

Abstract
Castration-resistant prostate cancer (CRPC) is an advanced-stage prostate cancer (PC) associated with high mortality. We reported that G-1, a selective agonist of G protein-coupled receptor 30 (GPR30), inhibited PC cell growth by inducing G2 cell cycle arrest and arrested PC-3 xenograft growth. However, therapeutic actions of G-1 and their relationships with androgen in vivo are unclear. Using the LNCaP xenograft to model PC growth during androgen-sensitive (AS) versus castration-resistant (CR) phase, we found that G-1 inhibited growth of CR but not AS tumors with no observable toxicity to the host. Substantial necrosis (~65%) accompanied by marked intratumoral infiltration of neutrophils was observed only in CR tumors. Global transcriptome profiling of human genes identified 99 differential expressed genes with “interplay between innate and adaptive immune response” as the top pathway. Quantitative-PCR confirmed upregulation of neutrophil-related chemokines and inflammation-mediated cytokines only in the G-1-treated CR tumors. Expression of murine neutrophil-related cytokines also was elevated in these tumors. GPR30 expression was significantly higher in CR than AS tumors. In cell-based experiments, androgen repressed GPR30 expression, a response reversible by anti-androgen or siRNA-induced androgen receptor silencing. Finally, in clinical specimens, 80% of CRPC metastases (n=123) express a high level of GPR30 whereas only 54% of the primary PCs (n=232) showed high GPR30 expression. Together, these results provide the first evidence that GPR30 is an androgen-repressed target and G-1 mediates the anti-tumor effect via neutrophil infiltration-associated necrosis in CRPC. Additional studies are warranted to firmly establish GPR30 as a therapeutic target in CRPC.
Keywords: Androgen-deprivation therapy, androgen-repressed gene, metastases, tumor-infiltrating neutrophils
Introduction
Androgen-ablation therapies are mainstay treatments for advanced prostate cancer (PC) (de Bono et al. 2011,Higano et al. 2009,Tannock et al. 2004). Unfortunately, almost all patients ultimately fail to respond to these therapies and develop castration-resistant PC (CRPC) that grows in the presence of castration levels of circulating testosterone (de Bono et al. 2011). Although chemotherapy (docetaxel or cabazitaxel) (de Bono et al. 2010, Tannock et al. 2004), immunotherapy (e.g., sipuleucel-T) (Higano et al. 2009, Kantoff et al. 2010), or complete androgen blockade (e.g., abiraterone) (de Bono et al. 2011) may extend the lives of some patients, these treatments all have documented side effects and a relatively short duration of response. Hence, the development of new CRPC therapies with durable efficacy and low toxicity is warranted.
Estrogens have a long history of efficacy for advanced PC (Oh 2002). Huggins and Hodges first reported the use of diethylstilbestrol for advanced PC in 1941 (Huggins and Hodges 2002). However, severe cardiovascular toxicity of oral estrogens limited their use in PC (Norman et al. 2008). The early efficacy of parenteral estrogen in recent studies (Schellhammer et al. 2012, Langley et al. 2013;) and especially the lower toxicity profiles owing to hepatic bypass (Norman et al. 2008) reinvigorated interest in the use of estrogens as a therapy for PC. In addition to the suppression of testosterone effects by estrogens, estrogens are also directly cytotoxic to PC cells (Ho et al. 2011). The actions of parenteral estrogens are believed to be mediated by the classical estrogen receptors (ERs), ESR1 and ESR2. However, the exact roles of the two ERs and its isoforms on PC growth and metastases may vary according to cellular contexts (Claessens and Tilley 2014, Nelson et al. 2014). We recently reported that G-1 (1(1-[4-(6-bromobenzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone), which selectively activates the third ER, G protein–coupled receptor 30 (GPR30 or GPER) (Bologa et al. 2006), inhibited the growth of multiple PC cell lines and PC-3 xenografts, and exerted few or no adverse effects on the animals (Chan et al. 2010). These findings suggest that G-1, by targeting GPR30, might offer a new treatment option for PC.
GPR30 is structurally unrelated to the classical ERs (ESR1 and ESR1). It is a seven–transmembrane receptor localized at the cell surface (Bologa et al. 2006, Funakoshi et al. 2006), endoplasmic reticulum (Otto et al. 2008, Prossnitz, Arterburn, and Sklar 2007, Thomas et al. 2005), perinuclear compartment (Cheng et al. 2011), and nucleus (Madeo and Maggiolini 2010). The successful development of a highly selective non-steroidal agonist, G-1, for GPR30, provides a tool for studying the action of GPR30 independent of the actions mediated by ESR1 and ESR2 (Blasko et al. 2009, Bologa et al. 2006). Activation of GPR30 was found to play opposite roles in the regulation of the growth of various normal and neoplastic tissues, promoting growth of breast, endometrium, and ovarian tissues (Albanito et al. 2007, Filardo et al. 2000,Pandey et al. 2009, Vivacqua et al. 2006) but inhibiting growth of thymocytes, urothelial cells, vascular smooth muscle cells, and ER-positive breast cancer cells (Albanito et al. 2007). The dual action of GPR30 could be related in part to its differential activation of downstream mediators, including EGFR, PI3K, Erk1/2, cAMP, and intracellular Ca2+ (reviewed in (Maggiolini and Picard 2010, Prossnitz and Barton 2011)). We demonstrated that in PC cells, the activation of GPR30 by G-1 leads to growth inhibition via an ERK/p21-mediated cell-cycle arrest at the G2 phase (Chan et al. 2010). In addition, we found that G-1 inhibited the growth of PC-3 xenografts that lack the androgen receptor. Still unknown are the mode of action of G-1 in vivo and the potential link between its efficacy and the androgen status in prostate cancer.
The present study evaluated the efficacy of G-1 in inhibiting the growth of LNCaP xenografts during the androgen-sensitive (AS) or the castration-resistant (CR) phase. We here report that G-1 inhibited the growth of the xenograft in castrated (low testosterone) animals but not in intact, androgen-supported animals (high testosterone). The G-1-induced growth inhibition in the CR xenograft was associated with massive necrosis, neutrophil infiltration, upregulation of a set of cell-mediated immune-response genes, and enhanced expression of GPR30. Cell-based experiments revealed that GPR30 is repressed by androgen, whereas immunohistochemical studies found a larger proportion of human CRPC metastases than primary PC express high GPR30 level. Collectively, these data provide support for targeting GPR30 with G-1 as a possible new approach for the treatment of CRPC.
