

Abstract
A flexible, highly sensitive sensor of oxygen in non-aqueous solvents is described. It consists of CdSe/ZnS nanoparticles decorated with a considerable number of pyrene units, thus making the formation of the pyrene excimer possible. The emission of the pyrene excimer and that of the nanoparticle are suitably separated from each other and also from the excitation wavelength. This sensor can be applied as a ratiometric oxygen sensor by using the linear response of the pyrene excimer lifetime combined with the linear response of the nanoparticle excited state lifetime. This nanohybrid has been assayed in seven media with different dielectric constants and viscosities over the whole oxygen concentration range. In addition, the sensor versatility provides an easy way for monitoring oxygen diffusion through systems.
Keywords: nanohybrids, organic solvents, oxygen, ratiometric, sensors
Introduction
Sensing of oxygen in aqueous solvents has been extensively studied, but few studies have been performed on organic solvents.1–3 The quantitative determination of dissolved oxygen in non-aqueous media is of great importance in the evaluation of chemical processes which can require the presence of oxygen or may be influenced by a small amount of such reagent.4–6 Consequently, there is an increasing interest in the development of optical sensors for detecting the presence of different oxygen concentrations in non-aqueous solvents in a faster and easier way than conventional amperometric electrodes. The latter are problematic in low conductivity media, apart from their consumption of oxygen and potential poisoning of electrodes.7, 8
Oxygen optrode sensors are generally based on the reduction of the luminescent intensity of a dye.9–14 In this respect, optical oxygen sensors based on the unique properties of photoactive nanoparticles have recently been developed, and, for example, metalloporphyrin-doped polyfluorene nanoparticles have been used as sensitive oxygen sensors in biological imaging.15 Many oxygen sensors are encapsulated within a matrix13, 16–21] which can sometimes cause a loss, or even variability, in their sensitivity, such as when using polymeric matrices and when they are applied in organic media.
Ratiometric sensors are based on the measurement of two different emission bands and permit signal rationing, which can increase the dynamic range and provide a built-in correction for environmental effects.2–24 With regards to ratiometric oxygen sensors, metalloporphyrin-capped (CdxZn1−xSe)CdyZn1−yS and CdSe core-shell nanoparticles,21, 25, 26 have been devised as versatile ratiometric, oxygen sensors with two-photon excitation in biological samples. It has to be taken into account that semiconductor-based nanoparticles, such as those comprising CdSe, are particularly relevant since they exhibit a broad absorption spectrum, a size-tuneable and narrow emission, and high photostability.
Regarding detection of oxygen in organic solvents, to our knowledge, there is only one example reported by Credi et al. of a ratiometric oxygen sensor in chloroform.27 It is a pyrene-capped core-shell CdSe/ZnS nanoparticle and its performance in oxygen sensing originates from the different sensitivity to the presence of oxygen of the CdSe nanoparticle self-emission, which is insensitive when compared to that of the pyrene monomer. However, this remarkable sensor was only tested in chloroform and its response deviated from linearity at the highest concentration of oxygen. In this sensor, the potential formation of a high local concentration of the fluorophore (pyrene) units at their periphery was not exploited. In fact, we have previously demonstrated that both pyrene emissive species, the monomer (M*, band 370–430 nm) and the excimer (E*, at ca. 480 nm), can be obtained using amazingly small pyrene concentrations by attaching pyrene units to CdSe core nanoparticles via a long flexible chain ending in a thiolate group, which exhibits high affinity for the nanoparticle surface.28, 29 However, the emission of the core in this nanohybrid was almost totally quenched due to the anchored thiolate and, consequently, this system exhibited a dual-fluorescence derived from both pyrene species.
We now envisaged that the exchange of the amine ligand of commercially available CdSe/ZnS (CS) nanoparticles with 4-(pyren-4-yl)butyl-11-mercaptoundecanoate (Py-L-SH) could be advantageous for the development of sensitive oxygen optrodes due to the unique properties of the resulting nanohybrids. Thus, it was expected that the CS intrinsic emission would be greater than that of core CdSe nanoparticles; in addition, the ZnS shell ensured a higher stability of their luminescence properties and also made them less toxic. Moreover, the nanohybrids would present three highly emissive channels with small overlapping spectra: the CS intrinsic emission and the two pyrene emissive species (M* and E*), which would increase its flexibility in sensing. In addition, the emission signals of two of these channels, specifically that of the E* and CS*, can be chosen to be suitably separated from each other and also from the excitation (large fluorescence Stokes shifts).
We report here on the suitability of pyrene-capped CdSe/ZnS nanohybrids (CS@Py) as oxygen sensors in non-aqueous solvents (Figure 1). The CS was chosen with an emission that did not overlap that of the pyrene species. These studies were performed using seven organic solvents, with dielectric constants (ɛ) ranging from 1.88 to 8, and viscosity values (η) ranging from 0.313 to 1.439 cP. Our data revealed that these nanohybrids can be used as highly efficient ratiometric oxygen sensors in organic solvents and in the whole range of oxygen concentrations by using the linear response of the E* emission lifetime, combined with the linear response (though with low sensitivity) of the CS* lifetime to oxygen.
Figure 1.
