SOS-inducible DNA polymerase IV (43).
The genetic requirements for adaptive mutation are those for DSB repair: recA, recBCD and ruvABC. In one model for adaptive mutation, incubation of FC40 on lactose minimal plates does not prevent occasional replication of the F-factor from its vegetative origin. The replication fork collapses at the F-factor conjugal origin of replication, which is nicked persistently under starvation conditions. The collapsed fork is reconstructed using homologous recombination. DNA replication by error-prone polymerase IV to extend the 3′-end of the invading strand during recombination, could produce base mismatches (43) which might not be corrected by mismatch repair due to its down-regulation under starvation conditions (see MutS above and VSP Repair below). Other models involving amplification of the lac operon have also been proposed (e.g., (170,187)), and reduced mismatch repair editing capacity would explain the mutation induction. As noted under Specificity of Mismatch Repair above, frameshift mutations are the most frequent type of mutational change in mutHLS bacteria, and this correlates with MutS having the highest affinity for heteroduplex DNA with a single base insertion/deletion (152). This is the correlation with the observed high frequency of frameshift mutation in adaptive mutagenesis. The reduced level of MutS coupled with the increased level of Vsr in stationary phase cells (see VSP and Mismatch Repair below) may explain the observation that MutL is limiting for adaptive mutation correction (60). On the other hand, Vsr has been shown to have no effect on adaptive mutation (44)
At present, although a role for mismatch repair in preventing adaptive mutation can be predicted on theoretical grounds, no convincing experimental data supports a direct role in the process.
Visualization of Mismatch Repair in Bacillus subtilis and E. coli
Although the details of mismatch repair in B. subtilis are less well understood than in E. coli, it is the only bacterium in which mismatch repair proteins have been visualized in vivo using GFP fusions. MutS-GFP and MutL-GFP fusion strains were constructed with their fusion proteins at their normal chromosomal location. Only the MutS-GFP was mismatch repair-proficient, although the MutL-GFP was able to participate in protein-protein interaction (188). MutS-GFP fluorescence was found associated with the nucleoid in exponentially growing cells, and about 5% showed a single focus. The same result was obtained in a mutL mutant strain showing that MutS-GFP localization is MutL independent. Similarly, MutL-GFP formed foci in growing cells but this did not occur in a mutS mutant background indicating that MutL foci were dependent on MutS localization. Growth of cells in the presence of 2-aminopurine (2-AP) stimulated focus formation fivefold for MutS-GFP and twofold for MutL-GFP and also increased the number of foci per cell. This increase was dependent on DNA replication and was not due to increased synthesis of the two fusion proteins. In growing cells exposed, or not, to 2-AP there was no continuous co-localization of the Mut-GFP fusions with the DNA polymerase III DnaX-GFP subunit (Recall that DnaX is an essential subunit of DNA polymerase III which is required in the re-synthesis step of mismatch repair). This study also suggests that since more than one protein is needed to form a focus, multiple copies of MutL and MutS are present at the mismatch.
The MutS protein of E. coli interacts with the beta sliding clamp (see Beta Clamp above) and the B. subtilis MutS does the same (185). MutS-GFP defective for clamp interaction has a mild mutator phenotype and reduced foci in response to 2-AP treatment suggesting that considerable mismatch repair can occur without MutS-clamp interaction (185). Surprisingly, MutS-GFP containing only the C-terminal 58 amino acids was able to form foci, indicating that interaction with the clamp, but not the mismatch, is necessary for focus formation. This result also suggests that MutS-clamp interaction is normally inhibited until a conformational change occurs in MutS upon binding to a mismatch. It was concluded that the beta-clamp aids in stabilizing MutS at a mismatch.
Fluorescent MutS and MutL proteins have also been visualized in growing E. coli cells. Elez et al (36) used MutL-GFP to show that the number of foci was increased in mutH and mutD5 (defective in 3′ to 5′ proofreading and saturated for mismatch repair) cells relative to wildtype. In the mutH strain, foci that appeared subsequently disappeared 40 to 50 min later. This time course is consistent with a replication error being generated and the resulting mismatch being correctly base paired at the next replication. The frequency of foci was found to be consistent with the prediction that the mutation rate in a population is roughly uniform.
VERY SHORT PATCH (VSP) REPAIR
5-Methylcytosine and Mutagenesis
Spontaneous mutational hotspots creating amber nonsense mutations occur in the E. coli lacI gene at 5-methylcytosine (5-meCyt) residues. The 5-meCyt bases are produced by DNA cytosine methyltransferase (Dcm) at its recognition site -CCAGG-, and deamination of 5-meCyt alters it to -CTAGG- (27) (Module 4.4.5. DNA Methylation). A similar result was obtained in the cI gene of phage lambda in growing, but not in non-growing, bacteria (96). As expected, in dcm mutant strains which lack 5-meCyt residues, no mutational hotspots were detected (27,97). These genetic effects have been attributed to the action of a repair system designated VSP (Very Short Patch) which processes G-T mismatches produced by deamination of 5-meCyt (11,99). VSP repair can thus be viewed as counteracting the potential mutagenic effects of 5-meCyt deamination (Fig. 12) (reviewed in (11).
FIGURE 12.

VSP Repair. The asterisk above the second cytosine in the top sequence denotes a 5-meCyt residue that can deaminate to form a T-G mismatch (center). In the reverse reaction, VSP repair restores the original sequence, and Dcm methylates the appropriate cytosine. The middle sequence can undergo two additional non-reversible reactions. If the mismatched DNA is replicated before repair, one of the daughter chromosomes will inherit a CG to TA mutation. Alternatively, the mismatched substrate is acted upon by mismatch repair converting the CG into a TA base pair.
VSP Repair and Genetic Recombination
It is instructive to describe in detail the discovery of VSP repair (96) because it shows how genetic results can reveal a new biological process by logical deduction in a well characterized system; in this case the cI gene of phage lambda. In the description that follows, the genetic configuration of four-factor crosses between cI alleles and the genes N and O is shown in Table 4. M. Lieb found that when a set of amber mutations in the cI gene of phage lambda was used in genetic crosses, anomalous recombination frequencies were obtained for certain alleles. The am6 mutation was one of these and it gave the highest frequency of unexpected cI+ recombinants. It will become clear that am6 marker was being converted to the wildtype allele by VSP repair during the crosses. Lieb had shown previously that when two cI mutations (not subject to repair) were crossed with each other in configuration I of Table 4, almost all the cI+ recombinants were N+O+. This result is consistent with breakage of the two parental molecules occurring between the two cI mutations. In contrast, when am6 was one of these markers only about half the cI+ recombinants were N+O+. This reduction could be explained if the break did not occur between the cI mutations, but in the intervals encompassing Nam53 and am6 or between the other cI mutation and Oam29. Following the break, branch migration would produce cI heteroduplex molecules. Using IS element insertions in the cI gene to block branch migration during crosses with am6, Lieb showed that there was a reduction in cI+ recombinants among the N+O+ class thereby confirming the hypothesis.
Table 4.
Frequencies (%) of cI+ recombinants in crosses with am6a
| cI mutation crossed with am6b | Distance (bp) from am6 |
cI+ frequency configuration I
|
cI+ frequency configuration II
|
||
|---|---|---|---|---|---|
| Observed | Expected | Observed | Expected | ||
| 330 | 5 | 2.9 | 0.1 | 1.4 | <0.01 |
| 321 | 20 | 5.6 | 0.4 | 1.2 | <0.01 |
| am302 | 31 | 4.5 | 0.6 | 1.4 | <0.01 |
| CP135c | 45 | 4.9 | 0.9 | 1.7 | <0.01 |
This table has been modified from reference 96.
X denotes the allele in this row.
CP, clear plaque morphology.
With this background, the data in Table 4 will now be considered. The markers are arranged in two configurations: in configuration I a single crossover is required to form N+ cI+ O+ recombinants, but in configuration II three crossovers are needed. From a large number of previous crosses with cI mutant phages, a mapping function was known allowing the expected recombination frequency to be calculated. It is immediately obvious from Table 4 that the observed frequency of recombinants is higher than expected in all crosses. If VSP repair converts am6 to the wildtype in configuration II in heteroduplex DNA, then instead of 3 crossovers only one is required, thereby explaining the high recombination frequency. In configuration I, the frequency of recombinants is higher than in configuration II, which is consistent with preferential repair of one of the two possible base pair configurations (G-T and not A-C). In configuration I, the frequency of recombinants for am330 is lower than for the other markers. This can be explained by assuming that am330 is so close to am6 that the repair event includes both of them. This would produce an excess of am330 recombinants and reduce the number of cI+ recombinants. Given the short distance between am6 and am330, this places an upper limit of 10 bp for the repair tract.
The results above allowed Lieb to establish the basics of VSP repair. It was known that am6 was located in a Dcm recognition site and Lieb proposed that the mutation arose by deamination of 5-meCyt to produce an amber mutation. During genetic crosses with am6, branch migration would produce G-T and A-C mismatches of which the G-T was repaired preferentially. As the repair tracts are much shorter than those created by mismatch repair (3,000 bases on average (205)), Lieb described this as “very short patch” repair. It is of interest to note that in the E. coli lacI gene, amber mutations at 5-meCyt sequences recombine with unexpectedly high frequencies with other mutations as close as 6 bp (27).