Materials and Methods
Human Specimens
Human tissue microarrays were obtained from Massachusetts General Hospital (primary PC) and the University of Washington (metastatic CRPC). Samples were de-identified; only those with complete clinical information, follow-up data, and good tissue quality were included. The primary PC cohort comprised one specimen each from 232 patients with PC (i.e. 232 specimens) taken at prostatectomy (Leung et al. 2010). The metastatic CRPC cohort comprised patients who participated in the Rapid Autopsy Program in 1999–2006; it consists of 123 CRPC specimens, including 75 bone (spine, ribs, pelvis, sternum, ischium, iliac, and sacrum), 29 lymph node, 14 liver, and 5 lung metastases tissues from 24 patients. The use of the specimens was reviewed and approved by the Institutional Review Board committees of the respective universities.
Cell Culture and siRNA Experiments
Human prostate cancer cell lines LNCaP and PC-3 were obtained from the American Type Culture Collection (ATCC, Manassas, VA), and passaged for <3 months after resuscitation. Both LNCaP and PC-3 were retro-authenticated by ATCC with short tandem repeat profiling (March 13, 2013) and confirmed to be the original cell line. LNCaP cells were maintained in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS; sodium pyruvate, 1 mmol/L; L-glutamine, 2 mmol/L; and D-glucose, 1.25 g/L. PC-3 cells were maintained in F-12K medium (ATCC) supplemented with 10% FBS. Cells were cultured at 37 °C and 5% CO2. For androgen treatment (R1881 and dihydrotestosterone, DHT), LNCaP (2.5 × 105) and PC-3 (2 × 105), cells were seeded in phenol red-free RPMI-1640 (with supplements) and F-12K medium, respectively, supplemented with 10% charcoal-stripped FBS. For drug treatment, drugs were added daily for 4 days, and medium was changed every 2 days. For siRNA-AR (siAR) transfection, cells were replenished with 1.6 ml of fresh medium and 400 μl of siAR-DharmaFECT mixture (50 nM Stealth RNAi siAR, Invitrogen; DharmaFECT3 for LNCaP and DharmaFECT2 for PC-3 cells, Dharmacon, Lafayette, CO, USA) the next day. At day 3, cells were recovered with respective medium containing 10% charcoal-stripped FBS, and drugs were added daily for 4 days. Transfection was repeated on day 2 of drug treatment. At the end of the experiments, cells were collected for RNA extraction. For the transfection-negative control, cells were treated with DharmaFECT and siRNA-non-targeting (siNT, Dharmacon).
Chromatin Immunoprecipitation (ChIP) Assay
ChIP-sequencing and ChIP were carried out as previously described using an antibody to androgen receptor (AR) (ab74272, Abcam, Cambridge, MA) (Decker et al. 2012). The site-specific qPCR primers for the AR binding site at the GPER (GPR30) locus were forward, 5′-CTGGGACAACGTGAGCAGTAAG-3′; reverse, 5′-CCAACTACTTTACCAGCCAGCA-3′. The primers for prostate-specific antigen (PSA) enhancer and control regions have been described previously (Zheng et al. 2013).
Microarray Experiment and Analysis
RNA was extracted from LNCaP xenografts with TRIzol Reagent (Invitrogen); RNA integrity numbers >8 (four animals in each group), as measured by Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA), were used for microarray analysis. The detailed microarray study is available in Supplementary Methods. The data are accessible through the NCBI Gene Expression Omnibus Series accession number GSE54974.
Xenograft Study
In the first set of experiments, GPR30 mRNA expression was compared in tumors grown before and after the castration of mice. Male athymic nude mice (4–6 weeks old, 20–25 g, Taconic, Hudson, NY) were implanted subcutaneously with one 2 cm-long silastic capsule containing ~15 mg of testosterone (Sigma, St Louis, MO) while the animals were under general anesthesia using isoflurane. After two days, LNCaP cells (5 × 106 cells) in 150 μl of Matrigel (BD Biosciences, Franklin Lakes, NJ) were injected subcutaneously into the flanks of the mice, and the tumors that developed were measured twice weekly (Chan et al. 2010). When the tumors reached 150–300 mm3, mice were divided into two groups: intact and castrated animals. Tumors growing in the intact mice are referred as androgen-sensitive (AS) tumors. For the castrated group, the silastic capsules were removed and the mice were surgically castrated under general anesthesia using isoflurane. Tumors regressed and then regrew after castration (~3 weeks post-castration); these tumors are referred as castration-resistant (CR). AS or CR tumors at ~1,000 mm3 were collected to determine the expression of GPR30 mRNA.
In the second set of experiments, the therapeutic efficacy of G-1 on AS and CR tumors was evaluated and compared. LNCaP xenografts were developed as described in the first set of experiments. Both AS and CR tumors were enrolled when tumors reach ~300–400 mm3. Mice were injected daily with vehicle (95% PBS, 2.5% DMSO, 2.5% ethanol; v/v) or G-1 (4 mg/kg) for 16 days. Tumors and body weight were measured twice weekly. Mice were sacrificed and weighed after removal of the xenografts. The protocol for animal use was approved by the Institutional Animal Care Committee at the University of Cincinnati.
Serum Enzyme Assays
Serum obtained from the mice was assayed for creatine kinase (CK), lactate dehydrogenase (LDH), alanine transaminase (ALT), and aspartate transaminase (AST) with IDTox enzyme assay kits (ID Labs, London, ON, Canada) following the manufacturer's protocols.
Quantitative Real-time PCR
Total RNA was treated with RNase-free DNase (Qiagen, Germantown, MD, USA) and reverse-transcribed (Chan et al. 2010). Real-time PCR was carried out as described previously (Chan et al. 2010). Species-specific primer sequences are presented in Supplementary Table S1. PCR reactions with SYBR GreenER PCR Master-Mix (Invitrogen) were monitored with the 7900HT Fast Real-time PCR System (Applied Biosystems). Individual mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Histopathology and Immunohistochemistry Analyzes
Formalin-fixed xenograft samples were processed for hematoxylin and eosin (H&E) staining and subjected to histologic examination for necrosis and inflammation; the thickness of the tumor capsule was determined by the surgical pathologist (JW). Immunohistochemistry (IHC) analysis of paraffin-embedded human PC and LNCaP xenograft sections was performed as previously described (Leav et al. 2001). Antibodies and quantification of necrosis and markers are described in Supplementary Methods and Table S2.
