

Abstract
Current antibiotics tend to be broad spectrum, leading to indiscriminate killing of commensal bacteria and accelerated evolution of drug resistance. Here, we use CRISPR-Cas technology to create antimicrobials whose spectrum of activity is chosen by design. RNA-guided nucleases (RGNs) targeting specific DNA sequences are delivered efficiently to microbial populations using bacteriophage or bacteria carrying plasmids transmissible by conjugation. The DNA targets of RGNs can be undesirable genes or polymorphisms, including antibiotic resistance and virulence determinants in carbapenem-resistant Enterobacteriaceae and enterohemorrhagic Escherichia coli. Delivery of RGNs significantly improves survival in a Galleria mellonella infection model. We also show that RGNs enable modulation of complex bacterial populations by selective knockdown of targeted strains based on genetic signatures. RGNs constitute a class of highly discriminatory, customizable antimicrobials that enact selective pressure at the DNA level to reduce the prevalence of undesired genes, minimize off-target effects and enable programmable remodeling of microbiota.
There is mounting concern over the emergence and proliferation of multidrug-resistant bacterial pathogens and the dwindling treatment options for these organisms1. Recently, carbapenem-resistant Enterobacteriaceae, a group of intestinal Gram-negative bacteria known to cause life-threatening opportunistic infections, were highlighted by the US Centers for Disease Control and Prevention as one of three most urgent threats among antibiotic-resistant bacteria1. Carbapenems have traditionally been reserved as a last resort treatment for Gram-negative infections, but the spread of extended-spectrum β-lactamases has necessitated the increased usage of carbapenems and favored the emergence of carbapenem-resistant strains refractory toward most or all current treatment options. The responsible enzymes, including New-Delhi metallo-β-lactamase 1 (NDM-1), may confer pan-resistance to β-lactam antibiotics and are frequently co-harbored with additional resistance determinants on mobile plasmids that facilitate rapid dissemination within and beyond Enterobacteriaceae2. The diversity of multidrug-resistant bacteria compounds the difficulty of developing treatments that target pathogens and commensal reservoirs but avoid nonspecific broad-spectrum activity.
Here, we introduce an alternative antimicrobial approach that imposes direct evolutionary pressure at the gene level by using efficiently delivered, programmable RGNs. We engineered the clustered, regularly interspaced, short palindromic repeats (CRISPR)–CRISPR-associated (Cas) system, naturally employed in bacteria as a defense strategy against mobile elements3,4, to effect cell death or plasmid loss upon detection of genetic signatures associated with virulence or antibiotic resistance. The Type II CRISPR-Cas system of Streptococcus pyogenes is an effective, programmable tool for genome editing and gene expression in a wide variety of organisms5. The specificity of CRISPR-Cas is dictated by short, spacer sequences flanked by direct repeats encoded in the CRISPR locus, which are transcribed and processed into CRISPR RNAs (crRNA)6. With the aid of a trans-activating small RNA (tracrRNA), crRNAs enable the Cas9 endonuclease to introduce double-stranded breaks in target DNA sequences6,7. Through simple modifications of spacers in the CRISPR locus, an RGN can direct cleavage of almost any DNA sequence, with the only design restriction being a requisite NGG motif immediately 3′ of the target sequence7. By packaging RGNs into bacteriophage particles or harnessing mobilizable plasmids, we implemented conditional-lethality devices with high specificity, modularity and multiplexability against undesired DNA sequences (Fig. 1a and Supplementary Fig. 1).
Figure 1.

RGN constructs delivered by bacteriophage particles (ΦRGN) exhibit efficient and specific antimicrobial effects against strains harboring plasmid or chromosomal target sequences. (a) Bacteriophage-delivered RGN constructs differentially affect host cell physiology in a sequence-dependent manner. If the target sequence is: (i) absent, the RGN exerts no effect; (ii) chromosomal, RGN activity is cytotoxic; (iii) episomal, the RGN leads to either (iiia) cell death or (iiib) plasmid loss, depending on the presence or absence of toxin-antitoxin (TA) systems, respectively. (b) Treatment of EMG2 wild-type (WT) or EMG2 containing native resistance plasmids, pNDM-1 (encoding blaNDM-1) or pSHV-18 (encoding blaSHV-18), with SM buffer, ΦRGNndm-1, ΦRGNshv-18, or multiplexed ΦRGNndm-1/shv-18 at a multiplicity of infection (MOI) ~20 showed sequence-dependent cytotoxicity as evidenced by a strain-specific reduction in viable cell counts (n = 3). CFU, colony-forming units. (c) E. coli EMG2 WT or EMG2 gyrAD87G populations were treated with SM buffer, ΦRGNndm-1 or ΦRGNgyrAD87G at MOI ~20, and viable cells were determined by plating onto Luria-Bertani agar (n = 3).
To establish RGN functionality in mediating sequence-specific cytotoxicity, we designed RGNs to induce double-stranded breaks in blaSHV-18 or blaNDM-1, which encode extended-spectrum and pan-resistance to β-lactam antibiotics, respectively (Supplementary Table 1)8,9. Transformation of E. coli by plasmid-borne RGNs (pRGNs) containing a chromosomal copy of these target genes resulted in nearly a 1,000-fold decrease in transformation efficiency as compared to wild-type cells lacking the target (Supplementary Fig. 2a). These results corroborate the mutual exclusivity between a functional crRNA and a cognate target locus10,11. Sequence analysis of 30 escape mutants (cells that receive and maintain an RGN plasmid despite the presence of a target sequence) revealed that tolerance was exclusively due to a defective construct that frequently arose from a spacer deletion within the CRISPR locus (Supplementary Fig. 3). Furthermore, deletion of the tracrRNA as well as inactivation of the RuvC-like nuclease domain of Cas9 (D10A)7 abrogated the loss of transformation efficiency in cells that harbored a target sequence. Thus, a catalytically active endonuclease, tracrRNA and crRNA are necessary and sufficient to mediate sequence-specific cytotoxicity in E. coli (Fig. 1a).
