

Abstract
Smith–Lemli–Opitz Syndrome (SLOS) is a congenital, autosomal recessive metabolic and developmental disorder caused by mutations in the enzyme which catalyzes the reduction of 7-dehydrocholesterol (7DHC) to cholesterol. Herein we show that dermal fibroblasts obtained from SLOS children display increased basal levels of LC3B-II, the hallmark protein signifying increased autophagy. The elevated LC3B-II is accompanied by increased beclin-1 and cellular autophagosome content. We also show that the LC3B-II concentration in SLOS cells is directly proportional to the cellular concentration of 7DHC, suggesting that the increased autophagy is caused by 7DHC accumulation secondary to defective DHCR7. Further, the increased basal LC3B-II levels were decreased significantly by pretreating the cells with antioxidants implicating a role for oxidative stress in elevating autophagy in SLOS cells. Considering the possible source of oxidative stress, we examined mitochondrial function in the SLOS cells using JC-1 assay and found significant mitochondrial dysfunction compared to mitochondria in control cells. In addition, the levels of PINK1 which targets dysfunctional mitochondria for removal by the autophagic pathway are elevated in SLOS cells, consistent with mitochondrial dysfunction as a stimulant of mitophagy in SLOS. This suggests that the increase in autophagic activity may be protective, i.e., to remove dysfunctional mitochondria. Taken together, these studies are consistent with a role for mitochondrial dysfunction leading to increased autophagy in SLOS pathophysiology.
Keywords: 7-Dehydrocholesterol, β-hydroxysterol-Δ7-reductase (DHCR7), Birth defects, Lysosomes, Mitophagy
1. Introduction
Smith–Lemli-Opitz Syndrome (SLOS) is an autosomal recessive disorder of cholesterol biosynthesis caused by mutations in the gene that encodes 3β-hydroxysterol-Δ7-reductase (DHCR7), the final enzyme in the cholesterol biosynthetic pathway. Affected individuals typically exhibit multiple anatomic malformations and intellectual disability, though the phenotypic expression of this condition is extremely variable. The clinical features of SLOS are thought to be primarily related to cholesterol deficiency and/or accumulation of cholesterol precursors and their metabolites. The primary metabolite that accumulates in SLOS is the immediate precursor to cholesterol in the Kandutsch–Russell cholesterol synthesis pathway, 7-dehydrocholesterol (7-DHC) [1], [2]. 7-DHC contains a double bond at carbon seven, which is reduced by DHCR7 to form unesterified cholesterol, but is otherwise structurally identical to cholesterol (Fig. 1). Tint et al. [2] first described the biochemical defect in SLOS patients by virtue of the accumulation of 7-DHC in the plasma of affected individuals [2]. This finding has become diagnostic for SLOS and has led to the detailed description of a large variety of DHCR7 mutations with over 154 mutations reported to date which include 130 missense, 8 nonsense, 8 deletions, 2 insertions, 1 indel, and 5 splice site mutations [3] and which may explain the large phenotypic variation observed for this disorder [4], [5]. In contrast with the genetics of SLOS, relatively little work has been done to address the cell biology of this debilitating disease. The discovery that 7-DHC accumulation might participate in the pathogenesis of SLOS stems from the early work of Honda et al. [6] who demonstrated that 7-DHC accumulates in skin fibroblasts cultured from patients with SLOS. This observation was confirmed by Wassif et al. [7] and extended by us in studies demonstrating that cell membranes from SLOS fibroblasts contain 7-DHC which alters membrane structure/function [8], [9].
Fig. 1.
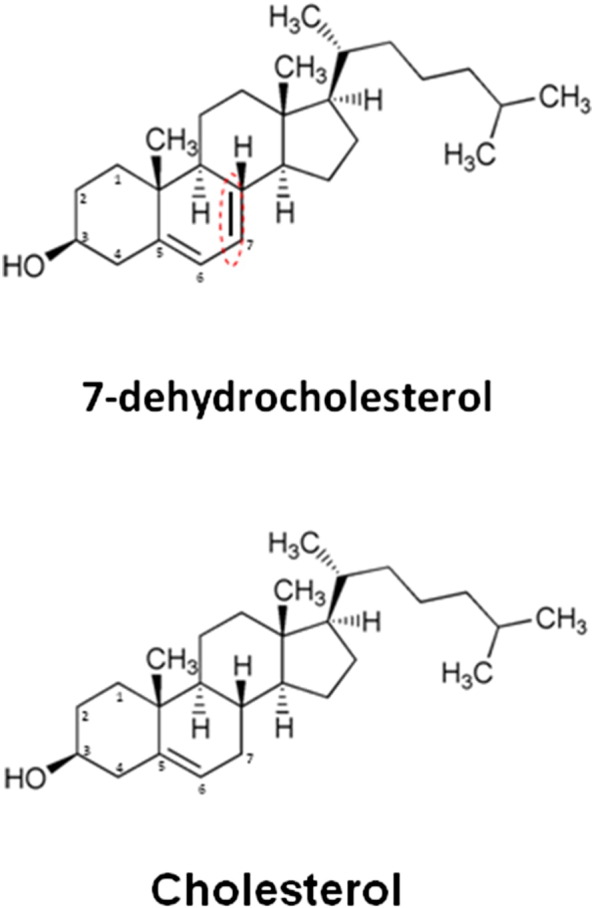
Chemical structures of cholesterol and 7- dehydrocholesterol (7DHC). Note that the only difference between the two molecules is the presence of a double bond between carbons 7 and 8 in 7-DHC.
Autophagy is an ancient cellular degradation pathway for long-lived and excess proteins, lipids, nucleotides, etc., along with unneeded or damaged cellular organelles including mitochondria, peroxisomes and endoplasmic reticulum. The autophagosomes are formed from a double membrane precursor phagophore and delivers their cargo to lysosomes by fusion where they are degraded to biologically active monomers, e.g., amino acids for cellular recycling [10]. In this way, autophagy continually “refreshes” the cytoplasm and thus plays a homeostatic role which is particularly important in terminally differentiated cells like neurons. A well defined set of autophagy-related genes (ATG 1–35) are required for autophagy and its related processes which are highly conserved among eukaryotes, and numerous studies have revealed a variety of physiologic roles of autophagy [11]. Autophagic activity has both selective and nonselective features which vary by how substrate cargo is delivered to the lysosome. For example, the chaperone-mediated class of autophagy is highly selective targeting proteins containing a KFERQ motif while the microautophagy class is largely nonselective and involves continuous degradation of cytosolic materials close to lysosomes by inward budding of the lysosomal membrane. Lastly, macroautophagy, the most widely studied autophagy class, can be largely selective specifically targeting defective proteins and organelles for engulfment into the phagophore which fuses with lysosomes for cargo degradation. However, microautophagy can also be nonselective, for example during nutrient starvation whereby autophagosomes envelop random cytosolic proteins and organelles for lysosomal degradation to re-supply the cell with essential amino acids and carbohydrates for protein, energy and neosynthesis. An important function of microautophagy is cargo-specific and responsible for the clearance of defective organelles and its specificity has been delineated along functional lines. Hence, “mitophagy” clears dysfunctional mitochondria, “pexophagy” clears peroxisomes, “xenophagy” clears invading bacteria, etc. In this way, selective autophagy serves as an important and essential cellular quality control preserving the steady-state content of functional organelles thereby maintaining a healthy cytosolic milieu. Importantly, while autophagy was initially thought to be a pro-survival mechanism, it is now appreciated that defective autophagy contributes to cellular pathology, notably in cancer, neurodegenerative diseases and defective immunity [10].