For clinical specimens, GPR30 expression was graded independently by two investigators (HML and JW) in a blinded fashion. Signal intensity (0–3) and percentage of signal coverage (0–100) of each section were scored, and the product of the intensity and coverage was represented as an H-score (0–300) (Huang et al. 2005). For the metastatic CRPC cohort, H-scores were an average of duplicated cores in a specified metastatic site of each patient. In all cases of bone metastases, two to three sites were acquired per patient and an average H-score was calculated. The distribution of the H-score showed bi-modal or multi-modal in the clinical data: 45% of the specimens showed H-scores <100, about 32% of the specimens showed H-scores 100–199 and 23% of the specimens amassed H-scores 200–300. In the present study, we used a dichotomous variable of H-score group (i.e. H-score ≥100 vs. H-score <100) in the analysis to reduce possible bias due to the distribution of the original H-score and to improve the statistical power. In order to assess the sensitivity of using different definition to the H-score variables, the same analysis was repeated after replacing the dichotomous variable with a three-level category variable (i.e. H-score 0–99 vs. 100–199 vs. 200–300) as well as the original H-score. Those results were found consistent to the main analysis using the dichotomous variable which was presented in this study.
Statistical analyses
Numerical dependent variables were analyzed by one-way ANOVA and post hoc Bonferroni tests to compare means if more than two groups were involved. T-tests were used if means of two groups were compared. Categorical dependent variables were compared among groups using Chi-square tests. All statistical tests were considered significant when p <0.05.
Results
G-1 inhibits growth and induces necrosis in CR tumors with no apparent toxicity to the host
We compared the inhibitory effect of G-1 in AS or CR tumors growing in intact or castrated (low testosterone) mice, respectively. Administration of G-1 significantly inhibited the growth of CR tumors after 16 days of treatment (p<0.05, Fig. 1). Similar results were obtained in CR tumors including C4-2 and PC-3 (Supplementary Fig. S3). Massive necrosis and inflammation were observed only in the G-1-treated LNCaP CR tumors (in seven of eight mice). Inflammation was attended by considerable neutrophil infiltration to the necrotic area as well as to the healthy area of these tumors (Fig. 2A). This intratumoral neutrophil infiltration was not seen in either vehicle-treated CR tumors or vehicle/G-1-treated AS tumors that displayed only ischemic necrotic foci with no inflammation/neutrophils (Fig. 2A). We did not examine T cells in this study because nude mice are deficient in these cells (Pelleitier and Montplaisir 1975). B cells and macrophages were found exclusively in the intratumoral stroma and the tumor capsule, respectively, in all treatment groups (Supplementary Fig. S1). Notably, G-1-induced necrosis occupied an average of 65% of the tumor volume (p=0.0003, Fig. 2B). Furthermore, G-1 significantly reduced the intratumoral microvessel density in CR tumors but not in AS tumors (Fig. 2C, left panel). No significant alteration of microvessel density was observed in the tumor capsule with G-1 treatment (Fig. 2C, right panel). In the viable area of the tumors, cell proliferation (Ki67 staining) remained relatively constant in the four treatment groups except for an increase of 10%–20% as compared with vehicle-treated counterparts in Ki67-staining cells in G-1-treated CR tumors (Fig. 2D, left panel). G-1 induced a slight but significant increase in apoptosis (cleaved caspase-3 staining) in the CR tumors (Fig. 2D, right panel).
Figure 1.
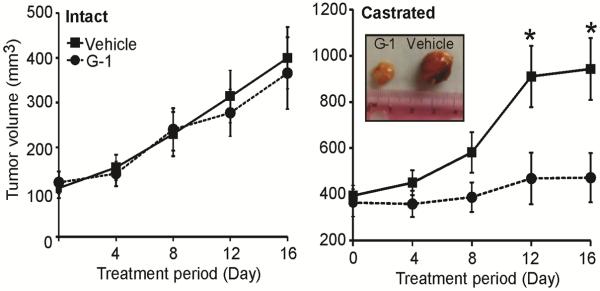
G-1 inhibited growth and induced necrosis in the CR tumor. G-1 inhibited growth of the CR (right panel) but not the AS tumor (left panel). When LNCaP xenografts grew to 150 mm3, mice were divided into two groups: intact and castrated. Intact animals were injected subcutaneously with vehicle (2.5% DMSO, 5% ethanol) or G-1 (4mg/kg) daily for 16 days. For the castrated group, mice were castrated and when the tumor re-emerged, they were treated with vehicle or G-1 daily for 16 days. Error bars represent mean ± SEM, n=6–8/group, *p<0.05. CR, castration-resistant; AS, androgen-sensitive.
Figure 2.

G-1 induced unique changes in gene expression in castrated animals. A) Heat map of hierarchically clustered differential gene expression in intact or castrated animals treated with vehicle or G-1 Blue, downregulated; yellow, upregulated; n=4 per group. A scheme for gene selection for Ingenuity Pathway Analysis is shown. B) Quantitative real-time PCR analyses of the G-1-induced human and mouse genes in intact and castrated animals. Data were normalized to the levels of housekeeping genes: human-specific GAPDH (for human genes) or ActB (for mouse genes). Error bars represent mean ± SEM, n=6. #p<0.05 compared with intact-vehicle treatment; *p <0.05 compared with castrated-vehicle treatment; ns, not significant.
Our previous work demonstrated that G-1 did not have general toxicity (body weight and tissue histology) in the animals (Chan et al. 2010). Here, we further report that G-1 did not induce any changes in body weight or cause functional damages to the heart or the liver in mice after 16 days of treatment with G-1, as indicated by the levels of injury biomarkers in the serum (CK and LDH for heart injuries; AST and ALT for liver injuries, Fig. 2E).