Further studies showed that VSP repair was reduced in polA and lig, mutant strains and thus suggested that this pathway uses some of the biochemical steps in base excision repair. Additionally, the vsr gene, that was absolutely required for VSP repair, was identified immediately downstream of the dcm gene (189). VSP was also reduced in mutS and mutL, but not mutH, mutant strains indicating an interaction with the mismatch repair machinery, that is described in more detail below (VSP and MutHLS-mismatch repair). Finally, it was shown that there was some degeneracy in regard to the Dcm site and the frequency of recombination, in that TTAGG, GTAGG and CTAGA mutations yielded 60%, 32% and 39% recombinants relative to the canonical sequence (100). Vsr turned out to be a sequence-specific endonuclease and this degeneracy correlates with Vsr nicking of substrates containing these and similar sequences (49).
Vsr
Vsr acts at the same DNA sequence (-CCWGG-) as the Dcm methyltransferase. This association is reflected in the adjacent location of their genes and their presumed regulation. There is an overlap at the 5′ end of the vsr gene by the last six codons of the dcm gene and it is in a +1 register relative to the dcm gene (189). Such an overlap is uncommon in E. coli and in this case may serve to link the expression of these genes. Both genes appear to be transcribed into a single mRNA, and translation of vsr appears to be dependent upon translation of the upstream dcm coding sequence (30). The mechanism by which this is achieved is not known. The location of the promoter(s) and its mode of regulation are also unknown.
As a consequence of the gene arrangement dcm mutations can also affect vsr. The most widely used dcm allele, dcm-6 (124), is defective in both methylation and VSP repair and shows mutational changes in codons 26 and 45 (30). The polar effect of the nonsense codon (TGA) at position 45 in dcm would most easily explain the effect on vsr. Mutations dcm-9 and dcm-10 are also Vsr− but dcm-1, dcm-4 and dcm-7 are Vsr+ (189). In addition to dcm-6, the mec mutant allele of dcm (61) has been used to research the function of the gene, although the location of the mutation in the gene is not known. Two large deletions which remove dcm and additional genes have been shown to lack Dcm methylation (6,12).
5-MeCyt is not a mutation hotspot in non-dividing cells indicating that VSP repair is much more efficient in resting cells than dividing cells (101). This phenotype correlates with the level of Vsr, which is barely detectable in growing cells but increases in stationary phase cells (115). The low Vsr level in dividing cells suggests that the T-G mismatch resulting from 5-meCyt deamination is more frequently replicated than repaired. Surprisingly, overproduction of Vsr in growing cells increases the mutation frequency but the resulting mutations are not just at Vsr recognition sites and the mutation spectrum is indistinguishable from that of mismatch repair-deficient bacteria (31). The mutator phenotype is due to Vsr interfering with mismatch repair and is the probable reason Vsr is down-regulated in growing cells. This interference is discussed in more detail below (VSP and MutHLS-mismatch repair).
T-G mismatches occur in non-replicating DNA by the deamination of 5-meCyt (Fig. 12). This reaction is analogous to the deamination of cytosine to form a uracil-guanine mismatch, which is a substrate for uracil-N-glycosylase. In contrast, the T-G mismatch is a substrate for the strand- and sequence-specific Vsr endonuclease, followed by conventional DNA polymerase I-dependent excision repair (63) and finally by DNA ligase (11). This simple reaction scheme is not complete because it does not explain the requirement for the MutS and MutL proteins and the Dam methyltransferase. The possible roles these additional requirements may play are described in more detail below.
The Vsr protein has 156 amino acid residues; the molecular structures of the truncated free enzyme and the full-length protein bound to DNA have been solved (16,198–200). The catalytic site has Asp51 and Glu25 residues conserved in type II restriction endonucleases, but unlike them Vsr produces extensive distortion of DNA. Unlike glycosylases and methyltransferases, Vsr does not flip the target base out of the helix but inserts three aromatic amino acids (Trp68, Trp86 and Phe67) into the major groove to promote endonucleolytic cleavage 5′ to the T in the mismatch. The cleaved DNA-Vsr complex is stable suggesting that some other protein is required to release Vsr from DNA. A likely candidate is DNA polymerase I, which is the next enzyme in the reaction sequence. The crystal structure does not explain how Vsr can incise T-G mismatches at –CCWGG-sequences that have four out of the five base pairs (49,98). Overall there is a resemblance between the way Vsr and MutS intercalate residues at the mismatch compared to enzymes that flip out bases.
VSP and Mismatch Repair
The T-G mismatch resulting from 5-meCyt deamination at Dcm recognition sites should also be recognized by the MutS protein (Fig. 12). Indeed overproduction of MutS decreases VSP repair and increases C to T mutations at Dcm sites (100,102) while, as noted above, overproduction of Vsr in growing cells produces an mismatch repair-deficient phenotype (31). These results indicate that MutS and Vsr compete for the same mismatches. VSP repair is severely reduced in mutS and mutL mutant bacteria (97) suggesting that Vsr interaction with MutL is necessary for repair. In vitro studies indicated that MutL promotes Vsr binding to substrate (32) and this was confirmed in yeast two-hybrid assays (118). Thus the mutagenicity of Vsr overproduction can be explained by titration of MutL making it unavailable to the mismatch repair system. In addition, overproduction of MutL and MutH, but not MutS, overcomes the mutagenic effects of Vsr overproduction (114). This result suggests that MutH and Vsr compete for binding to MutL.
The model of MutS and Vsr competing for mismatches is also supported by the finding that some dominant mutS mutations prevent VSP repair, while other dominant mutations increase it over the wildtype level (102). The first class may be MutS molecules tightly bound to the mismatch thus preventing Vsr access. The second mutS class may not normally interact with MutL and/or MutH but retain the ability to enhance VSP repair. The ability of MutS to enhance VSP repair is separate from the competition of binding substrate, as mutS mutant bacteria are deficient in VSP repair (97). In this context MutS and MutL act in concert to enhance VSP repair.
From the above, it is clear that the relative concentrations of Vsr and MutS in the cell are critical to the outcome of mismatch and VSP repair. Vsr is not detectable in growing cells, but increases dramatically in stationary-phase cells, while the MutS level decreases fourfold in stationary phase cells. In other words, during logarithmic growth when replication errors are most likely, mismatch repair is maximally efficient, while in stationary phase, when DNA synthesis is minimal and 5-meCyt deaminations accumulate, VSP repair is most active. The mechanism whereby MutS and MutL enhance VSP repair remains unresolved. One model is that MutS and MutL enhance Vsr binding at the mismatch through an alteration of DNA secondary structure (200). However, it is difficult to see how this could occur given that MutS and Vsr cannot together bind to the same mismatch. Another model is based on the observation that ATP hydrolysis after MutS binding leads to movement of the protein away from the mismatch (11). If repeated binding events lead to a concentration of MutS-MutL complexes near the mismatch, then Vsr binding at the mismatch could be stabilized by interaction through the MutL bridge preventing the recruitment of MutH and subsequent steps in mismatch repair.
VSP Repair and O6-Methylguanine Repair
O6-Methylguanine (O6meG) is formed by the action of methylating agents and promotes mutagenesis by miscoding with thymine (106) and may also be toxic to the cell if not removed (153). The methyl group of O6meG is rapidly removed by the Ada and Ogt methyltransferases (103). If not removed before replication, however, an O6meG -thymine pair is formed with high frequency. After removal of the methyl group, the resulting G-T mismatch can be a substrate for mismatch repair or VSP repair. It is assumed that mismatch repair processes the G-T mispair to G-C in most sequence contexts but what happens in Vsr recognition sequences? Rye et al (174) constructed single O6meG residues in the sequence –CCTO6meGG- in an M13 vector which was then transfected into mismatch repair, dam and vsr mutant strains. There was a substantial difference in the results between mutS, mutL and vsr strains but not wildtype, dam and mutH strains. These results are compatible with preferential correction by VSP repair in the given sequence context. Since the Vsr recognition sequence context is not absolute, correction by VSP could also occur at related sequences.
VSP Repair and Dam Methylation
Dam methylation is used by the E. coli mismatch repair system to differentiate the parental from the daughter strands, which is consistent with the mutator phenotype of dam mutant strains (Module 4.4.5. DNA Methylation). These mutant bacteria, however, have other phenotypes that indicate a role for Dam methylation in controlling the level of transcription initiation (105,109). This effect is usually manifested in the promoter region either directly at the −10 or −35 hexamers or indirectly at nearby recognition sequences for transcription factors. The effect of Dam methylation at a particular promoter sequence can have one of three outcomes: an increase, a decrease or no change in transcription initiation (120).
The efficiency of VSP repair is reduced in dam mutant cells but, unlike in wildtype cells, the Vsr level does not increase in stationary phase (10). Although the vsr mRNA level was not measured in wildtype and dam mutant cells, the authors concluded that the reduction of Vsr was due to a post-transcriptional mechanism.