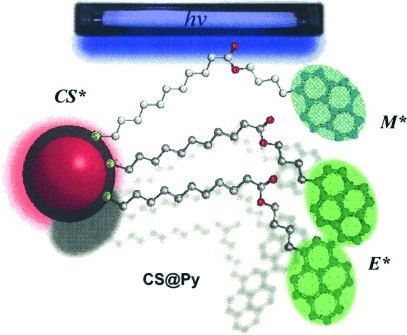
Pyrene-capped CdSe/ZnS nanohybrids (CS@Py) exhibiting three emissive species under UV light.
Results and Discussion
Synthesis and characterisation of CS@Py
Thiolate-stabilized CdSe/ZnS nanoparticles decorated with pyrene moieties (CS@Py) were prepared by the reaction of commercially available amine-capped CdSe/ZnS nanoparticles with 4-(pyren-4-yl)butyl-11-mercaptoundecanoate (Py-L-SH,28 [thiol]/[CS] proportion equal to 5000 molar ratio) under chloroform reflux for 48 h (see details in the Experimental Section). The exchange of the amine ligands by thiolates is an efficient process which preserves most of the emission of the nanoparticle.30, 31 After purification, the UV/Vis absorption spectrum of CS@Py showed the first exciton peak of the CS at 622 nm, as well as the presence of the pyrene chromophore in an average amount of 200 pyrene molecules per CS. High-resolution transmission electron microscopy (HRTEM) showed CS@Py nanoparticles with an average size of 8.3±1.6 nm (Figure 2).
Figure 2.
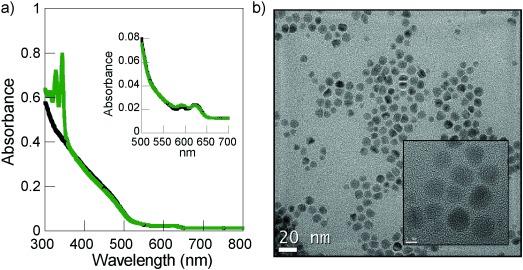
a) Comparison between the absorption spectrum of a THF solution of CS (—) and CS@Py (—), nanoparticle concentration: 40 nm; inset: enlargement of the CS@Py spectrum between 500 and 700 nm. b) HRTEM image of CS@Py, scale bar of 20 nm; inset: image with scale bar of 5 nm.
Excitation of deaerated tetrahydrofuran (THF) solutions of CS@Py (45 nm) at λexc 450 nm, where only the nanoparticle absorbs, showed the emission of the CS* (a narrow band centred at 640 nm, Φf=21 %), while excitation at λexc 340 nm showed that the emission of CS* did not overlap those of M* (λem at 376, 396 and 417 nm) and E* (λem at 480 nm), see Figure 3. Additionally, excitation spectra of CS@Py (λem 640 nm) indicated that the energy transfer from the pyrene singlet excited state to the CS was negligible in all the solvents (spectra not shown).
Figure 3.
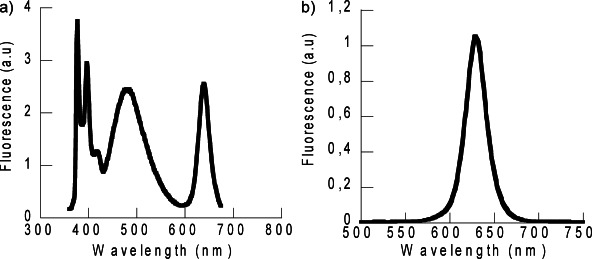
Emission spectra of deaerated THF solutions of CS@Py at a) λexc=340 nm (A340=0.665) and b) λexc=450 nm (A450=0.183).
In accordance with our previous results on pyrene-capped CdSe nanoparticles,28 the M* fluorescence decay of CS@Py was not a single exponential but fitted to two lifetimes (τ1 and τ2), one ascribed to a short-lived M* (Ms*) and the other to a long-lived M* (ML*), respectively. The decay kinetics of Ms* was coincident with the rise time of E* (τg). These findings are compatible with a nonhomogeneous distribution of Py among the remaining amine ligands on the nanoparticle surface. Consequently, some pyrenes can form the pyrene excimer, while others are too isolated to form the excimer.32, 33 These studies were performed using seven solvents: hexane, dioxane, toluene, chloroform, ethyl acetate, tetrahydrofuran, and dichloromethane. The physical properties of the solvents and the solubility of oxygen in these solvents are shown in Table 1.34
Table 1.
Physical properties of the solvents assayed through this study and the solubility of oxygen (at 0.21 and 1 atm) in these solvents.
| Solvent | ɛ[a] | μ[b] | η[c] | [O2][d] 0.21 atm | [O2][d] 1 atm |
|---|---|---|---|---|---|
| Hexane | 1.88 | – | 0.313 | 3.10 | 14.70 |
| Dichloromethane | 8.00 | 1.60 | 0.449 | 2.20 | 10.70 |
| Ethyl acetate | 6.02 | 1.78 | 0.451 | 1.90 | 8.89 |
| Tetrahydrofuran | 7.58 | 1.60 | 0.575 | 2.10 | 10.0 |
| Chloroform | 4.80 | 1.01 | 0.580 | 2.40 | 11.50 |
| Toluene | 2.38 | 0.36 | 0.586 | 2.10 | 9.88 |
| Dioxane | 2.2 | 0.40 | 1.439 | 1.30 | 6.28 |
[a] Dielectric constant. [b] Dipolar moment in Debye. [c] Viscosity (η) at 25 °C, cP. [d] O2 concentration in mm.