G-1 induced specific changes in gene expression exclusively in CR tumors
Global transcriptome profiling was performed on vehicle/G-1-treated AS and CR tumors (four groups of tumors, n=4). Overall, the profiling results identified 2,446 differentially expressed genes among the four treatment groups (FDR<0.1, p<0.01, n=4 per group). Unbiased hierarchical clustering analysis showed no significant differences in gene expression between the vehicle-treated and the G-1-treated AS tumors (Fig. 3, left side of heat-map). However, this analysis identified two clusters of genes (a total of 1,082) that were altered by G-1 exclusively in the CR tumors (Fig. 3, right side of heat map). Subsequent gene shaving using two additional criteria—p<0.01 and a difference of at least 1.5-fold between G-1-treated and vehicle-treated CR tumors—yielded a final set of 99 genes (Fig. 3A, grey panel). Ingenuity Pathway Analysis (IPA) of 99 genes showed an enrichment of the top biologic pathway `antigen presentation, cell-to-cell signaling and interaction, and inflammatory response', followed by `genetic disorder, neurological disease, skeletal and muscular disorders. Furthermore, the top canonical pathway shown in this specific set of G-1-associated genes is `communication between innate and adaptive immune cells' (Supplementary Table S3).
To focus on identifying molecular mediators of G-1-induced inflammation/neutrophil infiltration, we selected a set of genes from the 99-gene panel for confirmation based on a literature search showing their relatedness to cell-mediated immune responses. Quantitative real-time PCR analyses (n=6 per group) validated the upregulation of the expression of these genes in G-1-treated CR tumors but not in G-1-treated AS tumors as compared with their respective vehicle-treated controls. These include four chemokine genes CP, IL8, CCL2, and CXCL12; three interferon-induced antiviral genes IFIT2, IFIT3, IFIT4; and SOD2, an important oxidative stress response gene (Fig. 3B). Because human IL8 is a strong chemo-attractant for both human and mouse neutrophils (Geiser et al. 1993,Schaider et al. 2003), we analyzed murine neutrophil-related cytokine genes using quantitative real-time PCR. Expression of murine genes involved in neutrophil movement, accumulation, adhesion, activation, and phagocytic respiratory burst, including Il1b, Il6, Il18, TNFa, Cxcl12, Cxcl1, Cxcl3, S100a8, S100a9, and Cd14 (Cacalano et al. 1994,Eash et al. 2010,Harokopakis and Hajishengallis 2005,Leung et al. 2001,Ryckman et al. 2003), was elevated 1.8- to 50.9-fold in G-1 vs. vehicle-treated CR tumors (Fig. 3B). Interestingly, the expression of human IL1B was not altered in CR tumors with G-1 treatment.
Androgen represses GPR30 expression via androgen receptor, and castration increases GPR30 expression
In an attempt to explain why G-1 inhibited growth only in an androgen-deprived environment, we determined the effect of androgen on GPR30 expression. Androgen is the principal hormone regulating prostate function. Treatment of LNCaP cell cultures with R1881 (a synthetic androgen) or dihydrotestosterone (DHT, the physiologically active androgen) reduced the expression of GPR30 mRNA; the effects of these androgens were abolished by cotreatment with bicalutamide, an androgen receptor (AR) antagonist or by transduction of a siRNA against AR (Fig. 4A and B, upper panels). These responses were not observed in the AR-negative PC-3 cells (Fig. 4A and B, bottom panels). These data suggest that androgen represses GPR30 expression via mechanisms involving the AR. Furthermore, ChIP-sequencing analyses of LNCaP cells revealed a strong AR binding site ~3.5 kb downstream of the 3′ end of the GPR30 (GPER) gene after androgen stimulation (Supplementary Fig. S2, upper panel). This AR binding site on GPR30 was further validated by an independent site-specific ChIP-qPCR analysis (Supplementary Fig. S2, lower panel).
Figure 4.
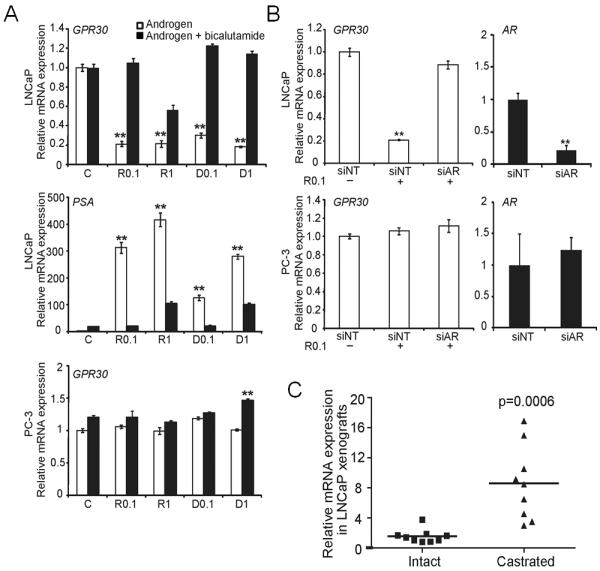
Androgen suppressed GPR30 expression via AR. A) Androgen (open bar, 0.1 and 1 nM) suppressed GPR30 expression, and suppression was reversed by bicalutamide (solid bar) in AR-positive LNCaP cells but not in AR-negative PC-3 cells. Cells were treated with androgen in the presence or absence of bicalutamide for 4 days. Prostate-specific antigen (PSA) was a positive control for the androgen-stimulated AR response gene. B) siAR abolished the androgen-suppressed GPR30 expression in LNCaP cells. Error bars represent mean ± SD of three independent experiments, **p<0.01. C) Castration upregulated GPR30 expression in vivo. RNA was extracted, and GPR30 expression of the LNCaP xenograft in intact mice (AS tumor, n=9) was compared with that after castration of the mice (CR tumor, n=9). Relative mRNA expression was compared to that of intact mouse #1. AR, androgen receptor; D, dihydroxytestosterone; PSA, prostate-specific antigen; R, R1881; siNT, siRNA-non-targeting; siAR, siRNA-AR.
In the LNCaP xenograft model, expression of GPR30 mRNA was significantly higher (~8-fold) in CR tumors grown in castrated mice than in AS tumors grown in intact mice (Fig. 4C). Expression of AR mRNA in CR tumors was increased 1.8–4.6-fold as compared with that in AS tumors (data not shown). These data are in concordance with data from cell-based studies that suggest GPR30 expression is repressed by androgen via AR-mediated signaling.
GPR30 expression is higher in metastatic CRPC than in primary PC
We reasoned that GPR30 in CRPC metastases needs to be expressed at significant levels before we can consider it as a new therapeutic target for CRPC. Hence, we used IHC to assess the level of GPR30 expression in specimens from two cohorts of patients. The first cohort includes only primary cancers from specimens obtained at prostatectomy (n=232) and the second comprises CRPC metastases (n=123). We found that 80% of the metastatic CRPC specimens expressed high levels of GPR30, with an H-score ≥100 as compared with 54% of primary PC specimens with an H-score ≥100 (p=0.001) (Fig. 5).
