

Abstract
The chromosome 8p11-p12 amplicon is present in 12% to 15% of breast cancers, resulting in an increase in copy number and expression of several chromatin modifiers in these tumors, including KAT6A. Previous analyses in SUM-52 breast cancer cells showed amplification and overexpression of KAT6A, and subsequent RNAi screening identified KAT6A as a potential driving oncogene. KAT6A is a histone acetyltransferase previously identified as a fusion partner with CREB binding protein in acute myeloid leukemia. Knockdown of KAT6A in SUM-52 cells, a luminal breast cancer cell line harboring the amplicon, resulted in reduced growth rate compared to non-silencing controls and profound loss of clonogenic capacity both in mono-layer and in soft agar. The normal cell line MCF10A, however, did not exhibit slower growth with knockdown of KAT6A. SUM-52 cells with KAT6A knockdown formed fewer mammospheres in culture compared to controls, suggesting a possible role for KAT6A in self-renewal. Previous data from our laboratory identified FGFR2 as a driving oncogene in SUM-52 cells. The colony forming efficiency of SUM-52 KAT6A knockdown cells in the presence of FGFR inhibition was significantly reduced compared to cells with KAT6A knockdown only. These data suggest that KAT6A may be a novel oncogene in breast cancers bearing the 8p11-p12 amplicon. While there are other putative oncogenes in the amplicon, the identification of KAT6A as a driving oncogene suggests that chromatin-modifying enzymes are a key class of oncogenes in these cancers, and play an important role in the selection of this amplicon in luminal B breast cancers.
Introduction
A significant step in breast cancer progression is activation of oncogenes via gene amplification and overexpression [1]. The chromosome 8p11-p12 amplicon, containing approximately 55 genes, is present in 12% to 15% of breast cancers and is correlated with poor prognosis in primary breast tumors. Amplification of 8p11-p12 is also correlated with histologic grade, increased Ki-67 proliferation index, and decreased 5-year metastasis-free survival [2], [3], [4], [5], [6], [7]. Due to its relevance in breast cancer, many studies have been aimed at characterizing this amplification and identifying the driving oncogenes in this region. Over the past several years, our laboratory and several others have analyzed the 8p11-p12 amplicon to identify possible driver oncogenes and determine the clinical relevance of these gene amplification events in breast cancer. These studies have resulted in the identification of a number of genes that play a role in breast cancer when the amplicon is present, including LSM-1, C8orf4 (TC-1), RAB11FIP1, WHSC1L1, ERLIN2, PROSC, PPAPDC, DDHD2 and others [2], [6], [8], [9], [10], [11], [12], [13]. Gelsi-Boyer et al. demonstrated that the amplicon can be sub-divided into four distinct regions that can be amplified independently of each other, and this partly explains the large number of candidate oncogenes identified to date from this region [3].
Recently, our lab has taken a genome-scale shRNA screening strategy to identify additional driving oncogenes in a panel of SUM breast cancer cell lines, with and without 8p11-p12 amplification, for which copy number and expression data are available. KAT6A was found to be a consistent hit in the shRNA screen and is significantly amplified and overexpressed in SUM-52 cells.
KAT6A, a gene commonly found in the 8p11-p12 amplicon, was first identified in 1996 as part of a chromosomal translocation t(8;16)(p11;p13) with CREB-binding protein in a subtype of acute myeloid leukemia (AML) [14]. Since then, additional translocations involving KAT6A in AML have been discovered, including translocations with the HATs p300 and TIF2 [15]. The KAT6A-TIF2 fusion is capable of immortalizing myeloid progenitor cells and inducing acute myeloid leukemia upon injection into irradiated mice [16]. Characterization of KAT6A in these gene rearrangements provides compelling evidence that KAT6A is an oncogene in leukemogenesis, much like c-myc, which is well documented in various chromosomal translocations promoting several forms of leukemia, and is also amplified in breast cancer.
In this study, we provide evidence that KAT6A functions as a novel oncogene in luminal breast cancers bearing the 8p11-p12 amplicon. We identified KAT6A as a top shRNA screen hit among genes both amplified and overexpressed in the SUM-52 breast cancer cell line. The influence of KAT6A knockdown on proliferation and viability of SUM-52 cells, and other cell lines with the 8p11-p12 amplicon was validated in separate experiments involving several short hairpin RNAs (shRNAs). Finally, data obtained with primary human breast cancers coupled with TCGA data support a role for KAT6A as a breast cancer oncogene.
Materials and Methods
Reagents
All chemicals were purchased from Sigma Aldrich unless otherwise noted. Antibodies against Actin and anti-acetyl Histone 3 were purchased from Millipore. The total-Histone 3 antibody was purchased from Abcam, and the KAT6A antibody from Novus Biologicals. The FGFR inhibitor PD170374 was purchased from Selleckchem. Expression Arrest GIPZ lentiviral KAT6A shRNA constructs and packaging mix were purchased from Open Biosystems. Primers used for RT-PCR reactions were ordered from IDT. The SUM breast cancer cell lines were maintained in Hams F-12 cell culture medium as described previously [17], [18], [19].
Cellecta Screening Method
Virus pools expressing shRNA constructs were prepared according to the Cellecta Pooled Lentiviral shRNA Libraries User Manual protocol (www.cellecta.com). HEK 293 T cells were transfected with each of the three Cellecta library plasmid DNA pools (Human Modules 1–3) and the Cellecta Ready-to-Use Packaging Mix (Cat #CPCP-K2A). For each module, virus was titered and used to transduce 5 × 107 SUM-52 cells at an MOI of 0.5 in the presence of 5 μg/ml polybrene. Following transduction, cells were cultured for 3 days to allow expression of the resistance marker. Non-transduced cells were eliminated from the culture by the addition of 2 μg/ml puromycin to the growth media. Three days after the addition of puromycin, cells were trypsinized and one-half of the total population was harvested for genomic DNA preparation. This DNA served as the reference time point DNA. The remaining cells were plated and grown for approximately 7 population doublings before being harvested for genomic DNA preparation. Genomic DNA was prepared by phenol:chloroform extraction according to the Cellecta Pooled Lentiviral shRNA Libraries User Manual protocol.
See Supplementary Methods for detailed descriptions of the shRNA screen and statistical analyses.
RT-PCR
For RT-PCR reactions, total RNA was isolated using the Qiagen RNeasy Plus Mini Kit. RNA was converted into cDNA using the iScript Advanced cDNA Synthesis Kit (BioRad). Following cDNA conversion, RT-PCR reactions were performed in 96-well white-walled plates using FastStart Universal SYBR Green Master mix (Roche). Reactions were performed on an Eppendorf Mastercycler Realplex2 under the following parameters: 1 cycle for 10 minutes at 95°C, 1 cycle for 15 seconds at 95°C, and 40 cycles at 53°C for 30 seconds. Primer sets included a control GAPDH primer set and a primer set specific to the KAT6A transcript.
Growth and Colony-Forming Assays
For growth assays, cells were plated in triplicate in 6-well plates at a density of 35,000 cells/well. Nuclei were harvested and cell counts obtained using a Beckman Coulter Counter as described previously [7].
For colony-forming assays, cells were plated in triplicate in 6-well plates at varying cell densities. Colonies formed for two weeks and then were fixed with 1 mL/well of 3.7% paraformaldehyde for 20 minutes at RT. Colonies were stained with 1 mL/well of 0.2% crystal violet for 15 minutes at RT and then de-stained with dH2O. For colony-forming assays including treatment with the FGFR inhibitor PD170374, cells were plated as stated above and treated with 0.1 μM of drug for 3 consecutive days. Colonies formed for an additional 10–14 days following drug treatment and were stained with crystal violet as described. Colony counts were generated using the GelCount System from Oxford Optronix.
