

Abstract
Hepatocellular carcinoma (HCC) often results from chronic liver injury and severe fibrosis or cirrhosis, but the underlying molecular pathogenesis is unclear. We previously reported that deletion of glucose regulated protein 94 (GRP94), a major endoplasmic reticulum chaperone, in the bone marrow and liver leads to progenitor/stem cell expansion. Since liver progenitor cell (LPC) proliferation can contribute to liver tumor formation, here we examined the effect of GRP94 deficiency on spontaneous liver tumorigenesis. Utilizing liver-specific Grp94 knockout mice driven by Albumin-Cre (cGrp94f/f), we discovered that while wild-type livers are tumor free up to 24 months, cGrp94f/f livers showed abnormal small nodules at 15 months and developed HCC and ductular reactions (DRs) by 21 months of age, associating with increased liver injury, apoptosis and fibrosis. cGrp94f/f livers were progressively repopulated by GRP94-positive hepatocytes. At 15 months, we observed expansion of LPCs and mild DRs, as well as increase in cell proliferation. In examining the underlying mechanisms for HCC development in cGrp94f/f livers, we detected increase in TGF-β1, activation of SMAD2/3, ERK, and JNK, and cyclin D1 upregulation at the premalignant stage. While epithelial-mesenchymal transition (EMT) was not evident, E-cadherin expression was elevated. Correlating with the recurrence of GRP94 positive-hepatocytes, the HCC was found to be GRP94-positive, whereas the expanded LPCs and DRs remained GRP94-negative. Collectively, this study uncovers that GRP94 deficiency in the liver led to injury, LPC expansion, increased proliferation, activation of oncogenic signaling, progressive repopulation of GRP94-positive hepatocytes and HCC development in aged mice.
Abbreviations: Alb, albumin; ALP, alkaline phosphatase; α-SMA, α-smooth muscle actin; ALT, alanine aminotransferase; CC, cholangiocarcinoma; CV, central vein; DR, ductular reaction; E-cadherin, epithelial cadherin; EMT, epithelial-mesenchymal transition; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; GRP94, glucose-regulated protein 94; GS, glutamine synthetase; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HepPar1, Hepatocyte Paraffin 1; HSP90, heat shock protein 90; IGF, insulin-like growth factor; IHC, immunohistochemistry; JNK, c-Jun N-terminal kinase; LPC, liver progenitor cell; panCK, pan-cytokeratin; PTEN, phosphatase and tensin homolog deleted on chromosome 10; PV, portal vein; TGF-β, transforming growth factor β; TIC, tumor initiating cell.
Introduction
Glucose-regulated protein (GRP94), encoded in humans by HSP90β1, shares 50% amino acid homolog with the heat shock protein HSP90 [1], [2]. It is an essential endoplasmic reticulum (ER) chaperone, assisting in protein folding, processing and secretion of selective clients. As such, GRP94 uniquely controls specific pathways critical for cell adhesion, proliferation, and organ homeostasis [3], [4], [5]. GRP94 also maintains Ca2+ homeostasis in the ER and protects cancer cells from apoptosis [6]. GRP94 can translocate to the cell surface serving additional functions, including modulating immune responses [2]. Recently, GRP94 has been implicated in tumorigenesis with differential outcomes in a context dependent manner [2], [7], [8], [9]. Thus, it is important to study the role of GRP94 in additional cancer models and how it affects oncogenic signaling in vivo.
Liver cancer is the fifth most common cancer worldwide, among which hepatocellular carcinoma (HCC) accounts for 70-85% of total cancer burden [10]. Late diagnosis, recurrence, and metastasis result in a poor prognosis of HCC, and the 5-year survival rate of patients undergoing surgical treatment is low [11]. HCC often occurs in steatohepatitis or cirrhosis background [11], [12] but the molecular pathogenesis of cancer development remains elusive. Thus, it is important to understand liver tumorigenic mechanisms so that new treatments can be developed.
Liver regeneration is normally carried out by hepatocytes and cholangiocytes (bile duct cells). However, during chronic liver injury and cirrhosis, the dividing ability of these parenchymal cells is compromised and activation of liver progenitor cells (LPCs) is observed [12]. Due to its bi-potential, LPCs residing in the niche, located in the most peripheral branches of the biliary tree, can differentiate into hepatocytes and cholangiocytes to restore the liver function [12]. Recently, the concept that liver cancer originates from liver tumor-initiating cells (TICs), which can be derived from normal LPCs, received much attention. Liver cancer with LPC characteristics is particularly aggressive [13] and deregulated LPCs have been shown to obtain tumor-initiating ability in vivo [14], [15], suggesting that LPCs might be the origin of at least part of liver cancer. Perturbation of oncogenic signaling is central to tumorigenesis through their effects on proliferation, apoptosis, and invasion. For example, human HCC overexpresses TGF-β [16], which has been shown to transform LPCs to TICs [17]. Other kinases reported to be upregulated in HCC include JNK, AKT and ERK, with the latter associated with increased proliferation of LPCs [18], [19], [20], [21].
We previously identified GRP94 as a novel regulator of cell adhesion, LPCs, and liver tumorigenesis [7]. By using Albumin-Cre (Alb-Cre) system, we generated the liver-specific Grp94 knockout mouse model (Grp94f/f; Alb-Cre or cGrp94f/f), and found GRP94 deficiency leads to hyperproliferation of LPCs with minor liver injury, correlating with impaired cell adhesion. Deletion of both Grp94 and Pten in the liver (cPtenf/fGrp94f/f) accelerates HCC and cholangiocarcinoma (CC) development, and selectively activates ERK. Since expanded LPCs might become TICs, it is interesting to determine whether GRP94 ablation alone is sufficient to induce spontaneous liver tumors. This study revealed liver injury and HCC formation in aged cGrp94f/f mice with progressive repopulation of GRP94-positive hepatocytes, upregulation of TGF-β1 concurrent with LPC expansion, as well as activation of SMAD2/3, ERK, and JNK prior to the onset of liver fibrosis and tumors. This study expands our knowledge of the biological functions of GRP94 in liver homeostasis and cancer progression.
Materials and Methods
Mice
Grp94f/f; Alb-Cre mice on a mixed C57BL/6; 129/Sv; 6xDBA2; 129 background were generated and genotyping was performed as previously described [7]. Littermates lacking Alb-Cre served as WT controls. Blood samples were collected through retro-orbital bleeding. All protocols for animal use were reviewed and approved by the USC Institutional Animal Care and Use Committee.
