

Abstract
Kabuki syndrome (KS) is a multiple congenital anomalies syndrome characterized by characteristic facial features and varying degrees of mental retardation, caused by mutations in KMT2D/MLL2 and KDM6A/UTX genes. In this study, we performed a mutational screening on 303 Kabuki patients by direct sequencing, MLPA, and quantitative PCR identifying 133 KMT2D, 62 never described before, and four KDM6A mutations, three of them are novel. We found that a number of KMT2D truncating mutations result in mRNA degradation through the nonsense-mediated mRNA decay, contributing to protein haploinsufficiency. Furthermore, we demonstrated that the reduction of KMT2D protein level in patients’ lymphoblastoid and skin fibroblast cell lines carrying KMT2D-truncating mutations affects the expression levels of known KMT2D target genes. Finally, we hypothesized that the KS patients may benefit from a readthrough therapy to restore physiological levels of KMT2D and KDM6A proteins. To assess this, we performed a proof-of-principle study on 14 KMT2D and two KDM6A nonsense mutations using specific compounds that mediate translational readthrough and thereby stimulate the re-expression of full-length functional proteins. Our experimental data showed that both KMT2D and KDM6A nonsense mutations displayed high levels of readthrough in response to gentamicin treatment, paving the way to further studies aimed at eventually treating some Kabuki patients with readthrough inducers.
Keywords: KMT2D, KDM6A, Kabuki syndrome, haploinsufficiency, readthrough
Introduction
Histone methylation is an epigenetic mechanism by which spatial and temporal expression of distinct genes and pathways are regulated at precise developmental stages. Different histone lysine methylation states (mono-, di-, or tri-methylation) are associated with gene transcriptional activation or repression depending on the location of the lysine residue.
In general, histone H3 lysine 4 (H3K4) di- and tri-methylation are linked to active transcription, whereas H3K27 di- and tri-methylation are associated with gene silencing [Santos-Rosa et al., 2002]. Aberrations in the histone modifiers have been associated with genetic diseases, such as Kleefstra syndrome, Sotos syndrome, Weaver syndrome, and Schinzel–Giedion syndrome [Berdasco and Esteller, 2013]. The discovery of histone methyltransferase (HMT) KMT2D (MIM #602113; RefSeq NM_003482.3, also known as MLL2, ALR/MLL4) and demethylase KDM6A (MIM #300128; RefSeq NM_021140.2, also known as UTX) genetic alterations in Kabuki syndrome (KS) patients expanded and highlighted the role of histone modifiers in causing congenital anomalies and intellectual disability [Bogershausen and Wollnik, 2013]. KS (MIM #147920) is an autosomal-dominant condition characterized by striking facial features, such as elongated palpebral fissures with eversion of the lateral third of the lower eyelid, short columella with depressed nasal tip, skeletal anomalies, dermatoglyphic abnormalities, mild-to-moderate mental retardation, and postnatal growth deficiency [Kuroki et al., 1981; Niikawa et al., 1981]. KS is commonly associated with congenital heart defects, genitourinary anomalies, cleft lip and/or palate, susceptibility to infections, gastrointestinal abnormalities, ophthalmologic defects, ptosis and strabismus, dental anomalies, including widely spaced teeth and hypodontia, and ear pits. Additionally, KS individuals might have a number of less frequent findings comprising visceral abnormalities and premature breast development in females.
In 2010, whole-exome sequencing successfully identified heterozygous mutations in the KMT2D gene as the major cause of KS [Ng et al., 2010]. Since then, 55%–65% of KS cases have been reported carrying a KMT2D mutation [Ng et al., 2010; Hannibal et al., 2011; Li et al., 2011; Micale et al., 2011; Paulussen et al., 2011; Banka et al., 2012, 2013; Makrythanasis et al., 2013; Miyake et al., 2013a]. The majority of mutations identified were de novo, although familial cases with autosomal-dominant inheritance have occasionally been described [Hannibal et al., 2011; Kokitsu-Nakata et al., 2012]. KMT2D gene maps to 12q13.12 and encodes a gigantic protein (5,537 residues) that belongs to the mixed lineage leukemia (MLL) family of HMTs. The MLL proteins are part of the SET (Su[var]3–9, enhancer-of-zeste, Trithorax) family of proteins [Dillon et al., 2005] that play important roles in the epigenetic control of active chromatin states [Issaeva et al., 2007]. They act as transcriptional coactivators and are involved in the expression control of genes essential for embryogenesis and development such as the HOX genes [Ansari and Mandal, 2010; Eissenberg and Shilatifard, 2010].
A subset of KS individuals was recently identified with either point mutations or microdeletions encompassing the X-linked gene, KDM6A [Lederer et al., 2012; Miyake et al., 2013a; Miyake et al., 2013b] that encodes a Histone H3 lysine-27 demethylase. KDM6A plays a crucial role in general chromatin remodeling [Hong et al., 2007; Lan et al., 2007] and interacts with KMT2D, in a conserved SET-1-like complex that trimethylates H3K4 [Issaeva et al., 2007]. The inactivation of the zebrafish kdm6a orthologue by morpholino is associated with severe and diverse structural defects and developmental abnormalities [Lindgren et al., 2013]; this inactivation resulted in the misregulation of HOX genes leading to a posterior developmental defect, whereas Kdm6a-deficient mice showed severe defects in heart development and embryonic lethality [Lee et al., 2012].
The majority of KMT2D and KDM6A nucleotide changes are truncating mutations (nonsense, frameshift, or splice site) that produce premature termination codons (PTCs), which are potentially deleterious. In the last few years, there have been several attempts to develop mutation-specific pharmacological approaches to restore sufficient levels of functional proteins. One approach that has been gaining prominence is that of using pharmacological agents to promote nonsense suppression or readthrough of PTCs thus enabling re-expression of full-length functional proteins [Nakamura et al., 2005; Bellais et al., 2010]. The potential of aminoglycosides and nonaminoglycosides as therapeutic tools has been demonstrated in several genetic disorders such as hemophilia, β-thalassemia, and spinal muscular atrophy, but most extensively in Duchenne muscular dystrophy and cystic fibrosis [Lee and Dougherty, 2012]. Interestingly, this treatment was successfully applied on an Ataxia-telangiectasia patient with heterozygous nonsense mutation, thereby demonstrating therapeutic ability despite the presence of a nonsense mutation in just one allele [Nakamura et al., 2011].