The contribution of each type of monomer was highly dependent on the solvent nature (Table 2). In addition, the growth and decay of the E* fluorescence fitted to two exponentials to yield the growth (kg) and decay (kd) components. The rise time of E* (τg) agreed well with the decay kinetics of the short-lived Ms* (compare the corresponding τg and τ2 in Table 2). In addition, Table 3 shows the dependence of CS* lifetime with the solvent nature.
Table 2.
Lifetime of the pyrene monomers, M*, and the rise time and decay of the E* of CS@Py registered for each solvent under nitrogen atmosphere.[a]
| Solvent | ML* | MS* | E* | |
|---|---|---|---|---|
| τ1 [ns] | τ2 [ns] | τg [ns] | τd [ns] | |
| Hexane | 65.5±5.0 (10) | 5.2±0.4 (90) | 5.6±0.6 (−120) | 46.9±1 (100) |
| Dichloromethane | 34.3±2 (11) | 3.7±0.3 (89) | 4.0±3 (−109) | 58.8±4.0 (100) |
| Ethyl acetate | 73.9±2 (29) | 13.2±1 (71) | 9.2±0.1 (−156) | 49.8±0.5 (100) |
| Tetrahydrofuran | 84.9±2 (30) | 14.3±1 (70) | 19.0±2 (−120) | 58.0±1 (100) |
| Chloroform | 34.8±1 (75) | 13.1±2 (25) | 13.0±0.3 (−127) | 39.0±1 (100) |
| Toluene | 89.8±6 (22) | 20.1±1 (78) | 22.6±1 (−105) | 58.6±2 (100) |
| Dioxane | 81.1±2 (46) | 21.8±1 (54) | 24.2±3 (−109) | 63.2±4 (100) |
[a] Relative contribution to the biexponential fit given in parentheses.
Table 3.
Lifetime of the nanoparticle, CS*, of CS@Py registered in each solvent under nitrogen atmosphere.
| Solvent | τ1 [ns][a] | τ2 [ns][a] | τav [ns][b] |
|---|---|---|---|
| Hexane | 36.1±1.4 (69) | 12.7±2.0 (31) | 32.9 |
| Dichloromethane | 43.4±3.0 (45) | 13.6±1.9 (55) | 35.1 |
| Ethyl acetate | 29.4±1.6 (17) | 6.2±1 (83) | 17.6 |
| Tetrahydrofuran | 45.0±3 (26) | 14.3±0.57 (4) | 30.4 |
| Chloroform | 36.1±1 (69) | 12.7±4 (31) | 32.9 |
| Toluene | 31.1±2 (37) | 9.4±1 (63) | 23.7 |
| Dioxane | 47.1±7 (46) | 12.5±2 (54) | 38.9 |
[a] Relative contribution to the biexponential fit given in parentheses. [b] Average lifetime calculated as τav=ΣAiτi2/ΣAiτi.
Monitoring of oxygen diffusion through intensity emission measurements
The effect of O2 concentration on the CS* luminescence as well as on that of the pyrene species was studied in the seven organic solvents using deaerated, aerated, and O2-saturated samples. The emission of pyrene species was highly sensitive to the presence of O2 (see Table 4). Regarding the decrease of the CS* emission in aerated samples, it varied from a negligible effect in THF and dioxane to a about 20 % decay in toluene (see Figure 4 a,b for THF and toluene, respectively, and figure S1 in the Supporting Information for the other solvents). Table 5 shows the comparatively small variation of the CS* average lifetime (τav) on the oxygen concentration for all the solvents.35, 6–39
Table 4.
Change in the I0/I and τ0/τ ratios for the emissive species in air and O2-saturated samples.
| Solvent | Emissive species | Air | Oxygen | ||
|---|---|---|---|---|---|
| I0/I | τ0/τ | I0/I | τ0/τ | ||
| Hexane | M* E* CS* | 5.4 6.3 1.2 | 5.5 3.1 1.0 | 11.4 41.5 1.5 | 8.4 11.1 1.1 |
| Dichloromethane | M* E* CS* | 2.2 2.5 1.2 | 3.0 1.9 1.2 | 4.6 11.2 1.9 | –[a] 5.5 1.3 |
| Ethyl acetate | M* E* CS* | 3.5 5.2 1.1 | 3.6 2.6 1.1 | 8.7 33.5 1.2 | 7.8 8.6 1.1 |
| Tetrahydrofuran | M* E* CS* | 3.1 4.7 1.0 | 4.0 2.7 0.93 | 7.6 30.0 1.0 | 8.0 7.2 0.97 |
| Chloroform | M* E* CS* | 2.2 3.7 1.2 | 1.7 1.7 1.0 | 4.1 16.9 1.5 | 3.8 4.2 1.3 |
| Toluene | M* E* CS* | 2.0 2.4 1.3 | 2.9 2.4 1.0 | 7.9 32.7 1.9 | 8.7 7.4 1.3 |
| Dioxane | M* E* CS* | 2.2 2.4 1.1 | 1.8 1.7 1.2 | 4.6 11.8 1.5 | 4.3 4.1 1.4 |
[a] The lifetime value was close to that of the detection limit of the instrument (<1 ns).