Antibiotic resistance genes are often present on large, multicopy plasmids capable of autonomous transfer in microbial populations, resulting in horizontal dissemination of drug resistance2. RGN activity against high-copy plasmids was verified using a GFP-expressing, ColE1-derived vector containing a standard β-lactamase selectable marker (pZE-blaz-gfp)12 or blaNDM-1 (pZE-blaNDM-1-gfp). Vectors bearing this ColE1 origin are reported to be present at copy numbers of 50–70 per cell12. Transformation of cells containing pZE-blaNDM-1- gfp with pRGNndm-1, a plasmid-borne RGN targeting blaNDM-1, led to a three-log10 reduction in transformants retaining resistance to the β-lactam antibiotic carbenicillin, whereas transformation of cells containing target-free pZE-blaz-gfp with pRGNndm-1 did not lead to a reduction in resistant transformants (Supplementary Fig. 2b). The activity of RGNs is therefore sufficient to exclude even high-copy antibiotic resistance plasmids from cells and can resensitize a resistant population to antibiotics. Similarly, transformation of cells possessing pZE-blaNDM-1-gfp with pRGNndm-1 led to an ~1,000-fold decrease in GFP-expressing cells, as measured by flow cytometry, but no decrease was found with transformation of cells possessing pZE-blaz-gfp with pRGNndm-1 (Supplementary Fig. 4).
The usefulness of RGNs for antimicrobial therapy hinges on high-efficiency delivery of genetic constructs to bacterial cells. We explored two mechanisms of horizontal gene transfer that are used by bacteria to acquire foreign genetic elements: plasmid conjugation and viral transduction. Although constrained by requirements for cell-cell contact, conjugative plasmids often have wide host ranges and no recipient factors necessary for DNA uptake have been identified13. Efficient transfer of RGNs was achieved using the broad-host-range plasmid R1162 mobilized by E. coli S17-1, which contains the conjugative machinery of plasmid RP4 integrated into its chromosome. In filter mating experiments, conjugative transfer of RGNs resulted in a 40- to 60-fold reduction in target carbenicillin-resistant recipient cells (Supplementary Fig. 1b). Under selection for transconjugants, transfer of RGNs into recipients yielded a 2- to 3-log10 reduction in target cells as compared to controls, suggesting that conjugation efficiency, as opposed to RGN activity, limited RGN efficacy in this context (Supplementary Fig. 1c). Future work will be necessary to further optimize the efficiency of conjugation-based delivery vehicles for antimicrobials based on mobilizable RGNs.
Bacteriophages are natural predators of bacteria and are highly adept at injecting DNA into host cells. To adapt phage for RGN delivery, we engineered phagemid vectors by pairing RGN constructs targeting blaNDM-1 or blaSHV-18 with an f1 origin to facilitate packaging into M13 particles. Phage-packaged RGNndm-1 (ΦRGNndm-1) was capable of comprehensively transducing a population of E. coli EMG2 (Supplementary Fig. 5). To test the ΦRGNs, we conjugated native plasmids containing blaNDM-1 (pNDM-1) or blaSHV-18 (pSHV-18) from clinical isolates into EMG2. Treatment of the EMG2 pNDM-1 or EMG2 pSHV-18 strains with the cognate ΦRGNs resulted in 2- to 3-log10 reductions in viable cells even in the absence of any selection (Fig. 1b). Furthermore, ΦRGNs engendered no toxicity against wild-type EMG2 or EMG2 containing noncognate plasmids (Fig. 1b).
In naturally occurring Type II CRISPR-Cas systems, the CRISPR locus may contain multiple spacers, each of which is processed into an independent crRNA molecule that, with tracrRNA, enables Cas9 to cleave cognate DNA sequences6. To explore the utility of a single ΦRGN exhibiting activity against more than one genetic signature, we engineered a construct containing two spacers encoding two different crRNAs for targeting the blaNDM-1 and blaSHV-18 resistance genes (ΦRGNndm-1/shv-18). ΦRGNndm-1/shv-18 generated 2- to 3-log10 reductions in viable cells of EMG2 pNDM-1 or EMG2 pSHV-18, but not of wild-type EMG2 (Fig. 1b). Thus, RGNs may be multiplexed against different genetic signatures, enabling simultaneous targeting of a variety of virulence factors and resistance genes that may exist in microbial populations.
In addition to antibiotic-modifying enzymes, such as β-lactamases, alterations in host proteins constitute a major antibiotic resistance mechanism in bacteria14. Owing to the specificity of the CRISPR-Cas system, we suspected that RGNs might discriminate between susceptible and resistant strains that differ by a single-nucleotide mutation in DNA gyrase (gyrA), which confers resistance to quinolone antibiotics (Supplementary Table 1)14. Indeed, ΦRGNgyrAD87G was specifically cytotoxic only for quinolone-resistant E. coli harboring the chromosomal gyrAD87G mutation and not for otherwise isogenic strains with the wild-type gyrA gene (Fig. 1c).