A recent report from our lab showed impairment of IP3 synthesis in SLOS cells [9]. Because impaired IP3 synthesis activates autophagy [12], [13], we set out to determine whether SLOS cells might demonstrate enhanced autophagy. Using skin fibroblasts obtained from children with confirmed diagnosis of SLOS, we found that numerous markers of autophagy are elevated under basal conditions including the hallmark protein of autophagy LC3B-II as well beclin-1 and increased cytoplasmic autophagosome inclusions. Taken together, we present for the first time data supporting the notion that autophagy is enhanced in SLOS cells, and that dysfunctional mitochondria likely stimulate mitophagy in SLOS cells. However, a defect in autophagic flux may also exist in SLOS cells thereby preventing the efficient clearance of defective mitochondria contributing further to cytopathology in children with SLOS.
2. Methods
Beclin-1 and LC3B rabbit polyclonal antibodies were from Cell Signaling (Danvers, MA). Actin mouse monoclonal antibody was from Chemicon (Billerica, MA). AY9944 (trans-1,4 bis-(2-dichlorobenzyl-aminomethyl)cyclohexane dihydrochloride) (1 μM as a working solution), N-acetylcysteine (NAC) and monodansylcadaverine (MDC) were from Sigma (St. Louis, MO). Lipoprotein-deficient serum was from Cocalico Biologicals, Inc. (Reamstown, PA). LC3-GFP was from Addgene and transfast transfection kits were from Promega. Lipofectamine 2000 and the antioxidant cocktail supplement B27 (50 × stock solution) were from Invitrogen (Grand Island, NY. JC-1 Mitochondrial Membrane Potential Assay Kit was from Cayman Chemical Company (Ann Arbor, MI).
2.1. Cell lines and culture
All fibroblast cell lines were obtained from our pediatric collection at OHSU (RDS PI). SLOS fibroblast cell lines were established from skin biopsy specimens obtained from SLOS patients with confirmed biochemical and genetic diagnosis. Table 1 lists the specific genotypes in fibroblasts isolated from children with SLOS. Control fibroblast cell lines were established from skin specimens obtained from healthy age-matched individuals during reconstructive surgery. The OHSU Institutional Review Board approved this study and a written consent to use their cell lines was obtained from the legal guardians of all subjects. All cell lines were maintained at 37 °C, 5% CO2 in DMEM supplemented with 10% FBS (Atlanta Biologicals), nonessential amino acids, 2 mM glutamine and a mix of penicillin (100 U) and streptomycin (100 U). MIA PaCa-2 cells were kindly provided by Drs Galina and Arafat at Thomas Jefferson University and cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 U penicillin and 100 U streptomycin/ml) and were used for their ease of transfection. For each experiment, both control and SLOS cells were incubated in lipoprotein-deficient serum (LPDS; Millipore) for 5–7 days to remove exogenous sources of cholesterol prior to sample collection as this results in elevated 7DHC and reduced cholesterol in SLOS cells similar to that which occurs in vivo [9], [14].
Table 1.
Patient genotypes. For each patient a clinical severity score was calculated using the published anatomical measure of severity [50], [51].
| Patient | Clinical severity score | Nucleotide | Amino acid | Nucleotide |
|---|---|---|---|---|
| 1 | 17 | c.529T>C | p.Trp177Arg | c.724C>T |
| 2 | 11 | c.906C>G | p.Phe302Leu | c.1409T>A |
| 3 | 44 | c.278C>T | p.Thr93Met | c.964-1G>C |
| 4 | 17 | c.461C>T | p.Thr154Met | c.292C>T |
| 5 | 30 | c.1384T>C | p.Tyr462His | c.964-1G>C |
| 6 | 20 | c.278C>T | p.Thr93Met | c.976G>T |
| 7 | 10 | c.1349G>T | p.Arg450Leu | c.964-1G>C |
| 8 | 11 | c.470C>T | p.Ala247Val | c.964-1G>C |
| 9 | 33 | c.1228G>A | P.Gly410Ser | Same mutation |
| 10 | 5 | c.1139G>A | p.Cys380Tyr | c.964-1G>C |
2.2. Monodansylcadaverine (MDC) staining
MDC is a fluorescent dye that preferentially stains autophagic vacuoles [15]. Cells were seeded onto 96-well plates, maintained overnight in DMEM containing 10% LPDS followed by incubation with 0.1 mM MDC in PBS for 1 h, and washed three times with PBS. Fluorescence was monitored using the GENios plate reader (Tecan US, Research Triangle Park, NC) (λex 335 nm, λem 525 nm). Cell protein concentrations were determined using the Bradford assay and used to normalize readings.
2.3. LC3-GFP transfection
MIA PaCa-2 cells were seeded onto six well plates one day prior to transfection. 1 μg LC3-GFP and 4 μl transfast reagent were used for each well. GFP fluorescence was examined two days after transfection. Fibroblasts were transfected by Xfect (following the vendor's guidelines) followed by growing cells on cover slips and fixing them in 4% paraformaldehyde at pH 7.4. Images were taken on a Zeiss LSM 510 confocal microscope using a X63 oil-immersion lens.
2.4. Western blotting
Cells were grown in 6-well plates, washed twice with PBS and lysed with 50 mM Tris–HCl buffer (pH 6.8) containing 20% glycerol, 1% sodium dodecyl sulfate (SDS), and a protease inhibitor cocktail (Sigma, St. Louis, MO). Protein concentration was determined using the DC protein assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein (40 μg) were subjected to SDS-PAGE for 1 h at 200 V and the resolved proteins were transferred to PVDF membranes at 100 V for 1 h. Membranes were blocked for 1 h in PBS containing 5% bovine serum albumin (BSA) and 0.1% Tween 20 (blocking buffer), then incubated overnight at 4 °C with the primary antibodies [anti-LC3B from Cell Signaling (Boston, MA) and anti-actin from Sigma at 1:1000 dilution. After several washings, the membranes were incubated with corresponding secondary antibodies (anti-rabbit-Alexa Fluor680 or anti-mouse-Alexa Fluor594) from Life Technologies (Grand island, NY) at 1:5000 dilution for 1 h. Bands were visualized and quantitated by scanning with the FluoChemi Q imager system (Protein Simple, Santa Clara, CA).