Mammosphere Formation
Cells were plated in triplicate for primary mammosphere formation in low-attachment 6-well plates. Spheres formed for one week, at which point two wells were transferred to standard 6-well plates in media supplemented with FBS. Attached spheres were fixed with 1 mL/well of 3.7% paraformaldehyde for 20 minutes at RT and stained with 1 mL/well of 0.2% crystal violet for 15 minutes at RT. For secondary mammosphere formation, spheres from one well of the primary mammosphere assay were harvested and centrifuged at 2500 RPM for 5 minutes. Spheres were re-suspended in 1 mL of trypsin and incubated at RT for 5 minutes. The trypsin was diluted with media and centrifuged as before. The cell pellet was washed with 5 mL of PBS and centrifuged, and the final cell pellet was re-suspended in 1 mL of 1 × Hanks buffer solution. Cells were re-plated in triplicate in 6-well low attachment plates. Spheres were allowed to form for 2 weeks and then transferred and stained with 0.2% crystal violet as described above. Images of primary and secondary spheres were taken using an EVOS Imaging System (Life Technologies). Mammosphere counts for both primary and secondary assays were generated using the GelCount System from Oxford Optronix.
RNA Sequencing
Total RNA was prepared using a Qiagen RNeasy Plus Mini Kit and processed by the MUSC Genomics core for 101-bp, paired-end RNA sequencing on an Illumina HiScanSQ. RNA integrity was verified on an Agilent 2100 TapeStation (Agilent Technologies, Palo Alto, CA). 100–200 ng of total RNA was used to prepare RNA-Seq libraries using the TruSeq RNA Sample Prep kit following the protocol described by the manufacturer (Illumina, San Diego, CA). Sequencing was performed on an Illumina HiScanSQ. Samples were demultiplexed using CASAVA (Illumina, San Diego, CA). Fastq files were used to map reads to the human genome (hg19, UCSC) utilizing Tophat2 with default settings. Bam files were imported into Partek Genomics Suite (St. Louis, MO) for analysis. Samples were filtered for ≥ 10 reads. Differential gene expression was performed by comparison of biological duplicate samples in Genomics Suite using advanced analysis of variance (ANOVA). Differential gene express values were then filtered by ± 1.5 fold change and by unadjusted P ≤ .05.
Results
Rationale for Studying the Oncogenic Potential of KAT6A
In an effort to comprehensively identify genes that control cell proliferation and survival in breast cancer cells, our laboratory is performing genome-scale, RNAi-based negative selection screens in our panel of SUM breast cancer cell lines. In this screen, the SUM-52 breast cancer cell line was transduced with a library of 82,000 lentiviral vectors that express shRNAs targeting 15,377 cellular genes with a minimum of 4 shRNAs per gene. Cells transduced with this library were harvested at either day 3 after infection or after approximately 7 population doublings, and the abundance of shRNAs at both time points was determined by PCR amplification, purification and sequencing of shRNA-associated barcode sequences. Fold depletion values were calculated by comparing the abundance of each shRNA at the final time point with the abundance at the initial time point. Fold depletion scores following statistical analysis for all of the shRNAs targeting each gene were used to identify genes that are necessary for the growth/survival of the SUM-52 breast cancer cell line. Integration of this list with the list of genes that are significantly copy number amplified in this cell line identified a small number of genes that are amplified, overexpressed and necessary for growth/survival of the cell line. KAT6A was found to be a consistent hit in this screen, as well as an additional screen using an early generation lower complexity library, and is significantly amplified and overexpressed in SUM-52 cells (Figure 1A).
Figure 1.
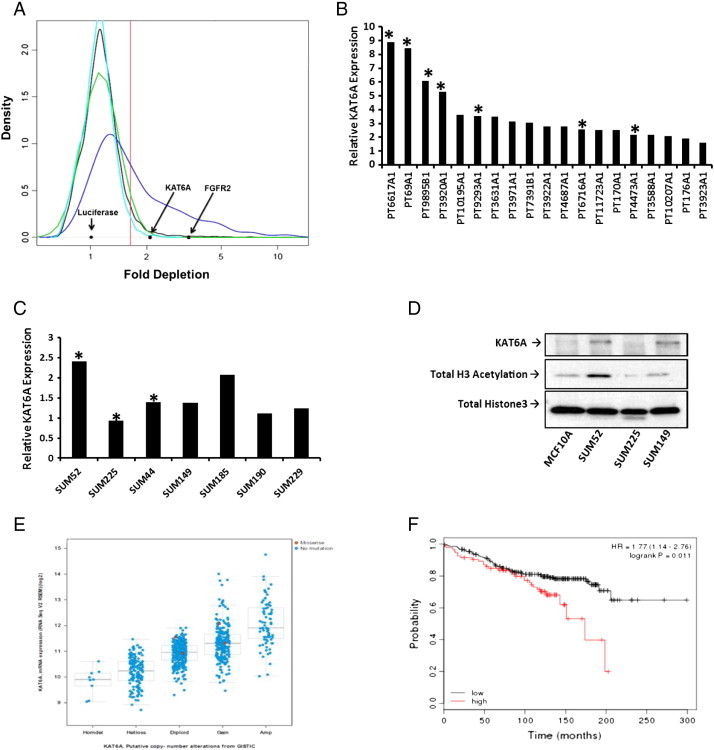
Rationale for studying the oncogenic potential of KAT6A. A. Fold depletion scores from the genome-wide shRNA screen were used to generate probability density plots. The black plot was generated using depletion scores for all genes, the blue plot was generated using depletion scores for commonly essential genes, the turquoise plot was generated using depletion scores for the negative control gene set, and the green plot is a normal distribution whose mean was determined by the median of the fold depletion scores for all genes. The vertical red line indicates the 95th percentile of the normal distribution that was used as a cutoff for determining hits in the screen. Black arrows indicate gene scores for Luciferase, KAT6A and FGFR2. B. Microarray data showing relative KAT6A expression in a panel of primary breast tumors suspected to harbor the 8p11-p12 amplicon. Primary tumors with the highest KAT6A expression were also positive for possessing the amplicon, as denoted by the asterisk. C. Microarray data showing that relative KAT6A expression is highest in 8p11-p12 amplicon bearing SUM breast cancer cell lines. D. Western blot for KAT6A protein expression in a panel of SUM breast cancer cell lines. Total Histone 3 acetylation levels were highest in SUM-52 cells, an 8p11-p12 amplicon bearing cell line. E. TCGA data showing a correlation between KAT6A copy number and mRNA expression. F. Kaplan-Meier analysis with overall survival in over 4,000 breast cancer cases showing a statistically significant difference in survival in patients with KAT6A amplification/overexpression, with curves diverging after approximately five years of follow-up.
Based on the RNAi screen data, we performed microarray analysis to examine the expression level of KAT6A in a small panel of primary breast cancers available in our lab that potentially carry the 8p11-p12 amplicon. As seen in Figure 1B, several primary tumor samples from this panel exhibited high levels of KAT6A gene expression. Additionally, tumors with the highest expression of KAT6A also had a KAT6A gene amplification. Likewise, a series of SUM breast cancer cell lines were analyzed by microarray, and KAT6A expression was highest in cells harboring the 8p11-p12 amplicon, particularly SUM-52 cells (Figure 1C). The SUM-52 cell line was generated from a patient with luminal B breast cancer who presented with an ER-positive primary tumor. The data in Figure 1D shows that KAT6A protein expression was highest in SUM-52 cells compared to normal MCF10A cells, SUM-149 and SUM-225 cell lines. Whereas SUM-225 cells have two small regions of 8p11 amplification, KAT6A is not amplified in these cells, and SUM-149 cells do not harbor the 8p11-p12 amplicon. As expected, KAT6A protein expression in SUM-225 cells was barely detectable; however, SUM-149 cells, although lacking the 8p11-p12 amplicon, express KAT6A protein at levels similar to SUM-52 cells. Because KAT6A is known as a histone acetyltransferase (HAT) with HAT activity on histone 3, we examined the acetylation status of histone 3 in cells with amplification of KAT6A. Total histone 3 acetylation was highest in SUM-52 cells compared to MCF10A, SUM-225, and SUM-149 cells (Figure 1D); however, the difference in histone 3 acetylation was most interesting between SUM-52 and SUM-149 cells. Although both cell lines express comparable levels of KAT6A protein, histone 3 acetylation was only increased in SUM-52 cells.
