

Abstract
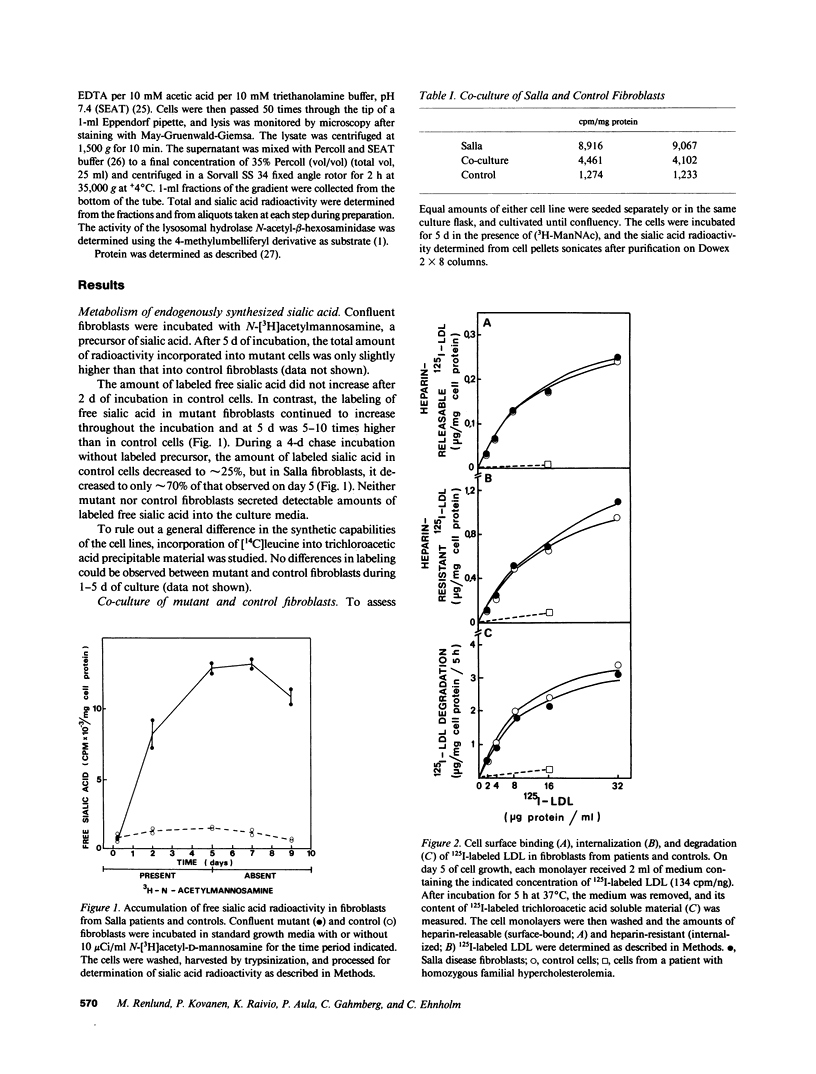
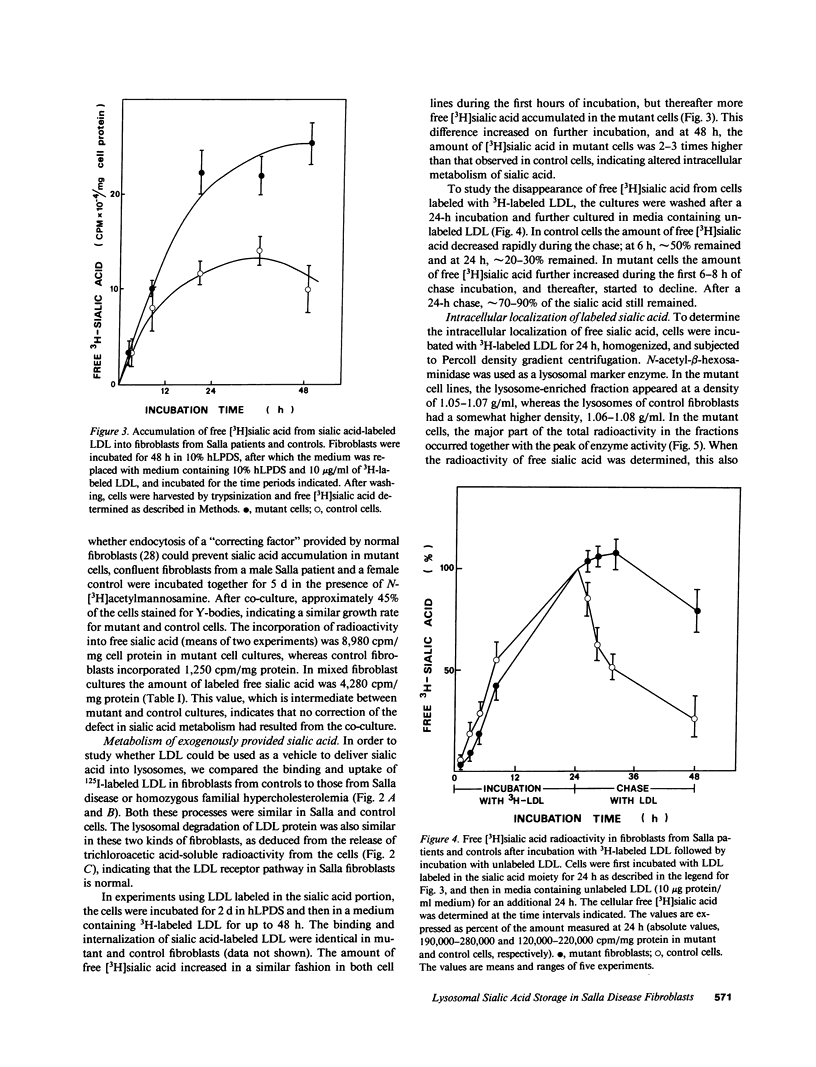
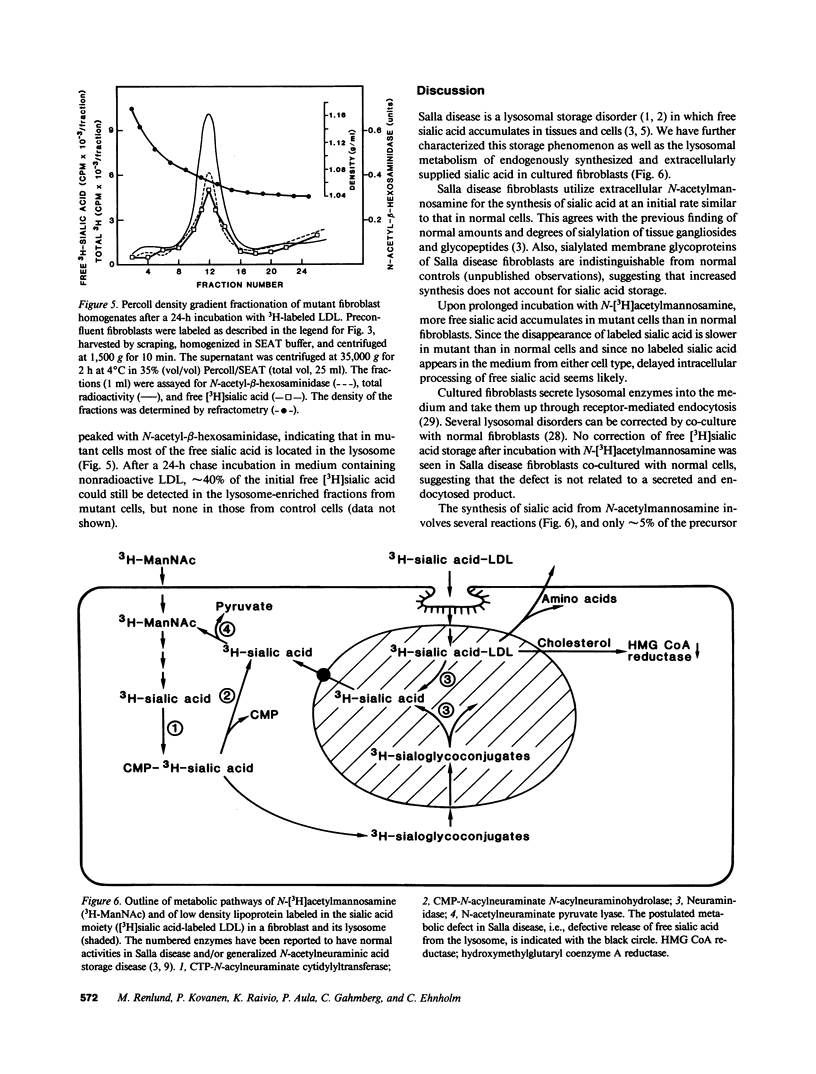
Salla disease is a lysosomal storage disorder characterized by mental retardation and disturbed sialic acid metabolism. To study endogenous synthesis and breakdown of sialic acid, fibroblasts were incubated for 5 d in the presence and then in the absence of N-[3H]acetylmannosamine. Labeling of free sialic acid was 5-10 times higher in mutant than in normal cells. Radioactivity decreased in 4 d by 75% in normal but only by 30% in mutant fibroblasts. The labeling pattern was not normalized upon coculture of mutant and normal cells. To study the metabolism of extracellular sialic acid, low-density lipoprotein (LDL) was labeled in the sialic acid moiety (periodate-NaB3H4) or in the protein moiety (125I). Binding, internalization, lysosomal degradation, and exit of products of protein catabolism were similar in normal and mutant fibroblasts. Upon incubation with LDL labeled in the sialic acid moiety, mutant cells accumulated 2-3 times more free sialic acid radioactivity than normal fibroblasts, mostly in the lysosomal fraction. After a 24-h chase incubation, radioactivity in free sialic acid decreased by 70-80% in normal but only by 10-30% in mutant cells. In mutant fibroblasts, 40% of the radioactivity remained in lysosomes, whereas no labeled free sialic acid was detected in lysosomes from normal fibroblasts. We conclude that in Salla disease, fibroblast endogenous synthesis of sialic acid and lysosomal cleavage of exogenous glycoconjugates is normal, but free sialic acid cannot leave the lysosome. These findings suggest that the basic defect in Salla disease is deficient transport of free sialic acid through the lysosomal membrane.
Full text
PDF



Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Aula P., Autio S., Raivio K. O., Rapola J., Thodén C. J., Koskela S. L., Yamashina I. "Salla disease": a new lysosomal storage disorder. Arch Neurol. 1979 Feb;36(2):88–94. doi: 10.1001/archneur.1979.00500380058006. [DOI] [PubMed] [Google Scholar]