2.5. Immunostaining
SLOS and control cells were cultured on cover slips and fixed using 10% formalin buffer. After 30 min of blocking in 3% BSA in PBS, the cover slips were incubated overnight at 4 °C with primary antibodies for LC3B & actin at 1:200 or primary antibodies for Parkin or Pink-1 (Abcam, Cambridge, MA) at 1:300 overnight at 4 °C. Cells were washed three times in PBS and incubated with corresponding secondary antibodies (Cy3 or FITC conjugated anti-rabbit or anti-mouse antibodies) at 1:400 for 1 h. Cells were washed again with PBS and mounted in a medium containing 4′-6-diamidino-2-phenylindole (DAPI) to stain the nuclei (Vector Laboratory, Burlington, Ontario, Canada). Slides were viewed using a Confocal Laser Scanning Biological Microscope (Olympus, Center Valley, PA). Images were captured using Fluoview FV1000 software. A negative control to confirm the specificity of the immunostaining was prepared using non-immune goat serum in place of the primary antibody.
2.6. Cellular sterol assay
Both control and SLOS cells were cultured in LPDS medium to confluence. Cells were collected in 1 × PBS and pelleted by centrifugation at 500 ×g for 10 min. Cell pellets were solubilized in 1.0 ml of 0.05% SDS. One aliquot was used for protein measurement (BCA™ Protein Assay Kit, Pierce, Rockford IL). The sterols were extracted from the remaining cell pellet using chloroform:methanol (2:1), after the internal standard (epicoprostanol, EPIC, Sigma C-2882) had been added. The solvent was removed by evaporation under nitrogen at 40 °C. Sterols were saponified by the addition of ethanol/KOH and incubated at 37 °C for 1 h. Sterols were extracted 2 times with hexane. The combined hexane extracts were evaporated to dryness and derivatized with BSTFA at 80 °C for 30 min. Concentrations of the trimethylsilyl ether derivatives of cholesterol and 7-dehydrocholesterol were measured using a ZB1701 column (30 m, 0.25 mm ID, 0.25 μm film thickness: Phenomenex, Torrance, CA) and selected-ion mass spectrometry (Agilent GC 6890N and MS 5975). The mass spectra data were collected in selected ion mode m/z = 355 and 370 for EPIC; m/z = 329 and 458 for cholesterol, and m/z = 325 and 351 for 7-dehydrocholesterol. Internal standard (EPIC) and authentic standards of cholesterol and 7-dehydrocholesterol (Steraloids, Newport, RI C3000-000) were used for calibration.
2.7. JC-1 mitochondrial membrane potential assay
Mitochondrial membrane potential was assayed using JC-1 (tetraethylbenzimidazolylcarbocyanine iodide) and the Pierce Mitochondrial Membrane Potential Assay kit according to the manufacturer's protocol (Abcam, Cambridge, MA.). Briefly, cells were cultured in 3-cm glass bottom dishes for microscopy studies and 96-well black culture plates for quantitative fluorescence measurements. For microscopy, 100 μl of the JC-1 staining solution was added per ml of culture medium for a 30-minute incubation followed by replacement with fresh medium. Photographic images were then taken using an Olympus confocal microscope. In cells with normal mitochondrial membrane potentials, JC-1 forms aggregates that are detected as red fluorescence, whereas cells with impaired mitochondrial membrane potential, the dye stays in a monomeric form that is detected as green fluorescence. For quantitative fluorescence studies, 5 μl of the JC-1 staining solution was used per 100 μl of medium with a 30-minute incubation. The cells were then washed twice and lysed with 100 μl of the kit assay buffer. Fluorescence measurements were performed with a SpectraMax M3 Microplate Reader (Molecular Devices, Sunnyvale, CA) set at 560 nm (λex) and 595 nm (λem) (red fluorescence) and at 485 nm (λex) and 535 nm (λem) (green fluorescence). The green-to-red fluorescence ratio was used as a measure of mitochondrial membrane potential [16], [17].
2.8. Isolation of neural stem cells from SLOS transgenic mice
Brains from wild-type mice harboring the SLOS mutation Dhcr7Δ3–5/Δ3–5 and heterozygous mice were a kind gift of Dr. F.D. Porter of the NIH [14]. Whole brains were subjected to papain digestion and the resulting cell suspension was cultured in Neurobasal A medium (Invitrogen, cat. # 10888-022) supplemented with l-glutamine, penicillin/streptomycin, B-27® Supplement (Gibco, cat. # 12587-010), human recombinant EGF (Millipore, cat. #: 01-107) and human recombinant FGF (Prepotech, cat. #: 100-1B) (complete Neurobasal A). After a few days, neurospheres formed and the cells were transferred to tissue culture dishes coated with poly-dl-ornithine and further cultured in a complete Neurobasal A medium. The resulting adherent cells were subjected to whole cell protein isolation and the protein was probed for LC3B-II using immunoblots as described above.
2.9. Dhcr7-deficient neuroblastoma cell line
The mouse neuroblastoma cell line Neuro2a was purchased from American Type Culture Collection (Rockville, MD). For the generation of Dhcr7-deficient Neuro2a cells, Neuro2a cells were cultured for 2 days before transfections. Cells were transfected with three different Dhcr7 siRNA oligonucleotides (Qiagen) using a Nucleofector instrument and Nucleofector Kit V (Amaxa GmbH, Cologne, Germany) optimized for use with Neuro2a cells. Briefly, 2 × 106 cells were resuspended in 100 μl transfection buffer, siRNA was added, and cells were electroporated using program T-24. The cells were grown for 24 h following transfection, and the expression of Dhcr7 was monitored by quantitative RT-PCR. To establish stable down-regulation of Dhcr7, after transfection of cells with pGIPZ plasmids, cells were grown in the presence of puromycin. The cell lines were maintained in DMEM supplemented with l-glutamine, 10% fetal bovine serum (FBS; Thermo Scientific HyClone, Logan, UT), and penicillin/streptomycin at 37 °C and 5% CO2. All cells were subcultured once a week, and the culture medium was changed every two days. For LC3 expression measurements, cells were grown for 48 h in cholesterol-deficient serum (Thermo Scientific HyClone Lipid Reduced FBS). The harvested cells were washed with cold PBS (pH 7.4) and centrifuged at 1000 rpm for 10 min at 4 °C. Cells were lysed in RIPA lysis buffer (with protease and phosphatase inhibitors), and protein measurement was done using Bio-Rad DC Protein Assay kit. The Western blotting was done using anti-LC3A/B/C (Abcam) and anti-LC3B (Abcam) antibodies. The expression level of the proteins against which the primary antibodies were directed was determined using enhanced chemiluminescence reagent (SuperSignal West Pico Chemiluminescent Substrate, Pierce) and exposing membranes to X-ray film (Kodak X-omat). Bands were analyzed using ScionImage 4.02b (ScionCorp, Frederick, MD, USA).
2.10. Statistics
All data are reported as the mean ± SEM. Statistical analyses were conducted using Student's two-tailed t test (paired or unpaired where appropriate) for comparisons between groups and ANOVA with Bonferroni's multiple comparison test using SPSS v11.0 software. Data with p ≤ 0.05 were considered statistically significant.
3. Results
3.1. Increased autophagosome formation in SLOS
LC3B-II is a signature protein of elevated autophagy in mammalian cells [18]. Using a LC3-GFP transfection protocol increased autophagosome formation was clearly visualized in SLOS cells (Fig. 2, panel A). The green fluorescent puncta indicates the location of LC3B-II protein in control and SLOS cells, with the SLOS cells showing greater puncta accumulation compared to control cells. In addition, MDC staining was performed to evaluate the abundance of autophagic vacuoles in SLOS and control cells as illustrated in Fig. 2B. The quantitative result of MDC staining in both cell lines demonstrates a 3.2-fold (p < 0.05) increase of MDC staining in SLOS cells. The results in Fig. 2A & B are consistent with an increased accumulation of autophagosomes in SLOS cells.
Fig. 2.

Increased autophagy in SLOS human dermal fibroblasts. (A) LC3-GFP transfection in control and SLOS fibroblasts to show autophagosome formation. (B) MDC staining of control and SLOS fibroblasts, normalized to total protein amounts (mean ± SEM). *p < 0.05, n = 5.
3.2. Up-regulated LC3B-II in SLOS cells
There are three forms of LC3 in mammalian cells: Pro LC3B (60KD), LC3B-I (17KD) and LC3B-II (13KD). LC3B-I is conjugated to phospholipid to form LC3-II, which is associated with the autophagosome membrane. Therefore, LC3B-II is widely used as an autophagy marker [19]. Western-blot analysis and confocal microscopy were used to assess LC3B-II expression and localization. Fig. 3A is a representative Western-blot image demonstrating LC3B-I (upper band), LC3B-II (lower band), and actin (bottom band) as internal loading controls. Fig. 3B shows the quantitative results of normalized LC3B-II expression in fibroblasts from 4 SLOS and 4 healthy control patients in which LC3B-II is increased 1.5-fold in SLOS cells (p < 0.05). In Fig. 3C are representative confocal images showing LC3B-II as red fluorescence, actin as green fluorescence and nuclei as DAPI blue fluorescence. LC3B-II appears primarily perinuclear in location and is increased in SLOS cells.
Fig. 3.

Increased LC3B-II and beclin-2 protein expression in SLOS. LC3B-II: Cell lysates from control (lanes 1–4) and SLOS human fibroblasts (lanes 5–8) were collected from LPDS cultured cells. (A) Representative Western blot, red fluorescent for LC3 and green fluorescent for β-actin. (B) Quantitation of LC3B-II amount by normalizing with β-actin, n = 7, p < 0.05. (C) Confocal image of LC3B localization in control and SLOS fibroblasts. Red fluorescence for LC3, green for actin and blue for nuclei. Beclin-1: Cell lysates from control (lanes 1–4) and SLOS human fibroblasts (lanes 5–8) were collected from LPDS cultured cells. (D) Representative Western blot, red fluorescent for beclin and green fluorescent for β-actin. (E) Quantitation bar graph of beclin expression, n = 6, p < 0.05.
3.3. Increased beclin-1 in SLOS
Another important protein in autophagy regulation is beclin-1 which is part of PI3kinase complex which stimulates autophagy [18]. Since our previous publication demonstrated a decrease in IP3 in SLOS fibroblasts [9], we measured the expression of beclin-1 in control and SLOS cells. Beclin-1 expression (upper band) was increased in SLOS cells as illustrated in Fig. 3D. Fig. 3E is the quantitative analysis of the Western-blot data showing a 2.7-fold increase in beclin-1 expression in SLOS cells compared to healthy control cells, consistent with our previous observation of reduced IP3 levels in the SLOS cells.
3.4. Correlation of the cell LC3B-II level with cellular 7DHC concentration
The results in Fig. 2, Fig. 3 strongly support the conclusion that autophagy is activated in SLOS cells when compared to controls. Because we hypothesized that this activation was related to the accumulation of 7DHC in cells, the primary metabolic defect in SLOS cells [1], [2], we evaluated the relationship between LC3B-II protein and 7DHC cellular concentrations & unesterified cholesterol level in SLOS and control cells. A total of 16 cell samples were collected for Western-blot to determine the expression level of LC3B-II and cellular sterol assay to determine 7DHC & cholesterol concentration. Fig. 4 shows a significant positive correlation (r2 = 0.6907, p < 0.01) between LC3B-II level and cellular 7DHC concentration in panel A, while no clear correlation between LC3B-II level and cellular cholesterol concentration in panel B. In this experiment, the cellular concentration of 7DHC is comparable to that reported in SLOS cells previously published by us and others [9], [14], [20].
Fig. 4.

Correlation of LC3B-II expression with cellular 7DHC and cholesterol concentration in SLOS and control cells. A total of 16 cell samples were collected from 4 SLOS cell lines and 4 control cell lines under two culture conditions (with FBS or with LPDS). For each cell sample, half was used for Western-blot to determine the expression level of LC3B-II and the other half was used for 7DHC and cholesterol measurement. Panel A showed the expression level of autophagy protein LC3B-II correlating with cellular 7DHC concentration, r2 = 0.6907. Panel B showed no correlation between the expression of LC3B-II and cellular cholesterol concentration.
Fig. 4B shows the lack of correlation between unesterified cholesterol and LC3B-II and thus supports the suggestion that LC3B-II correlates with cellular 7DHC concentration rather than with cellular cholesterol concentration.
3.5. Induced autophagy formation by inhibition of DHCR7 and the protective effect of antioxidants
The correlation between 7DHC and LC3B-II levels prompted us to use the specific DHCR7 inhibitor, AY9944 [21] to increase 7DHC levels followed by the assessment of autophagosome formation and LC3B-II levels. Fig. 5A shows MIA PaCa-2 cells transfected with LC3-GFP with and without AY9944 treatment showing an increase in autophagosomes in AY9944 treated cells. Immunoblots revealed an increased LC3B-II expression compared to cells with no AY9944 treatment (Fig. 5B). This result is consistent with the correlation between 7DHC and LC3B-II. Furthermore, the antioxidant cocktail/supplement/ B27 had a protective effect in control cells treated with AY9944 (Fig. 5, panel C), a similar effect as that seen with the single antioxidant N-acetylcysteine in Fig. 5, panel D. These results demonstrate that the antioxidant cocktail B27 and the single antioxidant N-acetylcysteine have very similar effects thereby supporting the suggestion that oxidative stress underlies the activation of autophagy in SLOS cells.
Fig. 5.

Inhibition of DHCR7 with AY9944 increases autophagic markers and the protective effect of antioxidants. (A) MIA PaCa-2 cells were transfected with LC3-GFP followed by treatment with AY9944 for 48 h. Autophagic vacuole formation was visualized by the accumulation of LC3-GFP. (B) LC3 Western-blot for AY9944 treated control and SLOS cells showing increased LC3B-II in control fibroblasts and even greater expression in SLOS fibroblasts. (C) B27, a nutritional supplement with antioxidants, decreased LC3B-II expression in AY9944 treated fibroblasts. Lanes 1–3, AY9944 treated control fibroblasts, and lanes 4–6, effects of B27 in AY9944 treated control fibroblasts. (C) LC3B-II expression before and after B27 and (D), LC3B-II expression before and after N-acetylcysteine (NAC), a potent antioxidant.
3.6. Impaired mitochondrial function in SLOS cells
Autophagy is important in mitochondrial quality control (QC) since it is the only cellular mechanism for disposing dysfunctional mitochondria. Moreover, oxidative stress as well as defective autophagy affect mitochondrial health and promote the accumulation of dysfunctional mitochondria [22]. To assess mitochondrial function we used the dye JC-1 to measure mitochondrial function spectrophotometrically and by confocal microscopy. In healthy cells JC-1 monomers aggregate on the mitochondrial surface and can be detected as red fluorescence while in unhealthy (i.e., depolarized) cells the JC-1 monomers do not aggregate and are detected as green fluorescence. Fig. 6A shows greater green fluorescence in SLOS cells compared to control cells which emit a predominantly red fluorescence signal. The ratio of fluorescent intensity of JC1-aggregates to fluorescent intensity of monomers is used as an indicator of mitochondrial membrane potential and thus mitochondrial health. As shown in Fig. 6, panel B we found a 25% increase in the ratio of green-to-red fluorescence in SLOS cells compared to control cells indicating the accumulation of significantly more dysfunctional mitochondria in SLOS cells under steady-state conditions.
Fig. 6.

Defective mitochondria in SLOS cells. Mitochondrial function measurements using JC-1 staining in SLOS and control cells. (A) Confocal images of JC-1 staining in SLOS and control cells. Red fluorescence indicates healthy cells and green indicates cells with mitochondrial dysfunction. (B) Quantitative analysis of JC-1 staining by a microplate reader, n = 5, p < 0.05.
3.7. Accumulation of Pink-1 on mitochondria in SLOS
The activation of mitophagy by dysfunctional mitochondria is regulated by the accumulation of Pink-1 on the mitochondrial outer membrane [23]. To determine whether the presence of dysfunctional mitochondria in SLOS cells serves to stimulate mitophagy, we assessed the level of Pink-1 in SLOS cells. Confocal images revealed an obvious increase in Pink-1 staining in SLOS cells compared with control (Fig. 7A), while a difference in Parkin between SLOS and control was not observed (Fig. 7B). Using nuclei staining as the internal control, the relative intensity of Pink1 was remarkably increased by 40% in SLOS vs control cells, and there was no clear change of Parkin as shown in Fig. 7D and confirmed in immunoblots (Fig. 7C) from control and SLOS cell protein isolates (n = 3). These data support the conclusion that defects in mitochondrial health contribute to the stimuli elevating autophagy in SLOS cells.
Fig. 7.

Activation of Pink-1 in SLOS cells. Immunostaining of Pink-1 and Parkin in SLOS and control cells. Panel A: representative images from immunostaining of Pink-1 in SLOS and control cells, red fluorescence for Pink-1 and blue fluorescence for nuclei, top row for control cells and the lower row for SLOS cells. Panel B: representative images from immunostaining of Parkin in SLOS and control cells, red fluorescence for Parkin and blue fluorescence for nuclei, top row for control cells and lower row for SLOS cells. Panel C: Western-blot of Pink-1 and Parkin for SLOS and control cells; β-actin was used as an internal control to normalize protein loading. Panel D: Image analysis of Pink-1 and Parkin immunostaining. The relative intensity of Pink-1 or Parkin was normalized by nuclei staining. Pink-1 was significantly increased in SLOS cells (n = 3, p < 0.05), while there was no significant change of the ratio of red/blue fluorescence in Parkin immunostaining.
3.8. Elevated autophagy in neural stem cells isolated from SLOS transgenic mice and Dhcr7 siRNA treated neuroblastoma cells
Lastly, since among the most common and devastating defects in SLOS children derive from CNS dysfunction including intellectual disability, we examined neural stem cells cultured from the brains of transgenic mice harboring the Dhcr7Δ3–5/Δ3–5 mutation, i.e., similar to mutations in homozygotic SLOS fetuses. As shown in Fig. 8, panel A, 1 heterozygote and 2 homozygotes have elevated LC3B-II protein supporting the suggestion that autophagy is increased in the CNS similar to that seen in dermal fibroblasts isolated from SLOS children. Further supporting a role for autophagy in the CNS, silencing Dhcr7 in neuroblastoma cells resulted in an increase in the LC3B-II/I ratio (Fig. 8B), indicative of increased autophagy in neuronal cells in which Dhcr7 is deleted. The cellular 7-DHC level was monitored in control Neuro2a (0.21 ± 0.01 ng/μg protein) and in Dhcr7-deficient cells (33.3 ± 6.2 ng/μg protein).
Fig. 8.

Increased autophagy in cell models on SLOS neurons. (A) Increased expression of LC3B-II in neuronal stem cells obtained from the brains of Dhcr7 knockout mice. Mutational defects of one of the SLOS genotypes (Dhcr7Δ3–05/Δ3–5) were generated in mice using genetic engineering. Whole cell protein extracts from heterozygote (+/−) and homozygote (−/−) cells were subjected to immunoblotting with LC3B-II antibody. (B) Increased LC3B-II in Dhcr7-deficient Neuro2a cells. The cellular 7-DHC was monitored in control Neuro2a (0.21 ± 0.01 ng/μg protein) and in Dhcr7-deficient cells (33.3 ± 6.2 ng/μg protein). Increased LC3B-II in neuroblastoma cells in which Dhcr7 was silenced with siRNA.
4. Discussion
The results from the present study demonstrate that LC3B-II and beclin-1, two important markers of autophagy, are elevated in skin fibroblasts from children with the Smith–Lemli–Opitz Syndrome, suggesting increased autophagy in this devastating disease. Further support for elevated autophagy in SLOS comes from our demonstration of increased autophagosome content in SLOS cells along with the finding that treating control cells obtained from healthy children with the selective DHCR7 inhibitor AY9944 demonstrates similar results. In addition, since the primary metabolic defect in SLOS is a mutation in DHCR7 which leads to decreased cholesterol content and elevated 7DHC levels, our observation that the increase in LC3B-II levels correlates tightly with cellular 7DHC, but not cholesterol content is consistent with the notion that 7DHC, or perhaps one of its auto-oxidation products, of which there are several [24], [25], causes the elevation in autophagic activity. Supporting a role for a 7DHC-derived reactive oxysterols, we found that the elevated autophagy can be reduced back toward control levels with the anti-oxidant/growth supplement B27 as well as the antioxidant N-acetylcysteine, consistent with a role for oxidative stress participating in the elevated autophagy. Since mitochondria are the primary source of reactive oxygen species, we also examined the degree to which dysfunctional mitochondria might occur in SLOS cells and found evidence for cytosolic accumulation of dysfunctional mitochondria. Since 7DHC, which accumulates in SLOS cells, is known to localize in membranes in general [20] and in the mitochondrial membrane in particular [26], we suggest that 7DHC, or one of its auto-oxidative sterol derivatives has a deleterious role in SLOS pathology by virtue of its ability to induce mitochondrial dysfunction by oxidative stress. However, our data of elevated autophagy markers in SLOS cells may reflect a combination of both an increased stimulation of autophagic flux, likely from dysfunctional mitochondria, i.e., mitophagy, but also an autophagic defect in which autophagic flux is at least partially impaired. We propose therefore that mitochondrial dysfunction, along with impaired autophagic flux is a significant component of SLOS pathophysiology.
Autophagy is a highly conserved cellular pathway which functions across species to degrade long-lived and dysfunctional cytoplasmic proteins and organelles and elegantly recycles the degradative products through the lysosomal apparatus back to the cellular metabolic machinery [18]. It was originally described as an adaptive response to starvation to provide nutrients and energy by “self eating,” [27] but has ultimately emerged as a highly efficient pathway to rid the cell of expired, misfolded and damaged proteins and organelles as well [28]. Autophagy is responsible for both nonselective “bulk” autophagy and selective degradation of specific proteins and organelles. A key player which is part of the “core autophagic machinery” is a member of the three MAP1 light chain 3 subfamilies (LC3A, B and C), of which LC3B-I is formed by the action of Atg 4 on Atg8 in mammals. LC3B-I is then conjugated to the membrane lipid phosphatidylethanolamine (PE) to form LC3B-II. This lipidation reaction is mediated by the interaction of Atg 3 with Atg 7, and LC3B-II thus becomes firmly bound to the early phagophore membrane and accompanies it throughout its transit to the final degradation events in the autolysosome. Hence LC3B-II, which is essential for the formation and expansion of the phagophore [28], serves as the autophagic hallmark protein [19]. In this study, the increase in LC3B-II is consistently seen in the SLOS cells and healthy control cells incubated in AY9944 which mimics the SLOS phenotype in cell culture by inhibiting DCHR7 [29], [30]. In addition, since LC3B-II levels correlate with cell 7DHC content, this finding supports the notion that autophagy is elevated in SLOS. However, elevation in autophagy markers can result either from activation of autophagy, a pro-survival process, or alternatively, from a block in autophagic flux, i.e., defective autophagy which is a deleterious process.
Autophagy is controlled by 2 separate signaling pathways, the Akt–mTOR–p70 S6K and beclin-1 pathways. While the mTOR pathway is described as the predominant pathway for activating autophagy [31], beclin-1, a member of the class III PI3K complex which plays a critical role in the formation of autophagosomes has been shown to activate autophagy independent of mTOR [32]. This pathway for controlling autophagy has been shown to contribute to the pathobiology of several sphingolipid storage diseases (LSDs) which are characterized by aberrant cholesterol and sphingolipid trafficking [33], [34]. Our observation that beclin-1 is elevated in lysates of SLOS cells which also harbor aberrant cholesterol metabolism suggests that the activation of autophagy in SLOS may be by the beclin-1 pathway. Moreover, our earlier finding of decreased IP3 levels in SLOS cells is consistent with this interpretation.
An interesting observation in this study was the reduction in autophagic activity with antioxidants. Since mitochondrion is a primary site of ROS generation, we examined mitochondrial functional status in SLOS cells using JC1 staining which identifies mitochondria with impaired bioenergetics, i.e., depolarized mitochondria. JC1 is a cationic dye that accumulates in healthy mitochondria [16], [17]. At low, i.e., depolarized mitochondrial transmembrane potentials (low ∆ψm), JC-1 binds predominantly as a monomer that yields a green fluorescence emission (530 ± 15 nm). At high transmembrane potentials (high ∆ψm) the dye aggregates more abundantly and yields a red to orange colored emission (590 ± 17.5 nm). Therefore an increase in the green to red fluorescent ratio is indicative of depolarization. Since mitochondrial transmembrane potential is an important parameter of mitochondrial function, JC1 fluorescence can be used as an indicator of mitochondrial functionality [35], [36] reporting collapse of the mitochondrial transmembrane potential which uncouples oxidative phosphorylation and ATP synthesis and thus mitochondrial dysfunction. We observed a 25% increase in green fluorescence in SLOS cells compared to control cells indicating the presence of dysfunctional mitochondria. Since dysfunctional mitochondria are cleared from the cytoplasm by mitophagy, it is reasonable to conclude that their presence in SLOS cells stimulates autophagy in an attempt to rid the cells of these dysfunctional organelles. To shed light on this, we examined the expression of Pink-1 and Parkin on mitochondrial membranes. A model for Pink-1/Parkin-mediated QC of mitochondria has been proposed by Youle's group [8], [37], [38], [39] and validated by others [23], [40], [41], [42], [43]. According to this model, mitochondria that have accumulated damage to the point where they can no longer maintain an adequate membrane potential are targeted for removal to lysosomes using the Pink-1/Parkin QC quality control pathway. Pink-1 (PTEN-induced putative kinase 1) is a cytosolic protein that is constitutively expressed and enters all mitochondria where in the presence of healthy (high ∆ψm) mitochondria, it reaches the inner mitochondrial membrane (IMM). At this location, it's amino-terminal end is rapidly cleaved by mitochondrial proteases, and the truncated Pink-1 is degraded by the proteosome. Since Pink-1 cleavage in mitochondria requires a high ∆ψm, in damaged mitochondria, i.e., with a low ∆ψm, Pink-1 fails to reach the IMM and remains localized to the outer mitochondrial membrane (OMM) resulting in its robust accumulation. Pink-1 present on the OMM triggers the migration of Parkin, an E3 ubiquitin ligase from the cytosol to the mitochondrion, likely by phosphorylation events on either Pink-1 [23] or Parkin [44], or both. Parkin thus polyubiquitinates various mitochondrial surface proteins thereby targeting them for delivery to the autophagosome by aggregation and binding to P62's cargo recognition domain [23], [40], [41]. Accordingly, the Pink-1/Parkin QC pathway is regulated at the bioenergetic level of the individual mitochondrion and is thus an upstream event that identifies individual damaged mitochondria for disposal. In this study we observed a 40% increase in mitochondrial Pink-1 consistent with the activation of mitophagy, i.e., autophagic removal of dysfunctional mitochondria in SLOS cells.
The nature of the stimulus for mitochondrial depolarization however is obscure at this time and needs further exploration. However, at least 3 potential mechanisms could account for this condition. One would be the presence of 7DHC-derived oxysterol derivatives in the mitochondrial membrane. We and Porter [45] have demonstrated several such lipid reactive species in AY9944 treated healthy cells and in SLOS cells. Excess oxidative stress from lipid peroxidation products could explain this. Consistent with oxidative stress in SLOS, Vaughn et al. [46] also found that antioxidant treatment with dimethylthiourea abrogated retinal dysfunction in rats fed the DHCR7 inhibitor AY9944. Likewise, Korade et al. [47] found that antioxidants reduce oxysterols in Dhcr7 mutant mice and in fibroblasts from SLOS patients, a maneuver that also normalized lipid gene expression in SLOS fibroblasts. Second, work by our group has shown that 7DHC is known to alter membrane structure/function, both in model membranes and in SLOS cells [9], [20]. Appropriate registration of the membrane phospholipid bilayers in both the inner and outer mitochondrial membranes is essential for coupling of oxidative phosphorylation reactions, and disturbances of the membrane bilayer physical state by the presence of 7DHC would be expected to uncouple this complex reaction pathway and lead to the generation of oxidative stress. Third, under a variety of stresses, mitochondria generate reactive oxygen species (ROS) from a number of different redox centers in the respiratory chain with the predominate ROS generated being O2− which can then be converted to H2O2 and ONOO− which at high levels can oxidize lipids and redox-reactive proteins thereby amplifying the oxidative damage. Hence, any or all of these mechanisms may account for the presence of mitochondrial dysfunction and contribute to SLOS pathology. The complexity of these pathways and the intricate interplay between reactive lipids, mitochondrial dysfunction and mitophagy need further attention to delineate the pathological role and possible development of new antioxidant therapies, particularly those targeting dysfunctional mitochondria [48], [49] in SLOS and other diseases caused by aberrant sterol metabolism.
