

Significance
Many natural systems are structured by the ordering of repeated, distinct shapes. Understanding how this happens is difficult because shape affects structure in two ways. One is how the shape of a cell or nanoparticle, for example, affects its surface, chemical, or other intrinsic properties. The other is an emergent, entropic effect that arises from the geometry of the shape itself, which we term “shape entropy,” and is not well understood. In this paper, we determine how shape entropy affects structure. We quantify the mechanism and determine when shape entropy competes with intrinsic shape effects. Our results show that in a wide class of systems, shape affects bulk structure because crowded particles optimize their local packing.
Keywords: entropy, self-assembly, colloids, nanoparticles, shape
Abstract
Entropy drives the phase behavior of colloids ranging from dense suspensions of hard spheres or rods to dilute suspensions of hard spheres and depletants. Entropic ordering of anisotropic shapes into complex crystals, liquid crystals, and even quasicrystals was demonstrated recently in computer simulations and experiments. The ordering of shapes appears to arise from the emergence of directional entropic forces (DEFs) that align neighboring particles, but these forces have been neither rigorously defined nor quantified in generic systems. Here, we show quantitatively that shape drives the phase behavior of systems of anisotropic particles upon crowding through DEFs. We define DEFs in generic systems and compute them for several hard particle systems. We show they are on the order of a few times the thermal energy () at the onset of ordering, placing DEFs on par with traditional depletion, van der Waals, and other intrinsic interactions. In experimental systems with these other interactions, we provide direct quantitative evidence that entropic effects of shape also contribute to self-assembly. We use DEFs to draw a distinction between self-assembly and packing behavior. We show that the mechanism that generates directional entropic forces is the maximization of entropy by optimizing local particle packing. We show that this mechanism occurs in a wide class of systems and we treat, in a unified way, the entropy-driven phase behavior of arbitrary shapes, incorporating the well-known works of Kirkwood, Onsager, and Asakura and Oosawa.
Nature is replete with shapes. In biological systems, eukaryotic cells often adopt particular shapes, for example, polyhedral erythrocytes in blood clots (1) and dendritic neurons in the brain (2). Before the development of genetic techniques, prokaryotes were classified by shape, as bacteria of different shapes were implicated in different diseases (3). Virus capsids (4, 5) and the folded states of proteins (6) also take on well-recognized, distinct shapes. In nonliving systems, recent advances in synthesis make possible granules, colloids, and nanoparticles in nearly every imaginable shape (7–12). Even particles of nontrivial topology now are possible (13).
The systematic study of families of idealized colloidal and nanoscale systems by computer simulation has produced overwhelming evidence that shape is implicated in the self-assembly* of model systems of particles (14–17). In these model systems, the only intrinsic forces between particles are steric, and the entropic effects of shape (which we term “shape entropy”†) can be isolated. Those works show that shape entropy begins to be important when systems are at moderate density (21).
In laboratory systems, however, it is not possible to isolate shape entropy effects with as much control, and so the role of shape entropy in experiment is less clear. However, intuition suggests that shape entropy becomes important when packing starts to dominate intrinsic interactions and therefore should be manifest in crowded systems in the laboratory.
Unlike other interactions, shape entropy is an emergent‡ quantity that is expected to become important as systems become crowded. Although entropy-driven phase behavior, from the crystallization of hard spheres (22–27) to the nematic transition in hard rods (28) to colloid–polymer depletion interactions (29), has been studied for decades, linking microscopic mechanisms with macroscopic emergent behavior is difficult in principle (30). Hence, even for idealized systems, despite the overwhelming evidence that shape entropy is implicated in phase behavior, understanding how shape entropy is implicated is only now starting to be distilled (14–17, 31–59). For example, the phase behavior of binary hard sphere mixtures (60–64) or polygons (40, 43, 65) can be deduced from global packing arguments, but for many other shapes (17), including “simple” platonic solids such as the tetrahedron (36) and its modifications (15), this is not the case.
One suggestion of how shape entropy is implicated in the phase behavior of systems of anisotropic particles is through the idea of directional entropic forces (DEFs). Damasceno et al. (15) inferred the existence of these forces by observing that in many idealized systems of convex polyhedral shapes, one tends to observe a high degree of face-to-face alignment between particles in crystals. However, the origin and strength of these forces are unclear.
Here we use computer simulations to address how these forces arise and construct a rigorous theoretical framework that enables this investigation. Our key results are as follows: (i) We quantify pairwise DEFs in arbitrary systems, compute them directly in several example systems, and show they are on the order of a few times the thermal energy () just before the onset of crystallization. (ii) We show that the microscopic mechanism underlying the emergence of DEFs is the need for particles to optimize their local packing in order for the system to maximize shape entropy. (iii) By computing quantities for DEFs that can be compared with intrinsic forces between particles, we determine when shape entropy is important in laboratory systems and suggest how to measure DEFs in the laboratory. (iv) We explain two notable features of the hard particle literature: the observed frequent discordance between self-assembled and densest packing structures (15, 17, 39, 41, 44, 53) and the high degree of correlation between particle coordination in dense fluids and crystals (17). (v) As we illustrate in Fig. 1, we show that the same local dense packing mechanism that was known to drive the phase behavior of colloid–depletant systems also drives the behavior of monodisperse hard particle systems, thereby allowing us to view—within a single framework—the entropic ordering considered here and in previous works (14–17, 21–29, 31–59, 66–81).
Fig. 1.

The general nature of entropic interactions treated in this work applies to a broad class of known systems. Here we represent them on three orthogonal axes. One axis represents, schematically, the shape of the constituent particles, with spheres at the origin. The other two axes concern the sea of particles that are being integrated out and provide the effective interaction. On one axis is the inverse of the strength of the interaction between them (where 0 represents hard steric exclusion). On the other axis is the ratio of the characteristic size of the particles of interest to that of the particle being integrated out. Other axes, not shown, represent the shape of the particle being integrated out, mixtures of particle shapes and types, etc. Examples of known experimental and model systems are sketched to illustrate their location on these axes (see references and description in text).
Methods
To compute statistical integrals, we used Monte Carlo (MC) methods. For purely hard particle systems, we used single-particle-move MC simulations for both translations and rotations for systems of 1,000 particles at fixed volume. Polyhedra overlaps were checked using the Gilbert–Johnson–Keerthi algorithm (82) as implemented in ref. 17. For penetrable hard sphere depletant systems, we computed the free volume available to the depletants using MC integration.
We quantified DEFs between anisotropic particles at arbitrary density by using the potential of mean force and torque (PMFT). Such a treatment of isotropic entropic forces was first given by De Boer (83) using the canonical potential of mean force (84); for aspherical particles, first steps were taken in this direction in refs. 85, 86.
Consider a set of arbitrary particles, not necessarily identical. Take the positions of the particles to be given by and the orientations of the particles to be given by . The partition function in the canonical ensemble, up to an overall constant, is given by
| [1] |
where U is the potential energy for the interaction among the particles. Suppose we are interested in only a single pair of particles, which we label with indices 1 and 2, and denote all the other (sea) particles with a tilde. The partition function is formally an integral over all the microstates of the system, weighted by their energies. Because we are interested in a pair of particles, we do not want to perform the whole integral to compute the partition function. Instead, we break up the domain of integration into slices in which the relative position and orientation of the pair of particles are fixed; we denote this by .