Figure 5.

GPR30 staining in primary PC and CRPC metastases. High level of GPR30 was detected in a larger proportion of metastatic CRPC specimens when compared to primary PC specimens. GPR30, G protein–coupled receptor 30; CRPC, castration-resistant prostate cancer.
GPR30 staining in PC was not correlated with age, Gleason score of primary cancer, final PSA level, type of androgen-deprivation therapy (ADT), or duration of ADT (Supplementary Table S4). Of interest, no difference in the H-scores of GPR30 was seen among the 75 bone metastases obtained from different locations (H-score ~162—165; pelvis/sternum/ischium/iliac/sacrum vs. rib/limb vs. the spine; Supplementary Table S5).
Discussion
Here we reported that G-1, a GPR30 agonist, inhibited the growth of CR tumors but not during their preceding AS growth phase, with no detectable toxicity to the host. The G-1-induced growth inhibitory response was manifested as massive necrosis attended by marked neutrophil infiltration in the affected tumors, associated with the activation of gene pathways involved in innate antitumor immunity. We also demonstrated that androgen suppressed GPR30 expression in an AR-dependent manner and that castration markedly upregulated its expression. Clinically, we observed an elevated prevalence of high levels of GPR30 in CRPC metastases when compared to that in primary PC. Taken together, these findings provide evidence for the effective preclinical targeting of GPR30 with G-1 for CRPC.
In this study, we aimed to examine the activation of GPR30 by G-1 in both an androgen-supported (intact) and an androgen-deprived (castrated) environment in vivo. We previously showed that G-1 inhibited growth in cell culture experiments and a hormone-independent PC-3 xenograft in castrated hosts (Chan et al. 2010). This study further demonstrated the efficacy of G-1 in the LNCaP xenograft model, which recapitulates the natural history of PC progression from AS to CR. We found that, in the LNCaP xenograft model, G-1 inhibited the growth of CR tumors but not AS tumors, suggesting that the androgen deprivation may favor the anti-tumor action of G-1.
Histologic examinations showed that G-1 induced massive tumor necrosis in the castrated mice and invasion of the viable region of G1-treated tumors with numerous tumor-infiltrating neutrophils (TINs). At the molecular level, upregulation of chemokine and inflammatory response genes, including CP, IL8, CCL2, CXCL12, and IFITs, were uncovered by transcriptome profiling and confirmed by qPCR. Thus, one hypothesis is that chemokines secreted by viable CRPC cells and/or additional tumor tissue remodeling factors stimulated by G-1 may direct the migration of neutrophils (illustrated in Fig. 6). Neutrophils have been implicated in tumor progression and antitumor response. Mild infiltration of neutrophils stimulates proliferation and metastasis in cancer (Gregory and Houghton 2011). However, high levels of TINs induce a destructive oncolytic response (Fu et al. 2011) and are associated with cytotoxicity and tumor regression (Di et al. 2001). Neutrophils produce cytotoxic mediators, including reactive oxygen species, proteases, membrane-perforating agents, and soluble cell-kill mediators (Di et al. 2001). Moderate or extensive levels of TINs are associated with reduced mortality in gastric cancer (Caruso et al. 2002), suggesting that neutrophils are active in immunosurveillance against cancer. Key TIN-associated cell-kill mediators, including IL1β and TNFα (Di et al. 2001), were detected in the G-1-induced tumor necrosis. Studies demonstrated that transgenic expression of tumor cells with IL8 and TNFα elicited prominent neutrophil-mediated antitumor activity (Hirose et al. 1995,Musiani et al. 1996). In addition, ceruloplasmin (CP) produced by the CR tumor cells attracts neutrophils and enhances phagocytosis of neutrophils (Saenko et al. 1994). In contrast to the systemic upregulation of cytokines, which may pose a health hazard to immunocompromised patients with cancer, local and specific recruitment of neutrophils may provide a new approach to the targeted treatment of cancer (Fu et al. 2011,Hirose et al. 1995).
Figure 6.
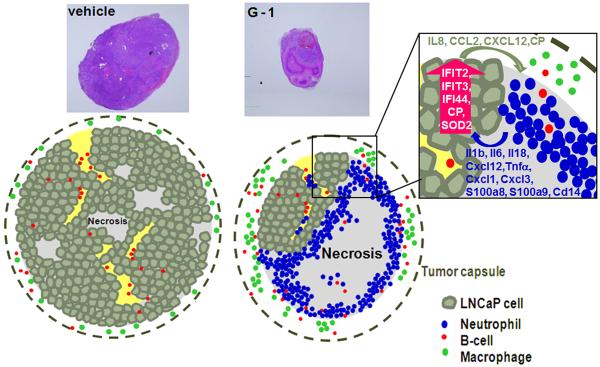
A schematic diagram showing G-1-induced innate antitumor response in castration-resistant LNCaP prostate cancer in vivo. For LNCaP xenografts in vehicle- or G-1-treated intact animals or vehicle-treated castrated animals, focal ischemic necrosis was detected in the tumor. However, in G-1-treated castrated animals, massive necrosis and neutrophil infiltration was detected in the necrotic area as well as within the viable area of the tumor. (Box) In human xenografts, the levels of expression of human-specific chemokine and inflammatory response genes were increased; in the mouse stroma, the levels of expression of a panel of murine-specific neutrophil-related cytokine genes were elevated. In both intact and castrated animals, macrophages resided in the tumor capsule and B cells localized in the intratumoral stroma of the LNCaP xenograft.
GPR30 expression has been reported to be upregulated by various growth factors, HIF-1α, and progestin (Ahola et al. 2002,Albanito et al. 2008,De et al. 2013,Recchia et al. 2011). However, only one report showed a decrease in GPR30 expression after estrogen treatment in the human internal mammary artery (Haas et al. 2007). This estrogen-induced suppression of GPR30 was not detected in neurons (Jacobi et al. 2007), suggesting that the regulation of GPR30 expression is cell context–specific. We showed here for the first time that androgen, the principal hormone in the prostate, inhibited GPR30 expression that was dependent on AR. Emerging interests have started to focus on the crosstalk between AR and ER signaling in PC (Yang et al. 2012, Claessens and Tilley 2014, Nelson et al. 2014). The goal of current treatments of CRPC is to maximally suppress androgen signaling, which may in turn remove the androgen suppression on GPR30 expression, resulting in a high level of GPR30 in late-stage CRPC. This study we provided convincing evidence that in both a preclinical model and in human specimens, reduced androgen level in CRPC enhanced GPR30 expression as compared with hormone-naïve PC. The wide expression and high levels of GPR30 may highlight an unprecedented opportunity to target this protein in clinical metastases of CRPC.