Killing curves revealed that ΦRGNs mediated rapid killing of target cells, with viable cell counts that decayed exponentially (t1/2 ~ 13 min) and maximal bactericidal effect was achieved by 2–4 h (Fig. 2a). Moreover, ΦRGN antimicrobial activity increased with phagemid particle concentration (Fig. 2b and Supplementary Fig. 6). To further characterize the cellular response to RGN-mediated targeting, we assessed treatment of cells that harbored a GFP reporter under SOS regulation. E. coli and other bacteria respond to chromosomal double-stranded breaks, including those artificially generated by the meganuclease I-SceI, by inducing DNA repair through the activation of the SOS response15. We observed a 2.6- or 4.0-fold increase in fluorescence in cells containing the reporter plasmid and a plasmid-borne (blaNDM-1) or chromosomal (gyrAD87G) target site, respectively, when treated with the cognate versus noncognate ΦRGNs (Supplementary Fig. 7). These results confirm that RGNs can induce DNA damage in target cells and demonstrate that they can be coupled with SOS-based reporters to detect specific genes or sequences, even at the single-nucleotide level.
Figure 2.

Characterization of ΦRGN-mediated killing of antibiotic-resistant bacteria. (a) Time-course treatment of EMG2 WT or EMG2 pNDM-1 with SM buffer, ΦRGNndm-1 or ΦRGNshv-18 at a multiplicity of infection (MOI) ~20. Data represent the fold change in viable colonies at indicated time points relative to time 0 h. (b) Dose-response curve of EMG2 WT and EMG2 gyrAD87G treated with various concentrations of ΦRGNgyrAD87G for 2 h. Data represent fold change in viable colonies relative to samples treated with SM buffer. Error bars (a, b), s.e.m. of three independent biological replicates (n = 3). (c) EMG2 E. coli containing the natural pNDM-1 plasmid or the blaNDM-1 gene in a synthetic expression vector (pZA-ndm1-gfp) were treated with either ΦRGNndm-1 or ΦRGNshv-18 at MOI ~ 20 and plated onto both nonselective LB and LB + carbenicillin (Cb) to select for blaNDM-1-containing cells. ΦRGNndm-1 treatment of cells harboring pNDM-1 resulted in a reduction in viability in the absence of selection, whereas ΦRGNndm-1 treatment of cells with pZA-ndm1-gfp demonstrated similar cytotoxicity only under selective pressure for maintenance of the pZA-ndm1-gfp plasmid. (d) EMG2 pSHV-18 complemented with the cognate antitoxin (pZA31-pemI) for the PemK toxin or a control vector (pZA31-gfp) was treated with SM buffer, ΦRGNndm-1 or ΦRGNshv-18. Cultures were plated on LB and LB + Cb and colonies were enumerated to assess cytotoxicity or plasmid loss.
We were intrigued to observe that targeted cleavage of blaNDM-1 with ΦRGNndm-1 in the context of the native plasmid was lethal to host cells, whereas targeted cleavage of the same gene in a standard cloning vector was not (‘pNDM-1’ versus ‘pZA-ndm1-gfp’, respectively, in Fig. 2c). Therefore, we hypothesized that ΦRGN-induced plasmid loss in itself does not elicit lethality, but rather results in cytotoxicity by means of other co-harbored plasmid-borne functions. Toxin-antitoxin systems are components of natural plasmids that ensure their persistence in bacterial populations by inhibiting the growth of daughter cells that do not inherit the plasmid. Toxin-antitoxin systems traditionally consist of a labile antitoxin that quenches the activity of a stable toxin. Owing to the differential stability of these two components, cessation of gene expression upon plasmid loss leads to depletion of the antitoxin pool faster than the toxin pool, resulting in de-repression of toxin activity and, ultimately, stasis or programmed cell death16. Analysis of the sequenced pSHV-18 plasmid revealed the presence of a single toxin-antitoxin module, pemIK, which is commonly found among isolates harboring extended-spectrum β-lactamases17. When complemented with the PemI antitoxin expressed constitutively in trans (pZA31-pemI), ΦRGNshv-18 treatment of EMG2 pSHV-18 abrogated cytotoxicity and instead resulted in resensitization of this multidrug-resistant strain to carbenicillin (Fig. 2d). We attributed this effect to PemI inactivation of the PemK toxin, thus enabling loss of the pSHV-18 plasmid without concomitant bacterial killing. Toxin-antitoxin systems can therefore dictate the outcome of ΦRGN activity on episomal targets, as their presence leads to cytotoxicity and their absence or neutralization to plasmid loss.
To further demonstrate the versatility of RGNs for specifically combating pathogens, we designed a ΦRGN to target intimin, a chromosomally encoded virulence factor of enterohemorrhagic E. coli O157:H7 (EHEC) necessary for intestinal colonization and pathology. Encoded by the eae gene, intimin is a cell-surface adhesin that mediates intimate attachment to the host epithelium, permitting subsequent disruption of intestinal tight junctions and effacement of microvilli18. Treatment of EHEC with ΦRGNeae resulted in a 20-fold reduction in viable cell counts; this cytotoxicity was increased an additional 100-fold under kanamycin selection for ΦRGNeae transductants (Fig. 3a). The increase in cytotoxicity with selection for cells receiving the construct suggests that the efficacy of ΦRGN treatment was limited by delivery in this strain. Furthermore, ΦRGN treatment was assessed in G. mellonella larvae, an infection model that yields virulence data often predictive for higher-order mammals19. This model has also been used to evaluate the efficacy of antimicrobials or phage therapy against various Gram-negative, Gram-positive and fungal pathogens19. Administration of ΦRGNeae to EHEC-infected G. mellonella larvae significantly improved survival over no treatment or an off-target ΦRGN control (log-rank test, P < 0.001) (Fig. 3b). Moreover, ΦRGNeae was significantly more effective than chloramphenicol treatment, to which the EHEC strain was resistant (log-rank test, P < 0.05) (Supplementary Fig. 8 and Supplementary Table 1). Although ΦRGNeae treatment was inferior to carbenicillin, to which the bacteria were susceptible (Supplementary Fig. 8 and Supplementary Table 1), these data support RGNs as viable alternatives for cases where bacteria are highly resistant to existing antibiotics. Improvements in delivery efficiency with ΦRGNeae would be expected to improve treatment efficacy and outcome.