We choose to work with coordinates that are invariant under translating and rotating the pair of particles. In two dimensions, a pair of particles has three scalar degrees of freedom. In three dimensions, for generic particles without any continuous symmetries, a pair of particles has six scalar degrees of freedom. In SI Appendix, we give an explicit form for these coordinates that is invariant under translations and rotations of the particle pair, and the interchange of their labels. Note that particles with continuous symmetry (i.e., spherical or axial) have fewer degrees of freedom. Separating out the integration over the sea particles gives the partition function as
| [2] |
We formally integrate over the degrees of freedom of the sea particles to write
| [3] |
where encodes the free energy of the sea particles with the pair of interest fixed, and J is the Jacobian for transforming from the absolute positions and orientations of the particles in the pair to their relative position and orientation. We define the PMFT for the particle pair implicitly through the expression
| [4] |
Equating the logarithms of the integrands on the right-hand sides of Eqs. 3 and 4 gives an expression for the PMFT :
| [5] |
In SI Appendix, we outline how to extract the forces and torques from Eq. 5 and give example calculations that determine the thermally averaged equations of motion for pairs of particles.
In cases in which the particle pair of interest has only excluded volume interactions, as in the remainder of this paper, it is convenient to combine the first two terms to cast expression Eq. 5 as
| [6] |
where H is the Heaviside step function that we use as a bookkeeping device to ensure that the effective potential is infinite for configurations that are sterically excluded, and is the minimum separation distance of the particle pair in their relative position and orientation, which is negative when the particles overlap and positive when they do not.
When the sea particles are penetrable hard sphere depletants, i.e., the sea particles are an ideal gas with respect to each other but hard with respect to the pair, the contribution of the sea particles to Eq. 5 can be evaluated directly. If we have N penetrable hard sphere depletants, we evaluate the sea contribution in Eq. 5 to be
| [7] |
where is the free volume available to the sea particles. If we consider two nearby configurations, we have that
| [8] |
where we have used the ideal gas equation of state for the depletant particles. Thus, up to an irrelevant additive constant,
| [9] |
The treatment of mixed colloid–polymer depletion systems as mutually hard colloids in the presence of noninteracting polymers is known in the literature as the penetrable hard sphere limit (66). Hence, Eq. 9 is the generalization of the Asakura–Oosawa (29) result for depletion interactions between spherical particles and particles of arbitrary shape.
Results
Entropic Forces in Monodisperse Hard Systems.
As argued in refs. 15, 17, we expect that for a pair of polyhedra, the DEFs favor face-to-face arrangements. In terms of the PMFT, we therefore expect face-to-face configurations to have the deepest well of effective attraction.
We compute the force components of the PMFT in Cartesian coordinates for polyhedra or polyhedrally faceted spheres and integrate over the angular directions, as described in SI Appendix, which also gives explicit results for torque components in an example monodisperse hard system. We then use a set of orthogonal coordinate systems for each face of the polyhedron (see SI Appendix for a schematic diagram of this for a tetrahedron) and linearly interpolate the PMFT to the coordinate frame of each facet.
In Fig. 2, we plot the PMFT in the direction perpendicular to the face for the three systems—hard tetrahedra (Fig. 2A), hard tetrahedrally faceted spheres (Fig. 2B), and hard cubes (Fig. 2C)—at various densities shown in the legend. We have chosen an axis that passes through the global minimum of the potential. Several independent runs were averaged to obtain these results. Because we are free to shift the PMFT by an additive constant, we have shifted the curves for each density for clarity. We show only points that lie within of the global minimum, because we can sample accurately at these points. As the density increases, the first minimum of the potential decreases and gets closer to contact. This indicates that the particles exhibit greater alignment at higher densities, as expected. Note that this is not merely an artifact of the decrease in average particle separation at higher densities because, as we show below and in SI Appendix, the alignment effect is concentrated near the center of the facet.
Fig. 2.

In monodisperse systems, we compute the PMFT by considering pairs of particles (A, C, and E). Density dependence of the PMFT along an axis perpendicular to the polyhedral face for a hard tetrahedron fluid (B), a fluid of tetrahedrally faceted hard spheres (D), and a hard cube fluid (F). Data are computed from the frequency histogram of the relative Cartesian coordinates of pairs of particles in MC simulations of monodisperse hard particles and correspond to the integration of the PMFT over relative orientation. We plot along the axis that contains the global minimum of the potential and plot only data that are within of the global minimum at each respective density, because we can sample such points reliably. The perpendicular distance z is given in units of the minimum separation between particles, which is twice the radius of the inscribing sphere of the given polyhedron. Error bars are smaller than the markers indicating data points, and a smooth curve through the data points in each series is used to guide the eye.
To understand what the PMFT is surrounding each shape, we do the following. In Fig. 3, we plot the PMFT in the plane parallel to the face that passes through the global minimum of the potential (which may be identified by the minimum of the respective curve in Fig. 2). The outline of one of the faces of one particle of the pair is indicated by the solid line. The second reference particle is allowed to have its center of mass anywhere on this plane. The orientation of the second particle may vary, and we integrate over all the angles. Because the particles cannot overlap, in practice not all orientations are allowed at close distances (we plot this effect for cubes in Fig. 4). Each row shows (from left to right) increasingly dense systems of tetrahedra (top row), tetrahedrally faceted spheres (middle row), and cubes (bottom row), respectively.
Fig. 3.

Directional entropic forces are emergent in systems of particles and, as such, cannot be imaged directly through microscopy. Here we show the location of potential wells by taking slices of the PMFT (computed from the frequency histogram of the relative Cartesian coordinates of pairs of particles in MC simulations of monodisperse hard particles) parallel to the faces of a tetrahedron (A–D), a tetrahedrally faceted sphere (E–H), and a cube (I–L) at various packing fractions , indicated at the head of the column. As the packing fraction increases from left to right, the potential well becomes stronger and its shape becomes more well-defined.
Fig. 4.
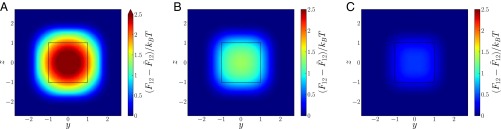
The density-independent contribution to the PMFT for cubes at distances of (A), 1.2 (B), and 1.3 (C), which arises intrinsically from the pair of colloids under consideration and contributes the first term factor on the right-hand side in Eq. 6. This contribution provides an effective repulsion between free particles, which is stronger at small separations (A) than at larger ones (B and C). The pattern of the repulsion is opposite that of the overall PMFT between cubes seen in Fig. 3, in which both factors in Eq, 6 have been taken together.