Figure 4.
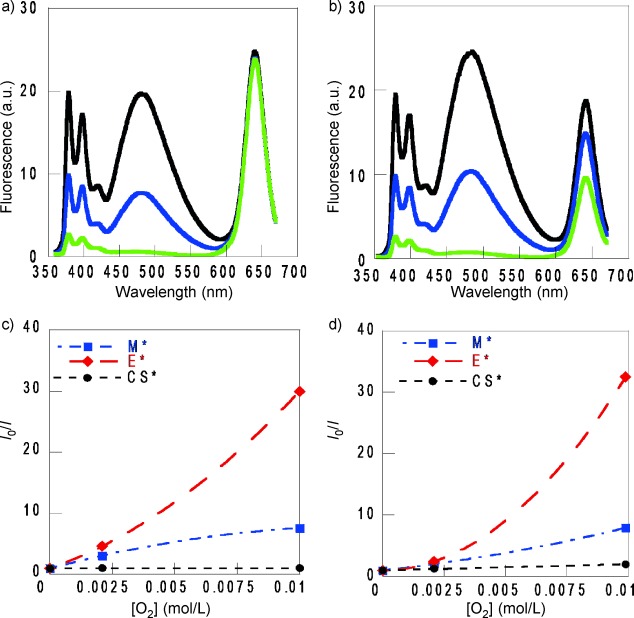
Emission spectra of CS@Py at λexc=340 nm (A340=0.15) in a) THF and b) toluene in the presence of increasing [O2]: the deaerated sample (—), the aerated sample (—) and the O2 saturated sample (—). I0/I dependence of [O2] for M*, E* and CS* of CS@Py in c) THF and d) toluene.
Table 5.
Lifetime of the pyrene monomer, M*, E* and CS * of CS@Py registered for the different solvents in the presence of increasing oxygen concentrations.
| Solvent | [O2] | ML* | MS* | E* | CS* | |
|---|---|---|---|---|---|---|
| mm | τ1 [ns] | τ2 [ns] | τg [ns] | τd [ns] | τCS [ns][a] | |
| Hexane | 0[b] 3.1 14.7 | 65.5±5 20.7±0.8 24.0±0.9 | 5.2±0.4 2.9±0.2 3.2±0.1 | 5.6±0.6 –[c] –[c] | 46.9±1 15.1±0.2 4.2±0.1 | 32.9±4 33.1±5 28.7±3 |
| Dichloromethane | 0[b] 2.2 10.7 | 34.3±2 25.2±0.3 –[c] | 3.7±0.3 1.8±0.1 –[c] | 4.0±3 9.7±1 –[c] | 58.8±4 30.4±0.5 11.0±0.3 | 35.1±8 29.6±3 27.4±5 |
| Ethyl acetate | 0[b] 1.9 8.89 | 73.9±2 24.5±2 23.1±1 | 13.2±1 8.2±0.7 3.3±0.1 | 9.2±0.1 6.7±1 4.1±0.2 | 49.8±0.5 18.9±0.6 5.8±0.2 | 17.6±4 16.3±3 16.1±5 |
| Tetrahydrofuran | 0[b] 2.1 10.0 | 84.9±2 23.5±1 28.1±1 | 14.3±1 11.0±2 4.8±0.2 | 19.0±2 10.5±0.4 3.1±0.1 | 58.0±1 21.2±0.5 8.1±0.2 | 30.4±5 32.6±8 31.5±9 |
| Chloroform | 0[b] 2.4 11.5 | 34.8±1 41.6±1 25.0±3 | 13.1±2 15.4±1 5.0±0.2 | 13.0±0.3 15.2±0.9 3.47±0.7 | 39.0±1 19.8±0.8 9.2±0.6 | 32.9±5 32.2±6 25.3±4 |
| Toluene | 0[b] 2.1 9.9 | 89.8±6 60.5±2 27.3±4 | 20.1±1 13.9±0.5 3.6±0.2 | 22.6±1 10.0±0.5 4.4±0.2 | 58.6±2 22.5±0.6 6.0±0.2 | 23.7±5 23.0±4 17.7±7 |
| Dioxane | 0[b] 1.3 6.3 | 81.1±2 39.9±1 26.3±3 | 21.8±1 13.2±2 11.3±1 | 24.2±3 14.7±0.6 6.3±0.5 | 63.2±4 38.4±0.8 15.5±0.8 | 38.9±4 33.3±8 27.9±7 |
[a] Average lifetime calculated as τav=ΣAiτi2/ΣAiτi. [b] Under nitrogen atmosphere. [c] The lifetime value was close to that of the detection limit of the instrument (<1 ns).