To explore further the relevance of KAT6A in breast cancer, we examined TCGA data derived from 1,000 primary breast cancers, and found that KAT6A expression is altered in 22% of breast cancer cases. Additionally, there is a strong correlation between copy number and expression at the mRNA level (Figure 1E). Kaplan-Meier analysis of more than 4000 breast cancer patients (http://kmplot.com/analysis/) showed that high level KAT6A expression (highest quartile) was associated with a statistically significant reduction in overall survival (Figure 1F). Interestingly, the Kaplan-Meier curves diverge after approximately 100 months of follow-up, suggesting that KAT6A plays a role in late recurrences, an important clinical problem in luminal breast cancer. These data support the hypothesis that KAT6A functions as a driving oncogene in breast cancer, and warrant further investigation to validate its activity as a transforming oncogene.
KAT6A Knockdown Reduces Growth in 8p11-p12 Amplicon-Bearing Breast Cancer Cells
To validate the shRNA screen results, KAT6A knockdown studies were performed with several cell lines. Lentiviral shRNA vectors were prepared by transfection of 293FT cells with five shRNA plasmids targeting different KAT6A sequences. Initially, only SUM-52 and MCF10A cells were transduced with the lentiviral shRNAs. Following lentiviral infection and puromycin selection, RNA was harvested to confirm knockdown of KAT6A by RT-PCR. The results showed that two shRNAs yielded the best knockdown, sh1-566 (sh1) and sh3-895 (sh3), compared to a non-silencing shRNA sequence, (NS) (Supplementary Figure 1). To measure the effects of reduced KAT6A expression on the growth of the SUM-52 and SUM-225 cell lines, the cells were plated in monolayer in 6-well plates, and their growth was assessed by cell counts on days 1, 4, and 7. Knockdown of KAT6A at the message and protein level in SUM-52 cells (Figure 2A) resulted in 50% reduction in cell growth (Figure 2C). KAT6A is not part of the 8p11-p12 amplicon in SUM-225 cells, and only a limited growth response to reduced KAT6A expression was observed (Figure 2D). Infection of SUM-149 cells, which lack the 8p11-p12 amplicon but still express the protein, did not affect proliferation (Figure 2E). Additionally, knockdown of KAT6A in MCF10A cells (Figure 2B) showed no effect on proliferation (Figure 2F). These data suggest that the effects of KAT6A on breast cancer cell proliferation are restricted to cells in which the gene is amplified and overexpressed.
Figure 2.
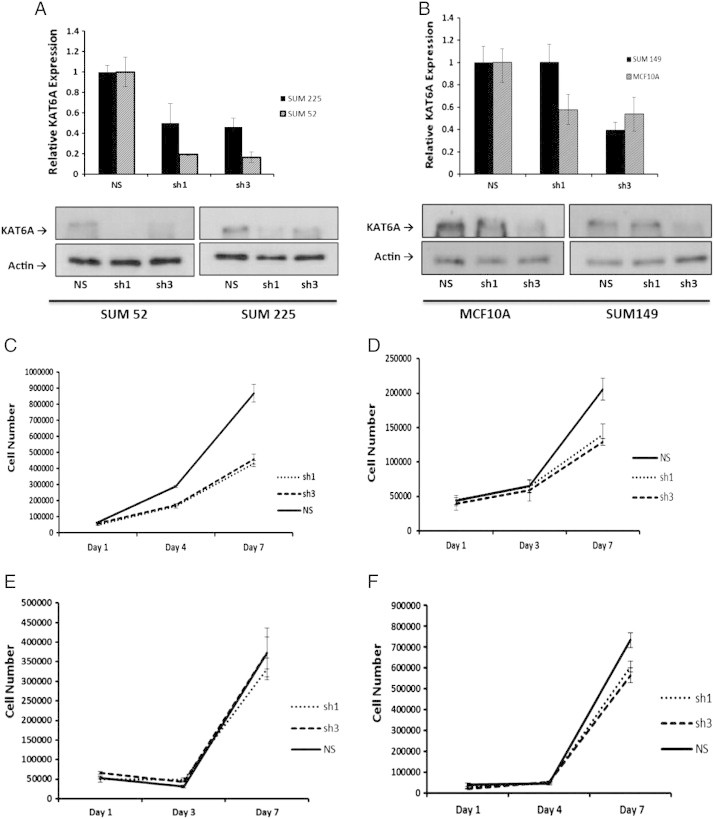
KAT6A knockdown reduces cell growth in 8p11-p12 amplicon bearing cell lines. A. RT-PCR results showing knockdown of KAT6A at the mRNA level in SUM-52 and SUM-225 cells transduced with lentivirus using shRNA1 and shRNA3. Below is a corresponding western blot showing knockdown of KAT6A at the protein level. B. RT-PCR data showing knockdown of KAT6A at the mRNA level in MCF10A and SUM-149 cells using lentiviral vectors shRNA1 and shRNA3. Below is a corresponding western blot showing knockdown of KAT6A protein. C - F. SUM-52, SUM-225, MCF10A, and SUM-149 control and KAT6A knockdown cells, respectively, were plated in triplicate and assayed for growth. Cell counts were taken on days 1, 4 and 7.
KAT6A Reduces the Clonogenic Capacity of SUM-52 Cells
Since reduction of KAT6A expression yielded growth inhibition in SUM-52 cells, we next addressed the colony-forming efficiency of SUM-52 cells after knockdown of KAT6A. As shown in Figure 3A, SUM-52 cells with depleted KAT6A levels exhibited a dramatic reduction in colony-forming ability compared to control cells. Moreover, colonies that did grow after KAT6A knockdown were not only smaller in size, but exhibited an abnormal morphology (Figure 3A and B). Cells with KAT6A knockdown produced small, scattered colonies composed of cells showing morphological signs of pyknosis. By contrast, non-silencing control cells produced tightly-packed colonies with cells displaying a normal appearance. Figure 3C shows the size distribution of colonies derived from KAT6A knockdown cells. The largest colonies from the knockdown cells were approximately 300 μm in diameter, whereas colonies from control cells were greater than 1000 μm in diameter. The colony-forming efficiency of MCF10A cells, however, was unaffected by knockdown of KAT6A (Figure 3D and E). In a separate experiment, we examined the role of KAT6A knockdown in anchorage-independent colony formation. KAT6A knockdown also inhibited the soft agar colony-forming ability of SUM-52 cells, as KAT6A knockdown cells formed fewer colonies compared to the non-silencing control (Supplementary Figure 2A and B). To extend these observations to other 8p11-p12 harboring cancer cell lines in which KAT6A is amplified and overexpressed, the ovarian cancer cell lines OAW28 and 59 M were assayed for colony-forming efficiency following KAT6A knockdown (Supplementary Figure 3A and B). These additional cell lines were identified using the TCGA Bioportal and the Broad Institute’s Cancer Cell Line Encyclopedia, which indicated a GISTIC copy number score of 1.563 for 59 M cells and 1.665 for OAW28 cells. As seen in Figure 3F and G, both OAW28 and 59 M cells, respectively, responded dramatically to knockdown of KAT6A, with both cell lines displaying a reduction in colony forming ability. Based on these results, KAT6A functions in regulating the clonogenic capacity of not only SUM-52 cells, but also of other cancer cell lines with the 8p11-p12 amplicon and KAT6A amplification/overexpression.
Figure 3.