Directional entropic forces originate from entropic patch sites (57)—geometric features that facilitate local dense packing—but as emergent notions, these patch sites cannot be imaged as, say, sticky patches created through gold deposition on the surface of a nanoparticle can be through electron microscopy. Instead, in Fig. 3 we plot the location and strength of entropic patches at different densities. These plots show that the entropic patches lie at the centers of the facets. The effect of the pressure of the sea particles may be seen by comparing the density-dependent PMFT from Eq. 6, plotted for cubes in Fig. 3 I–L, to the density-independent pair contribution for cubes in Fig. 4. The pair contribution clearly drives pairs of cubes away from direct face-to-face contact, and it is the other contribution from the sea particles that is responsible for driving them together. For the polyhedral shapes, the coordination of the patches corresponds to the locations of the vertices of the dual polyhedron, and the shapes of the patches themselves at high densities appear to reflect the symmetry of the dual. In ref. 57, we showed that systematic modification of the particle shape induces DEFs between particles that lead to the self-assembly of target crystal structures.
Note that although the tetrahedron and the tetrahedrally faceted sphere share the same point group symmetry, and the geometrical coordination of basins of attraction is the same in both cases, the shape of the effective potential and its strength are different. For example, contrasting the two shapes at packing fraction (Fig. 3), we see that the potential difference between the center of the facet of the tetrahedrally faceted sphere and the truncated vertex is more than a different from in the case of the actual tetrahedron. This indicates that small changes in particle shape may have dramatic effects on the structural coordination of the dense fluid.
The existence of the directionality in the PMFT in the dense fluid strongly suggests that DEFs provide the mechanism for crystallization. In Fig. 5, we show that DEFs persist, and increase, in the crystal. For concreteness, we study systems of cubically faceted spheres that are very close to perfect cubes at a packing fraction of (fluid) and (crystal). As we showed in ref. 57, at sufficiently high packing fractions, these particles self-assemble a simple cubic lattice. Upon increasing the packing density from the fluid to the crystal, the PMFT develops stronger anisotropy. For example, by comparing the PMFT at the center of the facet to the location of the vertex of the faceting cube, we note that the difference between identical points may increase by more than .
Fig. 5.

Directional entropic forces emerge in the fluid phase (Left) and persist into the crystal (Right). Here we plot the PMFT for cubically faceted spheres (from the family of shapes studied in ref. 57) that are nearly perfect cubes. In the dense fluid at (Left) and the crystal (Right), the PMFT has a similar form, but the strength of the interaction is at least stronger in the crystal.
Entropic Forces with Penetrable Hard Sphere Depletants.
We study DEFs as a function of colloid shape in systems with traditional weakly interacting, small depletants. One system consists of a pair of spherical particles that are continuously varying faceting with a single facet, to promote locally dense packing. The other is a system of spherocylinders of constant radius that are continuously elongated. In each case, the alteration creates a region on the surface of the particle with reduced spatial curvature.
As the amount of alteration to particle shape increases, it leads to stronger attraction between the sites of the reduced curvature, as encoded in the probability of observing particles with these entropic patches (57) adjacent. In the case of the faceted particle, we vary the depth of the facet linearly between zero (a sphere) and unity (a hemisphere), with the radius of the sphere fixed to 10 (Fig. 6E). In the case of spherocylinders, we studied cap radii fixed to 5 and cap centers interpolating linearly between 1 (nearly spherical) and 4 (an elongated spherocylinder), where all lengths are in units of the depletant radius (Fig. 6F). See SI Appendix for computational details. In Fig. 6 A and B, we show the probability that if a pair of particles is bound, then they are bound patch to patch entropically (specific binding) as a function of depletant pressure. Specific binding is depicted in the inset images and the left-hand particle pairs in Fig. 6 C and D, and is contrasted with patch-to-nonpatch (semispecific binding) and nonpatch-to-nonpatch (nonspecific binding) in the center and right-hand particle images in Fig. 6 C and D. Fig. 6 A and B show that we can tune binding specificity by adjusting the patch size and the depletant pressure. Different curves correspond to increasing patch size (more faceting) as the color goes from blue to red.
Fig. 6.

The probability of specific binding, for (A) particles with a single facet and (B) capsules in a bath of depletants as a function of depletant pressure. (C and D) Configurations that correspond to specific, semispecific, and nonspecific binding according to whether the binding occurs at sites of low curvature. (E and F) Singly faceted spheres and spherocylinders with various facet and cylinder sizes, respectively. Curves in A and B show the probability of specific binding at various faceting amounts; with increasing patch size (more faceting), the color goes from blue to red.
For spherocylinders, it is straightforward to show the effect of the entropic patches in generating torques that cause the particles to align. To isolate the part of the torque that comes from the patch itself, we rearrange Eq. 9 for nonoverlapping spherocylinders to get
| [10] |
Note that, conveniently, for ideal depletants, the expression on the right-hand side is independent of the depletant pressure. For clarity, we fix the separation distance R between the spherocylinders’ centers of mass to be larger than the spherocylinder diameter (which is the minimum separation distance) and fix the orientation of each spherocylinder to be normal to the separation vector between them ( in the coordinates in SI Appendix). Because it is always possible to shift the PMFT by a constant, for normalization purposes we define
| [11] |
where χ describes the angle between the spherocylinder symmetry axes. In Fig. 7, we plot for spherocylinders of different aspect ratios. For small side lengths of the spherocylinder, there is very weak dependence of the PMFT on χ. As the length of the cylinder increases (and therefore as the entropic patch gets larger), the χ dependence of the PMFT becomes more pronounced. This means that not only do the particles coordinate at their entropic patch sites, but there also exists a torque (75) that aligns the patches.
Fig. 7.

The angular (χ) dependence of the portion of the PMFT directly attributable to modification of particle shape (Ω; see Eq. 11), normalized to 0 at ) for spherocylinders in the presence of penetrable hard sphere depletants for different cylinder side lengths. For short side lengths (, light blue ■), there is very little angular variation, indicating very little entropic torque. As the side length, ℓ, increases from 1 to 5 (in units of the depletant radius, pink ▼), the strength of the dependence on χ increases, indicating greater torques, as expected.
Discussion
PMFT as an Effective Potential at Finite Density.
Extracting physical mechanisms from systems that are governed mainly by entropy is, in principle, a difficult task. This difficulty arises because entropic systems have many degrees of freedom that exist at the same energy or length scale. Typically, disparities in scale are used by physicists to construct “effective” descriptions of physical systems, in which many degrees of freedom are integrated out and only the degrees of freedom key to physical mechanisms are retained. Determining which degrees of freedom are key in entropic systems is difficult because of the lack of natural hierarchies, but to extract physical mechanisms, it still is necessary.