In this report, we have expanded the spectrum of mutations of KMT2D and KDM6A genes by analyzing our cohort of 303 Kabuki patients by direct sequencing, MLPA and quantitative PCR (qPCR). Based on KMT2D biological role, we designed functional studies that highlighted the haploinsufficiency of KMT2D as one of the mechanisms underlying the pathogenesis of the disease. Moreover, we evaluated the readthrough efficiency of 14 KMT2D and two KDM6A nonsense mutations and showed that 11 KMT2D and one KDM6A nonsense mutation responded to gentamicin treatment suggesting that this strategy can be effective to restore functional endogenous protein level and biological activity of KMT2D and KDM6A.
Material and Methods
Patients
In this study, 303 KS patients were included following the inclusions criteria reported in Micale et al. (2011). Patients were enrolled after obtaining appropriate informed consent by the physicians in charge and approval by the respective local ethics committees.
Cell Cultures, Nonsense-Mediated mRNA Decay Assay, and E2 Treatment
Lymphoblastoid cell lines (LCLs) were established from fresh peripheral blood leukocytes, infected by Epstein Barr Virus and cultured in RPMI 1640 supplemented with 10% of fetal bovine serum (FBS; Life Technologies, USA), l-glutamine, and 1% antibiotics mixture (penicillin and streptomycin 10,000 UI/ml; Life Technologies). Primary skin fibroblasts were grown in minimum essential medium supplemented with 1% l-glutamine, 10% FBS, and antibiotics. Nonsense-mediated mRNA decay (NMD) was assayed by treating fibroblast and lymphoblast cell lines with puromycin at a concentration of 200 μg/ml. After 8 hr of incubation, total RNA was obtained using the RNasy mini Kit (Qiagen, Germany) according to manufacturer instructions and Quantitect Reverse Transcription kit (Qiagen, Düsseldorf, Germany) was used for cDNA synthesis. Fibroblast cell lines were treated with 200 nM of 17B-estradiol (E2), incubated for 8 hr and then harvested for RNA and protein extraction.
MLPA, Long PCR, and Genomic Real-Time qPCR
Genomic DNAs were extracted from fresh and/or frozen peripheral blood leukocytes of the probands and their parents following standard procedures. MLPA analysis was performed as reported in Priolo et al. (2012) using probe mixture (SALSA MLPA KIT P389-A1 KMT2D; MRC-Holland, Amsterdam, The Netherlands) that contains 27 probes targeting exons across the KMT2D gene.
Long PCR was carried out with Expand Long Template PCR system (Roche, Mannheim, Germany) with combinations of primer pairs spanning the KMT2D exons not covered by MLPA. qPCR reactions were carried out with primers designed to amplify all the 29 KDM6A exons as described in Priolo et al. (2012).
Real-Time qPCR Assays
Total RNA was extracted from peripheral blood leukocytes using TRIZOL reagent (Life Technology) and reverse transcribed using the Quantitect Transcription kit (Qiagen), according to the manufacturer's instructions. Oligos for qPCR were designed using the Primer express program [Rozen and Skaletsky, 2000] with default parameters. EEF1A1 and GAPDH were used as references genes. qPCR reactions and calculations were made as reported in Ferrero et al. (2010). Significance was determined by a two-tailed unpaired t-test for means.
Dual Luciferase Reporter Vector System
A dual gene reporter pCRFL (gently provided by Prof. J-P Rousset) was used to quantify the effect of gentamicin on stop mutation readthrough in culture cells. Sequence to be analyzed spanning 27 nucleotides centered on the different stop mutations were inserted in-frame between the Renilla and Firefly Luciferase coding sequences through site-directed mutagenesis by using the QuickChange II kit (Stratagene, La Jolla, California, USA). All target DNA sequences were assessed by sequencing. Plasmids were purified using Qiagen Midiprep Kit according to the manufacturer's specifications. HEK293 cells were culture in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 1% antibiotics mixture, and incubated at 37% in humidity saturated 5% CO2. Cells were transfected using FuGene HD Transfection Reagent (Promega) with each pCRFL construct. Culture medium was replaced 24 hr later with fresh medium at a concentration of gentamicin (Hospira, Lake Forest, Illinois, USA) varying from 0 to 1,200 μg/ml. After 48 hr, the cells were lysed in passive lysis buffer and assayed for both Firefly and Renilla luciferase activity using the Dual-GLO® Luciferase Assay System (Promega, Madison, WI, USA).
This dual reporter allows the quantification of stop-codon readthrough, by measuring Firefly and Renilla activities, as previously described [Bidou et al., 2004].
Briefly, readthrough efficiency was estimated by comparing the Firelfly/Renilla luciferase ratio obtained for each nonsense mutation to an in-frame control. A 100% activity control was provided by a construct (TQ) with no stop codon between the coding sequences of the two reporters [Sermet-Gaudelus et al., 2007]. A pCRFL reporter vector harboring the 319d Duchenne muscular dystrophy mutation (pCRFL319), exhibiting the highest gentamicin-induced readthrough efficiency and the highest induction factor in NIH3T3-cultured cells assays [Bidou et al., 2004], was used as positive control. Values are the mean ± SEM of three experimental replicates from three independent transfections. Significance was determined by a two-tailed unpaired t-test for means.
PCR-Based Sequencing of KMT2D and KDM6A
Mutation screening of all 54 coding exons of the KMT2D gene was performed as reported in Micale et al. (2011). KDM6A (NM_021140.2) primers were designed to amplify exons and adjacent splice sites according to the reference sequences, using the Primer 3 Output program (http://frodo.wi.mit.edu/primer3/). A complete list of primer sequences and PCR conditions are available on request. The amplified products were subsequently purified and sequenced with a ready reaction kit (BigDye Terminator v1.1 Cycle; Warrington WA1 4SR, UK). The fragments obtained were purified using DyeEx plates (Qiagen) and resolved on an automated sequencer (3130xl Genetyc analyzer DNA Analyzer; ABI Prism). Sequences were analyzed using the Sequencer software (Gene Codes, Ann Arbor, MI). We resequenced all identified mutations in independent experiments. The following databases were used to obtain gene information: National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/), Ensembl Genome Server (http://www.ensembl.org/), UCSC Genome Bioinformatics (http://www.genome.ucsc.edu/), and 1000 genomes (http://browser.1000genomes.org/). All existing and new mutations were described following the recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen).