At low [O2], the dependence of the M* emission intensity was linear with the quencher concentration. However, in general, the I0/I intensity ratios did not increase linearly at high [O2], but they rather approached a plateau (Figure 4 c,d for THF and toluene, respectively; figure S2 in the Supporting Information for the other solvents). Likewise, time-resolved fluorescence studies showed that the τ0/τ lifetime ratios did not increase linearly at high [O2] either but also approached a plateau (figure S3 in the Supporting Information). In addition, the kinetic traces evidenced that M* formation showed negligible dependence with [O2] (see figure S4 in the Supporting Information for THF). All these results are consistent with the existence of a pre-equilibrium intermediate at high [O2], that is, the complex between M* and ground state oxygen (M*–O2 exciplexes), in the M* decay.40, 41, 2–44
Regarding the pyrene excimer, E* emission intensity was comparatively more sensitive to the presence of O2 in the saturated medium than in the aerated samples. For example, the I0/I intensity ratio changed from approximately 2.4 to 32.7 when [O2] changed from 2.10 mm (aerated sample) to 9.9 mm (O2-saturated sample) in toluene, as shown in Table 4. In addition, time-resolved fluorescence studies evidenced the decrease of E* formation with [O2] and, consequently, the E* emission intensity reflected the quenching of E* by O2 and that of its precursor (see figure S4 in the Supporting Information for THF samples).
Consequently, we found that CS@Py nanohybrids were not ideal sensors on the basis of the CS* and E* emission intensities. However, they could be used for monitoring the diffusion of O2 in different non-aqueous solvents or solvent mixtures under a specific oxygen pressure.
As an example, we studied the time-evolution of the E* emission intensity of deaerated samples under atmospheric air pressure. Figure 5 a compares the I0/I change in the E* emission versus time for each of the seven solvents. It showed that the profile was similar for THF, toluene, and chloroform, while the changes were faster in hexane and considerably slower in dioxane (for the profiles of the M* emission, see figure S5 in the Supporting Information). The maximum change in the emission (I0/I)max would correspond to [O2]0.21atm for each solvent.
Figure 5.
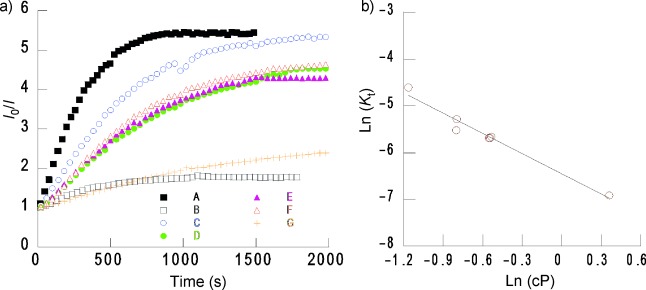
a) Change in excimer emission of deaerated CS@Py sample versus time under an air atmosphere, monitored for hexane (A), dichloromethane (B), ethyl acetate (C), tetrahydrofuran (D), chloroform (E), toluene (F) and dioxane (G). b) Regression between the Ln K and Ln cP for the pyrene excimer of CS@Py in the different solvents employed in this study.
A constant Kt was estimated from the dependence of the I0/I ratio versus time (t) in the linear part of the plots. A series of studies were made to evaluate the regression between the Kt value for the different solvents and their properties. Figure 5 b shows that there is a good linear regression between Ln Kt and Ln η (η, solvent viscosity) for E*, despite the fact that the solvents in this study were used as received without any further purification. The aforementioned correlation agrees well with the diffusion nature of the deactivation process.
Alternatively, the time-recovery of the E* emission of an O2-saturated sample in THF connected to a 1 L N2 container was also monitored. In this solvent, the comparative insensitivity of the nanoparticle emission to the removal of oxygen would allow the use of the nanohybrid as a dual sensor of the diffusion of nitrogen/removal of oxygen into/from the solution (see Figure 6 for THF). In general, CS@Py nanohybrids could be used to monitor the displacement of oxygen of saturated samples by other gases so long as they cannot be quenchers of the corresponding nanohybrid emissive species.
Figure 6.
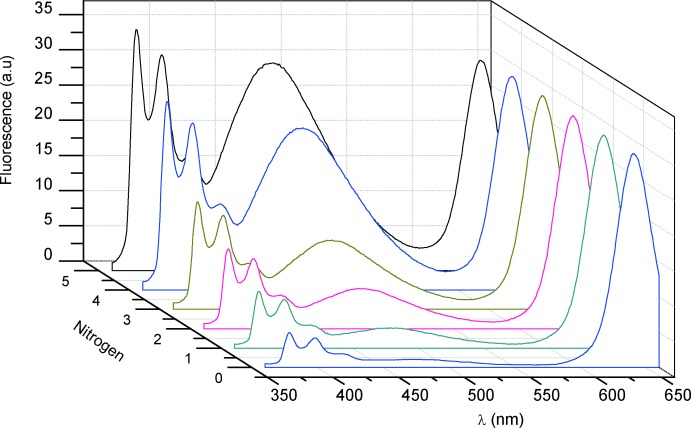
Evolution of the emissive species when oxygen saturated CS@Py samples (2 mL, 5 nm) in THF were connected to a container with nitrogen (1 L) in order to provide system deaeration.