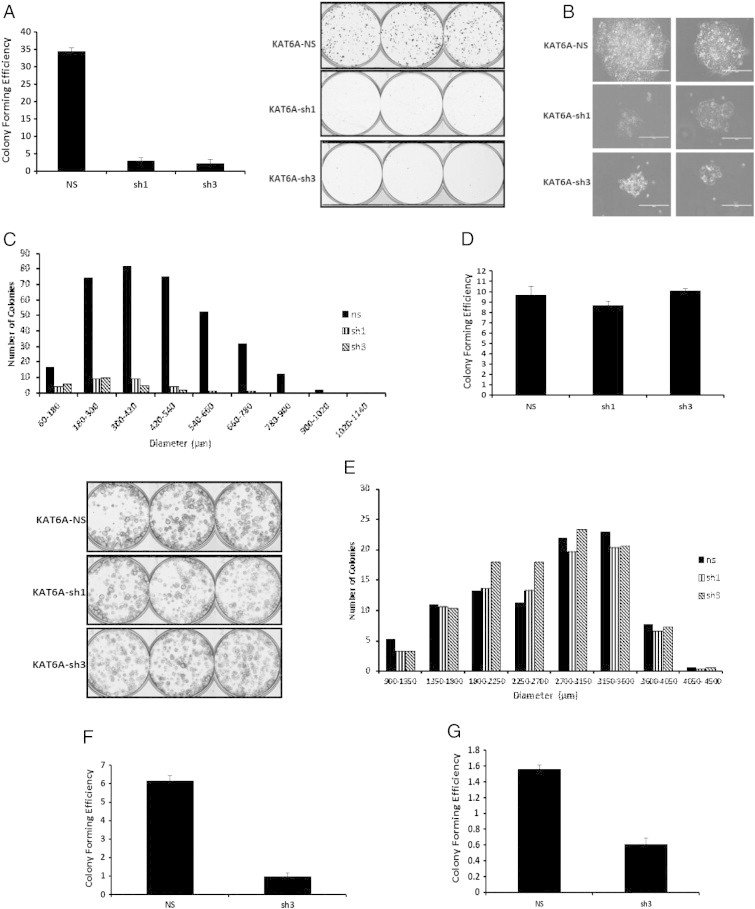
KAT6A reduces the clonogenic capacity of SUM-52 cells. A. SUM-52 control and KAT6A knockdown cells were plated in a colony-forming assay in triplicate in 6-well low-attachment plates. Colonies were allowed to grow for 2 weeks before staining with 0.2% crystal violet. Adjacent picture depicts the crystal violet-stained colonies. B. Images of SUM-52 control and KAT6A knockdown colonies taken using an EVOS Imaging System. C. Histogram showing colony size distribution for SUM-52 control and KAT6A knockdown colonies. D. MCF10A control and KAT6A knockdown cells were plated as above to assay colony-forming efficiency. Adjacent picture depicts 6-well plates with crystal violet-stained colonies. E. Histogram showing colony size distribution for MCF10A control and KAT6A knockdown colonies. F and G. OAW28 and 59 M control and KAT6A knockdown cells, respectively, were plated as stated above for colony forming assay. OAW28 and 59 M are ovarian cancer cell lines positive for both the 8p11-p12 amplicon and KAT6A amplification.
KAT6A Plays a Role in Breast Cancer Stem Cell Activity and Differentiation
In light of the effects of KAT6A knockdown on the growth and colony-forming efficiency of SUM-52 cells, the role of KAT6A in breast cancer stem cell activity and self-renewal was examined in a primary mammosphere assay. As shown in Figure 4A, approximately 2% of non-silencing control SUM-52 cells formed mammospheres. By contrast, less than 1% of KAT6A knockdown cells formed spheres.
Figure 4.
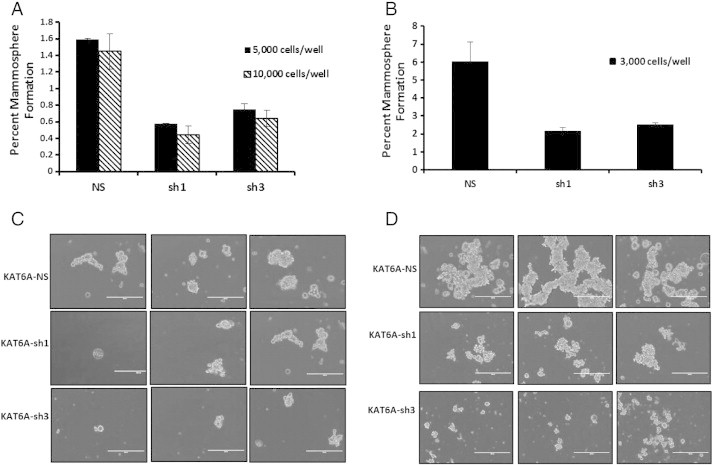
KAT6A plays a role in breast cancer stem cell activity. A. SUM-52 control and KAT6A knockdown cells were plated in triplicate in 6-well low-attachment plates for a primary mammosphere assay. After one week, mammospheres were transferred to standard 6-well plates overnight, stained with 0.2% crystal violet and counted. B. Cells from the primary mammosphere assay were harvested, trypsinized, and re-seeded in a secondary mammosphere assay at 3,000 cells/well. After one week, spheres were processed as above for staining. C. Images of primary mammospheres following one week of growth. D. Images of secondary mammospheres following two weeks of growth.
To confirm and extend these results, a secondary mammosphere assay was performed. The non-silencing control cells formed 4-fold more mammospheres than in the primary assay, suggesting that the cells plated for secondary mammosphere assay contained a higher percentage of cancer-initiating stem cells. Consistent with results in the primary mammosphere assay, non-silencing control cells again formed more mammospheres than KAT6A knockdown cells in the secondary mammosphere assay (Figure 4B). Images of mammospheres revealed striking differences not only between non-silencing and KAT6A knockdown spheres, but also between primary and secondary mammosphere assays. Mammospheres derived from control cells displayed classical sphere morphology. Although KAT6A knockdown spheres appeared similar, they were smaller in size compared to the control spheres (Figure 4C). Allowing secondary mammosphere cultures to grow over an extended period revealed a remarkable difference in morphology between the non-silencing and KAT6A knockdown mammospheres. Mammospheres derived from non-silencing control cells began to form three-dimensional duct-like structures after two weeks in culture (Figure 4D), which were never observed in mammosphere cultures derived from KAT6A knockdown cells. These results indicate that SUM-52 cells can form complex three dimensional structures after extended mammosphere culture, and that this capacity is lost upon KAT6A knockdown.
In many breast cancer cell types, expression of surface markers is a measure of stemness and self-renewal potential. In particular, the CD44+/CD24-/low expression pattern is associated with the breast cancer stem cell phenotype in some cell lines and tumors. Because knockdown of KAT6A resulted in reduced mammosphere formation, we investigated the expression levels of CD24 and CD44 in KAT6A knockdown and control cells by flow cytometry. Knockdown of KAT6A resulted in an increase in cells positive for both CD24 and CD44 to a level approximately three times above the non-silencing control. Perhaps more interesting was the increase in CD44 expression seen in KAT6A knockdown cells, as evidence now suggests that CD44 positivity is associated with differentiation [20]. Only 3.1% of non-silencing control cells were positive for CD44, while 8.7% of KAT6A-sh3-895 cells were positive for CD44 expression (Supplementary Figure 4A and B).
Influence of KAT6A on Gene Expression in SUM-52 Cells
To gain insight into the role of KAT6A as a breast cancer oncogene, we performed RNA sequencing in SUM-52 KAT6A knockdown and control cells. The heat map in Figure 5A was generated from four replicate experiments using the two different shRNAs described above. Since the replicates from each shRNA clustered together, we pooled the data resulting in four data sets for the bioinformatic analysis of the effect of KAT6A knock-down on gene expression. As can be seen from the figure, KAT6A knockdown had a significant effect on gene expression. We used NIH DAVID to analyze the differentially expressed genes that resulted from KAT6A knockdown, and identified several biological processes, molecular functions, and KEGG pathways associated with this gene set. The biological processes affected by KAT6A knockdown are listed in Table 1, with genes involved in Response to Hormone Stimulus and Gland Development processes as top hits. From this analysis, several well-known genes involved in regulating growth were highlighted. Some of the significantly down-regulated genes in KAT6A knockdown cells included ESR1, IRS1, IGF1R, and ERBB4 (Table 2 and Supplementary Table 1, 2). Interestingly, not only was the adenylate cyclase gene ADCY7 significantly down-regulated in response to KAT6A knockdown, but so were its two regulatory subunits, PRKAR1B and PRKAR2B. A number of interesting transcription factors were also identified using NIH DAVID analysis. As shown in Table 3 (and Supplementary Table 3), GATA3, TBX3, and MYC were all significantly down-regulated upon KAT6A knockdown. KEGG pathway analysis yielded additional insight into the role of KAT6A, showing signaling pathways effected by knockdown of KAT6A. As seen in Table 5, some of the major pathways enriched following KAT6A knockdown included Wnt, PI3K-Akt, Insulin/IGF-1, and ErbB signaling. Finally, merging the list of significantly regulated genes identified by RNA sequencing in KAT6A knockdown cells with hits from the genome-wide shRNA screen in SUM-52 cells identified several overlapping genes, including IGF1R, IRS1, IGF2BP3, and GATA3 (Table 5). The identification of genes that are both regulated in expression by KAT6A and hits in the shRNA screen provides functional evidence for the importance of these genes and pathways in breast cancer cells with KAT6A amplification. (See Table 4.)