In Silico Analysis
We analyzed the missense variants by the latest version of the server Polyphen-2 version 2.2.2 (http://genetics.bwh.harvard.edu/pph) (Adzhubei et al., 2010), Align GVGD (http://agvgd.iarc.fr/agvgd_input.php) [Tavtigian et al., 2006], PROVEAN v1.1 (http://provean.jcvi.org/index.php) [Choi et al., 2012], and SIFT v1.03 (http://sift.jcvi.org/) [Kumar et al., 2009], UMD-predictor (http://www.umd.be/) [Frederic et al., 2009], using default parameters. Splice-site variants were evaluated for putative alteration of regulatory process at the transcriptional or splicing level with NetGene2 (http://www.cbs.dtu.dk/services/NetGene2) [Brunak et al., 1991] and NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html) [Reese et al., 1997].
Protein Extraction and Western Blot Analysis
Normal and patient lymphoblastoid and fibroblast cell lines were treated with concentration of gentamicin varying from 800 to 1,200 μg/ml. After 48 hr, cells were lysed in 10-mM HEPES, 1.5-mM MgCl2, 10-mM KCl, 0.5-mM DTT, 1.5-mM Phenylmethylsulfonyl fluoride (PMSF), and 2 N KCl. Proteins were separated on a SDS-polyacrylamide gel. Western blots were prepared as reported in Caratozzolo et al. (2012) and probed with anti-KMT2D (Abnova, Taipei, Taiwan). Bound primary antibodies were visualized using ECL western blotting or ECL plus Western blotting detection reagents (GE Healthcare, UK).
Results
Mutation Screening of KMT2D and KDM6A
Since 2011, we performed a comprehensive mutational screening on 303 Kabuki patients by direct sequencing, MLPA, and qPCR on KMT2D and KDM6A genes, respectively. Of these 303 patients, 79 were described in previous studies where 53 pathogenic KMT2D mutations were identified in 51 patients (Supp. Table S1) [Micale et al., 2011; Makrythanasis et al., 2013; Ratbi et al., 2013].
Here, we extended the KMT2D mutational analysis to a new cohort of 224 individuals clinically diagnosed as KS identifying 82 (82/224, 36%) patients carrying causative KMT2D mutations.
Overall, in our whole KS cohort, we identified 133/303 (34%) patients with KMT2D mutations and 140 different KMT2D mutations; of them, 87/140 (62%) were identified in this study with 66/140 (47%) never described before. The following types of KMT2D mutations were identified: 37 nonsense (37/140, 26%), 42 frameshift (42/140, 30%), 46 missense (46/140, 33%), eight splice site (8/140, 6%), and seven indel (7/140, 5%) (Supp. Tables S1 and S2). Almost all identified mutations occurred de novo; only 16 missense and four indel variants were inherited from an apparently asymptomatic parent.
To explore the occurrence of intragenic deletions and duplications, we screened KMT2D exons by MLPA analysis on 207 KS patients: 164 KMT2D point mutation negative, 37 with KMT2D missense, and six indels. We identified the KB43 patient carrying a singleton deletion covering from exon 49 to a part of exon 51 (Supp. Fig. S1A). Long PCR, followed by direct sequencing, allowed us to map the boundaries of the deletion as c.15785-238_16168del2425insTTGTATCTCAA mutation (Supp. Fig. S1B). KMT2D mutations were submitted to the Leiden Open Variation Database (LOVD v.3.0; http://www.lovd.nl/3.0/home) public database.
To identify KDM6A mutations in KS patients, we first assessed KDM6A exon dosage by qPCR analysis on 139 samples with no KMT2D alteration without identifying any intragenic deletions and duplications. Then, we sequenced 29 coding exons of KDM6A along with its exon–intron boundaries in 98 patients (not enough DNA was available for the remaining 41 samples). We identified four KDM6A point mutations: one nonsense c.514C>T (p.Arg172X) [Banka et al., 2014], one frameshift c.1846_1849delACTC (p.Thr616TyrfsX8), one missense c.2939A>T (p.Asp980Val), and one splicing mutation c.3284+3_3284+6delAAGT (p.Asn1070_Lys1094del). Two of them occurred de novo; parental DNAs were unavailable for the others. KDM6A mutations were submitted to LOVD (http://www.lovd.nl/3.0/home) public database.
Characterization of KMT2D Splice-Site Mutations
We identified eight patients with KMT2D mutations in the proximity of the splice sites and we analyzed them by NetGene2 and Fruitfly softwares to predict the molecular consequences of the observed nucleotide changes. The availability of the RNA from those KS patients, but one, allowed us to experimentally determine the effect at the transcriptional or splicing level (Supp. Fig. S2). All identified intronic KMT2D variants cause aberrant splicing of the corresponding transcripts that result in a frameshift with the generation of a premature stop codon as reported below:
c.177-2A>C (p.Ser59ArgfsX86) affects the essential nucleotide −2 of the splice acceptor site of intron 2. Real-time PCR analysis and fragment sequencing of the carrier patient revealed skipping of the entire exon 3 (Supp. Fig. S2A).
c.400+1G>A (p.Ser59ArgfsX86) occurring within the GT splice donor site in intron 3 resulted in the disruption of the canonical splice site and again in skipping of exon 3 (Supp. Fig. S2B).
c.400-3A>G (p.Gly134GlufsX74) creates a novel AG splice acceptor site in intron 3. Sequencing of the normal-sized fragment amplified from patient's cDNA revealed a transcript with an insertion of an AG dinucleotide that corresponds to the effective splice-site acceptor of intron 3 (Supp. Fig. S2C).
c.13999+5G>A (p.Asn4614IlefsX5) occurs at nucleotide +5 of the splice donor site of intron 42. Sequencing analysis of the real-time PCR fragments confirmed that the observed shorter transcript results from a splicing failure and exclusion of exon 42 in the mRNA (Supp. Fig. S2D).
c.14252-6_14252-5insGAAA (p.Val4751_GlufsX22) consists of a four nucleotides insertion in intron 44 that causes skipping of the entire exon 45 (Supp. Fig. S2E).
c.14643+1G>A (p.Gln4882ProfsX36) occurring at nucleotide +1 of the splice donor site of intron 47 creates a novel splice acceptor site within exon 48. The PCR products from patient's cDNA showed two bands, one of the expected size, and an additional shorter band carrying a partial deletion of the exon 48 (Supp. Fig. S2F).
c.14644-3C>G (p.Glu4882ProfsX36) occurring at nucleotide −3 of the splice acceptor site of intron 47 determine the same molecular events caused by c.14643+1G>A mutation (Supp. Fig. S2G).
c.4693+1G>A (p.Val1561ArgfsX11) mutation was previously characterized and reported by Ratbi et al. (2013).