CS@Py as ratiometric oxygen sensors
Time-resolved measurements showed that, in general, the dependence of the M* lifetime on [O2] was not linear (Table 5 and figure S3 in the Supporting Infomation). This fact was consistent with the efficiency of O2 as the quencher, which deactivated both ML* and MS*.
Ideal fluorescent sensors are those that show a linear response and high sensitivity to the target quencher and, additionally, behave as ratiometric sensors. In this regard, the linear response of the E* lifetime to the whole range of [O2] (Table 5), from deaerated to aerated and O2-saturated samples, can be advantageously used for the detection and quantification of O2 in non-aqueous media. Moreover, the dynamic Stern–Volmer quenching constant of the E* emission by O2, KE*, in all the solvents, except dioxane, is more than 25 times greater than that of the nanoparticle, KCS* (see KE* and KCS* values in table S2 in the Supporting Infomation). The sensor response (R) with a linear-dependence on [O2] would be the ratio between the nanoparticle CS* lifetime (τCS) and that of excimer E* (τd), Table 6 (a plot of the dependence of R on the oxygen concentration for each solvent is shown in figure S6 of the Supporting Information).
Table 6.
Dependence of the CS@Py response (R) under nitrogen, air, and oxygen for different solvents.
| Solvent | R[a] N2 | R[a] [O2]0.21 atm | R[a] [O2]1 atm | QO2 [%] |
|---|---|---|---|---|
| Hexane | 0.70 | 2.19 | 6.83 | 91 |
| Dichloromethane | 0.59 | 0.97 | 2.49 | 81 |
| Ethyl acetate | 0.35 | 0.91 | 2.77 | 87 |
| Tetrahydrofuran | 0.52 | 1.53 | 3.88 | 87 |
| Chloroform | 0.82 | 1.39 | 2.75 | 76 |
| Toluene | 0.40 | 1.02 | 2.95 | 89 |
| Dioxane | 0.61 | 0.86 | 1.80 | 75 |
[a] R is the τCS/τd ratio. [b] QO2=(RN2−RO2)/RN2×100; RO2 at 1 atm.
The sensor sensitivity to oxygen (QO2) can thus be expressed by the overall quenching response to dissolved oxygen, defined by [Equation (1)].
| (1) |
where RN2 is the response in the fully deoxygenated sample and RO2 is that of the oxygenated sample. These values were remarkably high, varying from 75 % to 91 % (see Table 6).
Finally, regarding the pyrene-capped nanoparticles, their emissive properties (absorption and emission) preserved under air atmosphere for more than six months. Therefore, CS@Py nanohybrids fulfil the requirements to be used as ideal sensors of oxygen by using the linear response of the E* lifetime, as well as that of the ratio between the CS* and the E* lifetimes, to [O2].
Conclusion
In summary, pyrene capped CdSe/ZnS core-shell nanohybrids are useful in the monitoring and determination of dissolved oxygen in non-aqueous media. Their applicability as sensors is based on the unique emissive properties of the nanoparticles, namely high emission and photostability, as well as on their high surface-to-volume ratio, which makes the formation of the pyrene excimer possible, in addition to the pyrene monomer. By choosing the appropriate nanoparticle size, the emission signal of the pyrene excimer and that of the nanoparticle will be suitably separated from each other and also from the excitation light (large fluorescence Stokes shifts). The advantage of these nanohybrids as ratiometric oxygen sensors in organic solvents arises from the linear response of the pyrene excimer lifetime and that of the nanoparticle excited state lifetime. Our strategy reveals the key features for designing an efficient and flexible oxygen sensor in organic media within an ample range of dielectric constants and viscosities and over the whole oxygen concentration range.
Experimental Section
Reagents: All reagents were commercially available and used as received. 11-Mercaptoundecanol (MU) and 1-pyrenebutanol were purchased from Sigma–Aldrich. CHCl3, CH2Cl2, EtOAc, toluene, hexane and tetrahydrofuran (THF) of HPLC grade were purchased from Scharlau (Barcelona, Spain). Dioxane was purchased from Sigma–Aldrich. The core shell (CS) nanoparticles capped with a long-chain primary amine were purchased from Ocean Nanotech LLC (San Diego, CA, USA).
Synthesis of CS@Py: Amine-capped CdSe/ZnS nanoparticles (CS; 5.97×10−9 mol) and 4-(pyren-4-yl)butyl-11-mercaptoundecanoate (Py-L-SH; 3.13×10−5 mol, [thiol]/[CS]=5000 molar ratio) were mixed in of CHCl3 (25 mL). This mixture was heated to reflux under N2 atmosphere for 48 h. After almost total solvent evaporation (2 mL remaining) and the addition of MeOH (30 mL), the samples were centrifuged (8000 rpm, 20 min, 20 °C), and the supernatant was decanted. This process was repeated until the UV absorption spectrum of the supernatant did not detect the presence of the pyrene (five washing/centrifugation cycles). The nanoparticles (QD@Py) were dissolved in THF (2 mL). Spectroscopic measurements were performed at RT (T=22 °C).