A possible explanation for the Dam methylation effect on VSP repair might be through the action of a small, non-coding regulatory RNA (sRNA) (122). The sRNA molecules require the chaperone activity of the Hfq protein which is required to complex sRNA to mRNA. Such sRNAs have been implicated in a variety of regulatory mechanisms (192,204). To explain the effect of Dam methylation on VSP repair, it would be necessary to postulate that in dam mutant cells the level of transcription of an sRNA is altered due to the presence of one or more –GATC- sequences in the promoter region. This sRNA would act to control the level of translation of the vsr mRNA. For example, the 5′-UTR of the vsr message might be sequestered in growing cells but as they enter stationary phase, the binding of an sRNA would allow for unmasking translation sequences. In a dam mutant strain, transcription of the sRNA would be down-regulated in all phases of growth and no increase of Vsr is possible in stationary phase. Since sRNA molecules can act both positively and negatively to affect gene expression (192), other models involving sRNA are possible. The hypothesis involving sRNA in VSP can be tested experimentally and, if correct, the use of dam mutant strains might allow for additional examples of this type of posttranscriptional regulation.
CONCLUSION
The broad outline of mismatch repair in both prokaryotes and eukaryotes has been established. Mismatches are detected and bound by the MutS protein, which in turn recruits the MutL protein. Mismatch repair is bi-directional and can proceed from a strand nick either 3′ or 5′ to the mismatch that is introduced by an unknown mechanism. MutL introduces a nick on the opposite side of the mismatch. Exonuclease action begins at the nick allowing for 5′ to 3′ directionality and proceeds toward the mismatch and removes it. The resulting single-strand gap is filled by a replicative polymerase holoenzyme complex and the nick is sealed by DNA ligase.
The scheme is modified in E. coli and its relatives that use Dam methylation as an integral part of mismatch repair. Dam methylation is used to differentiate the parental strand from the newly synthesized strand. Mismatch binding by MutS is followed by the formation of a ternary complex of MutS, MutL and the MutH proteins. The MutH protein nicks either 3′ or 5′ to the mismatch at a hemimethylated Dam recognition sequence (-GATC-). The MutH protein is then removed from the complex and replaced by the UvrD helicase. The unwound single-strand is digested by one of several exonucleases with the appropriate 3′ or 5′ specificity. The single-strand gap is filled by a replicative polymerase holoenzyme and the nick is sealed by DNA ligase.
Future research on mismatch repair in non-E. coli bacteria will need to focus on at least four areas. First, how strand discrimination is achieved? That is, how is the initial nick near the mismatch introduced in the appropriate strand? Second, is there a helicase that binds to the nick and unwinds the DNA prior to the excision phase of mismatch repair? Third, how is the latent endonuclease activity of MutL controlled? Fourth, what is the importance of the interactions between the mismatch repair proteins and those of the replicative polymerase holoenzyme subunits?
Future research in E. coli and its relatives will be to unravel the details of the mechanism of mismatch repair. Although it might seem that the accumulated wealth of genetic and biochemical data have given us a detailed picture of the mechanism, the existence of three competing models to explain the initiation phase indicate the difficulties involved. How the repair system decides to choose the –GATC- either 5′ or 3′ to the mismatch is not known. At the ternary complex stage, the mechanism of replacement of MutH by UvrD has not been solved. How is exonuclease action terminated? What is the role of the beta sliding clamp? What is the significance of the interactions of the mismatch repair proteins and those of the DNA polymerase holoenzyme complex?
The MutS, MutL and MutH proteins can undergo conformational changes when bound to substrate and/or liganded with nucleotide. The MutS protein, for example, has been predicted to adopt eight conformational states and the challenge for the future will be to determine if a specific conformation is correlated with a specific step in mismatch repair. A similar argument goes for the conformational states of MutL and MutH. The role of mismatch repair proteins in antirecombination, mitosis and meiosis needs to be explored further. There is also evidence for eukaryotic MutS and MutL as determinants for cell cycle control but the details are lacking. Although much progress has been made in the mismatch repair area, there is still much more to be learned.
Table 2.
E. coli proteins required for mismatch repair
| Stage | Protein | Function |
|---|---|---|
| Initiation | MutS | Binds to base mismatches and 1–4 nt insertions or deletions |
| MutL | Molecular matchmaker: interacts with MutS in a mismatch-specific manner, and with MutH to activate endonuclease cleavage | |
| MutH | Nicks unmethylated strand 5′ to the G in hemimethylated to the G in hemimethylated GATC sequence | |
| Beta Sliding Clamp | Interacts with MutS and MutL; may enhance MutS binding and/or localization at a mismatch; processivity factor for polymerase III | |
| Excision | UvrD (helicase II) | Replaces MutH at nick to unwind DNA for excision |
| ExoI | 3′ to 5′ exonuclease | |
| ExoVII | 3′ to 5′ and 5′ to 3′ exonuclease | |
| ExoX | 3′ to 5′ exonuclease | |
| RecJ | 5′ to 3′ exonuclease | |
| Resynthesis | DNA polymerase III | Repair synthesis across gap |
| SsB | Single-strand binding protein; aids excision and re-synthesis | |
| Ligase | Seals nicks | |
| Methylation | Dam | Methylates A in hemimethylated GATC |
Acknowledgments
The research carried out in my laboratory described in this article was supported by the American Cancer Society, the National Science Foundation and the National Institutes of Health. The comments of the anonymous reviewers greatly improved the readability of this article.
References
- 1.Acharya S, Foster PL, Brooks P, Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol Cell. 2003;12:233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 2.Allen DJ, Makhov A, Grilley M, Taylor J, Thresher R, Modrich P, Griffith JD. MutS mediates heteroduplex loop formation by a translocation mechanism. EMBO J. 1997;16:4467–4476. doi: 10.1093/emboj/16.14.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aronshtam A, Marinus MG. Dominant negative mutator mutations in the mutL gene of Escherichia coli. Nucleic Acids Res. 1996;24:2498–2504. doi: 10.1093/nar/24.13.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arthur HM, Cavanagh DR, Finch PW, Emmerson PT. Regulation of the Escherichia coli uvrD gene in vivo. J Bacteriol. 1987;169:3435–3440. doi: 10.1128/jb.169.8.3435-3440.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avery OT, MacLeod CM, McCarty M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. J Exp Med. 1944;79:137–158. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bale A, d’Alarcao M, Marinus MG. Characterization of DNA adenine methylation mutants of Escherichia coli K12. Mutat Res. 1979;59:157–165. doi: 10.1016/0027-5107(79)90153-2. [DOI] [PubMed] [Google Scholar]
- 7.Ban C, Junop M, Yang W. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell. 1999;97:85–97. doi: 10.1016/s0092-8674(00)80717-5. [DOI] [PubMed] [Google Scholar]
- 8.Ban C, Yang W. Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell. 1998;95:541–552. doi: 10.1016/s0092-8674(00)81621-9. [DOI] [PubMed] [Google Scholar]
- 9.Ban C, Yang W. Structural basis for MutH activation in E. coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J. 1998;17:1526–1534. doi: 10.1093/emboj/17.5.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bell DC, Cupples CG. Very-short-patch repair in Escherichia coli requires the dam adenine methylase. J Bacteriol. 2001;183:3631–3635. doi: 10.1128/JB.183.12.3631-3635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhagwat AS, Lieb M. Cooperation and competition in mismatch repair: very short-patch repair and methyl-directed mismatch repair in Escherichia coli. Mol Microbiol. 2002;44:1421–1428. doi: 10.1046/j.1365-2958.2002.02989.x. [DOI] [PubMed] [Google Scholar]
- 12.Bhagwat AS, Sohail A, Roberts RJ. Cloning and characterization of the dcm locus of Escherichia coli K-12. J Bacteriol. 1986;166:751–755. doi: 10.1128/jb.166.3.751-755.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bignami M, Casorelli I, Karran P. Mismatch repair and response to DNA-damaging antitumour therapies. Eur J Cancer. 2003;39:2142–2149. doi: 10.1016/s0959-8049(03)00569-0. [DOI] [PubMed] [Google Scholar]