Histology and Immunostaining
Mouse liver tissues were fixed in 10% zinc formalin, embedded in paraffin, sectioned at 4 μm, and subjected to H&E staining and immunostaining as described [22]. Monoclonal rat anti-GRP94 (1:200) is from Enzo Life Sciences (Farmingdale, NY). Cell proliferation was evaluated by Ki67 staining (SP-6, 1:50, Thermo Scientific, Waltham, MA). Monoclonal mouse anti-glutamine synthetase (GS) (1:400, BD Biosciences, San Jose, CA) was used to examine liver zonation. Monoclonal mouse anti-hepatocyte paraffin 1 (HepPar1) (1:25, DakoCytomation, Denmark A/S) and polyclonal rabbit anti-wide spectrum cytokeratin (panCK) (1:75, Abcam, Cambridge, MA) antibodies were used to identify hepatocytes and cholangiocytes/LPCs, respectively. Mesenchymal cells were detected by monoclonal mouse anti-α-SMA (1:2000, Sigma, St. Louis, MO). Monoclonal rat anti-CD34 (1:100) is from BioLegend (San Diego, CA).
Plasma Biochemistry
Plasma alanine aminotransferase (ALT) was determined using ALT Reagent (Raichem, San Diego, CA). Plasma alkaline phosphatase (ALP) was measured following manufacturer’s instruction (Thermo Scientific, Waltham, MA).
TUNEL Assay
Apoptosis was determined using TUNEL staining (Roche Diagnostics, Mannheim, Germany).
Western Blot Analysis
Harvested tissues were frozen immediately, and homogenized in RIPA buffer with added protease and phosphatase inhibitor cocktail (Thermo Scientific, Waltham, MA). Cell lysates (25–30 μg) were subjected to SDS-PAGE and Western blot analysis as described [22]. The primary antibodies used are as follows. Monoclonal rat anti-GRP94 (1:5000) is from Enzo Life Sciences (Farmingdale, NY). Rabbit anti-ERK1/2 (1:1000), mouse anti-p-ERK1/2 (Thr202/Tyr204, E10, 1:1000), rabbit anti-AKT (1:1000), and rabbit anti-p-AKT (Ser473, 1:1000) are from Cell Signaling (Danvers, MA). Goat anti-SMAD2/3 (N-19, 1:1000), goat anti-p-SMAD2/3 (Ser423/425, 1:1000), rabbit anti-JNK (FL, 1:1000), mouse anti-p-JNK (Thr183/Tyr185, G7, 1:1000), and rabbit anti-β-catenin (H-102, 1:1000) are from Santa Cruz Biotechnology (Dallas, TX). Monoclonal rabbit anti-Cyclin D1 (SP4, 1:1000) is from Thermo Scientific (Waltham, MA). Monoclonal mouse anti-E-cadherin (1:1000) is from BD Biosciences (San Jose, CA). Monoclonal rabbit anti-integrin β1 (clone EP1041Y, 1:1000) and monoclonal mouse anti-active-β-catenin (anti-ABC) (clone 8E7, 1:1000) are from Millipore (Billerica, MA). Monoclonal rabbit anti-vimentin (clone EPR3776, 1:1000) is from Epitomics (Burlingame, CA). Mouse anti-β-actin (1:5000) is from Sigma (St. Louis, MO).
Fluorescent Microscopy
Immunofluorescence was visualized by Zeiss LSM 510 confocal microscope equipped with LSM 510 Version 4.2 SP1 acquisition software (Carl Zeiss). Confocal images were obtained with 40X oil lens. Images were then processed with LSM Image Browser R4.2 and Adobe Photoshop CS5.
Statistical Analysis
Statistical significance was assayed by 2-tailed Student’s t test, and the error bars reflect standard error (s.e.).
Results
Spontaneous Liver Tumor Development in Aged cGrp94f/f Mice
In following the effect of long-term GRP94 deficiency on the mouse liver, we observed that while wild-type (WT) livers were normal and tumor free at least up to 24 months, the livers of Grp94f/f; Alb-Cre mice (referred to as cGrp94f/f) developed small nodules at 15 months and prominent liver tumors by 21 months (Figure 1A and B). As noted previously [7], the cGrp94f/f liver weight relative to the whole body of mice at 2 and 9 months were lower than the WT. However, consistent with tumor development, this decrease was lower at 15 months and by 21 months, cGrp94f/f livers were 1.6-fold higher than the WT (Figure 1C). To examine liver injury in cGrp94f/f mice, we measured levels of plasma alanine aminotransferase (ALT) and alkaline phosphatase (ALP). While there was no increase in these injury markers at 9 months, a trend of mildly elevated ALT and ALP was detected in cGrp94f/f mice at 15 months, and substantial increase in ALT and ALP was observed at 21 months, concurrent with tumor formation (Figure 1D). cGrp94f/f livers exhibited an about 1.6-fold increase in apoptosis at 9 and 15 months, and a 2.7-fold increase at 21 months; however, the percentage of apoptotic cells remained below 0.6% at 21 months (Figure 1E).
Figure 1.
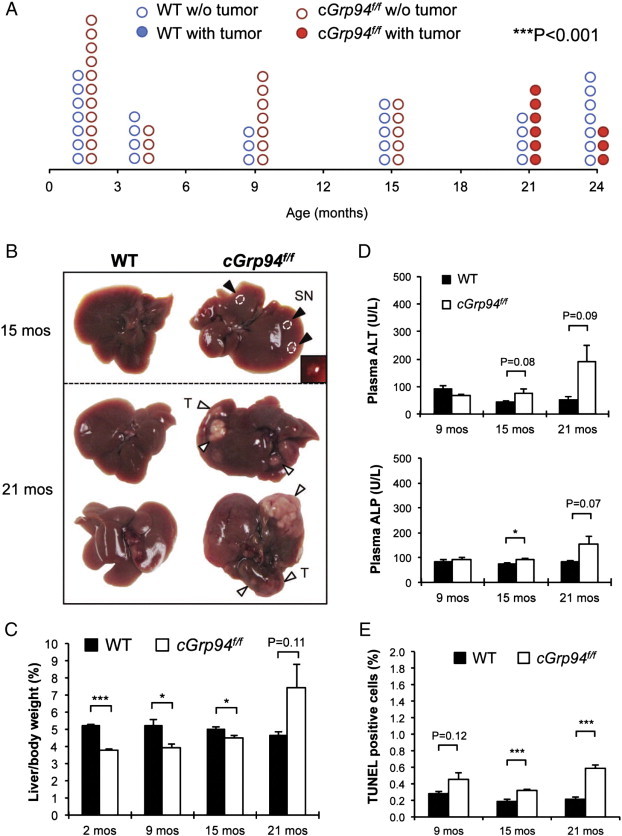
Spontaneous liver tumor formation in aged cGrp94f/f mice. (A) Liver tumor spectrum. Each circle represents one mouse. The solid and open circles represent mice with and without tumors, respectively (***P < 0.001, χ2 test). (B) Representative liver pictures at 15 and 21 months. Black arrowheads indicate small nodules (SN). The inset shows the enlarged SN. White arrowheads denote tumors (T) on the liver surface. (C) Liver weight to body weight percentage in WT and cGrp94f/f mice at indicated ages. (D) Plasma ALT and ALP measurements. (E) Quantitation of TUNEL-positive cells in cGrp94f/f livers at indicated ages. All data are presented as mean ± s.e. (*P < 0.05 and ***P < 0.001).