The number of Py units on each nanoparticle was estimated by firstly determining the extinction coefficient of Py in THF (ɛ=13 193 m−1 cm−1). The absorbance at 335 nm of a solution of CS@Py and that of CS, both at the same nanoparticle concentration, were registered (Figure 2). The difference between these absorbances was attributed to Py (APy(335 nm)=ACS@Py−ACS), thus allowing the estimation of Py concentration [Py]=APy(335 nm)/ɛcb, and, eventually, the ratio of Py units per nanoparticle.
Experiments at different oxygen pressures were carried out by saturating the solution of CS@Py with N2 (research grade) and/or O2 (ultrapure, 99 %) in all solvents.
Characterization: UV/Vis spectra of the samples were recorded in quartz cuvettes using an Agilent 8453E spectrophotometer. Steady-state fluorescence spectra (LPS-220B, motor driver (MD-5020), Brytebox PTI) were measured on a spectrofluorometer PTI, equipped with a lamp power supply and working at RT. The Felix 32 Analysis software was used to register the data. All the data were acquired using 1 cm×1 cm path length quartz cuvettes. Time-resolved measurements were made with a Time Master Fluorescence lifetime spectrometer TM-2/2003 from PTI. Sample excitation was afforded by PTIs own GL-3300 nitrogen laser. The kinetic traces were fitted by mono- or biexponential decay functions and reconvolution of the instrument response function. The accuracy of the fits was evaluated by the reduced χ2 values (0.8>χ2<F 1.2). QD images were obtained by high-resolution transmission electron microscopy (HRTEM, FEI Tecnai G2 F20) at an accelerating voltage of 200 kV. Samples were prepared by dropping the colloidal solution on a Lacey Formvar/carbon-coated copper grid. The digital analysis of the HRTEM micrographs was done using digital MicrographTM 1.80.70 for GMS by GATAN.
Acknowledgments
We thank the Ministry of Economy and Competitiveness (MINECO, Madrid, Spain) (Project CTQ2011–27758, contract granted to S.G-C), the Generalitat Valenciana (GVA, Spain) (ACOMP/2013/008) and the Fundació General de la Universitat de València (FGUV, Spain) (R.E.G contract).
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
References
- 1.Costa-Fernández JM, Diaz-García ME, Sanz-Medel A. Anal. Chim. Acta. 1998;360:17. [Google Scholar]
- 2.Xavier MP, García-Fresnadillo D, Moreno-Bondi MC, Orellana G. Anal. Chem. 1998;70:5184. [Google Scholar]
- 3.Quaranta M, Murkovic M, Klimant I. Analyst. 2013;138:6243. doi: 10.1039/c3an36782g. [DOI] [PubMed] [Google Scholar]
- 4.Mills A. Chem. Soc. Rev. 2005;34:1003. doi: 10.1039/b503997p. [DOI] [PubMed] [Google Scholar]
- 5.Klibanov AM. Nature. 2001;409:241. doi: 10.1038/35051719. [DOI] [PubMed] [Google Scholar]
- 6.Niederberger M, Garnweitner G. Chem. Eur. J. 2006;12:7282. doi: 10.1002/chem.200600313. [DOI] [PubMed] [Google Scholar]
- 7.Fisicaro P, Adriaens A, Ferrara E, Prenesti E. Anal. Chim. Acta. 2007;597:75. doi: 10.1016/j.aca.2007.06.032. [DOI] [PubMed] [Google Scholar]
- 8.Helm I, Jalukse L, Leito I. Sensors. 2010;10:4430. doi: 10.3390/s100504430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu H, Aylott JW, Kopelman R, Miller TJ, Philbert MA. Anal. Chem. 2001;73:4124. doi: 10.1021/ac0102718. [DOI] [PubMed] [Google Scholar]
- 10.Koo Y-EL, Cao Y, Kopelman R, Koo SM, Brasuel M, Philbert MA. Anal. Chem. 2004;76:2498. doi: 10.1021/ac035493f. [DOI] [PubMed] [Google Scholar]
- 11.Douglas P, Eaton K. Sens. Actuators B. 2002;82:200. [Google Scholar]
- 12.Reece J, Miller M, Arnold M, Waterhouse C, Delaplaine T, Cohn L, Cannon T. Appl. Biochem. Biotechnol. 2003;104:1. doi: 10.1385/abab:104:1:1. [DOI] [PubMed] [Google Scholar]
- 13.Achatz DE, Meier RJ, Fischer LH, Wolfbeis OS. Angew. Chem. 2011;123:274. doi: 10.1002/anie.201004902. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2011;50:260. [Google Scholar]
- 14.Zhang L, Su F, Buizer S, Lu H, Gao W, Tian Y, Meldrum D. Biomaterials. 2013;34:9779. doi: 10.1016/j.biomaterials.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu C, Bull B, Christensen K, McNeill J. Angew. Chem. 2009;121:2779. doi: 10.1002/anie.200805894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2009;48:2741. [Google Scholar]