Figure 5.
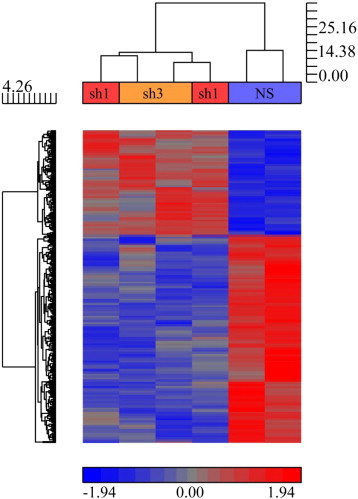
A. Heat map showing gene expression from four replicate RNA sequencing experiments in SUM-52 KAT6A knockdown and control cells. A gene list was generated using a 1.5 fold-change cut-off, 10 reads or more, and p-value of 0.05. A complete list of genes can be seen in Supplementary Table 1.
A. Heat map showing gene expression from four replicate RNA sequencing experiments in SUM-52 KAT6A knockdown and control cells. A gene list was generated using a 1.5 fold-change cut-off, 10 reads or more, and P-value of 0.05. A complete list of genes can be seen in Supplementary Table 1.
Table 1.
Top Biological Processes Identified by GO Analysis of Genes Whose Expression Is Regulated by KAT6A in SUM-52 Cells.
| GO Term | Count | % | P-value |
|---|---|---|---|
| Response to hormone stimulus | 19 | 5.5 | 2.9E-04 |
| Gland development | 11 | 3.2 | 2.9E-04 |
| Response to endogenous stimulus | 20 | 5.8 | 3.5E-04 |
| Response to organic substance | 29 | 8.4 | 3.6E-04 |
| Epithelium development | 14 | 4.0 | 4.9E-04 |
| Appendage morphogenesis | 9 | 2.6 | 6.7E-04 |
| Limb morphogenesis | 9 | 2.6 | 6.7E-04 |
Table 2.
Genes Regulating Growth Identified by GO Analysis in SUM-52 KAT6A Knockdown Cells.
| Official Gene Symbol | Fold Change | P-value |
|---|---|---|
| IGF1R | − 2.42513 | 0.000109472 |
| ESR1 | − 2.88771 | 0.00148708 |
| IRS1 | − 1.68545 | 0.00425694 |
| ERBB4 | − 2.21637 | 0.00832971 |
| PRKAR2B | − 1.6321 | 0.00885831 |
| PRKAR1B | − 1.50051 | 0.00958459 |
| WNT9A | 1.53544 | 0.0317711 |
| ADCY7 | − 1.63293 | 0.0345432 |
| WNT5A | − 2.06146 | 0.0407956 |
| WNT4 | − 1.77249 | 0.0467952 |
| DKK1 | − 1.57585 | 0.047577 |
Table 3.
Transcriptional Regulators Identified by GO Analysis in SUM-52 KAT6A Knockdown Cells.
| Official Gene Symbol | Fold Change | P-value |
|---|---|---|
| BTG2 | − 1.87652 | 0.000113777 |
| MYC | − 1.90468 | 0.000990004 |
| TBX3 | − 1.51777 | 0.00346325 |
| HOXA11 | − 1.54925 | 0.0162591 |
| GATA3 | − 1.70928 | 0.0319076 |
| HOXB13 | − 1.51992 | 0.0471189 |
Table 5.
Significantly Regulated Genes Identified by Merging the RNA Sequencing Gene List in SUM-52 KAT6A Knockdown Cells with Hits from the RNAi Screen Performed in SUM-52 Cells.
| Gene Symbol | P value | Fold Change |
|---|---|---|
| KAT6A | 1.22264E-05 | − 1.97641 |
| IGF1R | 0.000109472 | − 2.42513 |
| MTUS1 | 0.00234984 | − 1.62862 |
| ANTXR1 | 0.00258484 | − 1.82207 |
| CLEC7A | 0.00353284 | 2.66991 |
| IRS1 | 0.00425694 | − 1.68545 |
| TUBA1A | 0.00804264 | − 2.51687 |
| DLL3 | 0.0160265 | − 1.63383 |
| IGF2BP3 | 0.0180273 | 1.85286 |
| UST | 0.0222458 | − 1.55895 |
| MCF2L2 | 0.0316436 | − 2.41022 |
| GATA3 | 0.0319076 | − 1.70928 |
| TREX1 | 0.0476831 | 1.51602 |
Table 4.
Top Cancer-Relevant KEGG Pathways in KAT6A SUM-52 Knockdown Cells.
| SUM-52 |
|||
|---|---|---|---|
| pSize | P value | FDR | |
| Wnt signaling pathway | 143 | 0.00004 | 0.001415 |
| PI3K-Akt signaling pathway | 347 | 0.000010 | 0.002186 |
| Hippo signaling pathway | 156 | 0.000008 | 0.002186 |
| MAPK signaling pathway | 260 | 0.000086 | 0.006011 |
| Insulin signaling pathway | 140 | 0.000205 | 0.006011 |
| Calcium signaling pathway | 183 | 0.000184 | 0.010928 |
| Hedgehog signaling pathway | 51 | 0.000932 | 0.011069 |
| ErbB signaling pathway | 88 | 0.000944 | 0.014924 |
pSize indicates the number of genes in the KEGG pathway.
P value indicates the probability of obtaining the number of screen hits in the pathway by chance alone.
FDR indicates the global probability corrected for false discovery rate of obtaining the indicated number of pathway hits by chance alone.
KAT6A Interacts With FGFR2 to Enhance the Tumorigenicity of SUM-52 Cells
Previous work from our laboratory identified FGFR2 as a driving oncogene in SUM-52 cells. We have observed that while FGFR inhibition can inhibit the growth of these cells, this growth arrest is reversible, as washout of the drug results in prompt resumption of cell growth and no significant loss of colony forming ability (unpublished data and Figure 6A). To address the potential interaction of FGFR2 and KAT6A in SUM-52 cells, the colony-forming ability of SUM-52 non-silencing control cells or SUM-52 KAT6A knockdown cells was examined following treatment with the FGFR inhibitor PD170374. As shown in Figure 6A, exposure of non-silencing control cells to FGFR inhibition did not significantly affect the colony-forming ability of the cells after washout of the drug. As observed previously, the colony-forming efficiency of SUM-52 KAT6A knockdown cells was reduced compared to controls. However, unlike the control cells, the colony-forming efficiency of the KAT6A knockdown cells was decreased further following 72 hours of exposure to PD173074, and this decrease was statistically significant (P < .001). Images of colonies with KAT6A knockdown revealed a striking morphological difference compared to control colonies. As seen in Figure 6B, non-silencing control colonies appeared healthy in size and morphology after treatment and washout of PD170374; however, the KAT6A knockdown colonies that did grow after drug treatment were much smaller in size, with cells showing signs of pyknosis. MCF10A cells, however, were not negatively affected by either KAT6A knockdown or PD170374 treatment (seen in Figure 6C). These results suggest that the KAT6A oncogene interacts with the FGFR2 oncogene, a clear driver of transformed phenotypes in SUM-52 cells, and has an influence on the response of cells to a targeted receptor tyrosine kinase inhibitor.
Figure 6.