Figure 3.
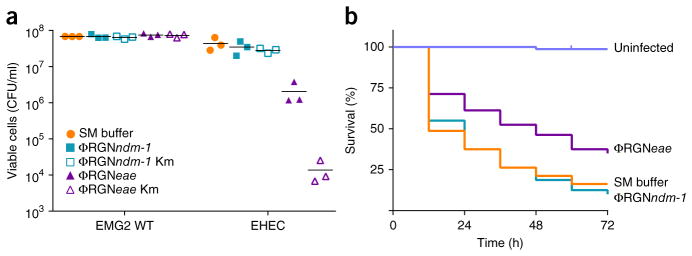
ΦRGN particles elicit sequence-specific toxicity against enterohemorrhagic E. coli in vitro and in vivo. (a) E. coli EMG2 wild-type (WT) cells or ATCC 43888 F′ (EHEC) cells were treated with SM buffer, ΦRGNndm-1 or ΦRGNeae at a multiplicity of infection (MOI) ~100 and plated onto LB agar to enumerate total cell number or LB+kanamycin (Km) to select for transductants with ΦRGNs (n = 3). (b) G. mellonella larvae were injected with either PBS or approximately 4 × 105 colony forming units (CFU) of EHEC. Subsequent administration of ΦRGNeae at MOI ~30 significantly improved survival compared to SM buffer or ΦRGNndm-1 treatment (Log-rank test, P < 0.001). Survival curves represent an aggregate of four independent experiments, each with 20 worms per treatment group (n = 80).
In addition to implementing targeted antimicrobial therapies, RGNs can be used to modulate the composition of complex bacterial populations (Fig. 4). Current therapies that use a prebiotic, probiotic or drug to modify the human microbiota have demonstrated potential for alleviating various disease states, but such therapies remain poorly characterized in terms of off-target effects and the specific mechanisms by which they act20. In concert with the host range of the delivery vehicle, RGN activity can selectively remove bacteria with specific genomic content. This could reduce the prevalence of unwanted genes, including antibiotic resistance and virulence loci, or metabolic pathways from bacterial communities without affecting bystanders.
Figure 4.

Programmable remodeling of a synthetic microbial consortium. A synthetic population composed of three different E. coli strains was treated with either SM buffer, ΦRGNndm-1, or ΦRGNgyrAD87G at an MOI ~100 and plated onto LB with chloramphenicol, streptomycin or ofloxacin to enumerate viable cells of E. coli CJ236, EMG2 pNDM-1 or RFS289 strains, respectively. ΦRGNndm-1 targets blaNDM-1 in EMG2 pNDM-1 and ΦRGNgyrAD87G targets the gyrAD87G allele in RFS289. Circle area is proportional to total population size and numbers represent viable cell concentrations (CFU/ml) of each strain after the indicated treatment. The s.e.m. based on three independent experiments is indicated in parentheses (n = 3).
As a proof-of-principle experiment for ‘bacterial knockdowns’ using RGNs, we constructed a synthetic consortium comprising three phage-susceptible E. coli strains with differential antibiotic resistance profiles. We used β-lactam–resistant E. coli EMG2 pNDM-1, quinolone-resistant RFS289 (gyrAD87G) and chloramphenicol- resistant CJ236. Application of ΦRGNndm-1 elicited >400-fold greater killing of EMG2 pNDM-1, compared to control, while leaving RFS289 and CJ236 cell populations intact. Treatment with ΦRGNgyrAD87G resulted in >20,000-fold greater killing of RFS289, compared to control, without a concomitant reduction in EMG2 pNDM-1 or CJ236 (Fig. 4). These results demonstrate that RGNs can selectively knockdown bacteria that contain target DNA sequences, thereby allowing the remaining nontarget bacteria to dominate the population. Adapting this approach for tuning endogenous microbiota could be accomplished by delivering RGNs in vivo by means of broad-host-range phages or phage cocktails, or with conjugative plasmids. An appropriately targeted bacterial knockdown approach could be used in functional studies of complex microbiota and to complement additive therapies, such as probiotics, for microbiome-associated diseases by clearing specific niches or removing defined genes from bacterial populations.