To extract the essential physics of shape for anisotropic colloidal particles, we computed the PMFT. In so doing, we described the PMFT as an effective potential, but we emphasize here that it is an effective potential in a restricted sense of the term. The restriction comes about because when we separate a system into a pair of reference colloids plus some sea particles, the sea particles may be identical to the reference colloids. Only the pair of reference colloids is described by the effective potential, which means it describes the behavior of a system of only two particles, with the rest implicit. Indeed, the rest of the colloids in the system are treated as an “implicit solvent” for the pair under consideration. Because of this fact, the PMFT for monodisperse colloids is not synonymous with the bare interaction potential for the whole system. Note that in systems of multiple species, there is some possibility of a broader interpretation of the PMFT for a single species having integrated out another species (87), but even in that case, difficulties arise (88). For example, Onsager (28) treated the nematic transition in hard rods by considering rods of different orientations as different “species,” but an effective potential between rods of the same species or orientation does not capture the physics of the system in the same way an effective potential between rods induced by polymer depletants would (21).
Interpreting the PMFT from Eq. 5 in the restricted sense, we see that it naturally exhibits three contributions: (i) The first term on the right-hand side is the bare interaction between the pair of particles, which originates from van der Waals, electrostatic, or other interactions of the system of interest. It encodes the preference for a pair of particles to be in a particular relative position and orientation. Here, we are concerned mostly with hard particles, so this term encodes the excluded volume interaction. (ii) The second term on the right-hand side is the logarithmic contribution from the Jacobian; it counts the relative number of ways the pair of particles can exist in a relative position and orientation. For a more technical discussion of this term, see SI Appendix. (iii) The third term is the free energy of the sea particles that are integrated out, keeping the relative position and orientation of the pair fixed. In purely hard systems, this last term is sea particle entropy, which is just the logarithm of the number of microstates available to the sea particles for that configuration of the pair.
The free energy minimizing configuration of the pair of particles is determined by a competition between the three terms in Eq. 5. Consider the contribution from the third term. For any nonattractive bare interactions between sea particles, if the pair of particles is separated by a distance that is less than the effective diameter of the sea particles (89) and adopts a configuration that packs more densely, the free energy of the sea particles will never increase. This is the case for bare interactions that are repulsive because of excluded volume (in which case, the system is dominated by entropy) and for sea particles that are soft and interpenetrable.
In the absence of the first two terms in Eq. 5, the third term will drive the pair of particles into denser local packing configurations. The driving force for local dense packing becomes stronger as the system density increases. To understand why this occurs, we note that DEFs arise by the pair of particles balancing the pressure of the sea particles. Rather than considering the effects of the sea on the pair, we momentarily consider the effects of the pair on the sea. The pair of interest forms part of the “box” for the sea particles. Density dependence arises because changes in the relative position and orientation are related to the local stress tensor. This contribution is on the order of , where P is the characteristic scale of the local stress tensor, which we expect to be related to the pressure, and σ is a characteristic length scale. Because this contribution is density dependent, the effective forces between particles are emergent. These emergent forces are directional, because some local packing configurations are preferred over others. For example, dense local packing configurations involve face-to-face contact for many particles, which is why face-to-face contact is observed in so many hard-particle crystals, and is borne out in Fig. 3. Of course, this also is akin to hard rods aligning axes in nematic liquid crystals (28). Indeed, in repulsive systems, only at sufficiently high densities is the system able to overcome any repulsion from the first two terms in Eq. 5, plotted in Fig. 4 for cubes, which drive the system away from locally dense packings. It is this competition between the drive toward local dense packing supplied by the sea particles’ entropy and the preferred local relative positions and orientations of the pair, which are induced by the maximization of shape entropy, that determines the self-assembly of entropic systems of repulsive (hard) shapes.
Two general lessons from the literature of hard particle self-assembly are intuitive in light of the mechanism we have described.
First, in a previous systematic study of shape and self-assembly using a family of highly symmetric convex polyhedra (17), it was shown that there is a remarkable correlation between the number of nearest neighbors in the crystal structure of a given polyhedron and the number of nearest neighbors in the dense fluid. Because systems at finite density exhibit a drive toward local dense packing, we would expect the organization of neighbor shells to be determined by the same local packing considerations in both the dense fluid and the crystal. Indeed, we might expect to predict crystal structure based on local packing considerations alone. In recent work, we designed particle shape to favor certain local dense packing arrangements to self-assemble targeted crystal structures (57). We viewed the induced anisotropy of the effective interactions between particles as the entropic analog of the intrinsic anisotropic interactions between enthalpically patchy particles (7, 90). Attractive entropic patches are features in particle shape that facilitate local dense packing.
Second, hard particle systems often assemble into their densest packing structure; however, some do not (15, 17, 39, 41, 44, 53). The present work helps highlight two key differences between self-assembly and packing: (i) Systems self-assemble because the second law of thermodynamics drives them to free energy minima, so like packing, self-assembly has a mathematical optimization problem at its root. In self-assembling systems, all three terms in Eq. 5 contribute, but in the infinite pressure limit relevant for packing, the entropy of the pair of particles does not contribute. This is because the first term provides the steric hindrance between particles, and the third term should scale with pressure, whereas the second term encodes the entropy of a set of relative positions and orientations of the pair, and this factor becomes irrelevant in the infinite pressure limit. (ii) Assembly is related to local dense packing, which we have characterized here with two-body potentials, whereas global dense packing ostensibly is an “all-body problem.” Given these two differences, it might be more surprising that densest packing solutions ever coincide with assembled structures than the fact that they frequently do not. However, the pair entropy term often only separates particles while preserving their alignment; for example, we showed that particles are observed more frequently with face-to-face alignment at small separations than in direct face-to-face contact. Moreover, in practice (e.g., ref. 91), the solution to the all-body packing problem often is given by the solution to a few-body problem.
Unification.
We saw above that directional entropic forces, as captured by the PMFT, arise when particles maximize shape entropy through local dense packing. We verified this drive to local dense packing with direct calculations for monodisperse hard shapes and for hard shapes in a sea of penetrable hard sphere depletants. Monodisperse hard-particle systems and colloid-depletant systems would, at first glance, seem to be different.
However, depletion systems (29) and monodisperse systems of hard anisotropic shapes are both entropy driven, as is the crystallization of hard spheres (22–27) and the nematic liquid crystal transition in hard rods (28). Indeed, since the original work by Asakura and Oosawa (29) on depletion interactions, it has been well known that the depletion-induced aggregation of colloids arises from an osmotic effect in which depletants liberate free volume for themselves by driving the colloids into dense packing configurations. However, the earlier work of de Boer (83) suggests that considerations of the potential of mean force in systems of isotropic particles lead to pairs of particles having certain preferred distances, because they balance their own repulsive forces with the sea particles’ preference for them to pack more densely.
In traditional depletion systems, the depletants are small compared with the colloids, spherical, and interpenetrable. In such systems, there is a substantial literature on the anisotropic binding of colloids by depletants starting with ref. 67 and followed by work on rough colloids (68–74), as well as more anisotropic shapes (42, 54, 55, 75, 80, 81) and lock-and-key systems (76–79). In all these systems, the anisotropic binding may be seen as resulting from a sea of depletants forcing colloids to adopt local dense packing configurations.