- BRUNETTI P., JOURDIAN G. W., ROSEMAN S. The sialic acids. III. Distribution and properties of animal N-acetylneuraminic aldolase. J Biol Chem. 1962 Aug;237:2447–2453. [PubMed] [Google Scholar]
- Bierman E. L., Stein O., Stein Y. Lipoprotein uptake and metabolism by rat aortic smooth muscle cells in tissue culture. Circ Res. 1974 Jul;35(1):136–150. doi: 10.1161/01.res.35.1.136. [DOI] [PubMed] [Google Scholar]
- Bilheimer D. W., Eisenberg S., Levy R. I. The metabolism of very low density lipoprotein proteins. I. Preliminary in vitro and in vivo observations. Biochim Biophys Acta. 1972 Feb 21;260(2):212–221. doi: 10.1016/0005-2760(72)90034-3. [DOI] [PubMed] [Google Scholar]
- Brown M. S., Goldstein J. L. Analysis of a mutant strain of human fibroblasts with a defect in the internalization of receptor-bound low density lipoprotein. Cell. 1976 Dec;9(4 Pt 2):663–674. doi: 10.1016/0092-8674(76)90130-6. [DOI] [PubMed] [Google Scholar]
- Ehnholm C., Garoff H., Renkonen O., Simons K. Protein and carbohydrate composition of Lp(a)lipoprotein from human plasma. Biochemistry. 1972 Aug 15;11(17):3229–3232. doi: 10.1021/bi00767a015. [DOI] [PubMed] [Google Scholar]
- Gahl W. A., Tietze F., Bashan N., Bernardini I., Raiford D., Schulman J. D. Characteristics of cystine counter-transport in normal and cystinotic lysosome-rich leucocyte granular fractions. Biochem J. 1983 Nov 15;216(2):393–400. doi: 10.1042/bj2160393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl W. A., Tietze F., Bashan N., Steinherz R., Schulman J. D. Defective cystine exodus from isolated lysosome-rich fractions of cystinotic leucocytes. J Biol Chem. 1982 Aug 25;257(16):9570–9575. [PubMed] [Google Scholar]
- Gahmberg C. G., Andersson L. C. Selective radioactive labeling of cell surface sialoglycoproteins by periodate-tritiated borohydride. J Biol Chem. 1977 Aug 25;252(16):5888–5894. [PubMed] [Google Scholar]
- Goldstein J. L., Brown M. S. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem. 1974 Aug 25;249(16):5153–5162. [PubMed] [Google Scholar]