KMT2D Mutant Transcripts Suffer NMD
KMT2D nonsense mutations may result in the partial transcripts degradation through NMD pathway, contributing to protein haploinsufficiency. To investigate this, we measured by qPCR the levels of KMT2D mRNA in three KS skin fibroblasts and four KS LCLs following treatment with puromycin, a known indirect NMD inhibitor [Brichta et al., 2008]. After puromycin treatment, we observed a significant 2.18-, 2.35-, and 4.01-fold increase in the KMT2D transcript levels in KB186, KB153, and KB3 fibroblast cell lines, respectively (Supp. Fig. S3A and Supp. Table S3E), and 1.93- and 1.69-fold for KB48 and KB83 LCLs, compared with untreated cells, respectively (Supp. Fig. S3C and Supp. Table S3F). The reduced amount of KMT2D mRNA levels in mutated cell lines and its recovery following puromycin treatment indicate that endogenous KMT2D mutant transcripts are subject to NMD. The physiological NMD substrate SC-35 1.7 Kb was included as positive control (Supp. Fig. S3B–D) [Brichta et al., 2008]. We confirmed these data by direct sequencing of real-time PCR on mRNA derived from fibroblasts of patients KB186, KB153, KB3, showing that NMD inhibition restores the expression of the KMT2D-mutated alleles (Supp. Fig. S3E–G).
KMT2D Mutations Affect Its Activity in Patients Cell Lines
The large prevalence of KMT2D protein truncating mutations suggests loss of function, and therefore the haploinsufficiency of KMT2D as the likely mechanism for the KS phenotype. To assess this, we measured the protein level of KMT2D in six KS LCLs, and three KS fibroblast cell lines carrying truncating mutations, and we found a significant reduction of KMT2D protein level when compared with control cell lines (Fig.1A–F and Supp. Table S3A).
Figure 1.

KMT2D methyltransferase activity is impaired in KS lymphoblastoid and fibroblast cell lines with KMT2D-truncating mutations. (A–F): Immunoblotting analysis by using anti-KMT2D antibody on whole protein lysate from six (A and B) KS lymphoblastoid (KB41, KB44, KB45, and KB82, KB83, KB153, and KB186) and three (C) fibroblast cell lines (KB186, KB153, and KB3) with nonsense mutations and frameshift mutations, compared with control cell lines. Pool: pooled protein lysates from two control cell lines. D–F: The density of each band was determined by densitometer. The expression level of KMT2D was determined by calculating the protein level for each sample, normalized to the corresponding GAPDH level. G: qPCR was performed to measure the expression of known target genes of KMT2D (HOXC6, S100A2, S100A4, S100A5, S100A6) in six KS–LCLs, compared with control cell lines. Scale bars represent standard errors. *P < 0.01.
To address whether truncated KMT2D mutations affect its transcriptional activity, we measured the mRNA level of direct known transcriptional target genes of KMT2D complex, including HOXC6 [Ansari et al., 2011] and some members of the S100A family genes, such as S100A2, S100A4, S100A5, and S100A6. As shown in Figure1G, KMT2D targets were downregulated in KS LCLs cells (Supp. Table S3B). Overall, these data show that the transcriptional activity of KMT2D is impaired in KS patients.
HOXC6 Expression Is Affected in Kabuki Patients
Many of the KMT2D mutations mapped before the LXXLL domain required for KMT2D binding to estrogen receptors (ERs), ERα, and ERβ. Previous studies demonstrated that KMT2D, in coordination with ERα and ERβ, transcriptionally regulates HOXC6 in an Estradiol (E2)-dependent manner. We thus hypothesized that the loss of the KMT2D–ER interaction affects ER gene expression. qPCR showed that HOXC6 transcriptional level decreased in KS fibroblast cells, a reduction that persists even after E2 treatment (Fig.2A and B and Supp. Table S3C). As KMT2D is associated with HOXC6 via binding to the estrogen receptor element (ERE), located in the HOXC6 promoter region, we assessed the ability of KMT2D to bind to the HOXC6 EREs in three KS patients by a luciferase reporter assay. We transfected the pGL3–ERE1–ERE2 luciferase-based reporter vector containing the HOXC6 EREs sequences (schematized in Fig.2C) in KS and healthy fibroblast cell lines treated with E2 for 8 hr, then we measured the luciferase activity. Using a two-tailed unpaired t-test, we detected a significant reduction of the luciferase activity in all patient fibroblast cell lines (Fig.2D and E and Supp. Table S3D), in both treated or not with E2, when compared with control cells transfected with pGL3 alone. Overall, these data indicate that KMT2D mutations impair the regulation of HOXC6 in Kabuki cell lines.
Figure 2.