However, one might ask how large or hard or aspherical the depletants can be before they cease to act as depletants. In the original work of Asakura and Oosawa (29), depletants were not restricted to be small; large depletants are the so-called protein limit (66). In experimental systems, as well as accurate models, depletants are not freely interpenetrable (66, 87, 92). In the original work of Asakura and Oosawa (29), depletants were imagined to be polymers, which certainly are not spherical, but were modeled as spheres.
Of course, it is well known that depletants may be aspherical or hard or large; in all cases, depletants exert an osmotic pressure that causes colloids to aggregate as the colloids adopt dense packing configurations. However, we argued above that consideration of the three contributions to the PMFT in Eq. 5 leads to a drive toward local dense packing in generic systems in which the sea particles may simultaneously be arbitrarily aspherical, hard, and large. Indeed, the sea particles may be the same species as the colloids and still behave as “depletants.” Hence, the classic works on the entropic behavior of systems as diverse as hard shapes from spheres (22–27), to rods (28), to tetrahedra (36), and colloid-depletant systems (29, 66) are similar not only because their phase behavior is driven by entropy, but also because entropy controls their phase behavior through a preference for local dense packing.
In Fig. 1, we show a schematic representation of the family of systems whose phase behavior is governed by the shape entropy-driven mechanism. We represent the family of systems on a set of three axes. One axis represents the shape of the colloidal pair of interest, in which we think of spheres as occupying the “zero” limit and rods infinity. A second axis is the softness of the sea particles; one limit represents hard-core particles (vanishing softness) and the other an interpenetrable ideal gas [for spherical sea particles, in the case that the sea particles may be considered depletants, this is the so-called penetrable hard sphere limit (66)]. A third axis is the inverse size of the sea particles, where a monodisperse system is at the origin, and the other limit has sea particles being very small compared with the “colloids.” One also might include other axes, such as the shape of the sea particles or the density of the system, but we omit them for simplicity.
As a guide, we have indicated the location of some well-studied model and experimental systems on these axes. Hard spheres sit at the origin (C) (22–27), hard rods (28) lie at the extreme of the shape axis (A), and Asakura and Oosawa’s model of depletants (29) lies in the plane of zero shape (between D and E). Some depletion systems are indicated (I, K, M); however, in experiments, particles are not all the same shape and sometimes are actually binary (J, L) or ternary (N) mixtures. Monodisperse hard particle systems lie along the shape axis (B, F) but are simply a limit of binary hard systems (G, H).
DEFs in Experimental Systems.
We have shown that DEFs arise from a mechanism that occurs in a wide variety of idealized systems. Do DEFs and shape entropy matter for experimental systems? Using systems in which shape entropy is the only factor that determines behavior, we have shown that in several example systems, DEFs are on the order of a few around the onset of crystallization. This puts DEFs directly in the relevant range for experimental systems. If in experiment there is sufficient control over the intrinsic forces between colloids that puts them at much less than the scale, systems will be shape entropy dominated. If the intrinsic forces are on the order of a , then there will be a competition between directional entropic forces and intrinsic forces. If the scale of the intrinsic forces is much more than a few , intrinsic forces will dominate shape entropy effects.
Can these forces be measured in the laboratory? In colloidal experiments, effective interaction potentials may be inferred from the trajectories of the colloids in situ, e.g., by confocal microscopy (93). Recently, it became possible to image anisotropic colloids in confocal microscopes and extract particle positions and orientations (94, 95). The determination of both particle positions and orientations makes it possible to directly extract the PMFTs computed here from experimental data. For isotropic potentials of mean force, this investigation already has been carried out by analyzing the pair correlation function (93). As in ref. 93, we may interpret this potential measurement as describing the thermally averaged time evolution of the relative position and orientation of pairs of particles. Furthermore, we have shown that pairs of particles are described by the PMFT regardless of the properties of the sea particles, so the PMFT describes the average time evolution of pairs of particles in monodisperse systems, as well as colloid-depletant systems.
Conclusions
In this paper, we show that shape entropy drives the phase behavior of systems of anisotropic shapes through directional entropic forces. We define DEFs in arbitrary systems and show that they are emergent and on the order of a few just below the onset of crystallization in example hard particle systems. By quantifying DEFs we put the effective forces arising from shape entropy on the same footing as intrinsic forces between particles that are important for self-assembly. In nano- and colloidal systems our results show that shape entropy plays a role in phase behavior when intrinsic forces between particles are on the order of a few or less. This figure guides the degree of control of intrinsic forces in particle synthesis required for controlling shape entropy effects in experiment. In microbiological systems, it facilitates the comparison between the effects of rigid shape in crowded environments and the elasticity of the constituents. We suggest how existing experimental techniques might be used to measure these forces directly in the laboratory. We show that the mechanism that generates DEFs is that maximizing shape entropy drives particles to adopt local dense packing configurations.
Finally, we demonstrate that shape entropy drives the emergence of DEFs, as particles adopt local dense packing configurations, in a wide class of soft matter systems. This shows that in a wide class of systems with entropy-driven phase behavior, entropy drives the phase behavior through the same mechanism (22–29).
Supplementary Material
Acknowledgments
We thank Daan Frenkel for helpful discussions concerning the early literature, Alexander Grosberg for helpful discussions concerning the interpretation of the PMFT and for comments on an earlier version of this work, and Ben Schultz for helpful suggestions. We are grateful to Robert Evans and Paul Chaikin for helpful feedback and encouragement. This material is based upon work supported by, or in part by, the US Army Research Office (ARO) under Grant Award W911NF-10-1-0518, the Department of Defense Assistant Secretary of Defense for Research and Engineering under Award N00244-09-1-0062, and the Department of Energy under Grant DE-FG02-02ER46000. The calculations for hard particle systems were supported by ARO. D.K. acknowledges funding by the FP7 Marie Curie Actions of the European Commission, Grant Agreement PIOF-GA-2011-302490 Actsa. The PMFT derivation and depletion calculations were supported by the Biomolecular Materials Program of the Materials Engineering and Science Division of Basic Energy Sciences at the US Department of Energy under Grant DE-FG02-02ER46000.
Footnotes
The authors declare no conflict of interest.
*We use “self-assembly” to apply to (thermodynamically) stable or metastable phases that arise from systems maximizing their entropy in the presence of energetic and volumetric constraints (temperature and pressure, i.e., spontaneous self-assembly) or other constraints (e.g., electromagnetic fields, i.e., directed self-assembly).
‡We use “emergent” to denote observed macroscopic behaviors that are not seen in isolated systems of a few constituents. At low pressures, hard particles behave like an ideal gas. Ordered phases arise only at reduced pressures of order one.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1418159111/-/DCSupplemental.