- Goldstein J. L., Brown M. S. The low-density lipoprotein pathway and its relation to atherosclerosis. Annu Rev Biochem. 1977;46:897–930. doi: 10.1146/annurev.bi.46.070177.004341. [DOI] [PubMed] [Google Scholar]
- HAVEL R. J., EDER H. A., BRAGDON J. H. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955 Sep;34(9):1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock L. W., Horwitz A. L., Dawson G. N-acetylneuraminic acid and sialoglycoconjugate metabolism in fibroblasts from a patient with generalized N-acetylneuraminic acid storage disease. Biochim Biophys Acta. 1983 Oct 4;760(1):42–52. doi: 10.1016/0304-4165(83)90122-8. [DOI] [PubMed] [Google Scholar]
- Hancock L. W., Thaler M. M., Horwitz A. L., Dawson G. Generalized N-acetylneuraminic acid storage disease: quantitation and identification of the monosaccharide accumulating in brain and other tissues. J Neurochem. 1982 Mar;38(3):803–809. doi: 10.1111/j.1471-4159.1982.tb08701.x. [DOI] [PubMed] [Google Scholar]
- Kamerling J. P., Strecker G., Farriaux J. P., Dorland L., Haverkamp J., Vliegenthart J. F. 2-Acetamidoglucal, a new metabolite isolated from the urine of a patient with sialuria. Biochim Biophys Acta. 1979 Mar 22;583(3):403–408. doi: 10.1016/0304-4165(79)90465-3. [DOI] [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Lloyd J. B. A study of permeability of lysosomes to amino acids and small peptides. Biochem J. 1971 Jan;121(2):245–248. doi: 10.1042/bj1210245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd J. B. Studies on the permeability of rat liver lysosomes to carbohydrates. Biochem J. 1969 Dec;115(4):703–707. doi: 10.1042/bj1150703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowden J. A., O'Brien J. S. Sialidosis: a review of human neuraminidase deficiency. Am J Hum Genet. 1979 Jan;31(1):1–18. [PMC free article] [PubMed] [Google Scholar]
- Maguire G. A., Docherty K., Hales C. N. Sugar transport in rat liver lysosomes. Direct demonstration by using labelled sugars. Biochem J. 1983 Apr 15;212(1):211–218. doi: 10.1042/bj2120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montreuil J., Biserte G., Strecker G., Spik G., Fontaine G., Farriaux J. P. Description d'un nouveau type de méliturie: la sialurie. Clin Chim Acta. 1968 Jul;21(1):61–69. doi: 10.1016/0009-8981(68)90011-9. [DOI] [PubMed] [Google Scholar]