- 14.Bjornson KP, Blackwell LJ, Sage H, Baitinger C, Allen D, Modrich P. Assembly and molecular activities of the MutS tetramer. J Biol Chem. 2003;278:34667–34673. doi: 10.1074/jbc.M305513200. [DOI] [PubMed] [Google Scholar]
- 15.Braun RE, O’Day K, Wright A. Autoregulation of the DNA replication gene dnaA in E. coli K-12. Cell. 1985;40:159–169. doi: 10.1016/0092-8674(85)90319-8. [DOI] [PubMed] [Google Scholar]
- 16.Bunting KA, Roe SM, Headley A, Brown T, Savva R, Pearl LH. Crystal structure of the Escherichia coli dcm very-short-patch DNA repair endonuclease bound to its reaction product-site in a DNA superhelix. Nucleic Acids Res. 2003;31:1633–1639. doi: 10.1093/nar/gkg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burdett V, Baitinger C, Viswanathan M, Lovett ST, Modrich P. In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc Natl Acad Sci USA. 2001;98:6765–6770. doi: 10.1073/pnas.121183298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cairns J, Overbaugh J, Miller S. The origin of mutants. Nature. 1988;335:142–145. doi: 10.1038/335142a0. [DOI] [PubMed] [Google Scholar]
- 19.Calmann MA, Evans JE, Marinus MG. MutS inhibits RecA-mediated strand transfer with methylated DNA substrates. Nucleic Acids Res. 2005;33:3591–3597. doi: 10.1093/nar/gki673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calmann MA, Marinus MG. MutS inhibits RecA-mediated strand exchange with platinated DNA substrates. Proc Natl Acad Sci USA. 2004 doi: 10.1073/pnas.0406104101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calmann MA, Nowosielska A, Marinus MG. Separation of mutation avoidance and antirecombination functions in an Escherichia coli mutS mutant. Nucleic Acids Res. 2005;33:1193–1200. doi: 10.1093/nar/gki263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calmann MA, Nowosielska A, Marinus MG. The MutS C terminus is essential for mismatch repair activity in vivo. J Bacteriol. 2005;187:6577–6579. doi: 10.1128/JB.187.18.6577-6579.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carraway M, Rewinski C, Wu TH, Marinus MG. Specificity of the Dam-directed mismatch repair system of Escherichia coli K-12. Gene. 1988;74:157–158. doi: 10.1016/0378-1119(88)90275-2. [DOI] [PubMed] [Google Scholar]
- 24.Carraway M, Youderian P, Marinus MG. Spontaneous mutations occur near dam recognition sites in a dam− Escherichia coli host. Genetics. 1987;116:343–347. doi: 10.1093/genetics/116.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Claverys JP, Lacks SA. Heteroduplex deoxyribonucleic acid base mismatch repair in bacteria. Microbiol Rev. 1986;50:133–165. doi: 10.1128/mr.50.2.133-165.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claverys JP, Mejean V. Strand targeting signal(s) for in vivo mutation avoidance by post- replication mismatch repair in Escherichia coli. Mol Gen Genet. 1988;214:574–578. doi: 10.1007/BF00330497. [DOI] [PubMed] [Google Scholar]
- 27.Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274:775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- 28.Cox EC. Bacterial mutator genes and the control of spontaneous mutation. Annu Rev Genet. 1976;10:548–555. doi: 10.1146/annurev.ge.10.120176.001031. [DOI] [PubMed] [Google Scholar]
- 29.Dao V, Modrich P. Mismatch-, MutS-, MutL-, and helicase II-dependent unwinding from the single-strand break of an incised heteroduplex. J Biol Chem. 1998;273:9202–9207. doi: 10.1074/jbc.273.15.9202. [DOI] [PubMed] [Google Scholar]
- 30.Dar ME, Bhagwat AS. Mechanism of expression of DNA repair gene vsr, an Escherichia coli gene that overlaps the DNA cytosine methylase gene, dcm. Mol Microbiol. 1993;9:823–833. doi: 10.1111/j.1365-2958.1993.tb01741.x. [DOI] [PubMed] [Google Scholar]
- 31.Doiron KM, Viau S, Koutroumanis M, Cupples CG. Overexpression of vsr in Escherichia coli is mutagenic. J Bacteriol. 1996;178:4294–4296. doi: 10.1128/jb.178.14.4294-4296.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drotschmann K, Aronshtam A, Fritz HJ, Marinus MG. The Escherichia coli MutL protein stimulates binding of Vsr and MutS to heteroduplex DNA. Nucleic Acids Res. 1998;26:948–953. doi: 10.1093/nar/26.4.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dupes NM, Walsh BW, Klocko AD, Lenhart JS, Peterson HL, Gessert DA, Pavlick CE, Simmons LA. Mutations in the Bacillus subtilis beta clamp that separate its roles in DNA replication from mismatch repair. J Bacteriol. 2010;192:3452–3463. doi: 10.1128/JB.01435-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Easton AM, Kushner SR. Transcription of the uvrD gene of Escherichia coli is controlled by the lexA repressor and by attenuation. Nucleic Acids Res. 1983;11:8625–8640. doi: 10.1093/nar/11.24.8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eisen JA. A phylogenomic study of the MutS family of proteins. Nucleic Acids Res. 1998;26:4291–4300. doi: 10.1093/nar/26.18.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elez M, Murray AW, Bi LJ, Zhang XE, Matic I, Radman M. Seeing mutations in living cells. Curr Biol. 2010;20:1432–1437. doi: 10.1016/j.cub.2010.06.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elez M, Radman M, Matic I. The frequency and structure of recombinant products is determined by the cellular level of MutL. Proc Natl Acad Sci USA. 2007;104:8935–8940. doi: 10.1073/pnas.0610149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ephrussi-Taylor H, Gray TC. Genetic studies of recombining DNA in pneumococcal transformation. J Gen Physiol. 1966;49:211–231. doi: 10.1085/jgp.49.6.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fabisiewicz A, Worth L., Jr Escherichia coli MutS,L Modulate RuvAB-dependent Branch Migration between Diverged DNA. J Biol Chem. 2001;276:9413–9420. doi: 10.1074/jbc.M005176200. [DOI] [PubMed] [Google Scholar]
- 40.Feinstein SI, Low KB. Hyper-recombining recipient strains in bacterial conjugation. Genetics. 1986;113:13–33. doi: 10.1093/genetics/113.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng G, Tsui HC, Winkler ME. Depletion of the cellular amounts of the MutS and MutH methyl-directed mismatch repair proteins in stationary-phase Escherichia coli K-12 cells. J Bacteriol. 1996;178:2388–2396. doi: 10.1128/jb.178.8.2388-2396.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fenton A, Marinus MG. Unpublished data. 2011 [Google Scholar]
- 43.Foster PL. Stress responses and genetic variation in bacteria. Mutat Res. 2005;569:3–11. doi: 10.1016/j.mrfmmm.2004.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foster PL, Rosche WA. Levels of the Vsr endonuclease do not regulate stationary-phase reversion of a Lac- frameshift allele in Escherichia coli. J Bacteriol. 1998;180:1944–1946. doi: 10.1128/jb.180.7.1944-1946.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fourrier L, Brooks P, Malinge JM. Binding discrimination of MutS to a set of lesions and compound lesions (base damage and mismatch) reveals its potential role as a cisplatin-damaged DNA sensing protein. J Biol Chem. 2003;278:21267–21275. doi: 10.1074/jbc.M301390200. [DOI] [PubMed] [Google Scholar]
- 46.Fram RJ, Cusick PS, Wilson JM, Marinus MG. Mismatch repair of cis-diamminedichloroplatinum(II)-induced DNA damage. Mol Pharmacol. 1985;28:51–55. [PubMed] [Google Scholar]
- 47.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger TE. DNA Repair and Mutagenesis. ASM Press; Washington DC: 2006. [Google Scholar]
- 48.Giraud A, Matic I, Tenaillon O, Clara A, Radman M, Fons M, Taddei F. Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science. 2001;291:2606–2608. doi: 10.1126/science.1056421. [DOI] [PubMed] [Google Scholar]
- 49.Glasner W, Merkl R, Schellenberger V, Fritz HJ. Substrate preferences of Vsr DNA mismatch endonuclease and their consequences for the evolution of the Escherichia coli K-12 genome. J Mol Biol. 1995;245:1–7. doi: 10.1016/s0022-2836(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 50.Glickman B, van den EP, Radman M. Induced mutagenesis in dam- mutants of Escherichia coli: a role for 6-methyladenine residues in mutation avoidance. Mol Gen Genet. 1978;163:307–312. doi: 10.1007/BF00271960. [DOI] [PubMed] [Google Scholar]
- 51.Glickman BW, Radman M. Escherichia coli mutator mutants deficient in methylation-instructed DNA mismatch correction. Proc Natl Acad Sci USA. 1980;77:1063–1067. doi: 10.1073/pnas.77.2.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grafstrom RH, Hoess RH. Nucleotide sequence of the Escherichia coli mutH gene. Nucleic Acids Res. 1987;15:3073–3084. doi: 10.1093/nar/15.7.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grilley M, Griffith J, Modrich P. Bidirectional excision in methyl-directed mismatch repair. J Biol Chem. 1993;268:11830–11837. [PubMed] [Google Scholar]
- 54.Grilley M, Holmes J, Yashar B, Modrich P. Mechanisms of DNA-mismatch correction. Mutat Res. 1990;236:253–267. doi: 10.1016/0921-8777(90)90009-t. [DOI] [PubMed] [Google Scholar]
- 55.Gross MD, Siegel EC. Incidence of mutator strains in Escherichia coli and coliforms in nature. Mutat Res. 1981;91:107–110. doi: 10.1016/0165-7992(81)90081-6. [DOI] [PubMed] [Google Scholar]