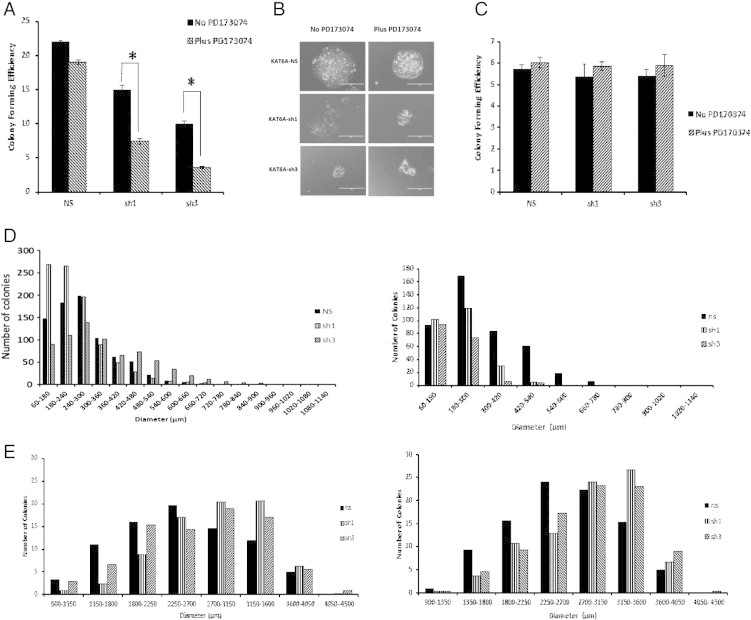
KAT6A interacts with FGFR2 in SUM-52 cells. A. Colony-forming efficiency of SUM-52 control and KAT6A knockdown cells treated with the FGFR inhibitor PD170374. Cells were treated with 0.1 μM of inhibitor for 3 consecutive days following cell plating, and colonies were allowed to grow for an additional 10–14 days before staining with 0.2% crystal violet. B. Images of SUM-52 control and KAT6A knockdown colonies treated with PD170374 taken using an EVOS Imaging System. C. Colony-forming efficiency of MCF10A non-silencing control and KAT6A knockdown cells treated with PD170374. Colonies were treated and stained as above. D. Histograms showing colony size distribution for SUM-52 cells treated with PD170374. The histogram on the left represents colonies formed from untreated cells, and the histogram on the right represents colonies formed from treated cells. E. MCF10A histograms showing colony size distribution following PD170374 treatment. The histogram on the left represents colonies formed from untreated cells, and the histogram on the right represents colonies from cells treated with inhibitor.
Discussion
Elucidation of novel candidate oncogenes from the 8p11-p12 amplicon has been the subject of on-going studies in our laboratory and a number of other laboratories over many years. The 8p11-p12 amplicon occurs in approximately 15% of breast cancers, the majority of which are luminal cancers. An interesting feature of this amplicon is the large number of candidate oncogenes already identified in this the region, and the consistent finding that many of these genes are coordinately amplified and overexpressed in breast cancer and lung cancer [2], [3], [4], [8], [9], [12], [13], [21]. In the present study, we demonstrate that KAT6A is another candidate oncogene from this region. Gelsi-Boyer et al. provided compelling evidence that the 8p11-p12 amplicon can be sub-divided into four distinct regions, each of which can be amplified independently of the others [3]. We have reported previously on candidate oncogenes from region III of this amplicon, which contains genes such as WHSC1L1, DDHD2, and FGFR1[5], [6], [7], [10], [11], [22]. The KAT6A oncogene is in region IV of the amplicon, which is closest to the centromere. KAT6A is co-amplified with region III oncogenes in approximately 7% of cases and amplified independently of this region in approximately 3% of breast cancer cases according to TCGA data.
We found KAT6A to be a consistent hit in an RNAi-based negative selection screen in SUM-52 breast cancer cells. Since KAT6A is both amplified and overexpressed in SUM-52 cells, the identification of KAT6A as a positive screen hit was suggestive of its importance as an oncogene in this cell line. Validation experiments using several KAT6A-specific shRNAs confirmed the screen result and demonstrated a significantly reduced growth rate and reduction in clonogenic potential in knockdown cells compared to non-silencing controls. Similar results were obtained using ovarian cancer cell lines that have KAT6A amplification, but normal mammary epithelial cells, or other breast cancer cells without the 8p11-p12 amplicon were not affected by KAT6A knockdown. Analysis of greater than 1000 primary breast tumors in the TCGA data base showed that KAT6A is amplified in 8.7% of invasive breast cancers, and this amplification is associated with overexpression at the mRNA level. Furthermore, overexpression of KAT6A was found to be associated with late recurrence of breast cancer. Thus, both functional genomics data obtained in our lab and data from the TCGA point to KAT6A as an important breast cancer oncogene.
Cancer cells are heterogeneous, and within any population of cancer cells in a cell line, only a relatively small fraction are considered clonogens based on their ability to form continuously expanding colonies when grown at clonal density, either in monolayer or in soft-agar. In the work reported here, we demonstrate a greater than 10-fold decrease in the clonogenic capacity of SUM-52 cells following knockdown of KAT6A. Colony size distribution shows that the majority of KAT6A knockdown colonies were smaller than the non-silencing control colonies. We interpret these results to indicate that reduction of KAT6A levels not only reduced the clonogenic fraction of SUM52 cells, but also reduced the growth rate of colonies that did form from the remaining clonogens.
We were intrigued by the relationship between the dramatic decrease in colony and mammosphere formation in SUM-52 cells upon knockdown of KAT6A, and the genes regulated by KAT6A. IGF1R, IRS1, and ERBB4 were among the genes reduced in expression as a result of KAT6A knockdown, which is consistent with a decrease in proliferation and colony formation by these cells. Interestingly, a recent publication by Heiser et al. showing the efficacy of 77 compounds in more than 50 breast cancer cell lines showed that the SUM-52 cell line was among the most sensitive to IGF1R inhibition [23], a result consistent with our association between KAT6A overexpression and the IGF1R.
Several interesting transcription factors were also down-regulated following knockdown of KAT6A, including MYC, TBX3, and GATA3. It is known that gene rearrangements involving KAT6A that occur in leukemia results in an inability of cells to differentiate, which is in keeping with the known roles of KAT6A and other MYST family genes in regulation of stem cell self-renewal and differentiation in hematopoiesis [24], [25], [26]. Thus, one mechanism by which KAT6A may affect breast tumorigenesis is by altering self-renewal capacity and differentiation of cells of the luminal mammary lineage. Several of our results are consistent with this hypothesis. Mammosphere formation in SUM-52 KAT6A knockdown cells was not only significantly reduced, but so too was the ability of the cells to form large, branching structures when cultured for extended periods in low-attachment plates in stem cell medium. The down-regulation of key transcription factors associated with mammary gland development in KAT6A knockdown cells is consistent with this observation. C-MYC is a well-known breast cancer oncogene and mechanisms that drive MYC expression in breast cancer cells play a key role in the growth and survival of breast cancer cells [27], [28], [29]. GATA3 is a key transcription factor in mammary gland development and plays a role in determining luminal cell fate [30], [31], and as we and others have shown, KAT6A and other oncogenes of the 8p11 amplicon are over expressed in luminal breast cancers like the SUM-52 cells. Finally, the finding of KAT6A regulation of TBX3 provides a link between breast cancer biology and leukemogenesis induced by this oncogene. KAT6A regulates expression of TBX1 in hematopoietic progenitor cells [26], while TBX3 has been shown to play an essential role in mammary gland development and ductal branching. Furthermore, TBX3 has been shown to be over expressed in a fraction of breast cancers, notably estrogen receptor positive, luminal breast cancers [32]. TBX3 has also been found to be an estrogen responsive gene in MCF-7 cells, and our results show that KAT6A regulates expression of ESR1 in SUM-52 cells. Thus, the genes regulated by KAT6A in SUM-52 cells are all key genes in the development of breast cancers of the luminal lineage, which further implicates KAT6A as a key driver of luminal breast cancer.