In light of the rising tide of antibiotic resistance, interest in engineered cellular and viral therapeutics as potential biological solutions to infectious disease has resurged. By repurposing parts developed by nature, synthetic biologists have designed artificial gene circuits for antimalarial production21 and engineered probiotics22 and phage therapeutics to eradicate biofilms23 or potentiate antibiotic activity24,25. Here, we demonstrate that transmissible CRISPR-Cas systems can act as a platform for programmable antimicrobials that harness site-specific cleavage to induce cytotoxicity, activate toxin-antitoxin systems, resensitize bacterial populations to antibiotics and modulate bacterial consortia. This work complements the recent finding that the Vibrio cholerae phage, ICP1, encodes its own CRISPR-Cas system to counteract a host-encoded phage defense locus26, and that CRISPR-Cas constructs transformed into electrocompetent cell populations are incompatible with cells that contain cognate target sequences11,27,28. In contrast to these other studies, we show that CRISPR-Cas technology can be applied both in situ for the removal of undesired genes from microbial populations and in vivo to treat infection in the absence of artificial selection. Moreover, we demonstrate that RGNs can be used to activate plasmid-borne toxin-antitoxin systems that have recently become an attractive antimicrobial strategy29. In addition to validating antimicrobial activity, we further demonstrate potential applications of RGNs in the deletion of plasmids from cells or the detection of DNA elements with up to single-nucleotide resolution using a DNA-damage-responsive reporter.
Because CRISPR-Cas systems are widely conserved in bacteria and archaea, the isolation, development and optimization of delivery vehicles will be required for the creation of RGNs capable of targeting additional strains, including multidrug-resistant pathogens as well as key members of endogenous microbiota. These next-generation phage systems should be designed with the goal of avoiding anticipated shortcomings, including host range limitations, phage resistance and concerns around immune reactions (Supplementary Discussion). Delivery systems that function in higher organisms could also enable RGNs to modulate the prevalence of specific genes in wild-type populations30.
Owing to the modularity and simplicity of CRISPR-Cas engineering, libraries of multiplexed ΦRGNs could be rapidly constructed to simultaneously target a plethora of antibiotic resistance and virulence determinants and to modulate the composition of complex microbial communities. The addition of facile, sequence-informed, rational design to a field that has been dominated by time- and cost-intensive screening for broad-spectrum, small-molecule antibiotics has the potential to reinvigorate the pipeline for new antimicrobials.
ONLINE METHODS
Strains and culture conditions
Unless otherwise noted, bacterial cultures were grown at 37 °C with Luria-Bertani (LB) medium (BD Difco). Bacterial strains used in this study are listed in Supplementary Table 2. Where indicated, antibiotics were added to the growth medium to the following final concentrations: 100 μg/ml carbenicillin (Cb), 30 μg/ml kanamycin (Km), 25 μg/ml chloramphenicol (Cm), 100 μg/ml streptomycin (Sm) and 150 ng/ml ofloxacin (Ofx).
Strain construction
E. coli EMG2 SmR was generated by plating an overnight culture of E. coli EMG2 onto LB+Sm. Spontaneous resistant mutants were re-streaked onto LB+Sm and an isolated colony was picked and used as the recipient for conjugation of the multidrug resistance plasmids. Overnight cultures of EMG2 SmR (recipient), E. coli CDC1001728 (donor for pNDM-1) and K. pneumoniae K6 (donor for pSHV-18) were washed in sterile PBS and 100 μl of donor and recipient were spotted onto LB agar plates and incubated at 37 °C overnight. Transconjugants were harvested by scraping the cells in 1 ml of sterile PBS and plating onto LB+Sm+Cb.
The chromosomal integrations of the blaNDM-1 and blaSHV-18 β-lactamase genes and generation of EMG2 gyrAD87G were performed by λ-Red recombineering using the pSIM9 system31. Templates for integration at the nonessential lacZYA locus were generated by amplifying the blaNDM-1 and blaSHV-18 genes from lysates of CDC1001728 and K6 using the primers rcD77/78 and rcD73/74, respectively. Templates for construction of EMG2 gyrAD87G were obtained by amplifying gyrA from RFS289 using primers mmD155/161.
Plasmid construction
To generate the RGN plasmids (Supplementary Fig. 2), we created an intermediate vector pZA-RGNØ, which lacks a CRISPR locus. The tracrRNA and PL(tetO-1) promoter were synthesized (Genewiz) and amplified using primers mmD98/99, cas9 was amplified from pMJ806 (ref. 7) using primers mmD74/75, and the vector backbone was amplified from pZA11G using primers mmD82/83. Each PCR product was purified and ligated by Gibson assembly32. To create the final backbone vector for the RGN plasmids, the pBBR1 origin, chloramphenicol resistance marker, tL17 terminator, and CRISPR locus cloning site were amplified from an intermediate vector pBBR1-MCS1-tL17 using mmD151/154, digested with NheI and SacI-HF, and ligated with pZA-RGNØ digested with SacI-HF and AvrII to create pZB-RGNØ. Digestion of this vector with PstI-HF and XbaI allowed for the insertion of assembled CRISPR loci. The tracrRNA pRGNndm-1 plasmid was created by amplification of pRGNndm-1 with mmD162/163, ClaI digestion, and self-ligation. The Cas9D10A mutant plasmid was constructed through site-directed mutagenesis of pRGNndm-1 with primers mmD108/109 and the KAPA HiFi PCR kit (KAPA Biosystems).
The CRISPR loci were constructed through isothermal annealing and ligation of short, single-stranded oligonucleotides (Integrated DNA Technologies). Each spacer and repeat piece was built by a corresponding oligo duplex connected to adjacent pieces by 6-bp overhangs. In addition, the terminal repeats were designed to contain a 17-bp extension comprised of a BsaI restriction site to generate an overhang that allowed insertion into the pUC57-Km-crRNAØ backbone vector synthesized by Genewiz. The oligos used to build each RGN are listed in Supplementary Tables 3 and 4.