- 56.Guarne A, Ramon-Maiques S, Wolff EM, Ghirlando R, Hu X, Miller JH, Yang W. Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. EMBO J. 2004;23:4134–4145. doi: 10.1038/sj.emboj.7600412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guild WR, Shoemaker NB. Mismatch correction in pneumococcal transformation: donor length and hex-dependent marker efficiency. J Bacteriol. 1976;128:291–301. doi: 10.1128/jb.125.1.125-135.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haber LT, Pang PP, Sobell DI, Mankovich JA, Walker GC. Nucleotide sequence of the Salmonella typhimurium mutS gene required for mismatch repair: homology of MutS and HexA of Streptococcus pneumoniae. J Bacteriol. 1988;170:197–202. doi: 10.1128/jb.170.1.197-202.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hall LM, Henderson-Begg SK. Hypermutable bacteria isolated from humans--a critical analysis. Microbiology. 2006;152:2505–2514. doi: 10.1099/mic.0.29079-0. [DOI] [PubMed] [Google Scholar]
- 60.Harris RS, Feng G, Ross KJ, Sidhu R, Thulin C, Longerich S, Szigety SK, Winkler ME, Rosenberg SM. Mismatch repair protein MutL becomes limiting during stationary-phase mutation. Genes Dev. 2007;11:2426–2437. doi: 10.1101/gad.11.18.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hattman S, Schlagman S, Cousens L. Isolation of a mutant of Escherichia coli defective in cytosine-specific deoxyribonucleic acid methylase activity and in partial protection of bacteriophage lambda against restriction by cells containing the N-3 drug-resistance factor. J Bacteriol. 1973;115:1103–1107. doi: 10.1128/jb.115.3.1103-1107.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hengge-Aronis R. Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev. 2002;66:373–95. doi: 10.1128/MMBR.66.3.373-395.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hennecke F, Kolmar H, Brundl K, Fritz HJ. The vsr gene product of E. coli K-12 is a strand- and sequence-specific DNA mismatch endonuclease. Nature. 1991;353:776–778. doi: 10.1038/353776a0. [DOI] [PubMed] [Google Scholar]
- 64.Holliday R. A mechanism for gene conversion in fungi. Genet Res. 1964;5:282–304. doi: 10.1017/S0016672308009476. [DOI] [PubMed] [Google Scholar]
- 65.Hong ES, Yeung A, Funchain P, Slupska MM, Miller JH. Mutants with temperature-sensitive defects in the Escherichia coli mismatch repair system: sensitivity to mispairs generated in vivo. J Bacteriol. 2005;187:840–846. doi: 10.1128/JB.187.3.840-846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Horst JP, Wu TH, Marinus MG. Escherichia coli mutator genes. Trends Microbiol. 1999;7:29–36. doi: 10.1016/s0966-842x(98)01424-3. [DOI] [PubMed] [Google Scholar]
- 67.Hsieh P. Molecular mechanisms of DNA mismatch repair. Mutat Res. 2001;486:71–87. doi: 10.1016/s0921-8777(01)00088-x. [DOI] [PubMed] [Google Scholar]
- 68.Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391–407. doi: 10.1016/j.mad.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang SN, Crothers DM. The role of nucleotide cofactor binding in cooperativity and specificity of MutS recognition. J Mol Biol. 2008;384:31–47. doi: 10.1016/j.jmb.2008.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huisman O, Fox MS. A genetic analysis of primary products of bacteriophage lambda recombination. Genetics. 1986;112:409–420. doi: 10.1093/genetics/112.3.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Inouye M, Inouye S. msDNA and bacterial reverse transcriptase. Annu Rev Microbiol. 1991;45:163–186. doi: 10.1146/annurev.mi.45.100191.001115. [DOI] [PubMed] [Google Scholar]
- 72.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 73.Jiang Y, Marszalek PE. Atomic force microscopy captures MutS tetramers initiating DNA mismatch repair. EMBO J. 2011;30:2881–2893. doi: 10.1038/emboj.2011.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 75.Jones M, Wagner R. N-Methyl-N’-nitro-N-nitrosoguanidine sensitivity of E. coli mutants deficient in DNA methylation and mismatch repair. Mol Gen Genet. 1981;184:562–563. doi: 10.1007/BF00352542. [DOI] [PubMed] [Google Scholar]
- 76.Joseph N, Duppatla V, Rao DN. Prokaryotic DNA mismatch repair. Prog Nucleic Acid Res Mol Biol. 2006;81:1–49. doi: 10.1016/S0079-6603(06)81001-9. [DOI] [PubMed] [Google Scholar]
- 77.JYSSUM K. Observations on two types of genetic instability in Escherichia coli. Acta Pathol Microbiol Scand. 1960;48:113–120. doi: 10.1111/j.1699-0463.1960.tb04747.x. [DOI] [PubMed] [Google Scholar]
- 78.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 79.Kang J, Huang S, Blaser MJ. Structural and functional divergence of MutS2 from bacterial MutS1 and eukaryotic MSH4-MSH5 homologs. J Bacteriol. 2005;187:3528–3537. doi: 10.1128/JB.187.10.3528-3537.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karran P, Marinus MG. Mismatch correction at O6-methylguanine residues in E. coli DNA. Nature. 1982;296:868–869. doi: 10.1038/296868a0. [DOI] [PubMed] [Google Scholar]
- 81.Klocko AD, Schroeder JW, Walsh BW, Lenhart JS, Evans ML, Simmons LA. Mismatch repair causes the dynamic release of an essential DNA polymerase from the replication fork. Mol Microbiol. 2011;82:648–663. doi: 10.1111/j.1365-2958.2011.07841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Konrad EB. Method for isolation of Escherichia coli mutants with enhanced recombination between chromosomal duplications. J Bacteriol. 1977;130:167–172. doi: 10.1128/jb.130.1.167-172.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kornberg A, Baker TA. DNA Replication. W.H. Freeman and Co; New York, N.Y: 1992. [Google Scholar]
- 84.Kramer B, Kramer W, Fritz HJ. Different base/base mismatches are corrected with different efficiencies by the methyl-directed DNA mismatch-repair system of E. coli. Cell. 1984;38:879–887. doi: 10.1016/0092-8674(84)90283-6. [DOI] [PubMed] [Google Scholar]
- 85.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 86.Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Molec Microbiol. 1995;16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- 87.Lacks S. Mutants of Diplococcus pneumoniae that lack deoxyribonucleases and other activities possibly pertinent to genetic transformation. J Bacteriol. 1970;101:373–383. doi: 10.1128/jb.101.2.373-383.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lahue RS, Au KG, Modrich P. DNA mismatch correction in a defined system. Science. 1989;245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- 89.Lahue RS, Modrich P. Methyl-directed DNA mismatch repair in Escherichia coli. Mutat Res. 1988;198:37–43. doi: 10.1016/0027-5107(88)90037-1. [DOI] [PubMed] [Google Scholar]
- 90.Lahue RS, Su SS, Modrich P. Requirement for d(GATC) sequences in Escherichia coli mutHLS mismatch correction. Proc Natl Acad Sci USA. 1987;84:1482–1486. doi: 10.1073/pnas.84.6.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lamers MH, Perrakis A, Enzlin JH, Winterwerp HH, de Wind N, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a GT mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 92.Langle-Rouault F, Maenhaut-Michel G, Radman M. GATC sequences, DNA nicks and the MutH function in Escherichia coli mismatch repair. EMBO J. 1987;6:1121–1127. doi: 10.1002/j.1460-2075.1987.tb04867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.LeClerc JE, Li B, Payne WL, Cebula TA. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science. 1996;274:1208–1211. doi: 10.1126/science.274.5290.1208. [DOI] [PubMed] [Google Scholar]
- 94.Leong PM, Hsia HC, Miller JH. Analysis of spontaneous base substitutions generated in mismatch-repair-deficient strains of Escherichia coli. J Bacteriol. 1986;168:412–416. doi: 10.1128/jb.168.1.412-416.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li F, Liu Q, Chen YY, Yu ZN, Zhang ZP, Zhou YF, Deng JY, Bi LJ, Zhang XE. Escherichia coli mismatch repair protein MutL interacts with the clamp loader subunits of DNA polymerase III. Mutat Res. 2008;637:101–110. doi: 10.1016/j.mrfmmm.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 96.Lieb M. Specific mismatch correction in bacteriophage lambda crosses by very short patch repair. Mol Gen Genet. 1983;191:118–125. doi: 10.1007/BF00330898. [DOI] [PubMed] [Google Scholar]
- 97.Lieb M. Bacterial genes mutL, mutS, and dcm participate in repair of mismatches at 5-methylcytosine sites. J Bacteriol. 1987;169:5241–5246. doi: 10.1128/jb.169.11.5241-5246.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lieb M, Allen E, Read D. Very short patch mismatch repair in phage lambda: repair sites and length of repair tracts. Genetics. 1986;114:1041–1060. doi: 10.1093/genetics/114.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lieb M, Bhagwat AS. Very short patch repair: reducing the cost of cytosine methylation. Mol Microbiol. 1996;20:467–473. doi: 10.1046/j.1365-2958.1996.5291066.x. [DOI] [PubMed] [Google Scholar]