- Neufeld E. F., Lim T. W., Shapiro L. J. Inherited disorders of lysosomal metabolism. Annu Rev Biochem. 1975;44:357–376. doi: 10.1146/annurev.bi.44.070175.002041. [DOI] [PubMed] [Google Scholar]
- Neufeld E. F., Sando G. N., Garvin A. J., Rome L. H. The transport of lysosomal enzymes. J Supramol Struct. 1977;6(1):95–101. doi: 10.1002/jss.400060108. [DOI] [PubMed] [Google Scholar]
- Pesonen M., Ansorge W., Simons K. Transcytosis of the G protein of vesicular stomatitis virus after implantation into the apical plasma membrane of Madin-Darby canine kidney cells. I. Involvement of endosomes and lysosomes. J Cell Biol. 1984 Sep;99(3):796–782. doi: 10.1083/jcb.99.3.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijngoud D. J., Tager J. M. The permeability properties of the lysosomal membrane. Biochim Biophys Acta. 1977 Nov 14;472(3-4):419–449. doi: 10.1016/0304-4157(77)90005-3. [DOI] [PubMed] [Google Scholar]
- Renlund M., Aula P., Raivio K. O., Autio S., Sainio K., Rapola J., Koskela S. L. Salla disease: a new lysosomal storage disorder with disturbed sialic acid metabolism. Neurology. 1983 Jan;33(1):57–66. doi: 10.1212/wnl.33.1.57. [DOI] [PubMed] [Google Scholar]
- Renlund M., Chester M. A., Lundblad A., Aula P., Raivio K. O., Autio S., Koskela S. L. Increased urinary excretion of free N-acetylneuraminic acid in thirteen patients with Salla disease. Eur J Biochem. 1979 Nov 1;101(1):245–250. doi: 10.1111/j.1432-1033.1979.tb04237.x. [DOI] [PubMed] [Google Scholar]
- Renlund M., Chester M. A., Lundblad A., Parkkinen J., Krusius T. Free N-acetylneuraminic acid in tissues in Salla disease and the enzymes involved in its metabolism. Eur J Biochem. 1983 Jan 17;130(1):39–45. doi: 10.1111/j.1432-1033.1983.tb07114.x. [DOI] [PubMed] [Google Scholar]
- Renlund M. Clinical and laboratory diagnosis of Salla disease in infancy and childhood. J Pediatr. 1984 Feb;104(2):232–236. doi: 10.1016/s0022-3476(84)80998-1. [DOI] [PubMed] [Google Scholar]