References
- 1.Cines DB, et al. Clot contraction: Compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood. 2014;123(10):1596–1603. doi: 10.1182/blood-2013-08-523860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Purves D, et al. Neuroscience. 5th Ed Sinauer; Sunderland, MA: 2011. [Google Scholar]
- 3.Madigan MT, Martinko JM, Stahl D, Clark DP. Brock Biology of Microorganisms. 13th Ed Benjamin Cummings; San Francisco: 2010. [Google Scholar]
- 4.Mannige RV, Brooks CL., 3rd Periodic table of virus capsids: Implications for natural selection and design. PLoS ONE. 2010;5(3):e9423. doi: 10.1371/journal.pone.0009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vernizzi G, Sknepnek R, Olvera de la Cruz M. Platonic and Archimedean geometries in multicomponent elastic membranes. Proc Natl Acad Sci USA. 2011;108(11):4292–4296. doi: 10.1073/pnas.1012872108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alberts B, et al. Molecular Biology of the Cell. 4th Ed Garland Science; New York: 2002. [Google Scholar]
- 7.Glotzer SC, Solomon MJ. Anisotropy of building blocks and their assembly into complex structures. Nat Mater. 2007;6(8):557–562. doi: 10.1038/nmat1949. [DOI] [PubMed] [Google Scholar]
- 8.Tao AR, Habas S, Yang P. Shape control of colloidal metal nanocrystals. Small. 2008;4(3):310–325. [Google Scholar]
- 9.Stein A, Li F, Wang Z. Synthesis of shaped particles and particle arrays by disassembly methods. J Mater Chem. 2009;19(15):2102–2106. [Google Scholar]
- 10.Sacanna S, Pine DJ. Shape-anisotropic colloids: Building blocks for complex assemblies. Curr Opin Colloid Interface Sci. 2011;16(2):96–105. [Google Scholar]
- 11.Cademartiri L, Bishop KJM, Snyder PW, Ozin GA. Using shape for self-assembly. Philos Trans R Soc A. 2012;370(1969):2824–2847. doi: 10.1098/rsta.2011.0254. [DOI] [PubMed] [Google Scholar]
- 12.Xia Y, Xia X, Wang Y, Xie S. Shape-controlled synthesis of metal nanocrystals. MRS Bull. 2013;38(4):335–344. [Google Scholar]
- 13.Senyuk B, et al. Topological colloids. Nature. 2013;493(7431):200–205. doi: 10.1038/nature11710. [DOI] [PubMed] [Google Scholar]
- 14.de Graaf J, van Roij R, Dijkstra M. Dense regular packings of irregular nonconvex particles. Phys Rev Lett. 2011;107(15):155501. doi: 10.1103/PhysRevLett.107.155501. [DOI] [PubMed] [Google Scholar]
- 15.Damasceno PF, Engel M, Glotzer SC. Crystalline assemblies and densest packings of a family of truncated tetrahedra and the role of directional entropic forces. ACS Nano. 2012;6(1):609–614. doi: 10.1021/nn204012y. [DOI] [PubMed] [Google Scholar]
- 16.Ni R, Gantapara AP, de Graaf J, van Roij R, Dijkstra M. Phase diagram of colloidal hard superballs: From cubes via spheres to octahedra. Soft Matter. 2012;8(34):8826–8834. [Google Scholar]
- 17.Damasceno PF, Engel M, Glotzer SC. Predictive self-assembly of polyhedra into complex structures. Science. 2012;337(6093):453–457. doi: 10.1126/science.1220869. [DOI] [PubMed] [Google Scholar]
- 18.LeRoux J. Settling velocity of ellipsoidal grains as related to shape entropy. Sediment Geol. 1996;101(1-2):15–20. [Google Scholar]
- 19.Ghochani M, et al. Tensile forces and shape entropy explain observed crista structure in mitochondria. Biophys J. 2010;99(10):3244–3254. doi: 10.1016/j.bpj.2010.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pineda-Urbina K, Guerrero RD, Reyes A, Gómez-Sandoval Z, Flores-Moreno R. Shape entropy’s response to molecular ionization. J Mol Model. 2013;19(4):1677–1683. doi: 10.1007/s00894-012-1725-4. [DOI] [PubMed] [Google Scholar]
- 21.Kamien RD. Entropic attraction and ordering. In: Gompper G, Schick M, editors. Soft Matter, Volume 3: Colloidal Order: Entropic and Surface Forces. Wiley-VCH; Weinheim, Germany: 2007. pp. 1–40. [Google Scholar]
- 22.Kirkwood JG. Molecular distribution in liquids. J Chem Phys. 1939;7(10):919–925. [Google Scholar]
- 23.Kirkwood JG, Monroe E. Statistical mechanics of fusion. J Chem Phys. 1941;9(7):514–526. [Google Scholar]
- 24.Kirkwood JG, Boggs EM. The radial distribution function in liquids. J Chem Phys. 1942;10(6):394–402. [Google Scholar]
- 25.Kirkwood JG, Maun EK, Alder BJ. Radial distribution functions and the equation of state of a fluid composed of rigid spherical molecules. J Chem Phys. 1950;18(8):1040–1047. [Google Scholar]
- 26.Alder BJ, Wainwright TE. Phase transition for a hard sphere system. J Chem Phys. 1957;27(5):1208–1209. [Google Scholar]
- 27.Wood WW, Jacobson JD. Preliminary results from a recalculation of the Monte Carlo equation of state of hard spheres. J Chem Phys. 1957;27(5):1207–1208. [Google Scholar]
- 28.Onsager L. The effects of shape on the interaction of colloidal particles. Ann N Y Acad Sci. 1949;51(4):627–659. [Google Scholar]
- 29.Asakura S, Oosawa F. Interaction between particles suspended in solutions of macromolecules. J Polym Sci. 1958;33(126):183–192. [Google Scholar]
- 30.Anderson PW. More is different. Science. 1972;177(4047):393–396. doi: 10.1126/science.177.4047.393. [DOI] [PubMed] [Google Scholar]
- 31.Frenkel D. Onsager’s spherocylinders revisited. J Phys Chem. 1987;91(19):4912–4916. [Google Scholar]
- 32.Stroobants A, Lekkerkerker HNW, Frenkel D. Evidence for one-, two-, and three-dimensional order in a system of hard parallel spherocylinders. Phys Rev A. 1987;36(6):2929–2945. doi: 10.1103/physreva.36.2929. [DOI] [PubMed] [Google Scholar]
- 33.Schilling T, Pronk S, Mulder B, Frenkel D. Monte Carlo study of hard pentagons. Phys Rev E. 2005;71(3):036138. doi: 10.1103/PhysRevE.71.036138. [DOI] [PubMed] [Google Scholar]
- 34.John BS, Juhlin C, Escobedo FA. Phase behavior of colloidal hard perfect tetragonal parallelepipeds. J Chem Phys. 2008;128(4):044909. doi: 10.1063/1.2819091. [DOI] [PubMed] [Google Scholar]
- 35.Triplett DA, Fichthorn KA. Monte Carlo simulation of two-dimensional hard rectangles: Confinement effects. Phys Rev E. 2008;77(1):011707. doi: 10.1103/PhysRevE.77.011707. [DOI] [PubMed] [Google Scholar]