Progressive Repopulation of cGrp94f/f Livers with GRP94-Positive Hepatocytes
Recent studies revealed that in conditional knockout models of ER chaperone in specific tissues, repopulation with non-recombined cells expressing the chaperone protein could occur [23], [24], [25]. To test this, we first examined the expression pattern of GRP94 in liver tissues from WT and cGrp94f/f mice by immunohistochemistry (IHC) staining. Our results showed that while GRP94 expression was largely ablated in cGrp94f/f livers at 2 months of age, by 9 months patches of the liver exhibited positive GRP94 staining which intensified progressively from 15 to 21 months, with most of the GRP94-positive cells being hepatocytes (Figure 2A). Western blot of liver lysates isolated from WT and cGrp94f/f mice confirmed gradual repopulation of GRP94-positive cells in cGrp94f/f livers as the mice aged, reaching about 40% of the WT level at 15 months (Figure 2B). At 21 months, the level of GRP94 in cGrp94f/f liver tumor lysates was further elevated at about 60% of the WT level (Figure 2B).
Figure 2.
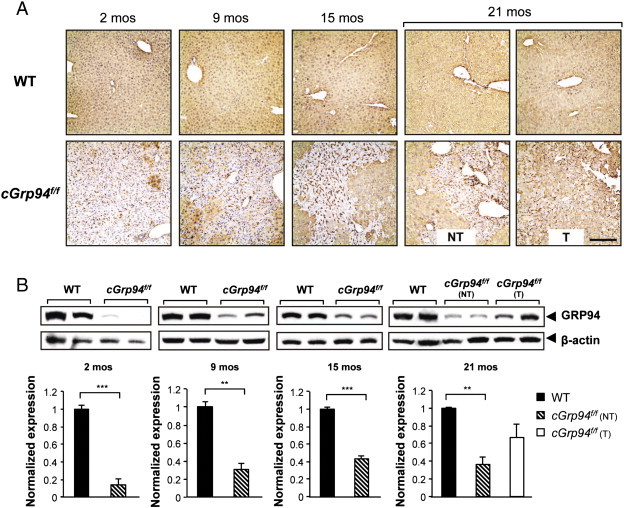
Progressive repopulation of GRP94-positive hepatocytes in cGrp94f/f livers. (A) GRP94 IHC in WT and cGrp94f/f livers at indicated ages. NT: non-tumor tissue. T: tumor tissue. Scale bar: 200 μm. (B) Western blot of liver lysates to detect GRP94 at indicated ages. NT: liver lysates from non-tumor tissues. T: liver lysates from tumor tissues. β-actin served as the loading control and corresponding quantifications are shown on the bottom. Data are presented as mean ± s.e. (**P < 0.01 and ***P < 0.001).
Increased Proliferation Preceding Hepatocellular Carcinoma Formation and Ductular Reactions in Livers of Aged cGrp94f/f Mice
Histological analysis of cGrp94f/f livers at 15 months showed mild fat accumulation, and the small nodules in these livers exhibited a combination of fat and hydropic change as evidenced by extensive cellular swelling (Figure 3A). IHC staining revealed patchy GRP94 expression in cGrp94f/f livers with even higher level of repopulation of GRP94-positive cells in the small nodules, correlating with increased proliferation as revealed by Ki67 staining (Figure 3A). Double staining of GRP94 and Ki67 confirmed that Ki67-positive cells were GRP94-positive (Figure 3B).
Figure 3.
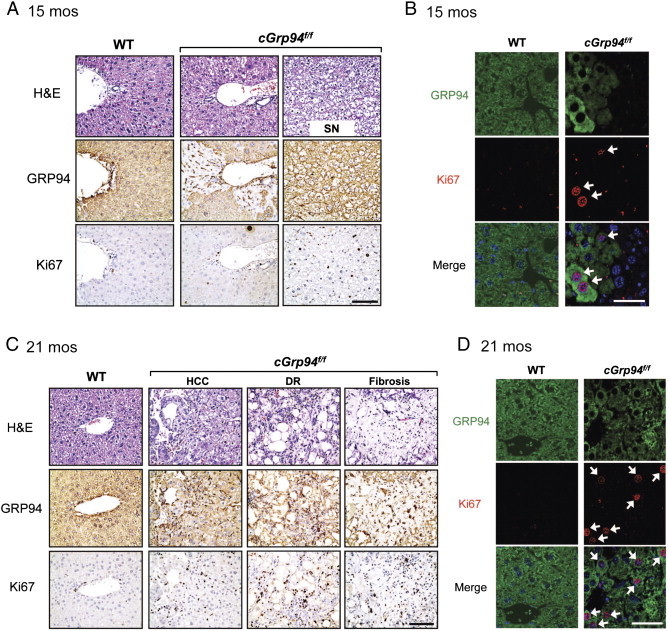
Increased proliferation and development of ductular reactions (DRs), fibrosis, and HCC in cGrp94f/f livers. (A) Liver H&E staining, GRP94, and Ki67 IHC at 15 months. SN: small nodule. Scale bar: 100 μm. (B) Immunofluorescent staining with GRP94 (green) and K67 (red) at 15 months. White arrows denote double-positive cells. Nuclei were stained with DAPI (blue). Scale bar: 25 μm. (C) Same as A but at 21 months. (D) Same as B but at 21 months.