- Rome L. H., Garvin A. J., Allietta M. M., Neufeld E. F. Two species of lysosomal organelles in cultured human fibroblasts. Cell. 1979 May;17(1):143–153. doi: 10.1016/0092-8674(79)90302-7. [DOI] [PubMed] [Google Scholar]
- SVENNERHOLM L. Quantitative estimation of sialic acids. II. A colorimetric resorcinol-hydrochloric acid method. Biochim Biophys Acta. 1957 Jun;24(3):604–611. doi: 10.1016/0006-3002(57)90254-8. [DOI] [PubMed] [Google Scholar]
- Schauer R. Characterization of sialic acids. Methods Enzymol. 1978;50:64–89. doi: 10.1016/0076-6879(78)50008-6. [DOI] [PubMed] [Google Scholar]
- Stevenson R. E., Lubinsky M., Taylor H. A., Wenger D. A., Schroer R. J., Olmstead P. M. Sialic acid storage disease with sialuria: clinical and biochemical features in the severe infantile type. Pediatrics. 1983 Oct;72(4):441–449. [PubMed] [Google Scholar]
- Thomas G. H., Scocca J., Libert J., Vamos E., Miller C. S., Reynolds L. W. Alterations in cultured fibroblasts of sibs with an infantile form of a free (unbound) sialic acid storage disorder. Pediatr Res. 1983 May;17(5):307–312. doi: 10.1203/00006450-198305000-00001. [DOI] [PubMed] [Google Scholar]
- Tondeur M., Libert J., Vamos E., Van Hoof F., Thomas G. H., Strecker G. Infantile form of sialic acid storage disorder: clinical, ultrastructural, and biochemical studies in two siblings. Eur J Pediatr. 1982 Oct;139(2):142–147. doi: 10.1007/BF00441499. [DOI] [PubMed] [Google Scholar]
- Van Lenten L., Ashwell G. Studies on the chemical and enzymatic modification of glycoproteins. A general method for the tritiation of sialic acid-containing glycoproteins. J Biol Chem. 1971 Mar 25;246(6):1889–1894. [PubMed] [Google Scholar]
- Virtanen I., Ekblom P., Laurila P., Nordling S., Raivio K. O., Aula P. Characterization of storage material in cultured fibroblasts by specific lectin binding in lysosomal storage diseases. Pediatr Res. 1980 Nov;14(11):1199–1203. doi: 10.1203/00006450-198011000-00010. [DOI] [PubMed] [Google Scholar]
- WARREN L. The thiobarbituric acid assay of sialic acids. J Biol Chem. 1959 Aug;234(8):1971–1975. [PubMed] [Google Scholar]