KAT6A, also known as monocytic leukemia zinc finger protein (MOZ) or MYST3, belongs to the highly conserved MYST family of histone acetyltransferases, important in post-translational modification of histones and chromatin structure. Currently, the MYST family includes Tip60, MOF, HBO1, MOZ (MYST3), and MORF [22]. The MYST family is known to acetylate lysine residues on histones H4, H3, H2A and H2A variants, and recent evidence suggests that MYST proteins regulate themselves by acetylation. Specifically, KAT6A has acetyltransferase activity towards lysine residues on histones H2B, H3 (K14) and H4 (K5, K8, K12, and K16) [22], [26]. Acetylation of histone H3K9 by KAT6A has been shown in vivo[33]. Although KAT6A is widely-known as an acetyltransferase, some HAT-independent functions have been reported. For example, KAT6A interacts with RUNX1 [34], [35], leading to transcription of hematopoietic genes. KAT6A also interacts with RUNX2 and plays a role in osteogenesis [36], [37] and these functions are independent of HAT activity. KAT6A also interacts with PU.1, important in the development of lymphoid and myeloid lineages, and with MLL, a histone methyl transferase that induces di-and tri-methylation of Histone H3 on lysine 4 [26]. ASH2L, another amplified and over expressed gene from the 8p11 amplicon has been shown to be the histone methylase in the MLL complex. The H3K4 di- and tri-methyl marks are associated with active gene expression, and KAT6A-MLL interactions have been associated with expression of homeobox genes in hematopoietic progenitor cells. Interestingly, two homeobox genes, HOXA11 and HOXA13 were regulated by KAT6A in SUM-52 cells. Thus, in breast cancer cells with the 8p11 amplicon, KAT6A may cooperate with other epigenome modifying oncogenes to aberrantly drive expression of a number of important cancer genes.
The significance of KAT6A in breast cancer is perhaps best reflected in the Kaplan-Meier plot shown in Figure 1F, which demonstrates a significant reduction in overall survival of patients with ER positive breast cancer and KAT6A amplification and overexpression. Not only is there a statistically significant divergence in mortality among patients with KAT6A overexpression, but this affect occurs following approximately 100 months of survival. This suggests that KAT6A plays a role in late recurrence of breast cancer, an issue of unique importance in ER-positive breast cancer. The results reported here suggest that amplification/overexpression of KAT6A could be a useful biomarker for predicting the risk of late recurrence, which could aid in the clinical decisions regarding the duration of anti-estrogen therapy. It is well-accepted that cancer is a disease driven by genetic abnormalities; however, it is becoming clear that alterations in epigenetic pathways play a role in oncogenesis. The identification of KAT6A as a driving oncogene in breast cancers with 8p11-p12 amplification is consistent with the emerging field of cancer epigenetics. Future studies will explore the role of KAT6A in breast cancer self-renewal and mechanisms governing clonogenicity. While there are other putative oncogenes in the amplicon, KAT6A has emerged as an oncogene that plays an important role in the selection of this amplicon in luminal breast cancers.
The following are the supplementary data related to this article.
Methods.
Supplementary Figure S1. RT-PCR data showing relative KAT6A expression in SUM-52 and MCF10A cells. Lentivirus was generated through transfection of 293FT cells using five lentiviral shRNA vectors, each targeting different KAT6A sequences. Cells were lentivirally transduced and selected in puromycin for 3 days before RNA was harvested to confirm knockdown of KAT6A. The shRNA vectors sh1 and sh3 yielded the best knockdown of KAT6A in both cell lines.
Supplementary Figure S2. (A) A soft agar assay with SUM52 non-silencing control and KAT6A knockdown cells was performed to assay for anchorage-independent growth. Results show the number of colonies is reduced upon knockdown of KAT6A. (B) Images of p-Iodonitrotetrazolium violet stained colonies were taken using an EVOS Imaging System. Colonies that formed under KAT6A knockdown are smaller compared to the non-silencing control.
Supplementary Figure S3. (A) RT-PCR data showing relative KAT6A expression in OAW28 and 59M cells. Cells were infected with only sh3, as this shRNA consistently yielded the best KAT6A knockdown. Following lentiviral transduction, cells were selected in puromycin for 3 days before RNA was harvested to confirm knockdown. (B) Western blot showing knockdown of KAT6A at the protein level.
Supplementary Figure S4. (A) CD24/CD44 flow cytometry analysis of SUM-52 non-silencing control and KAT6A knockdown down cells. (B) Table showing the percentage of SUM-52 cells (control and KAT6A knockdown) positive for CD24, CD44, and CD24/CD44 staining.
Complete gene list identified from four replicate RNA sequencing experiments, performed in SUM-52 KAT6A knockdown cells
Supplementary Table 2. Compare sh1 vs sh3 to NS
Supplementary Table 3. Sh1 and sh3
References
- 1.Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nat Genet. 2003;34:369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- 2.Garcia MJ, Pole JC, Chin SF, Teschendorff A, Naderi A, Ozdag H, Vias M, Kranjac T, Subkhankulova T, Paish C. A 1 Mb minimal amplicon at 8p11-12 in breast cancer identifies new candidate oncogenes. Oncogene. 2005;24:5235–5245. doi: 10.1038/sj.onc.1208741. [DOI] [PubMed] [Google Scholar]
- 3.Gelsi-Boyer V, Orsetti B, Cervera N, Finetti P, Sircoulomb F, Rouge C, Lasorsa L, Letessier A, Ginestier C, Monville F. Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol Cancer Res. 2005;3:655–667. doi: 10.1158/1541-7786.MCR-05-0128. [DOI] [PubMed] [Google Scholar]
- 4.Pole JC, Courtay-Cahen C, Garcia MJ, Blood KA, Cooke SL, Alsop AE, Tse DM, Caldas C, Edwards PA. High-resolution analysis of chromosome rearrangements on 8p in breast, colon and pancreatic cancer reveals a complex pattern of loss, gain and translocation. Oncogene. 2006;25:5693–5706. doi: 10.1038/sj.onc.1209570. [DOI] [PubMed] [Google Scholar]
- 5.Yang ZQ, Albertson D, Ethier SP. Genomic organization of the 8p11-p12 amplicon in three breast cancer cell lines. Cancer Genet Cytogenet. 2004;155:57–62. doi: 10.1016/j.cancergencyto.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 6.Yang ZQ, Streicher KL, Ray ME, Abrams J, Ethier SP. Multiple interacting oncogenes on the 8p11-p12 amplicon in human breast cancer. Cancer Res. 2006;66:11632–11643. doi: 10.1158/0008-5472.CAN-06-2946. [DOI] [PubMed] [Google Scholar]
- 7.Streicher KL, Yang ZQ, Draghici S, Ethier SP. Transforming function of the LSM1 oncogene in human breast cancers with the 8p11-12 amplicon. Oncogene. 2007;26:2104–2114. doi: 10.1038/sj.onc.1210002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chin SF, Teschendorff AE, Marioni JC, Wang Y, Barbosa-Morais NL, Thorne NP, Costa JL, Pinder SE, van de Wiel MA, Green AR. High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol. 2007;8:R215. doi: 10.1186/gb-2007-8-10-r215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prentice LM, Shadeo A, Lestou VS, Miller MA, deLeeuw RJ, Makretsov N, Turbin D, Brown LA, Macpherson N, Yorida E. NRG1 gene rearrangements in clinical breast cancer: identification of an adjacent novel amplicon associated with poor prognosis. Oncogene. 2005;24:7281–7289. doi: 10.1038/sj.onc.1208892. [DOI] [PubMed] [Google Scholar]
- 10.Yang ZQ, Moffa AB, Haddad R, Streicher KL, Ethier SP. Transforming properties of TC-1 in human breast cancer: Interaction with FGFR2 and beta-catenin signaling pathways. Int J Cancer. 2007;121:1265–1273. doi: 10.1002/ijc.22831. [DOI] [PubMed] [Google Scholar]