At 21 months of age, a trabecular pattern of HCC was evident and ductular reactions (DRs) were also detected in the tumor region of cGrp94f/f livers (Figure 3C). DR is the expansion of activated biliary epithelial cells in response to injury, and contains LPCs as well as a complex of ECM, inflammatory cells, and mesenchymal cells [26], [27]. Additionally, fibrosis was detected in the liver tumors in 50% of cGrp94f/f mice (Figure 3C). IHC staining revealed GRP94 expression and markedly higher Ki67 staining in cGrp94f/f livers (Figure 3C). GRP94 and Ki67 double staining further showed that most proliferating cells expressed GRP94 in cGrp94f/f livers (Figure 3D).
To characterize cGrp94f/f liver tumors, IHC staining of HepPar1, panCK, and α-SMA was performed on consecutive liver slides. While HepPar1 stained hepatocytes of WT livers at a low basal level, strong HepPar1 staining was detected in HCC which exhibited clear trabecular structures (Figure 4A). panCK staining marked bile ducts in the WT and highlighted DR areas in cGrp94f/f livers (Figure 4A). Interestingly, some HepPar1-labeled cells in HCC were also co-stained by panCK, raising the possibilities that they might come from the common progenitors, or hepatocytes might transdifferentiate into bile duct cells in the aged liver undergoing tumorigenesis. In WT livers, α-SMA stained smooth muscle cells in vessel walls. In contrast, many α-SMA-positive stellate cells were detected in cGrp94f/f tumors (Figure 4A) indicating stellate cell activation. Stellate cells represent the major cell type for matrix production, resulting in liver fibrosis [28] observed in cGrp94f/f livers at 21 months (Figure 3C). Furthermore, the development of HCC in cGrp94f/f livers was confirmed by strong CD34 staining, which showed endothelial cells surrounding the trabeculae (Figure 4B). Staining of glutamine synthetase (GS), a liver zonation marker expressed in about 50% of HCC [29], was also performed. GS staining labeled central vein (CV)-associated hepatocytes in the WT but revealed decreased spacing between GS-positive regions in cGrp94f/f livers at both 15 and 21 months. Increased GS expression with an irregular distribution was also observed in cGrp94f/f livers at 21 months, consistent with HCC formation (Figure 4C).
Figure 4.
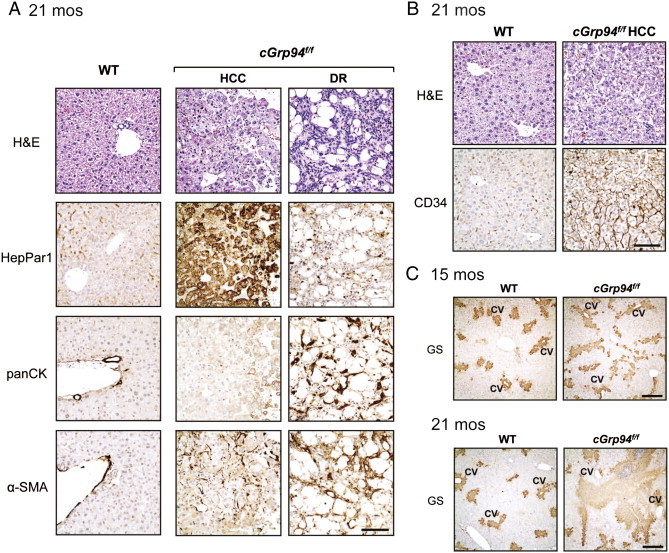
Characteristics of cGrp94f/f liver tumors. (A) H&E staining, HepPar1, panCK, and α-SMA IHC in WT livers and cGrp94f/f liver tumors at 21 months. Scale bar: 100 μm. (B) H&E and CD34 IHC in WT livers and cGrp94f/f HCC at 21 months. Scale bar: 100 μm. (C) GS IHC in livers at indicated ages. CV: central vein. Scale bar: 300 μm.
Activation of Oncogenic Signaling in Premalignant cGrp94f/f Livers
To identify potential molecular mechanisms contributing to tumorigenesis in aged cGrp94f/f mice, we examined key oncogenic pathways implicated in HCC in premalignant livers at 15 months, prior to the tumor onset. Our analysis showed prominent upregulation of TGF-β1 (Figure 5A), one of the most potent activators for stellate cell activation and liver fibrosis [28]. We also observed elevated level of its downstream target p-SMAD3 and a mild increase in p-SMAD2 (Figure 5A). This is in agreement with SMAD3 playing a more important role than SMAD2 in the development of liver fibrosis [30]. Additionally, we observed elevation of p-ERK and p-JNK, but not p-AKT in cGrp94f/f livers (Figure 5B). The expression of the cell cycle regulator cyclin D1 was strikingly increased (Figure 5B), consistent with higher proliferation of cGrp94f/f livers prior to tumor development.
Figure 5.
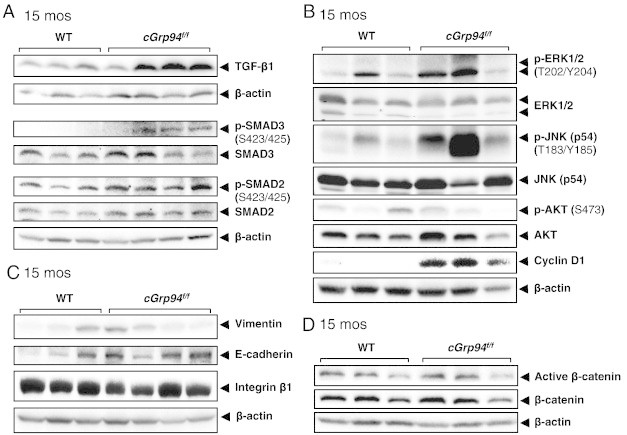
Activation of oncogenic signaling in premalignant cGrp94f/f livers at 15 months. (A) Representative Western blots of liver lysates for TGF-β1 and SMAD2/3. (B) Representative Western blots for the indicated kinases and cyclin D1. (C) Representative Western blots for vimentin, E-cadherin and integrin β1. (D) Representative Western blots for total and active β-catenin.