- 100.Lieb M, Rehmat S. Very short patch repair of T:G mismatches in vivo: importance of context and accessory proteins [published erratum appears in J Bacteriol 1995 May;177(9):2606] J Bacteriol. 1995;177:660–666. doi: 10.1128/jb.177.3.660-666.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lieb M, Rehmat S. 5-Methylcytosine is not a mutation hot spot in nondividing Escherichia coli. Proc Natl Acad Sci USA. 1997;94:940–945. doi: 10.1073/pnas.94.3.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lieb M, Rehmat S, Bhagwat AS. Interaction of MutS and Vsr: some dominant-negative mutS mutations that disable methyladenine-directed mismatch repair are active in very-short-patch repair. J Bacteriol. 2001;183:6487–6490. doi: 10.1128/JB.183.21.6487-6490.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lindahl T, Sedgwick B, Sekiguchi M, Nakabeppu Y. Regulation and expression of the adaptive response to alkylating agents. Annu Rev Biochem. 1988;57:133–157. doi: 10.1146/annurev.bi.57.070188.001025. [DOI] [PubMed] [Google Scholar]
- 104.Lloyd RG. lexA dependent recombination in uvrD strains of Escherichia coli. Mol Gen Genet. 1983;189:157–161. doi: 10.1007/BF00326069. [DOI] [PubMed] [Google Scholar]
- 105.Lobner-Olesen A, Skovgaard O, Marinus MG. Dam methylation: coordinating cellular processes. Curr Opin Microbiol. 2005;8:154–160. doi: 10.1016/j.mib.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 106.Loechler EL, Green CL, Essigmann JM. In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc Natl Acad Sci USA. 1984;81:6271–6275. doi: 10.1073/pnas.81.20.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Loh T, Murphy KC, Marinus MG. Mutational Analysis of the MutH Protein from Escherichia coli. J Biol Chem. 2001;276:12113–12119. doi: 10.1074/jbc.M007935200. [DOI] [PubMed] [Google Scholar]
- 108.Lopez de Saro FJ, Marinus MG, Modrich P, O’Donnell M. The beta sliding clamp binds to multiple sites within MutL and MutS. J Biol Chem. 2006;281:14340–14349. doi: 10.1074/jbc.M601264200. [DOI] [PubMed] [Google Scholar]
- 109.Low DA, Casadesus J. Clocks and switches: bacterial gene regulation by DNA adenine methylation. Curr Opin Microbiol. 2008;11:106–112. doi: 10.1016/j.mib.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 110.Lu AL, Clark S, Modrich P. Methyl-directed repair of DNA base-pair mismatches in vitro. Proc Natl Acad Sci USA. 1983;80:4639–4643. doi: 10.1073/pnas.80.15.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lundblad V, Kleckner N. Mismatch repair mutations of Escherichia coli K12 enhance transposon excision. Genetics. 1985;109:3–19. doi: 10.1093/genetics/109.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman AS, Helleday T. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005;33:3799–3811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Maas WK, Wang C, Lima T, Hach A, Lim D. Multicopy single-stranded DNA of Escherichia coli enhances mutation and recombination frequencies by titrating MutS protein. Mol Microbiol. 1996;19:505–509. doi: 10.1046/j.1365-2958.1996.392921.x. [DOI] [PubMed] [Google Scholar]
- 114.Macintyre G, Doiron KM, Cupples CG. The Vsr endonuclease of Escherichia coli: an efficient DNA repair enzyme and a potent mutagen. J Bacteriol. 1997;179:6048–6052. doi: 10.1128/jb.179.19.6048-6052.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Macintyre G, Pitsikas P, Cupples CG. Growth phase-dependent regulation of Vsr endonuclease may contribute to 5-methylcytosine mutational hot spots in Escherichia coli. J Bacteriol. 1999;181:4435–4436. doi: 10.1128/jb.181.14.4435-4436.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Maloy S, Zahrt T. Surrogate genetics: the use of bacterial hybrids as a genetic tool. Methods. 2000;20:73–79. doi: 10.1006/meth.1999.0907. [DOI] [PubMed] [Google Scholar]
- 117.Manelyte L, Urbanke C, Giron-Monzon L, Friedhoff P. Structural and functional analysis of the MutS C-terminal tetramerization domain. Nucleic Acids Res. 2006;34:5270–5279. doi: 10.1093/nar/gkl489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mansour CA, Doiron KM, Cupples CG. Characterization of functional interactions among the Escherichia coli mismatch repair proteins using a bacterial two-hybrid assay. Mutat Res. 2001;485:331–338. doi: 10.1016/s0921-8777(01)00071-4. [DOI] [PubMed] [Google Scholar]
- 119.Mao EF, Lane L, Lee J, Miller JH. Proliferation of mutators in A cell population. J Bacteriol. 1997;179:417–422. doi: 10.1128/jb.179.2.417-422.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marinus MG. DNA methylation in Escherichia coli. Annu Rev Genet. 1987;21:113–131. doi: 10.1146/annurev.ge.21.120187.000553. [DOI] [PubMed] [Google Scholar]
- 121.Marinus MG. Dr. Jekyll and Mr. Hyde: How the MutSLH repair system kills the cell. In: Higgins NP, editor. The Bacterial Chromosome. ASM Press; Washington DC: 2005. pp. 413–430. [Google Scholar]
- 122.Marinus MG, Casadesus J. Roles of DNA adenine methylation in host-pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol Rev. 2009;33:488–503. doi: 10.1111/j.1574-6976.2008.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marinus MG, Konrad EB. Hyper-recombination in dam mutants of Escherichia coli K-12. Mol Gen Genet. 1976;149:273–277. doi: 10.1007/BF00268528. [DOI] [PubMed] [Google Scholar]
- 124.Marinus MG, Morris NR. Isolation of deoxyribonucleic acid methylase mutants of Escherichia coli K-12. J Bacteriol. 1973;114:1143–1150. doi: 10.1128/jb.114.3.1143-1150.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Marinus MG, Morris NR. Biological function for 6-methyladenine residues in the DNA of Escherichia coli K12. J Mol Biol. 1974;85:309–322. doi: 10.1016/0022-2836(74)90366-0. [DOI] [PubMed] [Google Scholar]
- 126.Matic I, Ekiert D, Radman M, Kohiyama M. Generation of DNA-free Escherichia coli cells by 2-aminopurine requires mismatch repair and nonmethylated DNA. J Bacteriol. 2006;188:339–342. doi: 10.1128/JB.188.1.339-342.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Matic I, Radman M, Taddei F, Picard B, Doit C, Bingen E, Denamur E, Elion J. Highly variable mutation rates in commensal and pathogenic Escherichia coli [letter; comment] Science. 1997;277:1833–1834. doi: 10.1126/science.277.5333.1833. [DOI] [PubMed] [Google Scholar]
- 128.Matic I, Rayssiguier C, Radman M. Interspecies gene exchange in bacteria: the role of SOS and mismatch repair systems in evolution of species. Cell. 1995;80:507–515. doi: 10.1016/0092-8674(95)90501-4. [DOI] [PubMed] [Google Scholar]
- 129.Matson SW, Robertson AB. The UvrD helicase and its modulation by the mismatch repair protein MutL. Nucleic Acids Res. 2006;34:4089–4097. doi: 10.1093/nar/gkl450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McGraw BR, Marinus MG. Isolation and characterization of Dam+ revertants and suppressor mutations that modify secondary phenotypes of dam-3 strains of Escherichia coli K-12. Mol Gen Genet. 1980;178:309–315. doi: 10.1007/BF00270477. [DOI] [PubMed] [Google Scholar]
- 131.Mejean V, Claverys JP. Effect of mismatched base pairs on the fate of donor DNA in transformation of Streptococcus pneumoniae. Mol Gen Genet. 1984;197:467–471. doi: 10.1007/BF00329944. [DOI] [PubMed] [Google Scholar]
- 132.Mellon I, Champe GN. Products of DNA mismatch repair genes mutS and mutL are required for transcription-coupled nucleotide-excision repair of the lactose operon in Escherichia coli. Proc Natl Acad Sci USA. 1996;93:1292–1297. doi: 10.1073/pnas.93.3.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mendillo ML, V, Hargreaves V, Jamison JW, Mo AO, Li S, Putnam CD, Woods VL, Jr, Kolodner RD. A conserved MutS homolog connector domain interface interacts with MutL homologs. Proc Natl Acad Sci USA. 2009;106:22223–22228. doi: 10.1073/pnas.0912250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mendillo ML, Putnam CD, Kolodner RD. Escherichia coli MutS tetramerization domain structure reveals that stable dimers but not tetramers are essential for DNA mismatch repair in vivo. J Biol Chem. 2007;282:16345–16354. doi: 10.1074/jbc.M700858200. [DOI] [PubMed] [Google Scholar]
- 135.Meselson M. Methyl-Directed Repair of DNA Mismatches. In: Low KB, editor. The Recombination of Genetic Material. Academic Press; San Diego: 1988. pp. 91–113. [Google Scholar]
- 136.Miller JH. Spontaneous mutators in bacteria: insights into pathways of mutagenesis and repair. Annu Rev Microbiol. 1996;50:625–643. doi: 10.1146/annurev.micro.50.1.625. [DOI] [PubMed] [Google Scholar]
- 137.Miller JH. Perspective on mutagenesis and repair: the standard model and alternate modes of mutagenesis. Crit Rev Biochem Mol Biol. 2005;40:155–179. doi: 10.1080/10409230590954153. [DOI] [PubMed] [Google Scholar]
- 138.Modrich P. DNA mismatch correction. Annu Rev Biochem. 1987;56:435–466. doi: 10.1146/annurev.bi.56.070187.002251. [DOI] [PubMed] [Google Scholar]
- 139.Modrich P. Methyl-directed DNA mismatch correction. J Biol Chem. 1989;264:6597–6600. [PubMed] [Google Scholar]