- 11.Yang ZQ, Liu G, Bollig-Fischer A, Haddad R, Tarca AL, Ethier SP. Methylation-associated silencing of SFRP1 with an 8p11-12 amplification inhibits canonical and non-canonical WNT pathways in breast cancers. Int J Cancer. 2009;125:1613–1621. doi: 10.1002/ijc.24518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ray ME, Yang ZQ, Albertson D, Kleer CG, Washburn JG, Macoska JA, Ethier SP. Genomic and expression analysis of the 8p11–12 amplicon in human breast cancer cell lines. Cancer Res. 2004;64:40–47. doi: 10.1158/0008-5472.can-03-1022. [DOI] [PubMed] [Google Scholar]
- 13.Stec I, vOG J, den Dunnen JT. WHSC1L1, on human chromosome 8p11.2, closely resembles WHSC1 and maps to a duplicated region shared with 4p16.3. Genomics. 2001;76:5–8. doi: 10.1006/geno.2001.6581. [DOI] [PubMed] [Google Scholar]
- 14.Borrow J, Stanton VP, Jr., Andresen JM, Becher R, Behm FG, Chaganti RS, Civin CI, Disteche C, Dube I, Frischauf AM. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet. 1996;14:33–41. doi: 10.1038/ng0996-33. [DOI] [PubMed] [Google Scholar]
- 15.Chaffanet M, Gressin L, Preudhomme C, Soenen-Cornu V, Birnbaum D, Pebusque MJ. MOZ is fused to p300 in an acute monocytic leukemia with t(8;22) Genes Chromosomes Cancer. 2000;28:138–144. doi: 10.1002/(sici)1098-2264(200006)28:2<138::aid-gcc2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 16.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–596. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 17.Forozan F, Veldman R, Ammerman CA, Parsa NZ, Kallioniemi A, Kallioniemi O, Ethier SP. Molecular cytogenetic analysis of 11 new human breast cancer cell lines. Br J Cancer. 1999;81:1328–1334. doi: 10.1038/sj.bjc.6695007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ethier SP, Mahacek ML, Gullick WJ, Frank TJ, Weber BL. Cancer Res. 1993:627–635. [PubMed] [Google Scholar]
- 19.Ethier SP. Human breast cancer cell lines as models of growth regulation and disease progression. J Mammary Gland Biol Neoplasia. 1996;1:111–121. doi: 10.1007/BF02096306. [DOI] [PubMed] [Google Scholar]
- 20.Friedrichs K, Franke F, Lisboa BW, Kugler G, Gille I, Terpe HJ, Holzel F, Maass H, Gunthert U. CD44 isoforms correlate with cellular differentiation but not with prognosis in human breast cancer. Cancer Res. 1995;55:5424–5433. [PubMed] [Google Scholar]
- 21.Malchers F, Dietlein F, Schottle J, Lu X, Nogova L, Albus K, Fernandez-Cuesta L, Heuckmann JM, Gautschi O, Diebold J. Cell-autonomous and non-cell-autonomous mechanisms of transformation by amplified FGFR1 in lung cancer. Cancer Discov. 2014;4:246–257. doi: 10.1158/2159-8290.CD-13-0323. [DOI] [PubMed] [Google Scholar]
- 22.Yang XJ, Ullah M. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene. 2007;26:5408–5419. doi: 10.1038/sj.onc.1210609. [DOI] [PubMed] [Google Scholar]
- 23.Heiser LM, Sadanandam A, Kuo WL, Benz SC, Goldstein TC, Ng S, Gibb WJ, Wang NJ, Ziyad S, Tong F. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc Natl Acad Sci U S A. 2012;109:2724–2729. doi: 10.1073/pnas.1018854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perez-Campo FM, Borrow J, Kouskoff V, Lacaud G. The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood. 2009;113:4866–4874. doi: 10.1182/blood-2008-04-152017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez-Campo FM, Costa G, Lie ALM, Stifani S, Kouskoff V, Lacaud G. MOZ-mediated repression of p16 is critical for the self-renewal of neural and hematopoietic stem cells. Stem Cells. 2013;32:1591–1601. doi: 10.1002/stem.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez-Campo FM, Costa G, Lie-a-Ling M, Kouskoff V, Lacaud G. The MYSTerious MOZ, a histone acetyltransferase with a key role in haematopoiesis. Immunology. 2013;139:161–165. doi: 10.1111/imm.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borg A, Baldetorp B, Ferno M, Olsson H, Sigurdsson H. c-myc amplification is an independent prognostic factor in postmenopausal breast cancer. Int J Cancer. 1992;51:687–691. doi: 10.1002/ijc.2910510504. [DOI] [PubMed] [Google Scholar]
- 28.Roux-Dosseto M, Romain S, Dussault N, Desideri C, Piana L, Bonnier P, Tubiana N, Martin PM. c-myc gene amplification in selected node-negative breast cancer patients correlates with high rate of early relapse. Eur J Cancer. 1992;28A:1600–1604. doi: 10.1016/0959-8049(92)90050-c. [DOI] [PubMed] [Google Scholar]
- 29.Deming SL, Nass SJ, Dickson RB, Trock BJ. C-myc amplifi7cation in breast cancer: a meta-analysis of its occurrence and prognostic relevance. Br J Cancer. 2000;83:1688–1695. doi: 10.1054/bjoc.2000.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kouros-Mehr H, Bechis SK, Slorach EM, Littlepage LE, Egeblad M, Ewald AJ, Pai SY, Ho IC, Werb Z. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell. 2008;13:141–152. doi: 10.1016/j.ccr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kouros-Mehr H, Kim JW, Bechis SK, Werb Z. GATA-3 and the regulation of the mammary luminal cell fate. Curr Opin Cell Biol. 2008;20:164–170. doi: 10.1016/j.ceb.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Douglas NC, Papaioannou VE. The T-box transcription factors TBX2 and TBX3 in mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia. 2013;18:143–147. doi: 10.1007/s10911-013-9282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voss AK, Thomas T. MYST family histone acetyltransferases take center stage in stem cells and development. Bioessays. 2009;31:1050–1061. doi: 10.1002/bies.200900051. [DOI] [PubMed] [Google Scholar]
- 34.Kitabayashi I, Aikawa Y, Nguyen LA, Yokoyama A, Ohki M. Activation of AML1-mediated transcription by MOZ and inhibition by the MOZ-CBP fusion protein. EMBO J. 2001;20:7184–7196. doi: 10.1093/emboj/20.24.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida H, Kitabayashi I. Chromatin regulation by AML1 complex. Int J Hematol. 2008;87:19–24. doi: 10.1007/s12185-007-0004-0. [DOI] [PubMed] [Google Scholar]
- 36.Pelletier N, Champagne N, Stifani S, Yang XJ. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene. 2002;21:2729–2740. doi: 10.1038/sj.onc.1205367. [DOI] [PubMed] [Google Scholar]
- 37.Komori T. Regulation of bone development and maintenance by Runx2. Front Biosci. 2008;13:898–903. doi: 10.2741/2730. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods.
Supplementary Figure S1. RT-PCR data showing relative KAT6A expression in SUM-52 and MCF10A cells. Lentivirus was generated through transfection of 293FT cells using five lentiviral shRNA vectors, each targeting different KAT6A sequences. Cells were lentivirally transduced and selected in puromycin for 3 days before RNA was harvested to confirm knockdown of KAT6A. The shRNA vectors sh1 and sh3 yielded the best knockdown of KAT6A in both cell lines.
Supplementary Figure S2. (A) A soft agar assay with SUM52 non-silencing control and KAT6A knockdown cells was performed to assay for anchorage-independent growth. Results show the number of colonies is reduced upon knockdown of KAT6A. (B) Images of p-Iodonitrotetrazolium violet stained colonies were taken using an EVOS Imaging System. Colonies that formed under KAT6A knockdown are smaller compared to the non-silencing control.
Supplementary Figure S3. (A) RT-PCR data showing relative KAT6A expression in OAW28 and 59M cells. Cells were infected with only sh3, as this shRNA consistently yielded the best KAT6A knockdown. Following lentiviral transduction, cells were selected in puromycin for 3 days before RNA was harvested to confirm knockdown. (B) Western blot showing knockdown of KAT6A at the protein level.
Supplementary Figure S4. (A) CD24/CD44 flow cytometry analysis of SUM-52 non-silencing control and KAT6A knockdown down cells. (B) Table showing the percentage of SUM-52 cells (control and KAT6A knockdown) positive for CD24, CD44, and CD24/CD44 staining.
Complete gene list identified from four replicate RNA sequencing experiments, performed in SUM-52 KAT6A knockdown cells
Supplementary Table 2. Compare sh1 vs sh3 to NS
Supplementary Table 3. Sh1 and sh3