Since TGF-β is an inducer of epithelial-mesenchymal transition (EMT), which may contribute to tumorigenesis [31], EMT markers vimentin and E-cadherin were examined. We detected strong upregulation of the epithelial marker E-cadherin; however, there was no apparent difference in the levels of the mesenchymal marker vimentin, which displayed similar variation (Figure 5C). TGF-β signaling can also activate integrin β1 to induce EMT [32]. However, similar integrin β1 expression levels were detected in WT and cGrp94f/f livers (Figure 5C). Thus, despite increased TGF-β1 in premalignant cGrp94f/f livers, EMT was not evident. Recently, it has been reported that GRP94 is an essential chaperone of the Wnt co-receptor LRP6, required for Wnt/β-catenin signaling [4]. Nevertheless, the levels of both total and active β-catenin were similar in WT and cGrp94f/f livers (Figure 5D).
Expansion of GRP94-Negative Liver Progenitor Cells and Ductular Reactions in cGrp94f/f Mice
We previously demonstrated that liver-specific deletion of Grp94 leads to LPC proliferation [7], and bi-potential LPCs could give rise to liver tumors [12]. To address the origin of GRP94-positive HCC in cGrp94f/f mice, double staining of GRP94 and the LPC/bile duct marker panCK was performed on liver sections at 21 months. panCK labeled LPC/bile duct cells as well as DRs, and while WT bile duct cells co-expressed GRP94 and panCK, most LPC/bile ducts in cGrp94f/f livers were devoid of GRP94 expression (Figure 6A and B). Moreover, in agreement with DRs in cGrp94f/f livers at 21 months, we observed increased panCK-positive cells, many of which migrated away from portal veins, suggesting expansion of LPCs in cGrp94f/f livers (Figure 6A). LPC expansion was also detected in cGrp94f/f livers from 2 to 15 months of age, while DRs were observed at 15 months (Figure 6C). Quantitation of the stained cells confirmed that in contrast to the WT, almost all panCK-positive cells in cGrp94f/f livers were GRP94-negative (Figure 6D). Collectively, these results showed expansion of GRP94-negative LPCs in cGrp94f/f livers, giving rise to the GRP94-negative DRs in aged cGrp94f/f mice. Since HCC was mostly GRP94-positive and LPCs were mostly GRP94-negative, it is unlikely that HCC was primarily derived from LPCs.
Figure 6.
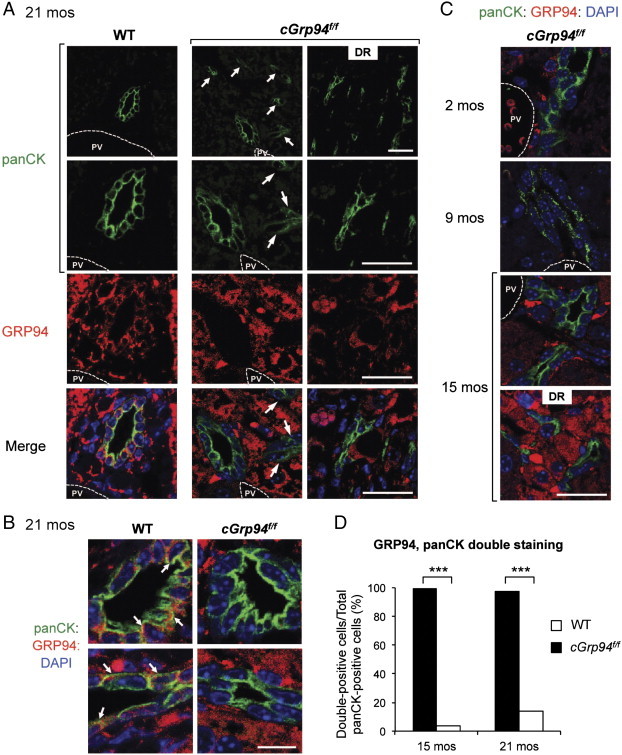
Expansion of GRP94-negative LPCs in cGrp94f/f livers. (A) Immunofluorescent staining of WT and cGrp94f/f livers at 21 months with panCK (green) and GRP94 (red). Nuclei were stained with DAPI (blue). White arrows denote panCK-positive cells migrating away from the portal vein (PV). Scale bars: 25 μm. (B) Magnified panCK-positive cells stained for GRP94 in WT and cGrp94f/f livers at 21 months. White arrows indicate examples of panCK, GRP94 double-positive cells. Scale bar: 10 μm. (C) Immunofluorescent staining of panCK (green) and GRP94 (red) in cGrp94f/f livers at 2, 9, and 15 months. Scale bar: 25 μm. (D) Quantitation of panCK, GRP94 double-positive cells (***P < 0.001, χ2 test).
Discussion
Based on the recent discovery that Grp94 knockout in the liver and bone marrow leads to progenitor/stem cell expansion [7], [33], [34], and such cells can fuel tumorigenesis, here we investigate whether GRP94 ablation alone is sufficient to induce spontaneous liver tumor formation. In this study, we provide the first demonstration that as cGrp94f/f mice aged, abnormal small nodules surfaced at 15 months and aggressive HCC was evident by 21 months, associating with liver injury, LPC expansion and repopulation of GRP94-positive hepatocytes. As summarized in Figure 7, we observed that at the early stage, GRP94 level was 10-30% of the WT and most hepatocytes and LPCs were GRP94-negative. At 15 months, GRP94 expression reverted back to 40% of the WT level. HCC developed at 21 months was mostly GRP94-positive, whereas most DRs remained GRP94-negative. This implies that the tumor development in cGrp94f/f livers is not due to the lack of GRP94 in tumor cells, but rather caused by some cell autonomous and/or non-autonomous events at the premalignant stage triggered by GRP94 deficiency.
Figure 7.
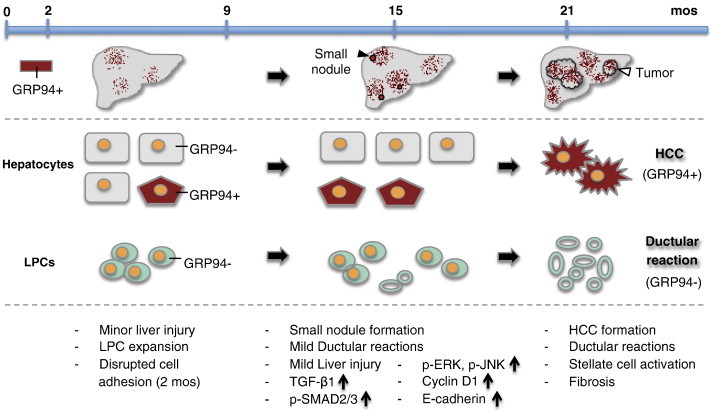
Schematic model of GRP94 expression pattern and liver tumor development in cGrp94f/f mice. The major phenotypes at each stage are listed below.