- 140.Modrich P. Mechanisms and Biological Effects of Mismatch Repair. Annu Rev Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- 141.Modrich P. Mismatch repair, genetic stability and tumour avoidance. Philos Trans R Soc Lond B Biol Sci. 1995;347:89–95. doi: 10.1098/rstb.1995.0014. [DOI] [PubMed] [Google Scholar]
- 142.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 143.Monti MC, Cohen SX, Fish A, Winterwerp HH, Barendregt A, Friedhoff P, Perrakis A, Heck AJ, Sixma TK, van den Heuvel RH, Lebbink JH. Native mass spectrometry provides direct evidence for DNA mismatch-induced regulation of asymmetric nucleotide binding in mismatch repair protein MutS. Nucleic Acids Res. 2011;39:8052–8064. doi: 10.1093/nar/gkr498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Morel P, Hejna JA, Ehrlich SD, Cassuto E. Antipairing and strand transferase activities of E. coli helicase II (UvrD) Nucleic Acids Res. 1993;21:3205–3209. doi: 10.1093/nar/21.14.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Natrajan G, Lamers MH, Enzlin JH, Winterwerp HH, Perrakis A, Sixma TK. Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Res. 2003;31:4814–4821. doi: 10.1093/nar/gkg677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nowosielska A, Marinus MG. Cisplatin induces DNA double-strand break formation in Escherichia coli dam mutants. DNA Repair (Amst) 2005;4:773–781. doi: 10.1016/j.dnarep.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 147.Nowosielska A, Marinus MG. DNA mismatch repair-induced double-strand breaks. DNA Repair (Amst) 2008;7:48–56. doi: 10.1016/j.dnarep.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nowosielska A, Smith SA, Engelward BP, Marinus MG. Homologous recombination prevents methylation-induced toxicity in Escherichia coli. Nucleic Acids Res. 2006;34:2258–2268. doi: 10.1093/nar/gkl222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.O’Donnell M, Kuriyan J. Clamp loaders and replication initiation. Curr Opin Struct Biol. 2006;16:35–41. doi: 10.1016/j.sbi.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 150.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 151.Pang PP, Lundberg AS, Walker GC. Identification and characterization of the mutL and mutS gene products of Salmonella typhimurium LT2. J Bacteriol. 1985;163:1007–1015. doi: 10.1128/jb.163.3.1007-1015.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Parker BO, Marinus MG. Repair of DNA heteroduplexes containing small heterologous sequences in Escherichia coli. Proc Natl Acad Sci USA. 1992;89:1730–1734. doi: 10.1073/pnas.89.5.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pauly GT, Hughes SH, Moschel RC. Comparison of mutagenesis by O6-methyl- and O6-ethylguanine and O4-methylthymine in Escherichia coli using double-stranded and gapped plasmids. Carcinogenesis. 1998;19:457–461. doi: 10.1093/carcin/19.3.457. [DOI] [PubMed] [Google Scholar]
- 154.Petit MA, Dimpfl J, Radman M, Echols H. Control of large chromosomal duplications in Escherichia coli by the mismatch repair system. Genetics. 1991;129:327–332. doi: 10.1093/genetics/129.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pillon MC, Lorenowicz JJ, Uckelmann M, Klocko AD, Mitchell RR, Chung YS, Modrich P, Walker GC, Simmons LA, Friedhoff P, Guarne A. Structure of the endonuclease domain of MutL: unlicensed to cut. Mol Cell. 2010;39:145. doi: 10.1016/j.molcel.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pillon MC, Miller JH, Guarne A. The endonuclease domain of MutL interacts with the beta sliding clamp. DNA Repair (Amst) 2011;10:87–93. doi: 10.1016/j.dnarep.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pinto AV, Mathieu A, Marsin S, Veaute X, Ielpi L, Labigne A, Radicella JP. Suppression of homologous and homeologous recombination by the bacterial MutS2 protein. Mol Cell. 2005;17:113–120. doi: 10.1016/j.molcel.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 158.Pitsikas P, Patapas JM, Cupples CG. Mechanism of 2-aminopurine-stimulated mutagenesis in Escherichia coli. Mutat Res. 2004;550:25–32. doi: 10.1016/j.mrfmmm.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 159.Pluciennik A, Burdett V, Lukianova O, O’Donnell M, Modrich P. Involvement of the beta clamp in methyl-directed mismatch repair in vitro. J Biol Chem. 2009;284:32782–32791. doi: 10.1074/jbc.M109.054528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pluciennik A, Modrich P. Protein roadblocks and helix discontinuities are barriers to the initiation of mismatch repair. Proc Natl Acad Sci USA. 2007;104:12709–12713. doi: 10.1073/pnas.0705129104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Priebe SD, Hadi SM, Greenberg B, Lacks SA. Nucleotide sequence of the hexA gene for DNA mismatch repair in Streptococcus pneumoniae and homology of hexA to mutS of Escherichia coli and Salmonella typhimurium. J Bacteriol. 1988;170:190–196. doi: 10.1128/jb.170.1.190-196.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Prieto AI, Ramos-Morales F, Casadesus J. Bile-induced DNA damage in Salmonella enterica. Genetics. 2004;168:1787–1794. doi: 10.1534/genetics.104.031062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Prieto AI, Ramos-Morales F, Casadesus J. Repair of DNA damage induced by bile salts in Salmonella enterica. Genetics. 2006;174:575–584. doi: 10.1534/genetics.106.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Prudhomme M, Martin B, Mejean V, Claverys JP. Nucleotide sequence of the Streptococcus pneumoniae hexB mismatch repair gene: homology of HexB to MutL of Salmonella typhimurium and to PMS1 of Saccharomyces cerevisiae. J Bacteriol. 1989;171:5332–5338. doi: 10.1128/jb.171.10.5332-5338.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Prudhomme M, Mejean V, Martin B, Claverys JP. Mismatch repair genes of Streptococcus pneumoniae: HexA confers a mutator phenotype in Escherichia coli by negative complementation. J Bacteriol. 1991;173:7196–7203. doi: 10.1128/jb.173.22.7196-7203.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pukkila PJ, Peterson J, Herman G, Modrich P, Meselson M. Effects of high levels of DNA adenine methylation on methyl-directed mismatch repair in Escherichia coli. Genetics. 1983;104:571–582. doi: 10.1093/genetics/104.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rasmussen LJ, Samson L. The Escherichia coli MutS DNA mismatch binding protein specifically binds O(6)-methylguanine DNA lesions. Carcinogenesis. 1996;17:2085–2088. doi: 10.1093/carcin/17.9.2085. [DOI] [PubMed] [Google Scholar]
- 168.Rayssiguier C, Thaler DS, Radman M. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature. 1989;342:396–401. doi: 10.1038/342396a0. [DOI] [PubMed] [Google Scholar]
- 169.Roberts JW, Shankar S, Filter JJ. RNA polymerase elongation factors. Annu Rev Microbiol. 2008;62:211–233. doi: 10.1146/annurev.micro.61.080706.093422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Roth JR, Kugelberg E, Reams AB, Kofoid E, Andersson DI. Origin of mutations under selection: the adaptive mutation controversy. Annu Rev Microbiol. 2006;60:477–501. doi: 10.1146/annurev.micro.60.080805.142045. [DOI] [PubMed] [Google Scholar]
- 171.Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J Mol Biol. 1968;31:291–304. doi: 10.1016/0022-2836(68)90445-2. [DOI] [PubMed] [Google Scholar]
- 172.Rydberg B. Bromouracil mutagenesis in Escherichia coli evidence for involvement of mismatch repair. Mol Gen Genet. 1977;152:19–28. doi: 10.1007/BF00264935. [DOI] [PubMed] [Google Scholar]
- 173.Rydberg B. Bromouracil mutagenesis and mismatch repair in mutator strains of Escherichia coli. Mutat Res. 1978;52:11–24. doi: 10.1016/0027-5107(78)90091-x. [DOI] [PubMed] [Google Scholar]
- 174.Rye PT, Delaney JC, Netirojjanakul C, Sun DX, Liu JZ, Essigmann JM. Mismatch repair proteins collaborate with methyltransferases in the repair of O(6)-methylguanine. DNA Repair (Amst) 2008;7:170–176. doi: 10.1016/j.dnarep.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schaaper RM. Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J Biol Chem. 1993;268:23762–23765. [PubMed] [Google Scholar]
- 176.Schaaper RM, Radman M. The extreme mutator effect of Escherichia coli mutD5 results from saturation of mismatch repair by excessive DNA replication errors. EMBO J. 1989;8:3511–3516. doi: 10.1002/j.1460-2075.1989.tb08516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sedgwick B. Repairing DNA-methylation damage. Nat Rev Mol Cell Biol. 2004;5:148–157. doi: 10.1038/nrm1312. [DOI] [PubMed] [Google Scholar]