Existing therapies for CRPC offer limited gains in survival and trigger adverse effects; thus attention has begun to call on the sequence for applying these treatments (Higano and Crawford 2011). The current LNCaP model represented a subtype of CRPC in which G-1 induced intra-tumoral neutrophil infiltration associated with tumor necrosis. Similarly, we reasoned that a subset of patients harboring CRPC may benefit from G-1 therapy if it is delivered before the patients receive chemotherapy which can compromise neutrophil production. In light of the most recent CRPC that failed second-generation ADT (i.e., abiraterone acetate and MDV3100), whether or not the expression of GPR30 or the population of patients expressing high levels of GPR30 is increased upon resistance is a clinically interesting question to further explore GPR30 as a novel targeted therapy for late-stage CRPC. Importantly, in all the animal studies reported to date, G-1 did not induce adverse effects (Blasko et al. 2009,Chan et al. 2010,Dennis et al. 2009,Gao et al. 2011). G-1 toxicity to the functions of vital organs including heart and liver has been further proved to be undetectable in the present study. One major concern of estrogen-related treatment in PC is the increase in the risk of venous thromboembolism (reviewed in (Cox and Crawford 1995)). Although G-1 is a specific GPR30 agonist that has been shown not to bind ERα at a concentration up to 10 μM (Bologa et al. 2006), definitive evidence for the absence of estrogen-mediated coagulopathy in vivo is warranted.
Our findings, taken together, showed that G-1 effectively inhibited preclinical CRPC growth with a low risk of toxicity, underscoring G-1 or other GPR30-specific agonists might serve as novel anticancer agents for CRPC that expresses GPR30. The upregulation of GPR30 expression after androgen ablation and the recruitment of neutrophils to the CR tumors both point to a potentially important therapeutic window for G-1/GPR30-targeted therapy preferably under the conditions of a low or ultra-low androgen level in CRPC before chemotherapy.
Supplementary Material
Figure 2.

G-1 induced massive necrosis and neutrophil infiltration in the CR tumors. A) G-1 triggered massive necrosis in CR tumors. Tumor sections were stained with H&E, and the necrotic area was quantified as described in Supplementary Methods. B) G-1 induced significant necrosis associated with massive inflammation associated with neutrophil infiltration, both surrounding the necrotic area and within the viable area, in CR tumors only. Yellow arrow represents massive inflammation. Magnification: 20× (H&E upper panel), 200× (H&E lower panel), 100× (neutrophil IHC, upper panel), and 200× (neutrophil IHC, lower panel). Scale bars represent 50 μm in all micrographs. C) G-1 reduced the microvessel area ratio in the intratumoral stroma region but not in the tumor capsule. Microvessel area ratio is calculated as the ratio of the microvessel area to the intratumoral stroma area or the capsule area. D) Ki67 and cleaved caspase-3 staining of tumor cells was used to determine proliferation and apoptosis, respectively. E) G-1 did not induce toxicity in castrated mice as determined by absence of change in body weight (left panel) and in serum assays of organ damage marker enzymes (right panel). Error bars represent mean ± SEM, n=6–8/group, *p<0.05; ns, not significant; CR, castration-resistant; H&E, hematoxylin and eosin; IHC, immunohistochemistry.
Acknowledgements
We thank the Genomics, Epigenomics and Sequencing Core at the University of Cincinnati for Affymetrix microarray experiments, Ms. Dan Song for her support in immunohistochemistry study, Dr. Yuet-Kin Leung for critical reading of the manuscript, and Ms. Nancy K. Voynow for her professional editing of this manuscript.
Funding This work was supported by Veteran Affairs (Merit Award I01BX000675 to SMH); the National Institutes of Health (grant numbers P30ES006096, U01ES019480, and U01ES020988 to SMH, P50CA097186 Pacific Northwest SPORE Career Development Award to HML); and Prostate Cancer Foundation (Young Investigator Award to HML).
Abbreviations
- ADT
androgen-deprivation therapy
- ALT
alanine transaminase
- AR
androgen receptor
- AS
androgen-sensitive
- AST
aspartate transaminase
- ATCC
American Type Culture Collection
- ChIP
chromatin immunoprecipitation
- CK
creatine kinase
- CR
castration-resistant
- CRPC
castration-resistant prostate cancer
- DHT
dihydrotestosterone
- ER
estrogen receptor
- GPR30
G protein–coupled receptor 30
- G-1
(1(1-[4-(6-bromobenzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c] quinolin-8-yl]-ethanone)
- H&E
hematoxylin and eosin
- IHC
immunohistochemistry
- LDH
lactate dehydrogenase
- PC
prostate cancer
- TINs
tumor-infiltrating neutrophils
Footnotes
Declaration of interest The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Ahola TM, Purmonen S, Pennanen P, Zhuang YH, Tuohimaa P, Ylikomi T. Progestin upregulates G-protein-coupled receptor 30 in breast cancer cells. European Journal of Biochemistry. 2002;269:2485–2490. doi: 10.1046/j.1432-1033.2002.02912.x. [DOI] [PubMed] [Google Scholar]
- Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Ando S, et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Research. 2007;67:1859–1866. doi: 10.1158/0008-5472.CAN-06-2909. [DOI] [PubMed] [Google Scholar]
- Albanito L, Sisci D, Aquila S, Brunelli E, Vivacqua A, Madeo A, Lappano R, Pandey DP, Picard D, Mauro L, et al. Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells. Endocrinology. 2008;149:3799–3808. doi: 10.1210/en.2008-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasko E, Haskell CA, Leung S, Gualtieri G, Halks-Miller M, Mahmoudi M, Dennis MK, Prossnitz ER, Karpus WJ, Horuk R. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. Journal of Neuroimmunology. 2009;214:67–77. doi: 10.1016/j.jneuroim.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nature Chemical Biology. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, Wood WI, Moore MW. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- Caruso RA, Bellocco R, Pagano M, Bertoli G, Rigoli L, Inferrera C. Prognostic value of intratumoral neutrophils in advanced gastric carcinoma in a high-risk area in northern Italy. Modern Pathology. 2002;15:831–837. doi: 10.1097/01.MP.0000020391.98998.6B. [DOI] [PubMed] [Google Scholar]