Since GRP94 possesses anti-apoptosis properties [6], [35] and is required for the processing and secretion of IGF-1, which is protective against liver injury [36], [37], we measured liver injury and apoptosis. We detected a limited elevation in ALT and ALP at 15 months prior to the onset of tumorigenesis, and a general increase in apoptosis spanning from 9 to 21 months. While these effects are modest, chronic exposure to mild liver injury during the aging process, when other mutations could accumulate, may trigger the adaptive response of expanding LPCs, which was observed in cGrp94f/f mice.
In examining the alteration of signaling pathways in premalignant cGrp94f/f livers, we observed concurrent increase in LPCs and TGF-β1 level, associating with SMAD2/3 activation. While activation of SMAD2/3 stimulates stellate cell activation and fibrosis [30], establishing a tumor reactive stroma [38], TGF-β in the microenvironment maintains TICs [31]. Furthermore, TGF-β exposure under chronic liver damage can induce transformation of LPCs, giving rise to liver TICs [17]. Thus, it is possible that TGF-β1 contributes to the generation of DRs from transformed LPCs in cGrp94f/f livers. Paradoxically, TGF-β is also anti-proliferative in most epithelial cells, including hepatocytes, but cancer cells can escape the tumor suppressor function of TGF-β [31]. Therefore, GRP94-positive hepatocytes might develop mechanisms to become resistant to TGF-β1-mediated growth inhibition. Concurrent gene alterations can also influence whether TGF-β promotes or suppresses tumor growth. For example, in the setting of p53 loss, TGF-β signaling enhances liver tumorigenesis [39]. It will be interesting to dissect the factors in aging cGrp94f/f mice that synergize with TGF-β signaling in liver tumor development.
Both ERK and JNK activation have been reported in HCC [18], [19]. JNK activity in HCC is significantly correlated with stem cell marker CD133 expression [20]. Similarly, ERK pathway is associated with aggressive tumor behaviors and promotes proliferation of Sca-1-positive LPCs [18], [21]. Although ERK activation was not detected in young cGrp94f/f mice at 2 months, we also observed increased p-ERK in cPtenf/fGrp94f/f livers, which formed mixed lineage tumors at 8–9 months [7]. Additionally, E-cadherin has been suggested as a tumor suppressor in various cancers and E-cadherin overexpression inhibits proliferation and invasiveness [40]. However, recent studies indicate that its role might be more complex, such that loss or upregulation of E-cadherin levels identify two distinct categories of HCC in both humans and transgenic mice [41], [42]. Overexpression of E-cadherin has been linked to invasive potential of tumor cells, including HCC [41], [43], [44], [45]. Further studies are required to address the causative roles of these pathways in GRP94-mediated liver tumors.
The differential GRP94 expression in HCC and DRs in cGrp94f/f mice raises the intriguing question on the cell origin of cGrp94f/f tumors. We speculate that since cells in the small nodules at 15 months were GRP94-positive hepatocytes with high proliferative activity, they could gain growth advantage, giving rise to the GRP94-positive HCC (Figure 7). Although LPCs also can contribute to HCC, due to GRP94 deletion in most cGrp94f/f LPCs, they are more likely to be the origin of GRP94-negative DRs in the liver tumors rather than the GRP94-positive HCC. Nonetheless, given the recent report that hepatocytes can undergo cellular conversion to biliary epithelial cells following injury [46], we cannot rule out that DRs might be derived from a subfraction of hepatocytes that are GRP94-negative. Likewise, a small fraction of GRP94-positive LPCs, which escape Cre at an earlier stage, might generate HCC. Future studies using specific cell lineage markers will be required to resolve this and our cGrp94f/f model may offer such an opportunity.
Lastly, in agreement with GRP94 expression in cGrp94f/f HCC cells, progressive repopulation of GRP94-positive cells was also observed in some cPtenf/fGrp94f/f HCC at 8–12 months (our unpublished results). GRP94 is upregulated in HBV-induced human HCC correlating with its progression [47], [48], and in Pten-null mediated mouse liver cancer (data not shown). Nonetheless, conditional knockout of GRP94 in the liver resulted in HCC in aged mice. How can these observations be reconciled? We hypothesize that in specific organs, such as the liver, where loss of GRP94 leads to LPC activation, tumorigenesis could occur in combination with other carcinogenic events activated during the aging process. Furthermore, there is generally a gain of GRP function in cancers in response to stress associated with tumorigenesis [2], and the anti-apoptotic function of GRP94 could confer survival advantage to cancer cells. Interestingly, GRP94-specific inhibitors have been identified [49], [50], and one of them reduces viability of HER2-overexpressing breast cancer cells and multiple myeloma cells in vitro[8], [49]. Given that hepatic GRP94 deficiency for 9 months in the mice leads to only minor liver injury and apoptosis, short-term treatment of cancers with these inhibitors might have minimal harmful effects on normal livers while reducing GRP94 activity in HCC.
Acknowledgments
We like to thank Dr. Bangyan Stiles, Dr. Keigo Machida, and members of the Lee laboratory for helpful discussions as well as Dr. Susan Groshen and Dr. Dongyun Yang for Chi-square test analysis. We also like to thank the USC Norris Comprehensive Cancer Center Translational Pathology Core Facility supported by NCI grant P30 CA014089 for histology and the Tissue Imaging Core Facility of the USC Research Center for Liver Diseases (P30 DK048522) for microscopy. This work was supported in part by funding from the National Institutes of Health (R01 CA027607 and P01 AG034906) to ASL.
Contributor Information
Wan-Ting Chen, Email: chenwant@usc.edu.
Dat Ha, Email: datha@usc.edu.
Gary Kanel, Email: gckanel@aol.com.
Amy S. Lee, Email: amylee@usc.edu, amylee@usc.edu.