- 178.Selby CP, Sancar A. Molecular mechanism of transcription-repair coupling. Science. 1993;260:53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 179.Shanabruch WG, Behlau I, Walker GC. Spontaneous mutators of Salmonella typhimurium LT2 generated by insertion of transposable elements. J Bacteriol. 1981;147:827–835. doi: 10.1128/jb.147.3.827-835.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Shanabruch WG, Rein RP, Behlau I, Walker GC. Mutagenesis, by methylating and ethylating agents, in mutH, mutL, mutS, and uvrD mutants of Salmonella typhimurium LT2. J Bacteriol. 1983;153:33–44. doi: 10.1128/jb.153.1.33-44.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shenoy S, Ehrlich KC, Ehrlich M. Repair of thymine.guanine and uracil.guanine mismatched base-pairs in bacteriophage M13mp18 DNA heteroduplexes. J Mol Biol. 1987;197:617–626. doi: 10.1016/0022-2836(87)90468-2. [DOI] [PubMed] [Google Scholar]
- 182.Siegel EC. Ultraviolet-sensitive mutator strain of Escherichia coli K-12. J Bacteriol. 1973;113:145–160. doi: 10.1128/jb.113.1.145-160.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Siegel EC, Vaccaro KK. The reversion of trp framshift mutations in mut, polA, lig and dnaE mutant strains of Escherichia coli. Mutat Res. 1978;50:9–17. [Google Scholar]
- 184.Simmons LA, Davies BW, Grossman AD, Walker GC. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol Cell. 2008;29:291–301. doi: 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Simmons LA, Davies BW, Grossman AD, Walker GC. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol Cell. 2008;29:291–301. doi: 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Skopek TR, Hutchinson F. Frameshift mutagenesis of lambda prophage by 9-aminoacridine, proflavin and ICR-191. Mol Gen Genet. 1984;195:418–423. doi: 10.1007/BF00341442. [DOI] [PubMed] [Google Scholar]
- 187.Slack A, Thornton PC, Magner DB, Rosenberg SM, Hastings PJ. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet. 2006;2:e48. doi: 10.1371/journal.pgen.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Smith BT, Grossman AD, Walker GC. Visualization of mismatch repair in bacterial cells. Mol Cell. 2001;8:1197–1206. doi: 10.1016/s1097-2765(01)00402-6. [DOI] [PubMed] [Google Scholar]
- 189.Sohail A, Lieb M, Dar M, Bhagwat AS. A gene required for very short patch repair in Escherichia coli is adjacent to the DNA cytosine methylase gene. J Bacteriol. 1990;172:4214–4221. doi: 10.1128/jb.172.8.4214-4221.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Springer B, Sander P, Sedlacek L, Hardt WD, Mizrahi V, Schar P, Bottger EC. Lack of mismatch correction facilitates genome evolution in mycobacteria. Mol Microbiol. 2004;53:1601–1609. doi: 10.1111/j.1365-2958.2004.04231.x. [DOI] [PubMed] [Google Scholar]
- 191.Stambuk S, Radman M. Mechanism and control of interspecies recombination in Escherichia coli. I. Mismatch repair, methylation, recombination and replication functions. Genetics. 1998;150:533–542. doi: 10.1093/genetics/150.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Storz G, Altuvia S, Wassarman KM. An abundance of RNA regulators. Annu Rev Biochem. 2005;74:199–217. doi: 10.1146/annurev.biochem.74.082803.133136. [DOI] [PubMed] [Google Scholar]
- 193.Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Su SS, Modrich P. Escherichia coli mutS-encoded protein binds to mismatched DNA base pairs. Proc Natl Acad Sci USA. 1986;83:5057–5061. doi: 10.1073/pnas.83.14.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sundin GW, Weigand MR. The microbiology of mutability. FEMS Microbiol Lett. 2007;277:11–20. doi: 10.1111/j.1574-6968.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- 196.TREFFERS HP, Spinelli V, Belser NO. A Factor (or Mutator Gene) Influencing Mutation Rates in Escherichia coli. Proc Natl Acad Sci USA. 1954;40:1064–1071. doi: 10.1073/pnas.40.11.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tsui HC, Feng G, Winkler ME. Negative regulation of mutS and mutH repair gene expression by the Hfq and RpoS global regulators of Escherichia coli K-12. J Bacteriol. 1997;179:7476–7487. doi: 10.1128/jb.179.23.7476-7487.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tsutakawa SE, Jingami H, Morikawa K. Recognition of a TG mismatch: the crystal structure of very short patch repair endonuclease in complex with a DNA duplex. Cell. 1999;99:615–623. doi: 10.1016/s0092-8674(00)81550-0. [DOI] [PubMed] [Google Scholar]
- 199.Tsutakawa SE, Morikawa K. The structural basis of damaged DNA recognition and endonucleolytic cleavage for very short patch repair endonuclease. Nucleic Acids Res. 2001;29:3775–3783. doi: 10.1093/nar/29.18.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tsutakawa SE, Muto T, Kawate T, Jingami H, Kunishima N, Ariyoshi M, Kohda D, Nakagawa M, Morikawa K. Crystallographic and functional studies of very short patch repair endonuclease. Mol Cell. 1999;3:621–628. doi: 10.1016/s1097-2765(00)80355-x. [DOI] [PubMed] [Google Scholar]
- 201.Veaute X, Delmas S, Selva M, Jeusset J, Le CE, Matic I, Fabre F, Petit MA. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 2005;24:180–189. doi: 10.1038/sj.emboj.7600485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Villarroya M, Perez-Roger I, Macian F, Armengod ME. Stationary phase induction of dnaN and recF, two genes of Escherichia coli involved in DNA replication and repair. EMBO J. 1998;17:1829–1837. doi: 10.1093/emboj/17.6.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Viswanathan M, Burdett V, Baitinger C, Modrich P, Lovett ST. Redundant exonuclease involvement in Escherichia coli methyl-directed mismatch repair. J Biol Chem. 2001;276:31053–31058. doi: 10.1074/jbc.M105481200. [DOI] [PubMed] [Google Scholar]
- 204.Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wagner R, Meselson M. Repair tracts in mismatched DNA heteroduplexes. Proc Natl Acad Sci USA. 1976;73:4135–4139. doi: 10.1073/pnas.73.11.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Walker GC. Mismatch Repair. In: Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger TE, editors. DNA Repair and Mutagenesis. 2. American Society for Microbiology; Washington DC: 2006. pp. 389–447. [Google Scholar]
- 207.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 208.Wang TC, Smith KC. Inviability of dam recA and dam recB cells of Escherichia coli is correlated with their inability to repair DNA double-strand breaks produced by mismatch repair. J Bacteriol. 1986;165:1023–1025. doi: 10.1128/jb.165.3.1023-1025.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Welsh KM, Lu AL, Clark S, Modrich P. Isolation and characterization of the Escherichia coli mutH gene product. J Biol Chem. 1987;262:15624–15629. [PubMed] [Google Scholar]
- 210.Westmoreland J, Porter G, Radman M, Resnick MA. Highly mismatched molecules resembling recombination intermediates efficiently transform mismatch repair proficient Escherichia coli. Genetics. 1997;145:29–38. doi: 10.1093/genetics/145.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Witkin EM. Time, temperature, and protein synthesis: a study of ultraviolet-induced mutation in bacteria. Cold Spring Harb Symp Quant Biol. 1956;21:123–140. doi: 10.1101/sqb.1956.021.01.011. [DOI] [PubMed] [Google Scholar]
- 212.Witkin EM. Radiation-induced mutations and their repair. Science. 1966;152:1345–1353. doi: 10.1126/science.152.3727.1345. [DOI] [PubMed] [Google Scholar]
- 213.Witkin EM, Sicurella NA. Pure clones of lactose-negative mutants obtained in Escherichia coli after treatment with 5-bromouracil. J Mol Biol. 1964;8:610–613. doi: 10.1016/s0022-2836(64)80017-6. [DOI] [PubMed] [Google Scholar]
- 214.Worth L, Clark S, Radman M, Modrich P. Mismatch repair proteins MutS and MutL inhibit RecA-catalyzed strand transfer between diverged DNAs. Proc Natl Acad Sci USA. 1994;91:3238–3241. doi: 10.1073/pnas.91.8.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wu TH, Marinus MG. Dominant negative mutator mutations in the mutS gene of Escherichia coli. J Bacteriol. 1994;176:5393–5400. doi: 10.1128/jb.176.17.5393-5400.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wyrzykowski J, Volkert MR. The Escherichia coli methyl-directed mismatch repair system repairs base pairs containing oxidative lesions. J Bacteriol. 2003;185:1701–1704. doi: 10.1128/JB.185.5.1701-1704.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Yang W. Lessons learned from UvrD helicase: mechanism for directional movement. Annu Rev Biophys. 2010;39:367–385. doi: 10.1146/annurev.biophys.093008.131415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zahrt TC, Maloy S. Barriers to recombination between closely related bacteria: MutS and RecBCD inhibit recombination between Salmonella typhimurium and Salmonella typhi. Proc Natl Acad Sci USA. 1997;94:9786–9791. doi: 10.1073/pnas.94.18.9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zdraveski ZZ, Mello JA, Marinus MG, Essigmann JM. Multiple pathways of recombination define cellular responses to cisplatin. Chem Biol. 2000;7:39–50. doi: 10.1016/s1074-5521(00)00064-8. [DOI] [PubMed] [Google Scholar]
- 220.Zhao J, Winkler ME. Reduction of GC --> TA transversion mutation by overexpression of MutS in Escherichia coli K-12. J Bacteriol. 2000;182:5025–5028. doi: 10.1128/jb.182.17.5025-5028.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]