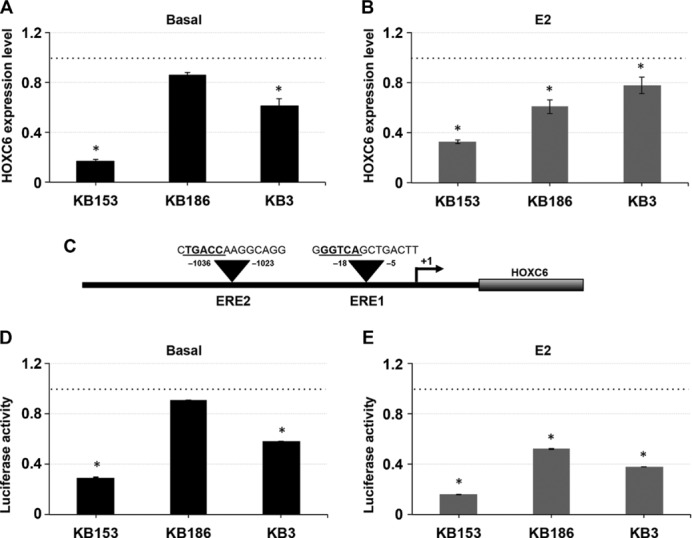
Effect of KMT2D haploinsufficiency on E2-induced expression of HOXC6. A and B: HOXC6 transcriptional level was determined in three KS patient fibroblast cell lines (KB153, KB186, and KB3) exposed to 100 nM E2 for 8 hr or not by using qPCR analysis, in comparison to a pool of two normal fibroblast cell lines. C–E: HOXC6 promoter fragment spanning the ERE1–ERE2 regions was cloned into a luciferase-based reporter construct (pGL3) and transfected into three KS patient fibroblast cell lines and into two normal fibroblast cell lines along with a Renilla luciferase construct used as an internal transfection control. Cells were then treated with 100 nM E2 (E) for 8 hr and subjected to luciferase assay. The luciferase activities (normalized to Renilla activity) in the presence or not of E2 over normal fibroblast cell lines were plotted. The experiment with three replicates was repeated at least thrice. Scale bars represent standard errors. *P < 0.01.
KMT2D and KDM6A Nonsense Mutations Are Responsive to Gentamicin-Induced Readthrough Treatment
A large number of KMT2D and KDM6A mutations are nonsense mutations that produce aberrant PTCs that are potentially deleterious. We evaluated the potential ability of gentamicin in suppressing KMT2D and KDM6A nonsense mutations by using an in vitro readthrough reporter system [Floquet et al., 2011]. Fourteen KMT2D and two KDM6A nonsense mutations were selected and tested. The mutations were selected, when possible, following the hypothetical rule of a uracil immediately upstream and a cytosine just downstream the stop codon as associated with an optimal gentamicin-induced readthrough, respectively [Floquet et al., 2012]. For each mutation, sequences spanning 27 nucleotides centered on the different stop mutations were inserted into the dual reporter vector pCRFL in-frame between Renilla and Firefly luciferase coding sequences [Floquet et al., 2011]. Readthrough levels were quantified in HEK293 cells transiently transfected by the dual reporter vector in the presence of increasing amounts of gentamicin. Cytotoxicity experiments demonstrated that no signs of cytotoxicity were detectable after 48 hr of treatment with up to 1,200 μg/ml of gentamicin (data not shown). The difference between basal and gentamicin-induced readthrough level was statistically significant (P < 0.01) for 11 KMT2D and one KDM6A nonsense mutations (Fig.3A and B and Supp. Table S4), although with a wide variability in strength. In agreement with the previous report [Floquet et al., 2012], we found that 11/12 (92%) of the mutations responsive to gentamicin have a uracil in −1 residue immediately upstream the stop codon and/or a cytosine in +4 position confirming that the presence of these residues promotes higher basal and gentamicin-induced readthrough than other nucleotides. To test the in vivo effectiveness of gentamicin, we evaluated the expression levels of two known targets of KMT2D, HOXC6, and S100A4 in LCLs from KS patients cultured in the presence of gentamicin. qPCR analysis revealed that KB41 LCL resulted in sixfold and threefold increase of HOXC6 and S100A4 mRNA levels, respectively (Fig.3C). In agreement with the in vitro assay, a significant increase of S100A4 mRNA levels was observed in KB45 cells, whereas we did not detect any significant increase of HOXC6 expression (Fig.3D). These findings are consistent with our in vitro observations about the effectiveness of readthrough that varies with respect to type of mutations and patient cell lines.
Figure 3.

Identification of KMT2D and KDM6A nonsense mutations responsive to gentamicin treatment. A and B: Comparison between basal and gentamicin-induced readthrough level. Readthrough efficiency for 14 nonsense mutations in the KMT2D gene and for two elsewhere published [Miyake et al., 2013b] nonsense mutations in the KDM6A gene was assessed in HEK293 cells exposed or not to 600–800–1,200 μg/ml of gentamicin for 48 hr. A pCRFL reporter vector harboring the 319d Duchenne muscular dystrophy mutation was used as positive control (CNT). Each value corresponds to the mean of four to six independent experiments. Scale bars represent standard errors. C and D: Gentamicin restored the expression of HOXC6 and S100A4 in patient cultured cells with different efficiency. qPCR analysis was performed to evaluated HOXC6 and S100A4 expression level in two patients LCLs, KB41 (C) and KB45 (D), and two control cell lines treated or not with 800 μg/ml of gentamicin for 48 hr. The results are the mean from at least two experiments performed in duplicate. *P < 0.01.
Overall, these studies suggest that readthrough strategies can be effective to restore functional KMT2D and KDM6A activity, although testing additional mutations, many other compounds and
different cell lines are mandatory to assess their potential therapeutic effectiveness.
Discussion
The orchestrated organization of epigenetic factors that control chromatin dynamism, including chromatin-remodeling proteins, is essential for the proper function of tissue homeostasis, cell identity, and development [Berdasco and Esteller, 2013]. KS is the most common of a growing group of multiple malformation syndromes associated to developmental delay that are caused by mutations in genes that encode proteins involved in histone methylation. In this study, we identified 133 KMT2D and four KDM6A mutations in 303 patients clinically diagnosed with KS. The mutation detection rates for KMT2D and KDM6A in KS suggest that other causative genes may be involved, although it can be taken into account that a number of these patients might have been misdiagnosed.
We demonstrated that KMT2D protein level is reduced and its activity impaired in KS cell lines. KMT2D through H3K4 methylation activity is a key epigenetic transcriptional regulator of the expression of multiple genes with related functions, including embryogenesis, development, and stem cell differentiation. Additionally, KMT2D-associated histone methyl transferase activity appears to be functional only in the context of its multiprotein complex, including ASH2L, RBBP5, WDR5, and other MLL family members [Issaeva et al., 2007; Dhar et al., 2012] and each KMT2D-interacting protein plays a distinct role in regulating MLL-mediated histone methylation and gene activation. In agreement with its biological function, the lack of KMT2D protein activity or of its ability to form a multiprotein complex might alter its role in histone methylation pathway with strong influences on global change in gene expression in many of the body's organs and tissues, resulting in some of the abnormalities of postnatal development of KS.