- 36.Haji-Akbari A, et al. Disordered, quasicrystalline and crystalline phases of densely packed tetrahedra. Nature. 2009;462(7274):773–777. doi: 10.1038/nature08641. [DOI] [PubMed] [Google Scholar]
- 37.Haji-Akbari A, Engel M, Glotzer SC. Phase diagram of hard tetrahedra. J Chem Phys. 2011;135(19):194101. doi: 10.1063/1.3651370. [DOI] [PubMed] [Google Scholar]
- 38.Evers WH, et al. Entropy-driven formation of binary semiconductor-nanocrystal superlattices. Nano Lett. 2010;10(10):4235–4241. doi: 10.1021/nl102705p. [DOI] [PubMed] [Google Scholar]
- 39.Agarwal U, Escobedo FA. Mesophase behaviour of polyhedral particles. Nat Mater. 2011;10(3):230–235. doi: 10.1038/nmat2959. [DOI] [PubMed] [Google Scholar]
- 40.Zhao K, Bruinsma R, Mason TG. Entropic crystal-crystal transitions of Brownian squares. Proc Natl Acad Sci USA. 2011;108(7):2684–2687. doi: 10.1073/pnas.1014942108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haji-Akbari A, Engel M, Glotzer SC. Degenerate quasicrystal of hard triangular bipyramids. Phys Rev Lett. 2011;107(21):215702. doi: 10.1103/PhysRevLett.107.215702. [DOI] [PubMed] [Google Scholar]
- 42.Rossi L, et al. Cubic crystals from cubic colloids. Soft Matter. 2011;7(9):4139–4142. [Google Scholar]
- 43.Avendano C, Escobedo FA. Phase behavior of rounded hard-squares. Soft Matter. 2012;8(17):4675–4681. [Google Scholar]
- 44.Henzie J, Grünwald M, Widmer-Cooper A, Geissler PL, Yang P. Self-assembly of uniform polyhedral silver nanocrystals into densest packings and exotic superlattices. Nat Mater. 2012;11(2):131–137. doi: 10.1038/nmat3178. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, et al. Reversible Kirkwood-Alder transition observed in Pt3Cu2 nanoctahedron assemblies under controlled solvent annealing/drying conditions. J Am Chem Soc. 2012;134(34):14043–14049. doi: 10.1021/ja304108n. [DOI] [PubMed] [Google Scholar]
- 46.Agarwal U, Escobedo FA. Effect of quenched size polydispersity on the ordering transitions of hard polyhedral particles. J Chem Phys. 2012;137(2):024905. doi: 10.1063/1.4734021. [DOI] [PubMed] [Google Scholar]
- 47.Smallenburg F, Filion L, Marechal M, Dijkstra M. Vacancy-stabilized crystalline order in hard cubes. Proc Natl Acad Sci USA. 2012;109(44):17886–17890. doi: 10.1073/pnas.1211784109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marechal M, Patti A, Dennison M, Dijkstra M. Frustration of the isotropic-columnar phase transition of colloidal hard platelets by a transient cubatic phase. Phys Rev Lett. 2012;108(20):206101. doi: 10.1103/PhysRevLett.108.206101. [DOI] [PubMed] [Google Scholar]
- 49.Marechal M, Kortschot RJ, Demirörs AF, Imhof A, Dijkstra M. Phase behavior and structure of a new colloidal model system of bowl-shaped particles. Nano Lett. 2010;10(5):1907–1911. doi: 10.1021/nl100783g. [DOI] [PubMed] [Google Scholar]
- 50.Marechal M, Dijkstra M. Phase behavior and structure of colloidal bowl-shaped particles: Simulations. Phys Rev E. 2010;82(3):031405. doi: 10.1103/PhysRevE.82.031405. [DOI] [PubMed] [Google Scholar]
- 51.Marechal M, Löwen H. Density functional theory for hard polyhedra. Phys Rev Lett. 2013;110(13):137801. doi: 10.1103/PhysRevLett.110.137801. [DOI] [PubMed] [Google Scholar]
- 52.Gantapara AP, de Graaf J, van Roij R, Dijkstra M. Phase diagram and structural diversity of a family of truncated cubes: Degenerate close-packed structures and vacancy-rich states. Phys Rev Lett. 2013;111(1):015501. doi: 10.1103/PhysRevLett.111.015501. [DOI] [PubMed] [Google Scholar]
- 53.Haji-Akbari A, Chen ER, Engel M, Glotzer SC. Packing and self-assembly of truncated triangular bipyramids. Phys Rev E. 2013;88(1):012127. doi: 10.1103/PhysRevE.88.012127. [DOI] [PubMed] [Google Scholar]
- 54.Young KL, et al. A directional entropic force approach to assemble anisotropic nanoparticles into superlattices. Angew Chem Int Ed Engl. 2013;52(52):13980–13984. doi: 10.1002/anie.201306009. [DOI] [PubMed] [Google Scholar]
- 55.Young KL, et al. Assembly of reconfigurable one-dimensional colloidal superlattices due to a synergy of fundamental nanoscale forces. Proc Natl Acad Sci USA. 2012;109(7):2240–2245. doi: 10.1073/pnas.1119301109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye X, et al. Competition of shape and interaction patchiness for self-assembling nanoplates. Nat Chem. 2013;5(6):466–473. doi: 10.1038/nchem.1651. [DOI] [PubMed] [Google Scholar]
- 57.van Anders G, Ahmed NK, Smith R, Engel M, Glotzer SC. Entropically patchy particles: Engineering valence through shape entropy. ACS Nano. 2014;8(1):931–940. doi: 10.1021/nn4057353. [DOI] [PubMed] [Google Scholar]
- 58.Millan JA, Ortiz D, van Anders G, Glotzer SC. Self-assembly of Archimedean tilings with enthalpically and entropically patchy polygons. ACS Nano. 2014;8(3):2918–2928. doi: 10.1021/nn500147u. [DOI] [PubMed] [Google Scholar]
- 59.Boles MA, Talapin DV. Self-assembly of tetrahedral CdSe nanocrystals: Effective “patchiness” via anisotropic steric interaction. J Am Chem Soc. 2014;136(16):5868–5871. doi: 10.1021/ja501596z. [DOI] [PubMed] [Google Scholar]
- 60.Sanders JV. Close-packed structures of spheres of two different sizes I. Observations on natural opal. Philos Mag A. 1980;42(6):705–720. [Google Scholar]
- 61.Murray MJ, Sanders JV. Close-packed structures of spheres of two different sizes II. The packing densities of likely arrangements. Philos Mag A. 1980;42(6):721–740. [Google Scholar]
- 62.Bartlett P, Ottewill RH, Pusey PN. Superlattice formation in binary mixtures of hard-sphere colloids. Phys Rev Lett. 1992;68(25):3801–3804. doi: 10.1103/PhysRevLett.68.3801. [DOI] [PubMed] [Google Scholar]
- 63.Eldridge MD, Madden PA, Frenkel D. Entropy-driven formation of a superlattice in a hard-sphere binary mixture. Nature. 1993;365(6441):35–37. [Google Scholar]