4.1. Study limitations
One limitation of this study is the use of dermal fibroblasts isolated from children with SLOS to represent defects in cell biology in tissues of the whole child, which by definition harbor both defective Dhcr7 alleles. Humane limitations prevent us from obtaining cells from other body tissues from these children. However, neuronal stem cells isolated from the brains of SLOS transgenic mice revealed an elevation of LCB3-II protein expression, similar to that seen in the dermal fibroblasts from SLOS children. In addition, neuroblastoma cells subjected to Dhcr7 silencing also show increased LC3B-II expression. These findings employing two different approaches support the idea that the derangements we report in autophagy are not likely limited to dermal fibroblasts and may also occur in the CNS of children with SLOS.
In conclusion, we present findings demonstrating for the first time elevated autophagy in SLOS cells. The data indicates that autophagic activity is increased due, at least in part, to the presence of excessive mitochondrial oxidative stress related to 7DHC accumulation which appears to initiate mitochondrial dysfunction thereby activating mitophagy to clear the cell of these dysfunctional organelles. However, the accumulation of dysfunctional mitochondria in SLOS cells points to the possibility of a concomitant defective autophagic machinery which would interfere with the ability of mitophagy to clear the defective proteins and organelles. In summary, we propose that dysfunctional mitochondria and defects in the autophagic machinery co-exist in SLOS cells which thereby impair cell function and abrogate the ability of the autophagy to perform its protective prosurvival function. Furthermore, these alterations may underlie the derangements in cell function that contribute to the pathophysiology in children with SLOS and further indicates that SLOS is, at least in part, a mitochondrial disease.
Acknowledgments
Support for this study was provided in part from NIH grant R01HL073980 (RDS/TNT) and DOD contract W81XW H08-2-00 (TNT).
References
- 1.Salen G., Shefer S., Batta A., Tint G., Xu G., Honda A., Irons M., Elias E. Abnormal cholesterol biosynthesis in the Smith–Lemli–Opitz syndrome. J. Lipid Res. 1996;37:1169–1180. [PubMed] [Google Scholar]
- 2.Tint G.S., Irons M., Elias E.R., Batta A.K., Frieden R., Chen T.S., Salen G. Defective cholesterol biosynthesis associated with the Smith–Lemli–Opitz syndrome. N. Engl. J. Med. 1994;330:107–113. doi: 10.1056/NEJM199401133300205. [DOI] [PubMed] [Google Scholar]
- 3.Waterham H., Hennekam R. Mutational spectrum of Smith–Lemli–Opitz syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2012;160C(4):263–284. doi: 10.1002/ajmg.c.31346. [DOI] [PubMed] [Google Scholar]
- 4.Correa-Cerro L.S., Porter F.D. 3b-hydroxysterol D7-reductase and the Smith–Lemli–Optiz syndrome. Mol. Genet. Metab. 2005;84(3):112–126. doi: 10.1016/j.ymgme.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 5.Yu H., Patel S. Recent insights into the Smith–Lemli–Opitz syndrome. Clin. Genet. 2005;68(5):383–391. doi: 10.1111/j.1399-0004.2005.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honda A., Tint G.S., Salen G., Kelley R.I., Honda M., Batta A.K., Chen T.S., Shefer S. Sterol concentrations in cultured Smith–Lemli–Opitz syndrome skin fibroblasts: diagnosis of a biochemically atypical case of the syndrome. Am. J. Med. Genet. 1997;68:282–287. [PubMed] [Google Scholar]
- 7.Wassif C., Vied D., Tsokos M., Connor W., Steiner R., Porter F. Cholesterol storage defect in RSH/Smith–Lemli–Opitz syndrome fibroblasts. Mol. Genet. Metab. 2002;75:325–334. doi: 10.1016/S1096-7192(02)00010-0. [DOI] [PubMed] [Google Scholar]
- 8.Narendra D.P., Youle R.J. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 2011;14(10):1929–1938. doi: 10.1089/ars.2010.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tulenko T.N., Boeze-Battaglia K., Mason R.P., Tint G.S., Steiner R.D., Connor W.E., Labelle E.F. A membrane defect in the pathogenesis of the Smith–Lemli–Opitz syndrome. J. Lipid Res. 2006;147(1):134–143. doi: 10.1194/jlr.M500306-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Yang Z., Klionsky D. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 2010;12(9):814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Z., Klionsky D. Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010;22(2):124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarkar S., Floto R., Berger Z., Imarisio S., Cordenier A., Pasco M., Cook L., Rubinsztein D. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005;170(7):1101–1111. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarkar S., Rubinsztein D. Inositol and IP3 levels regulate autophagy: biology and therapeutic speculations. Autophagy. 2006;2(2):132–134. doi: 10.4161/auto.2387. [DOI] [PubMed] [Google Scholar]
- 14.Wassif C., Zhu P., Kratz L., Krakowiak P., Battaile K., Weight F., Grinberg A., Steiner R., Nwokoro N., Kelley R., Stewart R., Porter F. Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith–Lemli–Opitz syndrome. Hum. Mol. Genet. 2001;10:555–564. doi: 10.1093/hmg/10.6.555. [DOI] [PubMed] [Google Scholar]
- 15.Rabinowitz J., White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Paepe B., Smet J., Vanlander A., Seneca S., Lissens W., De Meirleir L., Vandewoestyne M., Deforce D., Rodenburg R., Van Coster R. Fluorescence imaging of mitochondria in cultured skin fibroblasts: a useful method for the detection of oxidative phosphorylation defects. Pediatr. Res. 2012;72(3):232–240. doi: 10.1038/pr.2012.84. [DOI] [PubMed] [Google Scholar]
- 17.Keil V., Funke F., Zeug A., Schild D., Müller M. Ratiometric high-resolution imaging of JC-1 fluorescence reveals the subcellular heterogeneity of astrocytic mitochondria. Pflugers Arch. 2011;462(5):693–708. doi: 10.1007/s00424-011-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klionsky D. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 19.Mizushima N., Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 20.Ren G., Jacob R., Kaulin Y., Dimuzio P., Xie Y., Mason R., Tint G., Steiner R., Roullet J., Merkens L., Whitaker-Menezes D., Frank P., Lisant M., Cox R., Tulenko T. Alterations in membrane caveolae and BKCa channel activity in skin fibroblasts in Smith–Lemli–Opitz syndrome. Mol. Genet. Metab. 2011;104(3):346–355. doi: 10.1016/j.ymgme.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolf-Clauw M., Chevy F., Wolf C., Siliart B., Citadelle D., Roux C. Inhibition of 7-dehydrocholesterol reductase by the teratogen AY9944: a rat model for Smith–Lemli–Opitz syndrome. Teratology. 1996;54(3):115–120. doi: 10.1002/(SICI)1096-9926(199609)54:3<115::AID-TERA1>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 22.Lee J., Giordano S., Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem. J. 2012;441(2):523–540. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okatsu K., Oka T., Iguchi M., Imamura K., Kosako H., Tani N., Kimura M., Go E., Koyano F., Funayama M., Shiba-Fukushima K., Sato S., Shimizu H., Fukunaga Y., Taniguchi H., Komatsu M., Hattori N., Mihara K., Tanaka K., Matsuda N. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012;3:1016–1026. doi: 10.1038/ncomms2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu L., Korade Z., Porter N.A. Oxysterols from free radical chain oxidation of 7-dehydrocholesterol: product and mechanistic studies. J. Am. Chem. Soc. 2010;132(7):2222–2232. doi: 10.1021/ja9080265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korade Z., Xu L., Shelton R., Porter N.A. Biological activities of 7-dehydrocholesterol-derived oxysterols: implications for Smith–Lemli–Opitz syndrome. J. Lipid Res. 2010;51(11):3259–3269. doi: 10.1194/jlr.M009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slominski A., Zmijewski M., Semak I., Sweatman T., Janjetovic Z., Li W., Zjawiony J., Tuckey R. Sequential metabolism of 7-dehydrocholesterol to steroidal 5,7-dienes in adrenal glands and its biological implication in the skin. PLoS One. 2009;4(2):1–18. doi: 10.1371/journal.pone.0004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfeifer U. Cellular autophagy and cell atrophy in the rat liver during long-term starvation. A quantitative morphological study with regard to diurnal variations. Virchows Arch. B Cell Pathol. 1973;12(3):195–211. doi: 10.1007/BF02893998. [DOI] [PubMed] [Google Scholar]
- 28.Kraft C., Martens S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012;24(4):496–501. doi: 10.1016/j.ceb.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 29.Xu L., Sheflin L., Porter N., Fliesler S. 7-Dehydrocholesterol-derived oxysterols and retinal degeneration in a rat model of Smith–Lemli–Opitz syndrome. Biochim. Biophys. Acta. 2012;1821(6):877–883. doi: 10.1016/j.bbalip.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu L., Liu W., Sheflin L., Fliesler S., Porter N. Novel oxysterols observed in tissues and fluids of AY9944-treated rats: a model for Smith–Lemli–Opitz syndrome. J. Lipid Res. 2011;52(10):1810–1820. doi: 10.1194/jlr.M018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klionsky D., Emr S. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zalckvar E., Berissi H., Mizrachy L., Idelchuk Y., Koren I., Eisenstein M., Sabanay H., Pinkas-Kramarski R., Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10(3):285–292. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pacheco C., Kunkel R., Lieberman A. Autophagy in Niemann-Pick C disease is dependent upon beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007;16:1495–1503. doi: 10.1093/hmg/ddm100. [DOI] [PubMed] [Google Scholar]
- 34.Pacheco C., Lieberman A. Lipid trafficking defects increase beclin-1 and activate autophagy in Niemann-Pick type C disease. Autophagy. 2007;3:487–489. doi: 10.4161/auto.4586. [DOI] [PubMed] [Google Scholar]
- 35.Smiley S.T., Reers M., Mottola-Hartshorn C., Lin M., Chen A., Smith T.W., Steele G.D., Jr., Chen L.B. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. U. S. A. 1991;88(9):3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reers M., Smiley S.T., Mottola-Hartshorn C., Chen A., Lin M., Chen L.B. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995;260:406–417. doi: 10.1016/0076-6879(95)60154-6. [DOI] [PubMed] [Google Scholar]
- 37.Jin S., Lazarou M., Wang C., Kane L., Narendra D., Youle R. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narendra D.P., Jin S.M., Tanaka A., Suen D.F., Gautier C.A., Shen J., Cookson M.R., Youle R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Narendra D., Tanaka A., Suen D.F., Youle R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008;183(5):795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geisler S., Holmström K., Treis A., Skujat D., Weber S., Fiesel F., Kahle P., Springer W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010;6(7):871–878. doi: 10.4161/auto.6.7.13286. [DOI] [PubMed] [Google Scholar]
- 41.Okatsu K., Saisho K., Shimanuki M., Nakada K., Shitara H., Sou Y., Kimura M., Sato S., Hattori N., Komatsu M., Tanaka K., Matsuda N. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15(8):887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R.L., Kim J., May J., Tocilescu M.A., Liu W., Ko H.S., Magrane J., Moore D.J., Dawson V.L., Grailhe R., Dawson T.M., Li C., Tieu K., Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U. S. A. 2010;107(1):378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C.A., Sou Y.S., Saiki S., Kawajiri S., Sato F., Kimura M., Komatsu M., Hattori N., Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010;189(2):211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shiba-Fukushima K., Imai Y., Yoshida S., Ishihama Y., Kanao T., Sato S., Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012;2 doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu W., Xu L., Lamberson C., Merkens L., Steiner R., Elias E., Haas D., Porter N. Assays of plasma dehydrocholesteryl esters and oxysterols from Smith–Lemli–Opitz syndrome patients. J. Lipid Res. 2013;54(1):244–253. doi: 10.1194/jlr.M031732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaughan D., Peachey N., Richards M., Buchan B., Fliesler S. Light-induced exacerbation of retinal degeneration in a rat model of Smith–Lemli–Opitz syndrome. Exp. Eye Res. 2006;82(3):496–504. doi: 10.1016/j.exer.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korade Z., Xu L., Harrison F.E., Ahsen R., Hart S.E., Folkes O.M., Mirnics K., Porter N.A. Antioxidant supplementation ameliorates molecular deficits in Smith–Lemli–Opitz syndrome. Biol. Psychiatry. 2014;75(3):215–222. doi: 10.1016/j.biopsych.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith R., Adlam V., Blaikie F., Manas A., Porteous C., AM J., MF R., Logan A., Cochemé H., Trnka J., Prime T., Abakumova I., Jones B., Filipovska A., Murphy M. Mitochondria-targeted antioxidants in the treatment of disease. Ann. N. Y. Acad. Sci. 2008;1147:105–111. doi: 10.1196/annals.1427.003. [DOI] [PubMed] [Google Scholar]
- 49.Smith R., Hartley R., Cochemé H., Murphy M. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012;33:341–352. doi: 10.1016/j.tips.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 50.Kelley R.I., Hennekam R.C. The Smith–Lemli–Opitz syndrome. J. Med. Genet. 2000;37:321–335. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kratz L.E., Kelley R.I. Prenatal diagnosis of the RSH/Smith–Lemli–Opitz syndrome. Am. J. Med. Genet. 1999;82:376–381. [PubMed] [Google Scholar]