In our screening, we found 46 missense variants distributed across the entire length of the KMT2D gene including residues in PHD and SET domain. The in silico analysis predicts that many of them might be pathogenic, although the exact role they play in the disease has not been addressed. Recently, it has been shown that KS C1430R and C1471Y missense mutations [Hannibal et al., 2011] in PHD4–6 domains reduce PHD4–6 binding ability and abrogate the nucleosomal methylation activity of KMT2D, although they do not affect the interaction of KMT2D with ASH2L, RBBP5, and WDR5 [Dhar et al., 2012]. These results offer a valuable and feasible assay to test the pathogenicity of KMT2D missense mutations and implicate that the missense mutations described here that lies in PHD domains may contribute to KS by reducing KMT2D enzymatic activity and consequently its mediated transcriptional activation.
The majority of genetic alterations detected in Kabuki patients result in truncated proteins upstream the LXXLL domain, which is involved in KMT2D-complex binding to ERα. Rationally, the loss of the KMT2D–ERα interacting region might result in a dramatic alteration of the ERα-mediated pathways. Notably, some of the clinical features associated to deregulated ERα-signaling pathways are included in the large spectrum of KS phenotypes, comprising immunological defects and cardiac anomalies [Deroo and Korach, 2006]. Moreover, more than 20% of Kabuki patients show early breast development consistent with the biological role of ERα in mammary gland formation. In this regard, it was recently reported that the homeobox (HOX)-containing gene HOXC6, a critical player in mammary gland development and milk production, is transcriptionally activated via coordination of KMT2D and ER in an estrogen environment in breast cancer and placental choriocarcinoma cells. Specifically, the HMTs, KMT2D, and MLL3, in coordination with ERs, ERα, and ERβ, play critical roles in histone H3 lysine-4 trimethylation and in the recruitment of general transcription factors and RNA polymerase II in the EREs regions of the HOXC6 promoter during E2-dependent transactivation, leading to HOXC6 transcriptional activation. Interestingly, the knockdown of KMT2D by using specific antisense oligonucleotides suppressed E2-induced expression of HOXC6 [Ansari et al., 2011]. In agreement with all these findings, we found that HOXC6 expression is impaired in KS patients’ cell lines.
As Kabuki patients’ cell lines with KDM6A mutations were not available, it was not possible to study the activity of the endogenous KDM6A. However, it has been recently evaluated that the haploinsufficiency of Kdm6a in a zebrafish model. Kdm6a knockdown fish exhibited abnormal craniofacial structures that included the absence of the branchial arches and otoliths, as well as the absence, clefting, or inversion of the ceratohyal and abnormal patterning or clefting of Meckel's cartilages [Lindgren et al., 2013]. In addition, Kdm6a-deficient mice showed severe defects in heart development and embryonic lethality [Lee et al., 2012]. Very recently, it was demonstrated that Kdm6a knockdown affects expression of master regulatory genes involved in adipogenesis and osteogenesis [Hemming et al., 2014]. These observations further support the hypothesis that perturbation of a regulatory pathway shared by KDM6A is responsible for the clinical aspects of the KS.
The large size of the KMT2D cDNA hampers so far the development of any gene-therapy-based strategy. Mutation-based treatments are something of recent enough in genetic medicine, in which the nature of the mutation dictates the therapeutic strategy. As proof of principle, here we demonstrated the ability of gentamicin to induce the readthrough of naturally occurring stop mutations in the KMT2D and KDM6A genes using both in vitro and in vivo assays. Readthrough efficiency has been shown to also depend on the nature of the sequences surrounding the stop codon. Specifically, the consensus sequence U-STOP-C results in an optimal gentamicin-induced readthrough [Floquet et al., 2012]. However, the readthrough response to treatment is highly variable and little is known about the rules governing it and the response to different compounds. A wide variability in responsiveness to gentamicin was observed in our group of KMT2D and KDM6A mutations. Interestingly, the c.12844C>T mutation that exhibited the highest gentamicin-induced readthrough efficiency, follows the theoretical rule in that a timine residue is located immediately upstream and a cystein downstream from the stop codon.
After confirming in vitro nonsense suppression in KS, our study goes one step further to investigate the in vivo effectiveness of gentamicin on two cultured patient LCLs carrying nonsense mutations. Consistent with our in vitro data, we showed that gentamicin treatment is able to partially restore a functional endogenous KMT2D protein confirming the use of the in vitro assay as a good model for evaluating drug-induced readthrough and selecting patient carrying nonsense mutations that could benefit from treatment. Nevertheless, studies on a larger number of KS cell lines are mandatory.
Our analysis indicated that some of KMT2D-truncating transcripts suffer NMD process contributing to instability of mutant mRNA and haploinsufficiency of KMT2D protein, thus it is possible that NMD minimizes the effect of readthrough treatment for some KMT2D transcripts. To overcome this undesirable contingent issue, the readthrough strategy might be combined with inhibition of NMD by specific inhibitors and/or siRNA directed against NMD key factors as UPF1 or UPF2.
Several other aminoglycosides and nonaminoglycosides agents are used for their nonsense suppression activity in various types of cell culture with a different efficiency. Our preliminary results encourage further testing with other compounds including additional aminoglycoside such as G418 and amikacin, and nonaminoglycosides agents with improved biocompatibility, such as PTC124 and RTC13, RTC14, and NB30 [Bordeira-Carrico et al., 2012].
Conclusions
This study expanded the picture of KMT2D and KDM6A mutations that cause KS, adds some insight in the functional mechanisms that cause the disease, and finally provides the first preliminary proof-of-concept that naturally occurring nonsense mutations in KMT2D and KDM6A can be effectively suppressed providing a rational strategy for identifying patients likely to respond and therefore more likely to benefit from treatment with readthrough inducers.