- 64.Trizac E, Eldridge MD, Madden PA. Stability of the ab crystal for asymmetric binary hard sphere mixtures. Mol Phys. 1997;90(4):675–678. [Google Scholar]
- 65.Jiao Y, Stillinger FH, Torquato S. Optimal packings of superdisks and the role of symmetry. Phys Rev Lett. 2008;100(24):245504. doi: 10.1103/PhysRevLett.100.245504. [DOI] [PubMed] [Google Scholar]
- 66.Lekkerkerker HNW, Tuinier R. Colloids and the Depletion Interaction. Springer; Dordrecht, The Netherlands: 2011. [Google Scholar]
- 67.Mason TG. Osmotically driven shape-dependent colloidal separations. Phys Rev E. 2002;66(6 Pt 1):060402. doi: 10.1103/PhysRevE.66.060402. [DOI] [PubMed] [Google Scholar]
- 68.Zhao K, Mason TG. Directing colloidal self-assembly through roughness-controlled depletion attractions. Phys Rev Lett. 2007;99(26):268301. doi: 10.1103/PhysRevLett.99.268301. [DOI] [PubMed] [Google Scholar]
- 69.Zhao K, Mason TG. Suppressing and enhancing depletion attractions between surfaces roughened by asperities. Phys Rev Lett. 2008;101(14):148301. doi: 10.1103/PhysRevLett.101.148301. [DOI] [PubMed] [Google Scholar]
- 70.Badaire S, Cottin-Bizonne C, Woody JW, Yang A, Stroock AD. Shape selectivity in the assembly of lithographically designed colloidal particles. J Am Chem Soc. 2007;129(1):40–41. doi: 10.1021/ja067527h. [DOI] [PubMed] [Google Scholar]
- 71.Badaire S, Cottin-Bizonne C, Stroock AD. Experimental investigation of selective colloidal interactions controlled by shape, surface roughness, and steric layers. Langmuir. 2008;24(20):11451–11463. doi: 10.1021/la801718j. [DOI] [PubMed] [Google Scholar]
- 72.Yake AM, Snyder CE, Velegol D. Site-specific functionalization on individual colloids: Size control, stability, and multilayers. Langmuir. 2007;23(17):9069–9075. doi: 10.1021/la7011292. [DOI] [PubMed] [Google Scholar]
- 73.Snyder CE, Ong M, Velegol D. In-solution assembly of colloidal water. Soft Matter. 2009;5(11):1263–1268. [Google Scholar]
- 74.Kraft DJ, et al. Surface roughness directed self-assembly of patchy particles into colloidal micelles. Proc Natl Acad Sci USA. 2012;109(27):10787–10792. doi: 10.1073/pnas.1116820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roth R, van Roij R, Andrienko D, Mecke KR, Dietrich S. Entropic torque. Phys Rev Lett. 2002;89(8):088301. doi: 10.1103/PhysRevLett.89.088301. [DOI] [PubMed] [Google Scholar]
- 76.Odriozola G, Jiménez-Angeles F, Lozada-Cassou M. Entropy driven key-lock assembly. J Chem Phys. 2008;129(11):111101. doi: 10.1063/1.2981795. [DOI] [PubMed] [Google Scholar]
- 77.König P-M, Roth R, Dietrich S. Lock and key model system. Europhys Lett. 2008;84(6):68006. [Google Scholar]
- 78.Sacanna S, Irvine WTM, Chaikin PM, Pine DJ. Lock and key colloids. Nature. 2010;464(7288):575–578. doi: 10.1038/nature08906. [DOI] [PubMed] [Google Scholar]
- 79.Odriozola G, Lozada-Cassou M. Statistical mechanics approach to lock-key supramolecular chemistry interactions. Phys Rev Lett. 2013;110(10):105701. doi: 10.1103/PhysRevLett.110.105701. [DOI] [PubMed] [Google Scholar]
- 80.Triplett DA, Fichthorn KA. Entropic forces and directed alignment of hard squares in suspensions of rods and disks. J Chem Phys. 2010;133(14):144910. doi: 10.1063/1.3503975. [DOI] [PubMed] [Google Scholar]
- 81.Barry E, Dogic Z. Entropy driven self-assembly of nonamphiphilic colloidal membranes. Proc Natl Acad Sci USA. 2010;107(23):10348–10353. doi: 10.1073/pnas.1000406107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gilbert E, Johnson D, Keerthi S. A fast procedure for computing the distance between complex objects in three-dimensional space. IEEE J Robot Autom. 1988;4(2):193–203. [Google Scholar]
- 83.de Boer J. Molecular distribution and equation of state of gases. Rep Prog Phys. 1949;12(1):305–374. [Google Scholar]
- 84.Onsager L. Theories of concentrated electrolytes. Chem Rev. 1933;13(1):73–89. [Google Scholar]
- 85.Cole GHA. Pair distribution of simple non-spherical liquids. Proc Phys Soc. 1960;75(1):77–81. [Google Scholar]
- 86.Croxton C, Osborn T. The development of torque fields at the surface of molecular fluids. Phys Lett A. 1975;55(7):415–416. [Google Scholar]
- 87.Dijkstra M, van Roij R, Evans R. Phase diagram of highly asymmetric binary hard-sphere mixtures. Phys Rev E. 1999;59(5):5744–5771. doi: 10.1103/physreve.59.5744. [DOI] [PubMed] [Google Scholar]
- 88.Louis AA. Beware of density dependent pair potentials. J Phys Condens Matter. 2002;14:9187. [Google Scholar]
- 89.Malescio G, Pellicane G. Stripe phases from isotropic repulsive interactions. Nat Mater. 2003;2(2):97–100. doi: 10.1038/nmat820. [DOI] [PubMed] [Google Scholar]
- 90.Zhang Z, Glotzer SC. Self-assembly of patchy particles. Nano Lett. 2004;4(8):1407–1413. doi: 10.1021/nl0493500. [DOI] [PubMed] [Google Scholar]
- 91.Chen ER, Klotsa D, Engel M, Damasceno PF, Glotzer SC. Complexity in surfaces of densest packings for families of polyhedra. Phys Rev X. 2014;4(1):011024. [Google Scholar]
- 92.Baumgartl J, Dullens RPA, Dijkstra M, Roth R, Bechinger C. Experimental observation of structural crossover in binary mixtures of colloidal hard spheres. Phys Rev Lett. 2007;98(19):198303. doi: 10.1103/PhysRevLett.98.198303. [DOI] [PubMed] [Google Scholar]
- 93.Iacovella CR, Rogers RE, Glotzer SC, Solomon MJ. Pair interaction potentials of colloids by extrapolation of confocal microscopy measurements of collective suspension structure. J Chem Phys. 2010;133(16):164903. doi: 10.1063/1.3498746. [DOI] [PubMed] [Google Scholar]
- 94.Mohraz A, Solomon MJ. Direct visualization of colloidal rod assembly by confocal microscopy. Langmuir. 2005;21(12):5298–5306. doi: 10.1021/la046908a. [DOI] [PubMed] [Google Scholar]
- 95.Shah AA, Schultz B, Kohlstedt KL, Glotzer SC, Solomon MJ. Synthesis, assembly, and image analysis of spheroidal patchy particles. Langmuir. 2013;29(15):4688–4696. doi: 10.1021/la400317t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.