To assemble the CRISPR loci, 500 pmol of sense and antisense oligos in a given duplex were annealed by boiling for 10 min at 99 °C and cooled to room temperature. 300 pmol of each annealed duplex were combined with 15 U of T4 polynucleotide kinase (Affymetrix), 400 U of T4 DNA ligase (NEB), T4 ligase buffer (NEB) and ddH2O to a volume of 20 μl. Following incubation at 25 °C for 1 h, the reaction products were purified using a Qiagen QIAquick PCR Purification Kit. Purified products were digested for 3 h with BsaI-HF and re-purified using QIAquick. To prepare the crRNA backbone vector, pUC57-Km-crRNAØ was amplified using primers mmD104/105, subsequently digested with BbsI to generate compatible overhangs, and ligated with the assembled CRISPR loci. Positive clones of the CRISPR loci were digested from the entry vector using PstI-HF and XbaI and ligated into pZB-RGNØ digested with the same enzymes to create the final RGN plasmids.
Phagemid vector pZEf-gfp was created previously by adding the f1 origin amplified from the yeast shuttle pRS series33 into pZE22-gfp12. The RGN constructs consisting of the genes encoding the tracrRNA, Cas9 and a sequence-targeting crRNA were amplified as a single product from the respective pRGN vectors using KAPA HiFi polymerase (Kapa Biosystems) with primers rcD169/183 and digested with AvrII and XmaI (New England Biolabs). These inserts were ligated with a backbone derived from amplifying the kanamycin resistance cassette, ColE1 replication origin and the f1 origin required for packaging into M13 particles off of pZEf-gfp with primers rcD184/185 and digesting with the same enzymes. E. coli DH5αPro was transformed with ligated plasmids for sequence verification and plasmid purification.
The pZE-blaNDM-1-gfp and pZA-blaNDM-1-gfp vectors were constructed by swapping the antibiotic resistance cassette of the Lutz-Bujard vectors pZE12G and pZA12G12. The blaNDM-1 gene was amplified from a lysate of CDC1001728 using primers mmD8/9, and the PCR product was digested with SacI-HF and XhoI. The digested product was ligated into the Lutz-Bujard vectors digested with the same enzymes.
The PemI antitoxin complementation plasmid pZA31-pemI was created by first amplifying the pemI coding sequence from pSHV-18 with mmD253/254. The PCR product was digested with BamHI and KpnI and ligated with the large fragment of a pZA31G digest with the same enzymes. The SOS-responsive pZA3LG reporter plasmid was derived from pZE1LG34 by swapping the origin of replication and antibiotic resistance marker with pZA31G using AatII and AvrII as restriction enzymes.
Mobilizable RGNs were created by first amplifying the R1162 replication origin and oriT using mmD266/267. The chloramphenicol selection marker and RGN locus were amplified from pRGNndm-1 and pRGNshv-18 with mmD247/248. PCR products were digested with SpeI and XmaI, ligated and used to transform E. coli S17-1 λpir to create the donor cells used in matings.
Minimum inhibitory concentration (MIC) determination
MICs were determined by broth microdilution using LB broth according to the CLSI guidelines35.
Transformation assays
Overnight cultures were diluted 1:100 in fresh LB and grown to an optical density (OD600) of ~0.3–0.5. Following 15 min of incubation on ice, cultured cells were centrifuged at 3,200g, and pellets were resuspended in one tenth volume of TSS buffer (LB, 10% polyethylene glycol, 5% dimethyl sulfoxide, 50 mM Mg2+ at pH 6.5)36. A 100 μl aliquot of cells was incubated with 10 ng of RGN plasmid DNA. Plasmids were purified from the DH5αPro cloning host using a Qiagen QIAprep Spin Miniprep Kit and the concentration was determined using a Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen). Following 30 min of incubation on ice, cells were heat shocked at 42 °C for 30 s, returned to ice for 2 min and recovered for 1.5 h at 37 °C in 1 ml of SOC broth (HiMedia). For the chromosomal target assay, serial dilutions of cells were plated on LB+Cm to select for transformants. Plates were incubated overnight at 37 °C, and the number of colony-forming units (CFU) were enumerated the following day. Transformation efficiency was used to assess whether the given RGN plasmid was toxic to cells and was calculated as the CFU/ml per μg of DNA transformed (Supplementary Fig. 2a).
For the episomal target assay (Supplementary Fig. 2b), following recovery, cultures were washed in fresh LB, diluted 1:100 in LB supplemented with chloramphenicol to select for transformants and incubated for 16 h at 37 °C. Samples were washed in sterile PBS, serially diluted and plated on LB+Cm and LB+Cm+Cb or analyzed by flow cytometry (Supplementary Fig. 4). Colonies were enumerated the following day and plasmid loss was inferred by calculating the ratio of CbR+CmR CFUs to CmR CFUs.
Overnight cultures of RGN transformants were also diluted 1:100 in sterile PBS, aliquoted in duplicates in a 96-well plate and immediately assayed using a BD LSRFortessa cell analyzer. Cells were consistently gated by side scatter and forward scatter across independent biological replicates. Fluorescence measurements were performed using a 488-nm argon excitation laser. The GFP gate and laser voltages were initially determined using untreated pZE-blaZ-gfp and EMG2 cells as positive and negative controls, respectively, and implemented across biological replicates. BD FACSDIVA software was used for data acquisition and analysis.
Sequence analysis
Escape mutants from transformation assays were re-isolated by passaging surviving colonies onto LB+Cm+Cb. DNA isolation for escape sequencing analysis was done by either extracting plasmid DNA from isolated escape mutants using the Qiagen QIAprep Spin Miniprep Kit or by amplifying the integrated target locus using primers mmD9/234 or mmD3/4 for blaNDM-1 and blaSHV-18, respectively. Sequencing was performed by Genewiz using the primers mmD112–115/153 and rcD11 for analysis of the RGN plasmids and mmD3 or mmD234 for examination of the integrated resistance genes.