References
- 1.Ni M., Lee A.S. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee A.S. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eletto D., Dersh D., Argon Y. GRP94 in ER quality control and stress responses. Semin Cell Dev Biol. 2010;21:479–485. doi: 10.1016/j.semcdb.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu B., Staron M., Hong F., Wu B.X., Sun S., Morales C., Crosson C.E., Tomlinson S., Kim I., Wu D. Essential roles of grp94 in gut homeostasis via chaperoning canonical Wnt pathway. Proc Natl Acad Sci U S A. 2013;110:6877–6882. doi: 10.1073/pnas.1302933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maynard J.C., Pham T., Zheng T., Jockheck-Clark A., Rankin H.B., Newgard C.B., Spana E.P., Nicchitta C.V. Gp93, the Drosophila GRP94 ortholog, is required for gut epithelial homeostasis and nutrient assimilation-coupled growth control. Dev Biol. 2010;339:295–306. doi: 10.1016/j.ydbio.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reddy R.K., Lu J., Lee A.S. The endoplasmic reticulum chaperone glycoprotein GRP94 with Ca2+-binding and antiapoptotic properties is a novel proteolytic target of calpain during etoposide-induced apoptosis. J Biol Chem. 1999;274:28476–28483. doi: 10.1074/jbc.274.40.28476. [DOI] [PubMed] [Google Scholar]
- 7.Chen W.T., Tseng C.C., Pfaffenbach K., Kanel G., Luo B., Stiles B.L., Lee A.S. Liver-specific knockout of GRP94 in mice disrupts cell adhesion, activates liver progenitor cells, and accelerates liver tumorigenesis. Hepatology. 2014;59:947–957. doi: 10.1002/hep.26711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hua Y., White-Gilbertson S., Kellner J., Rachidi S., Usmani S.Z., Chiosis G., Depinho R., Li Z., Liu B. Molecular chaperone gp96 is a novel therapeutic target of multiple myeloma. Clin Cancer Res. 2013;19:6242–6251. doi: 10.1158/1078-0432.CCR-13-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morales C., Rachidi S., Hong F., Sun S., Ouyang X., Wallace C., Zhang Y., Garret-Mayer E., Wu J., Liu B. Immune chaperone gp96 drives the contributions of macrophages to inflammatory colon tumorigenesis. Cancer Res. 2014;74:446–459. doi: 10.1158/0008-5472.CAN-13-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 11.Bruix J., Boix L., Sala M., Llovet J.M. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5:215–219. doi: 10.1016/s1535-6108(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 12.Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818–3822. doi: 10.1038/sj.onc.1209558. [DOI] [PubMed] [Google Scholar]
- 13.Lee J.S., Heo J., Libbrecht L., Chu I.S., Kaposi-Novak P., Calvisi D.F., Mikaelyan A., Roberts L.R., Demetris A.J., Sun Z. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 14.Chiba T., Zheng Y.W., Kita K., Yokosuka O., Saisho H., Onodera M., Miyoshi H., Nakano M., Zen Y., Nakanuma Y. Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gastroenterology. 2007;133:937–950. doi: 10.1053/j.gastro.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 15.Chiba T., Seki A., Aoki R., Ichikawa H., Negishi M., Miyagi S., Oguro H., Saraya A., Kamiya A., Nakauchi H. Bmi1 promotes hepatic stem cell expansion and tumorigenicity in both Ink4a/Arf-dependent and -independent manners in mice. Hepatology. 2010;52:1111–1123. doi: 10.1002/hep.23793. [DOI] [PubMed] [Google Scholar]
- 16.Giannelli G., Mazzocca A., Fransvea E., Lahn M., Antonaci S. Inhibiting TGF-beta signaling in hepatocellular carcinoma. Biochim Biophys Acta. 2011;1815:214–223. doi: 10.1016/j.bbcan.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Wu K., Ding J., Chen C., Sun W., Ning B.F., Wen W., Huang L., Han T., Yang W., Wang C. Hepatic transforming growth factor beta gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology. 2012;56:2255–2267. doi: 10.1002/hep.26007. [DOI] [PubMed] [Google Scholar]
- 18.Schmitz K.J., Wohlschlaeger J., Lang H., Sotiropoulos G.C., Malago M., Steveling K., Reis H., Cicinnati V.R., Schmid K.W., Baba H.A. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J Hepatol. 2008;48:83–90. doi: 10.1016/j.jhep.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa H., Maeda S. Molecular mechanisms of liver injury and hepatocarcinogenesis: focusing on the role of stress-activated MAPK. Pathol Res Int. 2012;2012 doi: 10.1155/2012/172894. [Article ID 172894, 14 pages] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagiwara S., Kudo M., Nagai T., Inoue T., Ueshima K., Nishida N., Watanabe T., Sakurai T. Activation of JNK and high expression level of CD133 predict a poor response to sorafenib in hepatocellular carcinoma. Br J Cancer. 2012;106:1997–2003. doi: 10.1038/bjc.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin C., Samuelson L., Cui C.B., Sun Y., Gerber D.A. MAPK/ERK and Wnt/beta-Catenin pathways are synergistically involved in proliferation of Sca-1 positive hepatic progenitor cells. Biochem Biophys Res Commun. 2011;409:803–807. doi: 10.1016/j.bbrc.2011.05.094. [DOI] [PubMed] [Google Scholar]
- 22.Fu Y., Wey S., Wang M., Ye R., Liao C.P., Roy-Burman P., Lee A.S. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:19444–19449. doi: 10.1073/pnas.0807691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heijmans J., van Lidth de Jeude J.F., Koo B.K., Rosekrans S.L., Wielenga M.C., van de Wetering M., Ferrante M., Lee A.S., Onderwater J.J., Paton J.C. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep. 2013;3:1128–1139. doi: 10.1016/j.celrep.2013.02.031. [DOI] [PubMed] [Google Scholar]
- 24.Chen W.T., Zhu G., Pfaffenbach K., Kanel G., Stiles B., Lee A.S. GRP78 as a regulator of liver steatosis and cancer progression mediated by loss of the tumor suppressor PTEN. Oncogene. 2014 doi: 10.1038/onc.2013.437. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosekrans S.L., Heijmans J., Büller N.V., Westerlund J., Lee A.S., Muncan V., van den Brink G.R. ER stress induces epithelial differentiation in the mouse oesophagus. Gut. 2014 doi: 10.1136/gutjnl-2013-306347. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 26.Gouw A.S., Clouston A.D., Theise N.D. Ductular reactions in human liver: diversity at the interface. Hepatology. 2011;54:1853–1863. doi: 10.1002/hep.24613. [DOI] [PubMed] [Google Scholar]