- 16.Collier BB, Singh S, McShane M. Analyst. 2011;136:962. doi: 10.1039/c0an00661k. [DOI] [PubMed] [Google Scholar]
- 17.Jorge PAS, Mayeh M, Benrashid R, Caldas P, Santos JL, Farahi F. Appl. Opt. 2006;45:3760. doi: 10.1364/ao.45.003760. [DOI] [PubMed] [Google Scholar]
- 18.Liu L, Li B, Qin R, Zhao H, Ren X, Su Z. Nanotechnology. 2010;21:285701. doi: 10.1088/0957-4484/21/28/285701. [DOI] [PubMed] [Google Scholar]
- 19.Lu H, Jin Y, Tian Y, Zhang W, Holl MR, Meldrum DR. J. Mater. Chem. 2011;21:19293. doi: 10.1039/C1JM13754A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mills A, Tommons C, Bailey RT, Crilly P, Tedford MC. Anal. Chim. Acta. 2011;702:269. doi: 10.1016/j.aca.2011.06.059. [DOI] [PubMed] [Google Scholar]
- 21.McLaurin EJ, Greytak AB, Bawendi MG, Nocera DG. J. Am. Chem. Soc. 2009;131:12994. doi: 10.1021/ja902712b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kikuchi K, Takakusa H, Nagano T. TrAC Trends Anal. Chem. 2004;23:407. [Google Scholar]
- 23.Long L, Zhang D, Li X, Zhang J, Zhang C, Zhou L. Anal. Chim. Acta. 2013;775:100. doi: 10.1016/j.aca.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Mello JV, Finney NS. Angew. Chem. 2001;113:1584. [Google Scholar]
- Angew. Chem. Int. Ed. 2001;40:1536. [Google Scholar]
- 25.Lemon CM, Curtin PN, Somers RC, Greytak AB, Lanning RM, Jain RK, Bawendi MG, Nocera DG. Inorg. Chem. 2014;53:1900. doi: 10.1021/ic401587r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemon CM, Karnas E, Bawendi MG, Nocera DG. Inorg. Chem. 2013;52:10394. doi: 10.1021/ic4011168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amelia M, Lavie-Cambot A, McClenaghan ND, Credi A. Chem. Commun. 2011;47:325. doi: 10.1039/c0cc02163f. [DOI] [PubMed] [Google Scholar]
- 28.Agudelo-Morales CE, Galian RE, Pérez-Prieto J. Anal. Chem. 2012;84:8083. doi: 10.1021/ac302276j. [DOI] [PubMed] [Google Scholar]
- 29.Agudelo-Morales CE, Silva OF, Galian RE, Pérez-Prieto J. ChemPhysChem. 2012;13:4195. doi: 10.1002/cphc.201200637. [DOI] [PubMed] [Google Scholar]
- 30.Aguilera-Sigalat J, Rocton S, Sanchez-Royo JF, Galian RE, Perez-Prieto J. RSC Adv. 2012;2:1632. [Google Scholar]
- 31.Pong B-K, Trout BL, Lee J-Y. Langmuir. 2008;24:5270. doi: 10.1021/la703431j. [DOI] [PubMed] [Google Scholar]
- 32. The fluorescence decays of the pyrene species of CS@Py can be analysed using a Blob model, assuming that some pyrenes form excimer via diffusion, others are too isolated to form excimer, and a great deal of them are pre-associated and form an excimer after absorption of a photon; see ref. [33]
- 33.Siu H, Duhamel J. Macromolecules. 2004;37:9287. [Google Scholar]
- 34.Montalti M, Credi A, Prodi L, Gandolfi MT. Handbook of Photochemistry, 3rd ed. Boca Raton: CRC Press; 2006. [Google Scholar]
- 35. The emission lifetimes of the QDs are a subject of study, but the occurrence of multiphotonic processes leading to different decay processes is generally accepted. Thus, the decays were fitted to a multi-exponential model, as is commonly done in the case of QDs in solution, see ref. [36–39]
- 36.de Mello Donegá C, Bode M, Meijerink A. Phys. Rev. B. 2006;74:085320. [Google Scholar]
- 37.Ruedas-Rama MJ, Orte A, Hall EAH, Alvarez-Pez JM, Talavera EM. ChemPhysChem. 2011;12:919. doi: 10.1002/cphc.201000935. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, Qu L, Zhang J, Peng X, Xiao M. Nano Lett. 2003;3:1103. [Google Scholar]
- 39.Wadhavane PD, Galian RE, Izquierdo MA, Aguilera-Sigalat J, Galindo F, Schmidt L, Burguete MI, Pérez-Prieto J, Luis SV. J. Am. Chem. Soc. 2012;134:20554. doi: 10.1021/ja310508r. [DOI] [PubMed] [Google Scholar]
- 40.Kalb M, Gudipati MS. J. Phys. Chem. A. 1998;102:508. [Google Scholar]
- 41. For other examples of the influence of pre-equilibrium intermediates in the decay of excited states, see ref. [42–44]
- 42.Rathore R, Hubig SM, Kochi JK. J. Am. Chem. Soc. 1997;119:11468. [Google Scholar]
- 43.Hubig SM, Rathore R, Kochi JK. J. Am. Chem. Soc. 1999;121:617. [Google Scholar]
- 44.Pérez-Prieto J, Galian RE, Morant-Miñana MC, Miranda MA. Chem. Commun. 2005:3180. doi: 10.1039/b500697j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary