

Abstract
The intestinal epithelium is equipped with sensing receptor mechanisms that interact with luminal microorganisms and nutrients to regulate barrier function and gut immune responses, thereby maintaining intestinal homeostasis. Herein, we clarify the role of the extracellular calcium-sensing receptor (CaSR) using intestinal epithelium-specific Casr−/− mice. Epithelial CaSR deficiency diminished intestinal barrier function, altered microbiota composition, and skewed immune responses towards proinflammatory. Consequently, Casr−/− mice were significantly more prone to chemically induced intestinal inflammation resulting in colitis. Accordingly, CaSR represents a potential therapeutic target for autoinflammatory disorders, including inflammatory bowel diseases.
Keywords: Inflammation, calcium-sensing receptor, epithelial cells, gut microbiota, intestinal barrier function, colitis
Introduction
The single layer of epithelium composing the intestinal mucosa acts as a barrier to impede the passage of toxins, pathogens, and foreign antigens, while selectively allowing the transport of electrolytes, essential nutrients, and water from the intestinal lumen into peripheral circulation (1). Molecules pass through the intestinal epithelial monolayer by two routes: transcellular, through the apical and basolateral membranes of a cell; and paracellular, via the intercellular space between adjacent cells (2). It is the function of apical junctional complexes to seal off the paracellular pathway of transport. The tight junction (TJ) and the adherens junction (AJ) comprise the apical junctional complex. In addition, desmosomes, located basolaterally beneath the AJ, strengthen the bond between adjacent epithelial cells by facilitating cellular proximity and TJ assembly (3). These epithelial barriers are not absolutely impermeable, and intestinal epithelia have TJs with lower resistance than do other types of epithelia, such as that of the gallbladder, which compartmentalize irritating bile acids from the rest of the abdominal cavity (3). Unfortunately, these low resistance TJs are prone to leakage, and emerging data suggest that the pathogenesis of inflammatory bowel disease (IBD) is related to three fundamental and self-perpetuating circumstances: compromised intestinal barrier function; exposure of intestinal luminal contents to leukocytes in the lamina propria; and uncontrolled inflammatory immune responses (1).
Importantly, it has recently been shown that the gut epithelium serves as a “communicator” between the luminal flora and the subepithelial immune system comprised of innate and adaptive immune components, including dendritic cells (DCs) and lymphocytes (4). The epithelium expresses an array of pattern recognition receptors (PRRs), and disruption of these epithelium-derived bacteria-sensing/modulating mechanisms can result in uncontrolled immune responses (5–9). Resident gut commensal bacteria can also shape local mucosal and systemic immunity by providing critical signals that maintain gut homeostasis (10). Previous studies have demonstrated that nutrients can modulate the composition of the gut microbiota, epithelial cell function, and host immunity (11–19). For nutrient-dependent signaling, the gut epithelium is equipped with various nutrient-sensing mechanisms; however, their importance in gut bacteria-sensing, epithelial cell function, and immune homeostasis remains largely unknown.
One such nutrient-sensing receptor is the extracellular calcium-sensing receptor (CaSR) (20). CaSR is a G protein-coupled nutrient-sensing receptor that is widely expressed in a range of tissues and species (21, 22) to regulate calcium homeostasis (20) and osmotic balance (22–24). Epithelial cells along the entire gastrointestinal (GI) tract express the CaSR (25–30). Although previous investigations have studied the role of GI CaSR in fluid transport, intestinal epithelial differentiation, growth, and nutrient sensing (30, 31), local and systemic consequences of alterations in CaSR signaling have not been detailed.
Here, we investigated additional roles of the CaSR in the intestinal mucosae using intestinal epithelium-specific receptor knockout mice (Casr−/−). We show that CaSR plays a key role in the maintenance of intestinal barrier function, gut microbiome composition, and in the control of intestinal and systemic immune responses.
Materials and Methods
Mice
C57BL/6 mice lacking Casr expression in intestinal epithelial cells (Casr−/−) and their Casr+/+ littermates were bred and maintained in-house at the University of Florida Communicore Animal Facility. Casr−/− mice were generated as previously described (31). Briefly, Casrflox/flox mice were bred with transgenic mice expressing Cre Recombinase under the control of the villin 1 promoter and genotyped prior to all experiments after a minimum of two generations. Mice were used at 5–10 weeks of age in accordance with the Animal Welfare Act and the Public Health Policy on Humane Care. All procedures were approved by the Institutional Animal Case and Use Committee (IACUC) at the University of Florida.
Ex vivo transepithelial electrical resistance (TEER), Short-Circuit Current (ISC), Transepithelial Conductance (GT), and Permeability Measurements of Intestinal Tissues
Differences in electrogenic ion transport in the colons of Casr+/+ and Casr−/− mice were quantified by measuring the short circuit current responses of freshly isolated colonic tissues mounted in modified Ussing chambers (Physiologic Instruments, San Diego, CA), as previously described (32, 33). Intestinal permeability was assessed using 4 kDa fluorescein isothiocyanate dextran, as described previously (34).
Lamina Propria Leukocyte (LPL) Preparation
Colonic lamina propria (LP) cells were isolated as previously described (35), with minor modifications. Digestion buffer consisted of DMEM (GIBCO®, Life Technologies) containing 0.25 mg/mL collagenase type VII (Sigma-Aldrich), 0.125 U/mL Liberase TM Research Grade (Roche Applied Science, Indianapolis, IN), 10 mM HEPES, 0.1M CaCl2 (Sigma-Aldrich), and 5% FBS (3 × 10 min digestions). Cells obtained from the digestions were combined and stained for flow cytometry-based analyses.
Flow Cytometry and Antibodies
Single cell suspensions obtained from processed spleens and MLNs, and LP lymphocytes were stained with LIVE/DEAD Aqua Dead Cell Stain Kit® (Molecular Probes®, Life Technologies). Mouse Fc Blocking Reagent (Miltenyi Biotec, Auburn, CA) was used prior to staining with combinations of the following antibodies or their corresponding isotype controls from eBioscience (San Diego, CA), Biolegend (San Diego, CA), BD Pharmingen, or R&D Systems (Minneapolis, MN): CD45-(30-F11)-eFluor650NC, CD11c-(N418)-BV605, CD11b-(M1/70)-PE-Cy7, CD11b-(M1/70)-APC-Cy7, F4/80-(BM8)-PB, GR1-(RB6-8C5)-APC-Cy7, I-A/I-E MHCII-(2G9)-FITC, I-A/I-E MHCII-(2G9)-PE, CD3-(145-2C11)-APC-Cy7, CD4-(RM4-5)-BV605, CD8-(53-607)-PE-Cy7, PD-1-(29F.1A12)-BV421, Pro-IL-1β-(NJTEN3)-APC, TNFα-(MP6-XT22)-PerCP-eFluor710, IL-6-(MP5-20F3)-FITC, IFNγ-(XMG1.2)-PerCP-Cy5.5, IL-17A-(TC11.18H10.1)-PE, FoxP3-(FJK-16A)-PE, IL-1R-(JAMA-147)-PE. Prior to intracellular staining, cells were fixed and permeabilized with BD Cytofix/Cytoperm™ (BD Biosciences). A BD LSRFortessa™ (BD Biosciences) cell analyzer was used to acquire stained, fixed cells. Data were analyzed with FlowJo software (Tree Star, Ashland, OR).
Real-Time PCR
For global gene expression changes, RNA was isolated from the tissues specified with Aurum™ Total RNA Kit (Bio-Rad). iScript™Select cDNA Synthesis Kit (Bio-Rad) was used for reverse transcription and the obtained cDNA used for quantitative PCR by SYBR® Green Dye gene expression assay on a Bio-Rad CFX96™ Real time system; n=8/group. The reaction was carried out in 10μl final volume with an initial denaturation at 95°C, followed by temperature cycling of 95°C 30s, 60°C 30s and 72°C 30s; 40 cycles were totally performed. For microbiota composition, ZR Fecal DNA MiniPrep™ Kit (Zymo Research, Irvine, CA) was used to extract total fecal DNA per the manufacturer’s instructions. Real-time PCR analysis was performed on 2 ng of total DNA template (SsoAdvanced™ SYBR® Green Supermix, Bio-Rad) to target variable regions of bacteria group-specific 16S rRNA sequences (36); n=8/group. Groups were normalized to the housekeeper Eubacteria group to determine the relative abundance. Primers used can be found in Table S1.
16S ribosomal DNA Sequencing
Fecal DNA was amplified by Illumina Miseq compatible primers, targeting the 16S rDNA V4–V5 region for microbiome analyses. Amplicons were purified by QIAquick Gel extraction kit (Qiagene, Madison, WI) and quantified by Qubit® 2.0 Fluorometer (Invitrogen, Grand Island, NY) and Kapa SYBR fast qPCR kit (Kapa Biosystems, Inc., Woburn, MA). Equal amounts of amplicons were pooled with 10% of Phix control. Miseq v2 reagent kit (Illumina, Inc., San Diego, CA) was used to run the pooled samples on the Illumina Miseq. Data were analyzed as previously described (37).
Dextran Sulfate Sodium (DSS)-Induced Colitis
Casr+/+ and Casr−/− mice received 3% DSS in the drinking water for 7 days to induce colitis. Disease progression, including weight loss and diarrhea, was monitored throughout the study. Body condition scoring (BCS) of the mice, as determined by a veterinarian, was used as criteria for early termination of the experiment. Despite severe colitis, none of the mice reached a BCS requiring euthanasia. Mice were sacrificed at day 13 and the colons isolated for analyses. Tissues were fixed, sectioned, and stained with hematoxylin and eosin (H&E) by Histology Tech Services (Gainesville, FL). Sections were analyzed/scored blindly by a boarded veterinary pathologist (JLO). Colitis was graded based on 7 parameters (0–17). Namely, degree of inflammation in the LP (0–3), goblet cell loss (0–2), abnormal crypts (0–3), crypt abscess (0–1), mucosal erosion and ulceration (0–1), submucosal spread to transmural involvement (0–3), and neutrophil numbers at 40x magnification (0–4). Stool consistency was scored as follows: 0=normal, 1=pasty, 2=watery, 3=watery with perianal staining.
Statistical Analyses
Unless stated otherwise, representative data indicate mean ± SEM. Significance was determined by two-tailed unpaired t tests for two group comparisons (GraphPad Prism 6 for Mac OS X, La Jolla, CA).
Results
Intestinal CaSR contributes to intestinal barrier function integrity
The evaluation of intestinal permeability ex vivo using Ussing chambers revealed that Casr−/− mice showed reduced transepithelial resistance (TEER), and higher transepithelial conductance (GT) and passive transport of FITC-conjugated dextran (Figure 1A–C). Interestingly, significant alterations in short-circuit current (ISC) and ion transporter transcript measurements, indicative of defective transcellular transport, were not observed between the two groups (Figure 1D), suggesting that the CaSR exclusively affects the paracellular transport pathway. Apical junctional complexes, such as TJs, primarily maintain the epithelial barrier; TJs involve complex interactions between approximately 40 proteins, including the transmembrane proteins, occludins and claudins. These proteins are anchored to the actin filaments and myosin light chain through the zonula occludens (ZO) family. Consistent with a defective intestinal epithelial barrier, Casr−/− mice had a decreased colonic expression of TJ molecules, particularly occludin-2, a major component of TJs (Figure 1E). We then investigated a range of ion transporter-associated genes and found that Casr−/− mice also exhibited a significantly increased expression of myosin light-chain kinase-1, an enzyme that controls contractility of the perijunctional actomyosin rings and epithelial permeability (Figure 1F).
Figure 1. (2 columns). Gastrointestinal epithelial barrier dysfunction in Casr−/− mice.
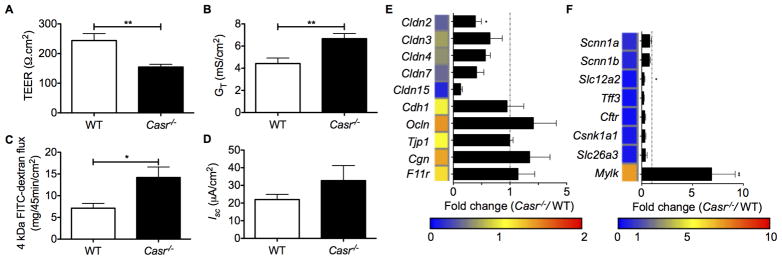
Intestinal barrier integrity (A–D) and gene expression measured by Real-Time PCR (E, F) of steady-state Casr−/− mice and wild-type controls; n=10 mice/group. Heat maps were developed from the mean fold change in expression (Casr−/−/WT) calculated. TEER (A), transepithelial conductance (B), passive transport of FITC-conjugated dextran (C), and short-circuit current (D) of colons measured ex vivo. Tight junction- and ion transporter-associated gene expression of isolated colon tissues measured by Real-Time PCR (E, F). Data are shown as mean +/− SEM. *P<0.05, **P<0.01 compared with controls.
Defect in epithelial CaSR signaling leads to gut microbe imbalance
A mutual interaction exists between the gut microbiota and the epithelial cells comprising the intestinal barrier, and both populations can influence the other. To assess if the breakdown of intestinal epithelial integrity in Casr−/− mice alters the distribution of microbiota between either side of the epithelial barrier, we used a combination of real-time PCR and Illumina Miseq to analyze the microbiota of steady-state Casr−/− and wild-type mice. No significant differences were observed between the overall richness and diversity of the two gut microbial communities (Figure 2A, B). However, deeper analyses indicated significant changes in composition. For instance, at the phylum level, we noted an outgrowth in the minor group Deferribacteraceae (Figure 2C), which was previously found to correlate with inflammatory responses in the colons of Citrobacter rodentium-infected mice (38), a model of bacterial colitis. Concurrently, beneficial lactobacilli were decreased in Casr−/− mice (Figure 2D). Moreover, the relative abundance and distribution of the Gram-positive Clostridium coccoides was significantly altered in Casr−/− mice, with depletion noted in the lumen and enrichment in the subepithelium (Figure 2E). Consistent with enhanced bacteria translocation and dissemination in host tissues, Casr−/− mice had significantly decreased epithelial expression of Reg3b and Reg3g, which encode secreted C-type lectins that bind and protect against translocation and dissemination of Gram-negative (39) and Gram-positive bacteria (40, 41), respectively (Figure 3B).
Figure 2. (2 columns). Gut microbiota composition of Casr−/− mice.

Microbiota composition alterations due to CaSR deficiency in healthy mice; n=10 control mice and n=20 Casr−/− mice. A. Microbial evenness, diversity, and species richness in mice tested. Left: The species evenness index was calculated using the formula J′= H′/H′max, where H′ is the Shannon diversity index and H′max is the maximal value of H′. Middle: The Shannon diversity index was used to estimate microbial diversity for each group. Right: The Chao richness index was used as a measure of species richness. B. Unweighted UniFrac analyses were used to calculate distances between samples obtained from the two groups and three-dimensional scatterplots were generated by using principal coordinate analysis (PCoA). C. Bacteria genera most enriched or depleted in Casr−/− mice, as measured by linear discriminant analysis (LDA). D, E. Changes in abundance of specific bacteria and potential dissemination were measured by Real-Time PCR. Data are shown as mean +/− SEM. *P<0.05, **P<0.01 compared with control mice.
Figure 3. (1.5 columns). Increased inflammation in the colons of Casr−/− mice.

Gene expression profile of the distal colons of Casr−/− and Casr+/+ littermate controls; n=10 control mice and n=20 Casr−/− mice. Data are shown as mean +/− SEM. *P<0.05, **P<0.01, ***P<0.001 compared with WT. Heat maps were developed from the mean fold change in expression (Casr−/−/WT) calculated.
Enhanced intestinal inflammation and immune cell activation in Casr−/− mice
Dysbiosis of the intestinal microbiota may lead to pathogenic inflammatory immune responses locally and systemically. Indeed, gene array analyses of the distal colon of wild-type and Casr−/− mice demonstrated a marked increase in the expression of a range of PRRs and cytokine-encoding genes in the colons of Casr−/− mice (Figure 3). To distinguish between proinflammatory responses in the intestinal epithelium due to attenuated CaSR signaling versus responses induced in resident immune cells, we examined colonic leukocytes by flow cytometry. Data show that colonic CD11b+ DCs upregulated their costimulatory molecules and proIL-1β and its receptor in Casr−/− mice (Figure 4A, 4B). As further evidence of chronic intestinal inflammation in Casr−/− mice, higher IL1R and programmed cell death (PD)1 were significantly expressed on CD4+ and CD8+ T cells (Figure 5A, 5B).
Figure 4. (1.5 columns). Activation of proinflammatory innate immune responses in Casr−/− mice.

A. Cell surface expression of CD40, CD80, CD86, and B7-H1 in CD45+MHCIIhiCD11c+F4/80−CD11b+ colonic DCs was analyzed by flow cytometry. Gray tinted line = isotype control; blue line = WT; red line = Casr−/− mice. B. Production of the proinflammatory cytokine IL-1β and expression of its receptor in colonic DCs. Data represent observations from two independent experiments and are shown as mean +/− SEM. *P<0.05, **P<0.01, ***P<0.001 compared with WT. n=5 mice/group.
Figure 5. (1.5 columns). Colonic T cell phenotype in Casr−/− mice.

Cell surface expression of IL1R (A) and PD-1 (B) on CD4+ and CD8+ T cells was analyzed by flow cytometry. Data represent observations from two independent experiments and are shown as mean +/− SEM. *P<0.05 compared to WT. n=5 mice/group.
We then investigated differences in systemic immune responses in these mice. We found a selective expansion of CD11b+ DCs in the mesenteric lymph nodes (MLNs) and spleens of Casr−/− mice (Figure S1A&B). Moreover, higher numbers of Gr-1+CD11b+ neutrophils were noted in the spleens of Casr−/− mice (Figure S1C). Similar to the colon, splenic CD11b+ DCs were significantly activated, as evident by proIL-1β production (Figure S2A&B). In addition, IL-10+ was significantly reduced in splenic DCs of Casr−/− mice (Figure S2B). DCs isolated from the MLNs of Casr−/− mice yielded similar trends, but no statistically significant differences (Figure S3). Also, as in the colon, CaSR deficiency resulted in IL1R and PD1 upregulation in splenic and mesenteric CD4+ T cells (Figure S4).
Intestinal IL1R signaling in CD4+ T cells has previously been found to promote Th17 responses (42). We found that IL-17A+ as well as IFNγ+ CD4+ T cells were increased in the MLNs of Casr−/− mice (Figure S5). Notably, despite the chronic intestinal inflammation observed in intestinal epithelium-specific Casr−/− mice, the frequency of FoxP3+ regulatory T cells (Tregs) was not decreased in these mice (Figure S6); in fact, the number of Tregs was significantly increased in the MLNs of Casr−/− mice (Figure S6B), perhaps as a compensatory mechanism to counteract the preexisting inflammation.
Casr−/− mice are more susceptible to DSS-induced colitis
Considering the notion that intestinal barrier dysfunction and imbalanced microbiota may contribute to enhanced susceptibility of Casr−/− mice to autoinflammatory diseases (e.g., IBD), the DSS acute induced colitis model was employed (43). DSS promotes mucosal destruction independent of the intestinal microbiota, but the intestinal microbiota is thought to modify responsiveness and susceptibility to this chemical (44, 45). Casr−/− mice demonstrated more severe colitis with delayed recovery compared to their littermate counterparts, as demonstrated by weight changes and stool consistency at the endpoint of the experiment (Figure 6A, B). Consistently, wild-type mice were able to recover from colitis, as shown by the gross morphology and histopathology of the colons at day 13 post-DSS (Figure 6C, D). In contrast, Casr−/− mice still displayed severe colonic inflammation and mucosal damage (Figure 6C, D), indicating that the intestinal CaSR plays a key role in the regulation of induced-inflammation.
Figure 6. (1.5 columns). Casr−/− mice are highly susceptible to DSS-induced colitis.

Casr−/− and Casr+/+ littermate controls were given 3% DSS in the drinking water for 7 days. Disease progression was scored by weight loss (A), diarrhea (B), gross inflammation of the colon (C), and histopathology (D). Data are shown as mean +/− SEM. *P<0.05, **P<0.01 compared to WT. n=5 mice/group. 20x magnification.
Discussion
We demonstrate that CaSR is a key molecule expressed in gut epithelial cells that contributes to the preservation of intestinal epithelial cell integrity, and maintenance of immune homeostasis in the gut, the disruption of which results in intestinal inflammation. Recently, it was shown that “global” Casr−/parathyroid hormone (Pth)− double knockout mice had chronic intestinal inflammation, with increased neutrophil infiltration, myeloperoxidase activity, TNFR1 expression (46), and susceptibility to DSS (47). However, given the global nature and multiple factors involved in that study, it was difficult to ascertain the cause of the inflammation. Nonetheless, the study suggested that intestinal CaSR may be anti-inflammatory. Indeed, activation of CaSR by its dietary agonists, calcium, spermine, or tryptophan, reduced inflammation (Tang & Cheng, unpublished observations), whereas inhibition of the receptor by depletion of dietary calcium enhanced gut inflammation in animal models of induced colitis (48). Interestingly, while gut-specific CaSR detects nutrients and is potentially anti-inflammatory, the CaSR in murine bone marrow-derived macrophages/monocytes is pro-inflammatory in that it detects the “danger signal” (i.e., Ca released as a result of tissue injury) and activates the NLRP3 inflammasome (49). Thus, role of CaSR in inflammation appears to be cell-type specific.
Nutrient availability and nutrient-sensing in the host significantly contribute to gut homeostasis and the immune responses induced (48, 50–61). In the present study, we demonstrate that a single deficiency in epithelial CaSR altered the composition of the gut microbiota, with Casr−/− mice having significantly increased Deferribacteraceae and reduced abundance of lactobacilli and C. coccoides (cluster XIVa of the genus Clostridium), despite identical environmental conditions. Clostridium cluster XIVa species are beneficial commensal bacteria that induce butyrate production, Treg development, maintenance of barrier function, and competition with pathogens in gut colonization. The specific depletion of commensal Clostridia, together with the outgrowth of small groups such as Deferribacteraceae, may thus contribute to the disruption of mucosal homeostasis and subsequent activation of local immune responses seen in Casr−/− mice. Indeed, depletion of Clostridia has been consistently associated with chronic inflammation in patients with IBD (62–65) and atopy (66), whereas Deferribacteraceae was shown to correlate with the level of inflammatory responses in C. rodentium-infected mice (38). Whether these changes are a cause or a consequence of inflammation, or consequent to the downregulation of the anti-inflammatory CaSR, requires further investigation.
The immune responses observed in Casr−/− mice suggest that intestinal epithelial CaSR deficiency leads to a shift in local and systemic innate and T cell immune responses from a status characterized by regulation to one that is highly stimulated. However, since the expression of PD1, a molecular signature of T cell exhaustion (67), was significantly enhanced on T lymphocytes, the activated immune phenotype displayed in the immune cells of Casr−/− mice may not necessarily indicate active, effective immunity, but evidence of chronic intestinal inflammation. In further support of chronic inflammatory responses, Th1 and Th17 responses were elevated in Casr−/− mice. Interestingly, FoxP3+ Tregs were not decreased, but were increased in Casr−/− mice, particularly in the MLNs. Nonetheless, the quality of these Tregs warrants further investigation, as studies have highlighted the plasticity of these cells toward a proinflammatory phenotype (68). Therefore, considering the heightened state of inflammation in Casr−/− mice, it is conceivable that Casr−/− mice-derived FoxP3+ Tregs may be functionally defective. This state of chronic inflammation rendered Casr−/− mice highly susceptible to DSS-induced colitis, indicating that, in addition to defective intestinal barrier function and increased inflammation, CaSR deficiency may also result in impaired wound healing in the gut.
In summary, the present study demonstrates that attenuated CaSR signaling in the gut epithelium leads to enhanced permeability of the epithelial barrier, resulting in translocation and dissemination of luminal bacteria and activation of local and systemic innate and adaptive proinflammatory immune responses. The subsequent excessive gut inflammation disrupts the intestinal milieu and impairs gut homeostasis, thereby contributing to susceptibility to autoinflammatory diseases, such as IBD. Thus, the CaSR may serve as a potential therapeutic target for a range of autoinflammatory intestinal disorders, including IBD and colon cancer.
Supplementary Material
Acknowledgments
Gut-specific Casr−/− mice were kindly provided by Dr. Wenhan Chang at Endocrine Research, VA Medical Center, University of California at San Francisco.
This work was supported in part by the National Institute of Allergy and Infectious Diseases RO1 AI093370 (MM), the Department of Defense CA111002 (MM), the Ocala Royal Dames for Cancer Research (MM), and the Gatorade Foundation (MM); and also by CDNHF/NASPGHAN award 00102979 (SXC), NICHD K08HD079674 (SXC), and The Children’s Miracle Network (SXC). SXC is a recipient of the year 2012–2013 NASPGHAN Foundation’s “Fellow to Faculty Transition Award in Inflammatory Bowel Disease”.
We would also like to extend our gratitude to Mr. Eric Li, Dr. Liangli Jiang, Ms. Ashley Mila, and Ms. Minzhi Peng for technical support.
Abbreviations
- CaSR
calcium-sensing receptor
- IBD
inflammatory bowel disease
- DCs
dendritic cells
- TJ
tight junction
- AJ
adherens junction
- PRRs
pattern recognition receptors
- GI
gastrointestinal
- DSS
dextran sodium sulfate
- TEER
transepithelial electrical resistance
- ISC
short-circuit current
- GT
trans-epithelial conductance
- LP
lamina propria
- Tregs
regulatory T cells
- PD1
programmed cell death 1
- PTH
parathyroid hormone
- TNFR1
tumor necrosis factor receptor 1
Footnotes
The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. 2009;124(1):3–20. doi: 10.1016/j.jaci.2009.05.038. quiz 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gunzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev. 2013;93(2):525–69. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marchiando AM, Graham WV, Turner JR. Epithelial barriers in homeostasis and disease. Annu Rev Pathol. 2010;5:119–44. doi: 10.1146/annurev.pathol.4.110807.092135. [DOI] [PubMed] [Google Scholar]
- 4.Pott J, Hornef M. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep. 2012;13(8):684–98. doi: 10.1038/embor.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greten FR, Eckmann L, Geten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 6.Zaph C, Troy AE, Taylor BC, Berman-Booty LD, Guild KJ, Du Y, Yost EA, Gruber AD, May MJ, Greten FR, et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;447(7135):552–6. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]
- 7.Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, Huth M, Nikolaev A, Neufert C, Madison B, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446(7135):557–61. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 8.Bashir ME, Louie S, Shi HN, Nagler-Anderson C. Toll-like receptor 4 signaling by intestinal microbes influences susceptibility to food allergy. J Immunol. 2004;172(11):6978–87. doi: 10.4049/jimmunol.172.11.6978. [DOI] [PubMed] [Google Scholar]
- 9.Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nature Rev Immunol. 2008;8:411–20. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- 10.Chervonsky AV. Intestinal commensals: influence on immune system and tolerance to pathogens. Curr Opin Immunol. 2012;24(3):255–60. doi: 10.1016/j.coi.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Borrelli O, Cordischi L, Cirulli M, Paganelli M, Labalestra V, Uccini S, Russo P, Cucchiara S. Polymeric diet alone versus corticosteroids in the treatment of active pediatric Crohn’s disease: a randomized controlled open-label trial. Clinical gastroenterology and hepatology. 2006;4(6):744–53. doi: 10.1016/j.cgh.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Dziechciarz P, Horvath A, Shamir R, Szajewska H. Meta-analysis: enteral nutrition in active Crohn’s disease in children. Alimentary pharmacology & therapeutics. 2007;26(6):795–806. doi: 10.1111/j.1365-2036.2007.03431.x. [DOI] [PubMed] [Google Scholar]
- 13.Grafors JM, Casswall TH. Exclusive enteral nutrition in the treatment of children with Crohn’s disease in Sweden: a questionnaire survey. Acta Paediatr. 2011;100(7):1018–22. doi: 10.1111/j.1651-2227.2011.02178.x. [DOI] [PubMed] [Google Scholar]
- 14.Heuschkel RB, Menache CC, Megerian JT, Baird AE. Enteral nutrition and corticosteroids in the treatment of acute Crohn’s disease in children. Journal of pediatric gastroenterology and nutrition. 2000;31(1):8–15. doi: 10.1097/00005176-200007000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Levine A, Milo T, Buller H, Markowitz J. Consensus and controversy in the management of pediatric Crohn disease: an international survey. Journal of pediatric gastroenterology and nutrition. 2003;36(4):464–9. doi: 10.1097/00005176-200304000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Lochs H, Dejong C, Hammarqvist F, Hebuterne X, Leon Sanz M, Schtz T, van Gemert W, van Gossum A, Valentini L, Lbke H, et al. ESPEN Guidelines on Enteral Nutrition: Gastroenterology. Clinical nutrition. 2006;25(2):260–74. doi: 10.1016/j.clnu.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 17.Ludvigsson JF, Krantz M, Bodin L, Stenhammar L, Lindquist B. Elemental versus polymeric enteral nutrition in paediatric Crohn’s disease: a multicentre randomized controlled trial. Acta paediatrica. 2004;93(3):327–35. [PubMed] [Google Scholar]
- 18.Zachos M, Tondeur M, Griffiths AM. Enteral nutritional therapy for induction of remission in Crohn’s disease. Cochrane Database of Systematic Reviews. 2007:CD000542. doi: 10.1002/14651858.CD000542.pub2. [DOI] [PubMed] [Google Scholar]
- 19.McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM. Mechanisms of obesity-induced inflammation and insulin resistance: insights into the emerging role of nutritional strategies. Front Endocrinol. 2013;4(52) doi: 10.3389/fendo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366(6455):575–80. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- 21.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81(1):239–97. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- 22.Nearing J, Betka M, Quinn S, Hentschel H, Elger M, Baum M, Bai M, Chattopadyhay N, Brown EM, Hebert SC, et al. Polyvalent cation receptor proteins (CaRs) are salinity sensors in fish. Proc Natl Acad Sci U S A. 2002;99(14):9231–6. doi: 10.1073/pnas.152294399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riccardi D, Park J, Lee WS, Gamba G, Brown EM, Hebert SC. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc Natl Acad Sci U S A. 1995;92(1):131–5. doi: 10.1073/pnas.92.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sands JM, Naruse M, Baum M, Jo I, Hebert SC, Brown EM, Harris HW. Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J Clin Invest. 1997;99:1399–405. doi: 10.1172/JCI119299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chattopadhyay N, Cheng I, Rogers K, Riccardi D, Hall A, Diaz R, Hebert SC, Soybel DI, Brown EM. Identification and localization of extracellular Ca(2+)-sensing receptor in rat intestine. Am J Physiol. 1998;274(1 Pt 1):G122–30. doi: 10.1152/ajpgi.1998.274.1.G122. [DOI] [PubMed] [Google Scholar]
- 26.Cheng SX, Geibel J, Hebert S. Extracellular polyamines regulate fluid secretion in rat colonic crypts via the extracellular calcium-sensing receptor. Gastroenterology. 2004;126(1):148–58. doi: 10.1053/j.gastro.2003.10.064. [DOI] [PubMed] [Google Scholar]
- 27.Cheng SX, Okuda M, Hall A, Geibel JP, Hebert SC. Expression of calcium-sensing receptor in rat colonic epithelium: evidence for modulation of fluid secretion. Am J Physiol. 2002;283:G240–50. doi: 10.1152/ajpgi.00500.2001. [DOI] [PubMed] [Google Scholar]
- 28.Gama L, Baxendale-Cox LM, Breitwieser GE. Ca2+-sensing receptors in intestinal epithelium. Am J Physiol. 1997;273(4 Pt 1):C1168–75. doi: 10.1152/ajpcell.1997.273.4.C1168. [DOI] [PubMed] [Google Scholar]
- 29.Hebert S, Cheng S, Geibel J. Functions and roles of the extracellular Ca2+sensing receptor in the gastrointestinal tract. Cell Calcium. 2004;35(3):239–47. doi: 10.1016/j.ceca.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 30.Geibel JP, Hebert SC. The functions and roles of the extracellular Ca2+-sensing receptor along the gastrointestinal tract. Annu Rev Physiol. 2009;71:205–17. doi: 10.1146/annurev.physiol.010908.163128. [DOI] [PubMed] [Google Scholar]
- 31.Rey O, Chang W, Bikle D, Rozengurt N, Young SH, Rozengurt E. Negative crosstalk between calcium-sensing receptor and β-catenin signaling systems in colonic epithelium. J Biol Chem. 2012;287(2):1158–67. doi: 10.1074/jbc.M111.274589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng SX. Calcium-sensing receptor inhibits secretagogue-induced electrolyte secretion by intestine via the enteric nervous system. Am J Physiol Gastrointest Liver Physiol. 2012;303(1):G60–70. doi: 10.1152/ajpgi.00425.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geibel J, Sritharan K, Geibel R, Geibel P, Persing JS, Seeger A, Roepke TK, Deichstetter M, Prinz C, Cheng SX, et al. Calcium-sensing receptor abrogates secretagogue- induced increases in intestinal net fluid secretion by enhancing cyclic nucleotide destruction. Proc Natl Acad Sci U S A. 2006;103(25):9390–7. doi: 10.1073/pnas.0602996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stenman LK, Holma R, Gylling H, Korpela R. Genetically obese mice do not show increased gut permeability or faecal bile acid hydrophobicity. Br J Nutr. 2013;110(6):1157–64. doi: 10.1017/S000711451300024X. [DOI] [PubMed] [Google Scholar]
- 35.Kathania M, Zadeh M, Lightfoot YL, Roman RM, Sahay B, Abbott JR, Mohamadzadeh M. Colonic immune stimulation by targeted oral vaccine. PLoS One. 2013;8(1):e55143. doi: 10.1371/journal.pone.0055143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76(3):907–15. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, Wang GP. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol. 2013;51(9):2884–92. doi: 10.1128/JCM.00845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffmann C, Hill DA, Minkah N, Kirn T, Troy A, Artis D, Bushman F. Community-wide response of the gut microbiota to enteropathogenic Citrobacter rodentium infection revealed by deep sequencing. Infect Immun. 2009;77(10):4668–78. doi: 10.1128/IAI.00493-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Ampting MT, Loonen LM, Schonewille AJ, Konings I, Vink C, Iovanna J, Chamaillard M, Dekker J, van der Meer R, Wells JM, et al. Intestinally secreted C-type lectin Reg3b attenuates salmonellosis but not listeriosis in mice. Infection and immunity. 2012;80(3):1115–20. doi: 10.1128/IAI.06165-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313(5790):1126–30. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J Exp Med. 2007;204(8):1891–900. doi: 10.1084/jem.20070563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, Maloy KJ. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med. 2012;209(9):1595–609. doi: 10.1084/jem.20111453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohamadzadeh M, Pfeiler EA, Brown JB, Zadeh M, Gramarossa M, Managlia E, Bere P, Sarraj B, Khan MW, Pakanati KC, et al. Regulation of induced colonic inflammation by Lactobacillus acidophilus deficient in lipoteichoic acid. Proc Natl Acad Sci U S A. 2011;108 (Suppl 1):4623–30. doi: 10.1073/pnas.1005066107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr Protoc Immunol. 2014;104:15.25.1–15.25.14. doi: 10.1002/0471142735.im1525s104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perše M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol. 2012;2012:718617. doi: 10.1155/2012/718617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Macleod RJ. Extracellular calcium-sensing receptor/PTH knockout mice colons have increased Wnt/β-catenin signaling, reduced non-canonical Wnt signaling, and increased susceptibility to azoxymethane-induced aberrant crypt foci. Lab Invest. 2013;93(5):520–7. doi: 10.1038/labinvest.2013.51. [DOI] [PubMed] [Google Scholar]
- 47.Turki R, Kelly J, Pacheco I, MacLeod R. Effect of increased dietary calcium on murine colonic CaSR expression and the effect of induced colitis on “rescued” CaSR-/PTH- (C-/P-) knockout mice. Bone. 2012:S23–S4. [Google Scholar]
- 48.Pele LC, Thoree V, Mustafa F, He S, Tsaprouni L, Punchard NA, Thompson RP, Evans SM, Powell JJ. Low dietary calcium levels modulate mucosal caspase expression and increase disease activity in mice with dextran sulfate sodium induced colitis. J Nutr. 2007;137:2475–80. doi: 10.1093/jn/137.11.2475. [DOI] [PubMed] [Google Scholar]
- 49.Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492(7427):123–7. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loscher CE, Draper E, Leavy O, Kelleher D, Mills KHG, Roche HM. Conjugated linoleic acid suppresses NF-kappa B activation and IL-12 production in dendritic cells through ERK-mediatedIL-10 induction. J Immunol. 2005;175:4990–8. doi: 10.4049/jimmunol.175.8.4990. [DOI] [PubMed] [Google Scholar]
- 51.Weatherill AR, Lee JY, Zhao L, Lemay DG, Youn HS, Hwang DH. Saturated and polyunsaturated fatty acids reciprocally modulate dendritic cell functions mediated through TLR4. J Immunol. 2005;174:5390–7. doi: 10.4049/jimmunol.174.9.5390. [DOI] [PubMed] [Google Scholar]
- 52.Zeyda M, Säemann MD, Stuhlmeier KM, Mascher DG, Nowotny PN, Zlabinger GJ, Waldhäusl W, Sulnig T. Polyunsaturated fatty acids block dendritic cell activation and function independently of NF-kappaB activation. J Biol Chem. 2005;280(14):14293–301. doi: 10.1074/jbc.M410000200. [DOI] [PubMed] [Google Scholar]
- 53.Cheng SX. Calcium-sensing receptor in the gut: evidence for its role in mediating known nutritional therapy for inflammatory bowel disease (abstract) JPGN. 2012;55(suppl 1):E70. [Google Scholar]
- 54.Bovee Oudenhoven IM, Wissink ML, Wouters JT, Van der Meer R. Dietary calcium phosphate stimulates intestinal lactobacilli and decreases the severity of a salmonella infection in rats. The Journal of nutrition. 1999;129(3):607–12. doi: 10.1093/jn/129.3.607. [DOI] [PubMed] [Google Scholar]
- 55.Bovee-Oudenhoven IMJ, Lettink-Wissink MLG, Van Doesburg W, Witteman BJM, Van Der Meer R. Diarrhea caused by enterotoxigenic Escherichia coli infection of humans is inhibited by dietary calcium. Gastroenterology. 2003;125(2):469–76. doi: 10.1016/s0016-5085(03)00884-9. [DOI] [PubMed] [Google Scholar]
- 56.van der Meer R, Welberg JWM, Kuipers F, Kleibeuker JH, Mulder NH, Termont DS, Vaonk RJ, De Vries HT, De Vries EG. Effects of supplemental dietary calcium on the intestinal association of calcium, phosphate, and bile acids. Gastroenterology. 1990;99:1653–9. doi: 10.1016/0016-5085(90)90471-c. [DOI] [PubMed] [Google Scholar]
- 57.Schepens MA, Vink C, Schonewille AJ, Dijkstra G, van der Meer R, Bovee-Oudenhoven IM. Supplemental calcium attenuates the colitis-related increase in diarrhea, intestinal permeability, and extracellular matrix breakdown in HLA-B27 transgenic rats. J Nutr. 2009;139:1525–33. doi: 10.3945/jn.109.105205. [DOI] [PubMed] [Google Scholar]
- 58.Matsumoto M, Kurihara S, Kibe R, Ashida H, Benno Y. Longevity in mice is promoted by probiotic-induced suppression of colonic senescence dependent on upregulation of gut bacterial polyamine production. PLoS One. 2011;6(8):e23652. doi: 10.1371/journal.pone.0023652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zarrabian S, Buts JP, Fromont G, Tran TU, Macry J, Mendy F, Roger L, Cèzard JP. Effects of alimentary intact proteins and their oligopeptide hydrolysate on growth, nitrogen retention, and small bowel adaptation in inflammatory turpentine rat. Nutrition. 1999;15:474–80. doi: 10.1016/s0899-9007(99)00054-4. [DOI] [PubMed] [Google Scholar]
- 60.Nieves CJ, Langkamp-Henken B. Arginine and immunity: a unique perspective. Biomed Pharmacother. 2002;56:471–82. doi: 10.1016/s0753-3322(02)00291-3. [DOI] [PubMed] [Google Scholar]
- 61.Ameho CK, Adjei AA, Harrison EK, Takeshita K, Morioka T, Arakaki Y, Ito E, Suzuki I, Kulkarni AD, Kawajiri A, et al. Prophylactic effect of dietary glutamine supplementation on interleukin 8 and tumour necrosis factor alpha production in trinitrobenzene sulphonic acid induced colitis. Gut. 1997;41:487–93. doi: 10.1136/gut.41.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104(34):13780–5. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55(2):205–11. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sokol H, Seksik P, Rigottier-Gois L, Lay C, Lepage P, Podglajen I, Marteau P, Dore J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12(2):106–11. doi: 10.1097/01.MIB.0000200323.38139.c6. [DOI] [PubMed] [Google Scholar]
- 65.Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. Journal of clinical microbiology. 2005;43(7):3380–9. doi: 10.1128/JCM.43.7.3380-3389.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Candela M, Rampelli S, Turroni S, Severgnini M, Consolandi C, De Bellis G, Masetti R, Ricci G, Pession A, Brigidi P. Unbalance of intestinal microbiota in atopic children. BMC microbiology. 2012;12:95. doi: 10.1186/1471-2180-12-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zinselmeyer BH, Heydari S, Sacristán C, Nayak D, Cammer M, Herz J, Cheng X, Davis SJ, Dustin ML, McGavern DB. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med. 2013;210(4):757–74. doi: 10.1084/jem.20121416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khazaie K, Zadeh M, Khan MW, Bere P, Gounari F, Dennis K, Blatner NR, Owen JL, Klaenhammer TR, Mohamadzadeh M. Abating colon cancer polyposis by Lactobacillus acidophilus deficient in lipoteichoic acid. Proc Natl Acad Sci U S A. 2012;109(26):10462–7. doi: 10.1073/pnas.1207230109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.