- Chan QK, Lam HM, Ng CF, Lee AY, Chan ES, Ng HK, Ho SM, Lau KM. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell Death and Differentiation. 2010;17:1511–1523. doi: 10.1038/cdd.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SB, Graeber CT, Quinn JA, Filardo EJ. Retrograde transport of the transmembrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPER) from the plasma membrane towards the nucleus. Steroids. 2011;76:892–896. doi: 10.1016/j.steroids.2011.02.018. [DOI] [PubMed] [Google Scholar]
- Claessens F, Tilley W. Androgen signaling and steroid receptor crosstalk in endocrine cancers. Endocrine-Related Cancer. 2014;21:E3–5. doi: 10.1530/ERC-14-0274. doi: 10.1530/ERC-14-0274. [DOI] [PubMed] [Google Scholar]
- Cox RL, Crawford ED. Estrogens in the treatment of prostate cancer. Journal of Urology. 1995;154:1991–1998. [PubMed] [Google Scholar]
- de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr., Saad F, et al. Abiraterone and increased survival in metastatic prostate cancer. New England Journal of Medicine. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–1154. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- De MP, Bartella V, Vivacqua A, Lappano R, Santolla MF, Morcavallo A, Pezzi V, Belfiore A, Maggiolini M. Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene. 2013;32:678–688. doi: 10.1038/onc.2012.97. [DOI] [PubMed] [Google Scholar]
- Decker KF, Zheng D, He Y, Bowman T, Edwards JR, Jia L. Persistent androgen receptor-mediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Research. 2012;40:10765–10779. doi: 10.1093/nar/gks888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, Bologa CG, Leitao A, Brailoiu E, Deliu E, et al. In vivo effects of a GPR30 antagonist. Nature Chemical Biology. 2009;5:421–427. doi: 10.1038/nchembio.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di CE, Forni G, Lollini P, Colombo MP, Modesti A, Musiani P. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood. 2001;97:339–345. doi: 10.1182/blood.v97.2.339. [DOI] [PubMed] [Google Scholar]
- Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. Journal of Clinical Investigation. 2010;120:2423–2431. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Molecular Endocrinology. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- Fu X, Tao L, Rivera A, Xu H, Zhang X. Virotherapy induces massive infiltration of neutrophils in a subset of tumors defined by a strong endogenous interferon response activity. Cancer Gene Therapy. 2011;18:785–794. doi: 10.1038/cgt.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochemical and Biophysical Research Communications. 2006;346:904–910. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- Gao F, Ma X, Ostmann AB, Das SK. GPR30 activation opposes estrogen-dependent uterine growth via inhibition of stromal ERK1/2 and estrogen receptor alpha (ERalpha) phosphorylation signals. Endocrinology. 2011;152:1434–1447. doi: 10.1210/en.2010-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser T, Dewald B, Ehrengruber MU, Clark-Lewis I, Baggiolini M. The interleukin-8-related chemotactic cytokines GRO alpha, GRO beta, and GRO gamma activate human neutrophil and basophil leukocytes. The Journal of Biological Chemistry. 1993;268:15419–15424. [PubMed] [Google Scholar]
- Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Research. 2011;71:2411–2416. doi: 10.1158/0008-5472.CAN-10-2583. [DOI] [PubMed] [Google Scholar]
- Haas E, Meyer MR, Schurr U, Bhattacharya I, Minotti R, Nguyen HH, Heigl A, Lachat M, Genoni M, Barton M. Differential effects of 17beta-estradiol on function and expression of estrogen receptor alpha, estrogen receptor beta, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension. 2007;49:1358–1363. doi: 10.1161/HYPERTENSIONAHA.107.089995. [DOI] [PubMed] [Google Scholar]
- Harokopakis E, Hajishengallis G. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. European Journal of Immunology. 2005;35:1201–1210. doi: 10.1002/eji.200425883. [DOI] [PubMed] [Google Scholar]
- Higano CS, Crawford ED. New and emerging agents for the treatment of castration-resistant prostate cancer. Urologic Oncology. 2011;29:S1–S8. doi: 10.1016/j.urolonc.2011.08.013. [DOI] [PubMed] [Google Scholar]
- Higano CS, Schellhammer PF, Small EJ, Burch PA, Nemunaitis J, Yuh L, Provost N, Frohlich MW. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115:3670–3679. doi: 10.1002/cncr.24429. [DOI] [PubMed] [Google Scholar]
- Hirose K, Hakozaki M, Nyunoya Y, Kobayashi Y, Matsushita K, Takenouchi T, Mikata A, Mukaida N, Matsushima K. Chemokine gene transfection into tumour cells reduced tumorigenicity in nude mice in association with neutrophilic infiltration. British Journal of Cancer. 1995;72:708–714. doi: 10.1038/bjc.1995.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SM, Lee MT, Lam HM, Leung YK. Estrogens and prostate cancer: etiology, mediators, prevention, and management. Endocrinology and Metabolism Clinics of North America. 2011;40:591–614. doi: 10.1016/j.ecl.2011.05.002. doi: 10.1016/j.ecl.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HJ, Neven P, Drijkoningen M, Paridaens R, Wildiers H, Van LE, Berteloot P, Amant F, Vergote I, Christiaens MR. Association between tumour characteristics and HER-2/neu by immunohistochemistry in 1362 women with primary operable breast cancer. Journal of Clinical Pathology. 2005;58:611–616. doi: 10.1136/jcp.2004.022772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins C, Hodges CV. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. Journal of Urology. 2002;168:9–12. doi: 10.1016/s0022-5347(05)64820-3. [DOI] [PubMed] [Google Scholar]
- Jacobi JS, Martin C, Nava G, Jeziorski MC, Clapp C, Martinez de la EG. 17-Beta-estradiol directly regulates the expression of adrenergic receptors and kisspeptin/GPR54 system in GT1-7 GnRH neurons. Neuroendocrinology. 2007;86:260–269. doi: 10.1159/000107770. [DOI] [PubMed] [Google Scholar]