Phagemid purification
Phagemids encoding the RGNs were purified using the Qiagen QIAprep Spin Miniprep Kit (Qiagen) and used to transform E. coli DH5αPro along with the m13cp helper plasmid for generation of phagemid-loaded M13 particles37. Strains were inoculated and grown overnight in 250 ml LB+Cm+Km to maintain M13cp and the phagemid, respectively. Cells were pelleted and the supernatant fluid containing the phagemid particles was passed through a 0.2-μm filter. For all purifications except the ΦRGNgyrAD87G purification for the dose-response curve, M13 phagemid particles were precipitated by the addition of 5% polyethylene glycol (PEG-6000) and 0.5M NaCl and incubation overnight at 4 °C (ref. 38) and pelleted at 12,000g for 1 h. Purified phagemid pellets were resuspended gently in 1/100th volume of SM buffer (50 mM Tris-HCl [pH 7.5]), 100 mM NaCl, 10 mM MgSO4) and stored at 4 °C. For the ΦRGNgyrAD87G purification for the dose-response curve (Fig. 2b), M13 phagemid particles were precipitated39 through the addition of concentrated HCl to pH 4.2 and subsequently pelleted at 13,000g for 15 min. The phagemid pellet was resuspended in 1/100th volume of water and concentrated NaOH was added to pH 7.0 to solubilize phagemid particles. Tris-HCl [pH 7.5], NaCl and MgSO4 were added to reconstitute the composition of SM buffer.
Titers were measured by incubating sample dilutions with E. coli EMG2 for 30 min and enumerating transductants by plating on LB and LB+Km. Titers were defined in TFU100/ml, which is the concentration of phagemid at which ~100% of a recipient population of an equivalent cell concentration would be transduced.
Phagemid kill assays
Cultures were inoculated and grown overnight in LB with appropriate antibiotics at 37 °C with shaking. The following day, overnights were subcultured 1:100 into 3 ml LB (no antibiotics) and grown at 37 °C with shaking until the OD600 reached ~0.8. Cultures were diluted into LB to ~108 CFU/ml for pNDM-1 and pSHV-18 assays (Fig. 1b, 2a) or ~106 CFU/ml for gyrAD87G (Fig. 1c) and EHEC assays (Fig. 3a) and 245 μl of the suspension was added to 5 μl of purified phagemid stock in a 96-well plate and incubated static at 37 °C. The number of viable cells in samples at each interval during the time-course or at 2 h for endpoint assays was determined by serial dilution and spot plating onto LB, LB+Cb, and LB+Km to analyze cytotoxicity, plasmid loss and phagemid delivery, respectively. Initial suspensions were also diluted and plated onto LB to quantify the initial bacterial inocula. Colonies were enumerated after 8–9 h incubation at 37 °C to calculate cell viability (CFU/ml) and averaged over three independent experiments. Nonlinear curve fitting of the time-course to an exponential decay curve was performed using GraphPad Prism.
G. mellonella model
Larvae of the model organism G. mellonella19 were purchased from Vanderhorst Wholesale, Inc. (St. Marys, OH, USA) and received in the final larval instar for survival assays. Larvae were removed from food source, allowed to acclimate for at least 24 h at room temperature in the dark, and used within 4 d of receipt. For all injections, a KDS100 (KD Scientific) or Pump 11 Elite (Harvard Apparatus) automated syringe pump was set to dispense a 10 μl volume at a flow rate of ~1 μl/s through a 1-ml syringe (BD) and 26 G needle (BD). To prepare bacteria for injection, an overnight culture of E. coli O157:H7 43888 F′ was subcultured in Dulbecco’s Modified Eagle Medium (Gibco) for 4 h at 37 °C with shaking until OD600 ~0.6. Cultures were washed twice in PBS and diluted to a concentration of ~4 × 105 CFU/ml. In accordance with other studies40, 20 larvae per treatment group were randomly selected based on size (150–250 mg) and excluded based on poor health as evidenced by limited activity, dark coloration or reduced turgor before experiments. Larvae were delivered injections without blinding of either PBS or bacteria behind the final left proleg. Approximately an hour after the first injection, SM buffer, antibiotic or ΦRGN treatment was administered behind the final right proleg (Fig. 3c, Supplementary Fig. 8). Larvae were incubated at 37 °C and survival was monitored at 12 h intervals for 72 h, with death indicated by lack of movement and unresponsiveness to touch19. Kaplan-Meier survival curves were generated and analyzed with the log-rank test using GraphPad Prism.
LexA reporter assay
Overnight cultures of EMG2 WT, EMG2 pNDM-1 and EMG2 gyrAD87G containing the SOS-responsive reporter plasmid pZA3LG were diluted 1:50 in LB and incubated with either SM buffer, ΦRGNndm-1 or ΦRGNgyrAD87G at MOI ~5 for 2 h at 37 °C (Supplementary Fig. 7). Cultures were diluted 1:5 in 250 μl of sterile PBS and analyzed using a BD LSR Fortessa cell analyzer, as above. BD FACSDIVA software was used for data acquisition and analysis was performed using FlowJo software.