- 27.Williams M.J., Clouston A.D., Forbes S.J. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology. 2014;146:349–356. doi: 10.1053/j.gastro.2013.11.034. [DOI] [PubMed] [Google Scholar]
- 28.Bissell D.M. Chronic liver injury, TGF-beta, and cancer. Exp Mol Med. 2001;33:179–190. doi: 10.1038/emm.2001.31. [DOI] [PubMed] [Google Scholar]
- 29.Di Tommaso L., Franchi G., Park Y.N., Fiamengo B., Destro A., Morenghi E., Montorsi M., Torzilli G., Tommasini M., Terracciano L. Diagnostic value of HSP70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology. 2007;45:725–734. doi: 10.1002/hep.21531. [DOI] [PubMed] [Google Scholar]
- 30.Inagaki Y., Okazaki I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56:284–292. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikushima H., Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 32.Mamuya F.A., Duncan M.K. aV integrins and TGF-beta-induced EMT: a circle of regulation. J Cell Mol Med. 2012;16:445–455. doi: 10.1111/j.1582-4934.2011.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo B., Lam B.S., Lee S.H., Wey S., Zhou H., Wang M., Chen S.Y., Adams G.B., Lee A.S. The endoplasmic reticulum chaperone protein GRP94 is required for maintaining hematopoietic stem cell interactions with the adult bone marrow niche. PLoS One. 2011;6:e20364. doi: 10.1371/journal.pone.0020364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo B., Tseng C.C., Adams G.B., Lee A.S. Deficiency of GRP94 in the hematopoietic system alters proliferation regulators in hematopoietic stem cells. Stem Cells Dev. 2013;22:3062–3073. doi: 10.1089/scd.2013.0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ostrovsky O., Ahmed N.T., Argon Y. The chaperone activity of GRP94 toward insulin-like growth factor II is necessary for the stress response to serum deprivation. Mol Biol Cell. 2009;20:1855–1864. doi: 10.1091/mbc.E08-04-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hijikawa T., Kaibori M., Uchida Y., Yamada M., Matsui K., Ozaki T., Kamiyama Y., Nishizawa M., Okumura T. Insulin-like growth factor 1 prevents liver injury through the inhibition of TNF-alpha and iNOS induction in D-galactosamine and LPS-treated rats. Shock. 2008;29:740–747. doi: 10.1097/shk.0b013e31815d0780. [DOI] [PubMed] [Google Scholar]
- 37.Aksu I., Baykara B., Kiray M., Gurpinar T., Sisman A.R., Ekerbicer N., Tas A., Gokdemir-Yazar O., Uysal N. Serum IGF-1 levels correlate negatively to liver damage in diabetic rats. Biotech Histochem. 2013;88:194–201. doi: 10.3109/10520295.2012.758311. [DOI] [PubMed] [Google Scholar]
- 38.Cadamuro M., Morton S.D., Strazzabosco M., Fabris L. Unveiling the role of tumor reactive stroma in cholangiocarcinoma: an opportunity for new therapeutic strategies. Transl Gastrointest Cancer. 2013;2:130–144. doi: 10.3978/j.issn.2224-4778.2013.04.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris S.M., Baek J.Y., Koszarek A., Kanngurn S., Knoblaugh S.E., Grady W.M. Transforming growth factor-beta signaling promotes hepatocarcinogenesis induced by p53 loss. Hepatology. 2012;55:121–131. doi: 10.1002/hep.24653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeanes A., Gottardi C.J., Yap A.S. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27:6920–6929. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei Y., Van Nhieu J.T., Prigent S., Srivatanakul P., Tiollais P., Buendia M.A. Altered expression of E-cadherin in hepatocellular carcinoma: correlations with genetic alterations, beta-catenin expression, and clinical features. Hepatology. 2002;36:692–701. doi: 10.1053/jhep.2002.35342. [DOI] [PubMed] [Google Scholar]
- 42.Calvisi D.F., Ladu S., Conner E.A., Factor V.M., Thorgeirsson S.S. Disregulation of E-cadherin in transgenic mouse models of liver cancer. Lab Invest. 2004;84:1137–1147. doi: 10.1038/labinvest.3700147. [DOI] [PubMed] [Google Scholar]
- 43.Osada T., Sakamoto M., Ino Y., Iwamatsu A., Matsuno Y., Muto T., Hirohashi S. E-cadherin is involved in the intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 1996;24:1460–1467. doi: 10.1053/jhep.1996.v24.pm0008938181. [DOI] [PubMed] [Google Scholar]
- 44.Tomlinson J.S., Alpaugh M.L., Barsky S.H. An intact overexpressed E-cadherin/alpha, beta-catenin axis characterizes the lymphovascular emboli of inflammatory breast carcinoma. Cancer Res. 2001;61:5231–5241. [PubMed] [Google Scholar]
- 45.Saha B., Chaiwun B., Imam S.S., Tsao-Wei D.D., Groshen S., Naritoku W.Y., Imam S.A. Overexpression of E-cadherin protein in metastatic breast cancer cells in bone. Anticancer Res. 2007;27:3903–3908. [PubMed] [Google Scholar]
- 46.Yanger K., Zong Y., Maggs L.R., Shapira S.N., Maddipati R., Aiello N.M., Thung S.N., Wells R.G., Greenbaum L.E., Stanger B.Z. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27:719–724. doi: 10.1101/gad.207803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lim S.O., Park S.G., Yoo J.H., Park Y.M., Kim H.J., Jang K.T., Cho J.W., Yoo B.C., Jung G.H., Park C.K. Expression of heat shock proteins (HSP27, HSP60, HSP70, HSP90, GRP78, GRP94) in hepatitis B virus-related hepatocellular carcinomas and dysplastic nodules. World J Gastroenterol. 2005;11:2072–2079. doi: 10.3748/wjg.v11.i14.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao D.F., Wu X.H., Su X.Q., Yao M., Wu W., Qiu L.W., Zou L., Meng X.Y. Abnormal expression of HSP gp96 associated with HBV replication in human hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2006;5:381–386. [PubMed] [Google Scholar]
- 49.Patel P.D., Yan P., Seidler P.M., Patel H.J., Sun W., Yang C., Que N.S., Taldone T., Finotti P., Stephani R.A. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013;9:677–684. doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duerfeldt A.S., Peterson L.B., Maynard J.C., Ng C.L., Eletto D., Ostrovsky O., Shinogle H.E., Moore D.S., Argon Y., Nicchitta C.V. Development of a Grp94 inhibitor. J Am Chem Soc. 2012;134:9796–9804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]