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. New England Journal of Medicine. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- Langley RE, Cafferty FH, Alhasso AA, Rosen SD, Sundaram SK, Freeman SC, Pollock P, Jinks RC, Godsland IF, Kockelbergh R, et al. Cardiovascular outcomes in patients with locally advanced and metastatic prostate cancer treated with luteinising-hormone-releasing-hormone agonists or transdermal oestrogen: the randomised, phase 2 MRC PATCH trial (PR09) Lancet Oncology. 2013;14:306–316. doi: 10.1016/S1470-2045(13)70025-1. doi: 10.1016/S1470-2045(13)70025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leav I, Lau KM, Adams JY, McNeal JE, Taplin ME, Wang J, Singh H, Ho SM. Comparative studies of the estrogen receptors beta and alpha and the androgen receptor in normal human prostate glands, dysplasia, and in primary and metastatic carcinoma. American Journal of Pathology. 2001;159:79–92. doi: 10.1016/s0002-9440(10)61676-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung BP, Culshaw S, Gracie JA, Hunter D, Canetti CA, Campbell C, Cunha F, Liew FY, McInnes IB. A role for IL-18 in neutrophil activation. Journal of Immunology. 2001;167:2879–2886. doi: 10.4049/jimmunol.167.5.2879. [DOI] [PubMed] [Google Scholar]
- Leung YK, Lam HM, Wu S, Song D, Levin L, Cheng L, Wu CL, Ho SM. Estrogen receptor beta2 and beta5 are associated with poor prognosis in prostate cancer, and promote cancer cell migration and invasion. Endocrine-Related Cancer. 2010;17:675–689. doi: 10.1677/ERC-09-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo A, Maggiolini M. Nuclear alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer Research. 2010;70:6036–6046. doi: 10.1158/0008-5472.CAN-10-0408. [DOI] [PubMed] [Google Scholar]
- Maggiolini M, Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. Journal of Endocrinology. 2010;204:105–114. doi: 10.1677/JOE-09-0242. [DOI] [PubMed] [Google Scholar]
- Musiani P, Allione A, Modica A, Lollini PL, Giovarelli M, Cavallo F, Belardelli F, Forni G, Modesti A. Role of neutrophils and lymphocytes in inhibition of a mouse mammary adenocarcinoma engineered to release IL-2, IL-4, IL-7, IL-10, IFN-alpha, IFN-gamma, and TNF-alpha. Laboratory Investigation. 1996;74:146–157. [PubMed] [Google Scholar]
- Nelson AW, Tilley WD, Neal DE, Carroll JS. Estrogen receptor beta in prostate cancer: friend or foe? Endocrine-Related Cancer. 2014;21:T219–234. doi: 10.1530/ERC-13-0508. doi: 10.1530/ERC-13-0508. [DOI] [PubMed] [Google Scholar]
- Norman G, Dean ME, Langley RE, Hodges ZC, Ritchie G, Parmar MK, Sydes MR, Abel P, Eastwood AJ. Parenteral oestrogen in the treatment of prostate cancer: a systematic review. British Journal of Cancer. 2008;98:697–707. doi: 10.1038/sj.bjc.6604230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh WK. The evolving role of estrogen therapy in prostate cancer. Clinical Prostate Cancer. 2002;1:81–89. doi: 10.3816/cgc.2002.n.009. [DOI] [PubMed] [Google Scholar]
- Otto C, Rohde-Schulz B, Schwarz G, Fuchs I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R, et al. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology. 2008;149:4846–4856. doi: 10.1210/en.2008-0269. [DOI] [PubMed] [Google Scholar]
- Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. The EMBO Journal. 2009;28:523–532. doi: 10.1038/emboj.2008.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelleitier M, Montplaisir S. The nude mouse: a model of deficient T-cell function. Methods and Achievements in Experimental Pathology. 1975;7:149–166. [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein-coupled receptor for estrogen. Molecular and Cellular Endocrinology. 2007;265:138–142. doi: 10.1016/j.mce.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nature Reviews Endocrinology. 2011;7:715–726. doi: 10.1038/nrendo.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recchia AG, De Francesco EM, Vivacqua A, Sisci D, Panno ML, Ando S, Maggiolini M. The G protein-coupled receptor 30 is up-regulated by hypoxia-inducible factor-1alpha (HIF-1alpha) in breast cancer cells and cardiomyocytes. The Journal of Biological Chemistry. 2011;286:10773–10782. doi: 10.1074/jbc.M110.172247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. Journal of Immunolology. 2003;170:3233–3242. doi: 10.4049/jimmunol.170.6.3233. [DOI] [PubMed] [Google Scholar]
- Saenko EL, Skorobogat'ko OV, Tarasenko P, Romashko V, Zhuravetz L, Zadorozhnaya L, Senjuk OF, Yaropolov AI. Modulatory effects of ceruloplasmin on lymphocytes, neutrophils and monocytes of patients with altered immune status. Immunological Investigations. 1994;23:99–114. doi: 10.3109/08820139409087792. [DOI] [PubMed] [Google Scholar]
- Schaider H, Oka M, Bogenrieder T, Nesbit M, Satyamoorthy K, Berking C, Matsushima K, Herlyn M. Differential response of primary and metastatic melanomas to neutrophils attracted by IL-8. International Journal of Cancer. 2003;103:335–343. doi: 10.1002/ijc.10775. [DOI] [PubMed] [Google Scholar]
- Schellhammer P. Life after failure of traditional androgen deprivation therapy. Urologic Oncology. 2012;30:S10–4. doi: 10.1016/j.urolonc.2012.01.009. doi: 10.1016/j.urolonc.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Tannock IF, de WR, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. New England Journal of Medicine. 2004;351:1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Ando S, Maggiolini M. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17beta-estradiol and hydroxytamoxifen in endometrial cancer cells. Molecular Endocrinology. 2006;20:631–646. doi: 10.1210/me.2005-0280. [DOI] [PubMed] [Google Scholar]
- Yang L, Ravindranathan P, Ramanan M, Kapur P, Hammes SR, Hsieh JT, Raj GV. Central role for PELP1 in nonandrogenic activation of the androgen receptor in prostate cancer. Molecular Endocrinology. 2012;26:550–561. doi: 10.1210/me.2011-1101. doi: 10.1210/me.2011-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Decker KF, Zhou T, Chen J, Qi Z, Jacobs K, Weilbaecher KN, Corey E, Long F, Jia L. Role of WNT7B-induced noncanonical pathway in advanced prostate cancer. Molecular Cancer Research. 2013;11:482–493. doi: 10.1158/1541-7786.MCR-12-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.