Acknowledgments
We acknowledge the family that agreed to participate and made this study possible. We also acknowledge Professor J-P Rousset for providing pCRFL vector. We thank The Genomic Disorder Biobank, member of the Telethon Network of Genetic Biobanks and of the EuroBioBank network for providing us with some specimen and Mariani Foundation, Milan for supporting the clinical activities of the UOS Genetica Clinica Pediatrica Fondazione, MBBM Monza.
Disclosure statement: The authors declare no competing interests.
Supporting Information
Disclaimer: Supplementary materials have been peer-reviewed but not copyedited.
Figure S1. MLPA analysis of KMT2D gene.
Figure S2. Schematic representation of KMT2D splicing mutations and their effect on cDNA patients.
Figure S3. KMT2D mutations are regulated by NMD.
Table S1. KMT2D and KDM6A mutations identified in our cohort of Kabuki patients.
Table S2. In silico prediction of pathogenic effect of KMT2D# and KDM6A missense variants.
Table S3. Values of the measured quantities and p-values.
Table S4. Basal and gentamic-induced readthrough for KB mutations.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari KI, Hussain I, Shrestha B, Kasiri S, Mandal SS. HOXC6 is transcriptionally regulated via coordination of MLL histone methylase and estrogen receptor in an estrogen environment. J Mol Biol. 2011;411:334–349. doi: 10.1016/j.jmb.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari KI, Mandal SS. Mixed lineage leukemia: roles in gene expression, hormone signaling and mRNA processing. FEBS J. 2010;277:1790–804. doi: 10.1111/j.1742-4658.2010.07606.x. [DOI] [PubMed] [Google Scholar]
- Banka S, Howard E, Bunstone S, Chandler K, Kerr B, Lachlan K, McKee S, Mehta S, Tavares A, Tolmie J, Donnai D. MLL2 mosaic mutations and intragenic deletion-duplications in patients with Kabuki syndrome. Clin Genet. 2013a;83:467–471. doi: 10.1111/j.1399-0004.2012.01955.x. [DOI] [PubMed] [Google Scholar]
- Banka S, Lederer D, Benoit V, Jenkins E, Howard E, Bunstone S, Kerr B, McKee S, Chris Lloyd I, Shears D, Stewart H, White SM. 2014. Novel KDM6A (UTX) mutations and a clinical and molecular review of the X-linked Kabuki syndrome (KS2). Clin Genet.[Epub ahead of print] [DOI] [PubMed]
- Banka S, Veeramachaneni R, Reardon W, Howard E, Bunstone S, Ragge N, Parker MJ, Crow YJ, Kerr B, Kingston H, Metcalfe k, Chandler K, et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2012;20:381–388. doi: 10.1038/ejhg.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellais S, Le Goff C, Dagoneau N, Munnich A, Cormier-Daire V. In vitro readthrough of termination codons by gentamycin in the Stuve-Wiedemann syndrome. Eur J Hum Genet. 2010;18:130–132. doi: 10.1038/ejhg.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdasco M, Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet. 2013;132:359–383. doi: 10.1007/s00439-013-1271-x. [DOI] [PubMed] [Google Scholar]
- Bidou L, Hatin I, Perez N, Allamand V, Panthier JJ, Rousset JP. Premature stop codons involved in muscular dystrophies show a broad spectrum of readthrough efficiencies in response to gentamicin treatment. Gene Ther. 2004;11:619–627. doi: 10.1038/sj.gt.3302211. [DOI] [PubMed] [Google Scholar]
- Bogershausen N, Wollnik B. Unmasking Kabuki syndrome. Clin Genet. 2013;83:201–211. doi: 10.1111/cge.12051. [DOI] [PubMed] [Google Scholar]
- Bordeira-Carrico R, Pego AP, Santos M, Oliveira C. Cancer syndromes and therapy by stop-codon readthrough. Trends Mol Med. 2012;18:667–678. doi: 10.1016/j.molmed.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Brichta L, Garbes L, Jedrzejowska M, Grellscheid SN, Holker I, Zimmermann K, Wirth B. Nonsense-mediated messenger RNA decay of survival motor neuron 1 causes spinal muscular atrophy. Hum Genet. 2008;123:141–153. doi: 10.1007/s00439-007-0455-7. [DOI] [PubMed] [Google Scholar]
- Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- Caratozzolo MF, Micale L, Turturo MG, Cornacchia S, Fusco C, Marzano F, Augello B, D'Erchia AM, Guerrini L, Pesole G, Sbisa E, Merla G, Tullo A. TRIM8 modulates p53 activity to dictate cell cycle arrest. Cell Cycle. 2012;11:511–523. doi: 10.4161/cc.11.3.19008. [DOI] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116:561–570. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X, Reinberg D, Lee MG. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26:2749–2762. doi: 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005;6:227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339:240–249. doi: 10.1016/j.ydbio.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero GB, Howald C, Micale L, Biamino E, Augello B, Fusco C, Turturo MG, Forzano S, Reymond A, Merla G. An atypical 7q11.23 deletion in a normal IQ Williams-Beuren syndrome patient. Eur J Hum Genet. 2010;18:33–38. doi: 10.1038/ejhg.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floquet C, Deforges J, Rousset JP, Bidou L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2011;39:3350–3362. doi: 10.1093/nar/gkq1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floquet C, Hatin I, Rousset JP, Bidou L. Statistical analysis of readthrough levels for nonsense mutations in mammalian cells reveals a major determinant of response to gentamicin. PLoS Genet. 2012;8:e1002608. doi: 10.1371/journal.pgen.1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederic MY, Lalande M, Boileau C, Hamroun D, Claustres M, Beroud C, Collod-Beroud G. UMD-predictor, a new prediction tool for nucleotide substitution pathogenicity—application to four genes: FBN1, FBN2, TGFBR1, and TGFBR2. Hum Mutat. 2009;30:952–959. doi: 10.1002/humu.20970. [DOI] [PubMed] [Google Scholar]
- Hannibal MC, Buckingham KJ, Ng SB, Ming JE, Beck AE, McMillin MJ, Gildersleeve HI, Bigham AW, Tabor HK, Mefford HC, Cook J, Yoshiura K. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet A. 2011;155A:1511–1516. doi: 10.1002/ajmg.a.34074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming S, Cakouros D, Isenmann S, Cooper L, Menicanin D, Zannettino A, Gronthos S. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells. 2014;32:802–815. doi: 10.1002/stem.1573. [DOI] [PubMed] [Google Scholar]
- Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci USA. 2007;104:18439–18444. doi: 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, Mazo A, Eisenbach L, Canaani E. Knockdown of ALRMLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol. 2007;27:1889–1903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokitsu-Nakata NM, Petrin AL, Heard JP, Vendramini-Pittoli S, Henkle LE, dos Santos DV, Murray JC, Richieri-Costa A. Analysis of MLL2 gene in the first Brazilian family with Kabuki syndrome. Am J Med Genet A. 2012;158A:2003–2008. doi: 10.1002/ajmg.a.35454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99:570–573. doi: 10.1016/s0022-3476(81)80256-9. [DOI] [PubMed] [Google Scholar]
- Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, Roberts TM, Chang HY, Shi Y. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449:689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, Ghariani SC, Maystadt I, Dallapiccola B, Verellen-Dumoulin C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet. 2012;90:119–124. doi: 10.1016/j.ajhg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HL, Dougherty JP. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol Ther. 2012;136:227–266. doi: 10.1016/j.pharmthera.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Lee S, Lee JW, Lee SK. UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev Cell. 2012;22:25–37. doi: 10.1016/j.devcel.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Bogershausen N, Alanay Y, Simsek Kiper PO, Plume N, Keupp K, Pohl E, Pawlik B, Rachwalski M, Milz E, Thoenes M, Albrecht B, et al. A mutation screen in patients with Kabuki syndrome. Hum Genet. 2011;130:715–724. doi: 10.1007/s00439-011-1004-y. [DOI] [PubMed] [Google Scholar]
- Lindgren AM, Hoyos T, Talkowski ME, Hanscom C, Blumenthal I, Chiang C, Ernst C, Pereira S, Ordulu Z, Clericuzio C, Drautz JM, Rosenfeld JA, et al. Haploinsufficiency of KDM6A is associated with severe psychomotor retardation, global growth restriction, seizures and cleft palate. Hum Genet. 2013;132:537–552. doi: 10.1007/s00439-013-1263-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makrythanasis P, van Bon BW, Steehouwer M, Rodríguez-Santiago B, Simpson M, Dias P, Anderlid BA, Arts P, Bhat M, Augello B, Biamino E, Bongers E, et al. MLL2 mutation detection in 86 patients with Kabuki syndrome: a genotype–phenotype study. Clin Genet. 2013;84:539–545. doi: 10.1111/cge.12081. [DOI] [PubMed] [Google Scholar]
- Micale L, Augello B, Fusco C, Selicorni A, Loviglio MN, Silengo MC, Reymond A, Gumiero B, Zucchetti F, D'Addetta EV, Belligni E, Calcagni A, et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J Rare Dis. 2011;6:38. doi: 10.1186/1750-1172-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake N, Koshimizu E, Okamoto N, Mizuno S, Ogata T, Nagai T, Kosho T, Ohashi H, Kato M, Sasaki G, Mabe H, Watanabe Y, et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am J Med Genet A. 2013a;161:2234–2243. doi: 10.1002/ajmg.a.36072. [DOI] [PubMed] [Google Scholar]
- Miyake N, Mizuno S, Okamoto N, Ohashi H, Shiina M, Ogata K, Tsurusaki Y, Nakashima M, Saitsu H, Niikawa N, Matsumoto N. KDM6A point mutations cause Kabuki syndrome. Hum Mutat. 2013b;34:108–110. doi: 10.1002/humu.22229. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Du L, Tunuguntla R, Fike F, Cavalieri S, Morio T, Mizutani S, Brusco A, Gatti RA. Functional characterization and targeted correction of ATM mutations identified in Japanese patients with ataxia-telangiectasia. Hum Mutat. 2011;33:198–208. doi: 10.1002/humu.21632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Toba S, Hirai M, Morishita S, Mikami T, Konishi M, Itoh N, Kurosaka A. Cloning and expression of a brain-specific putative UDPGalNAc: polypeptide N-acetylgalactosaminyltransferase gene. Biol Pharm Bull. 2005;28:429–433. doi: 10.1248/bpb.28.429. [DOI] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981;99:565–569. doi: 10.1016/s0022-3476(81)80255-7. [DOI] [PubMed] [Google Scholar]
- Paulussen AD, Stegmann AP, Blok MJ, Tserpelis D, Posma-Velter C, Detisch Y, Smeets EE, Wagemans A, Schrander JJ, van den Boogaard MJ, van der Smaqt J, van Haeringen A, et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum Mutat. 2011;32:E2018–E2025. doi: 10.1002/humu.21416. [DOI] [PubMed] [Google Scholar]
- Priolo M, Micale L, Augello B, Fusco C, Zucchetti F, Prontera P, Paduano V, Biamino E, Selicorni A, Mammi C, Lagana C, Zelante L, Merla G. Absence of deletion and duplication of MLL2 and KDM6A genes in a large cohort of patients with Kabuki syndrome. Mol Genet Metab. 2012;107:627–629. doi: 10.1016/j.ymgme.2012.06.019. [DOI] [PubMed] [Google Scholar]
- Ratbi I, Fejjal N, Micale L, Augello B, Fusco C, Lyahyai J, Merla G, Sefiani A. Report of the first clinical case of a Moroccan Kabuki patient with a novel MLL2 mutation. Mol Syndromol. 2013;4:152–156. doi: 10.1159/000346798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Sermet-Gaudelus I, Renouil M, Fajac A, Bidou L, Parbaille B, Pierrot S, Davy N, Bismuth E, Reinert P, Lenoir G, Lesure JF, Rousset JP, Edelman A. In vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC Med. 2007;5:5. doi: 10.1186/1741-7015-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. MLPA analysis of KMT2D gene.
Figure S2. Schematic representation of KMT2D splicing mutations and their effect on cDNA patients.
Figure S3. KMT2D mutations are regulated by NMD.
Table S1. KMT2D and KDM6A mutations identified in our cohort of Kabuki patients.
Table S2. In silico prediction of pathogenic effect of KMT2D# and KDM6A missense variants.
Table S3. Values of the measured quantities and p-values.
Table S4. Basal and gentamic-induced readthrough for KB mutations.