Bacterial conjugation
Donor and recipient strains grown overnight in LB with appropriate antibiotics were diluted 1:100 in fresh media and grown to an OD600 ~1. Cells were pelleted and resuspended in sterile PBS, and mating pairs were mixed at a donor to recipient ratio of 340 ± 66:1. Mating mixtures were pelleted, resus-pended in 20 μl of PBS and spotted onto nitrocellulose filters placed on LB agar plates. Initial bacterial suspensions were serially diluted and plated on LB agar plates to quantify the initial inocula. Matings proceeded at 37 °C for 3 h with a single mixing step. At 90 min, mating mixtures were collected by vigorously vortexing the filters in 1 ml sterile PBS. Cells were pelleted, resuspended in 20 μl PBS and re-seeded onto filters and incubated as above for the remaining 90 min. At the end of the 3 h mating, cells were again recovered by vigorously vortexing the filters in 1 ml sterile PBS. Mating mixtures were serially diluted in PBS and plated onto LB+Cb to select for total number of Cb-resistant recipient cells and LB+Cb+Cm to select for transconjugants. Colonies were enumerated following overnight incubation at 37 °C to determine viable cell counts and were averaged over nine independent biological replicates (Supplementary Fig. 1).
Synthetic consortia remodeling
E. coli CJ236, EMG2 pNDM-1 and RFS289 strains grown overnight in LB with appropriate antibiotics were diluted 1:100 into fresh LB (no antibiotics) and grown to OD600 ~0.8. Cultures were seeded into fresh LB such that the initial mixture contained ~1 × 106 CFU/ml of each strain and 245 μl of the suspension was added to 5 μl of SM buffer or purified ΦRGNndm-1 or ΦRGNgyrAD87G in triplicate in a 96-well plate and spotted onto LB +Cm, +Sm and +Ofx to quantify the initial concentration of CJ236, EMG2 pNDM-1 and RFS289, respectively. Samples were then incubated, plated and enumerated as in phagemid kill assays. The composition of the synthetic ecosystem under each treatment condition was determined by counting viable colonies on plates selective for each strain as above and data were calculated as viable cell concentration (CFU/ml) averaged over three biological replicates (Fig. 4).
Data analysis and statistics
All data were analyzed using GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA, USA, http://www.graphpad.com/).
Supplementary Material
Acknowledgments
We would like to thank R. Meyer (Institute for Cell and Molecular Biology, University of Texas Austin) for R1162, D.L. Court (Center for Cancer Research, National Cancer Institute at Frederick) for pSIM9, and A.R.M. Bradbury (Los Alamos National Laboratory) for M13cp. The authors thank J. Rubens for assistance with flow cytometry experiments and H. Gancz and D. Zurawski for assistance with the Galleria infection model. T.K.L. acknowledges support from the US National Institutes of Health (NIH) New Innovator Award (1DP2OD008435), an NIH National Centers for Systems Biology grant (1P50GM098792), the Defense Threat Reduction Agency (HDTRA1-14-1-0007), the US Army Research Laboratory and the US Army Research Office through the Institute for Soldier Nanotechnologies (W911NF13D0001) and the Henry L. and Grace Doherty Professorship in Ocean Utilization. R.J.C. is supported by funding from the NIH/National Institute of General Medical Sciences Interdepartmental Biotechnology Training Program (5T32 GM008334), and M.M. is a Howard Hughes Medical Institute International Student Research fellow and a recipient of a Fonds de recherche Santé Québec Master’s Training Award.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
R.J.C. and M.M. designed and performed experiments. R.J.C., M.M. and T.K.L. conceived this study, analyzed the data, discussed results and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States. 2013 [Google Scholar]
- 2.Nordmann P, Dortet L, Poirel L. Carbapenem resistance in Enterobacteriaceae: here is the storm! Trends Mol Med. 2012;18:263–272. doi: 10.1016/j.molmed.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 4.Garneau JE, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 5.Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deltcheva E, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rasheed JK, et al. Characterization of the extended-spectrum beta-lactamase reference strain, Klebsiella pneumoniae K6 (ATCC 700603), which produces the novel enzyme SHV-18. Antimicrob Agents Chemother. 2000;44:2382–2388. doi: 10.1128/aac.44.9.2382-2388.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasheed JK, et al. New Delhi metallo-β-lactamase-producing Enterobacteriaceae, United States. Emerg Infect Dis. 2013;19:870–878. doi: 10.3201/eid1906.121515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bikard D, Hatoum-Aslan A, Mucida D, Marraffini LA. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe. 2012;12:177–186. doi: 10.1016/j.chom.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Gomaa AA, et al. Programmable removal of bacterial strains by use of genome-targeting CRISPR-Cas systems. MBio. 2014;5:e00928–13. doi: 10.1128/mBio.00928-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1–I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pérez-Mendoza D, de la Cruz F. Escherichia coli genes affecting recipient ability in plasmid conjugation: are there any? BMC Genomics. 2009;10:71. doi: 10.1186/1471-2164-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacoby GA. Mechanisms of resistance to quinolones. Clin Infect Dis. 2005;41(suppl 2):S120–S126. doi: 10.1086/428052. [DOI] [PubMed] [Google Scholar]
- 15.Pennington JM, Rosenberg SM. Spontaneous DNA breakage in single living Escherichia coli cells. Nat Genet. 2007;39:797–802. doi: 10.1038/ng2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayes F. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science. 2003;301:1496–1499. doi: 10.1126/science.1088157. [DOI] [PubMed] [Google Scholar]
- 17.Mnif B, et al. Molecular characterization of addiction systems of plasmids encoding extended-spectrum beta-lactamases in Escherichia coli. J Antimicrob Chemother. 2010;65:1599–1603. doi: 10.1093/jac/dkq181. [DOI] [PubMed] [Google Scholar]
- 18.Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 19.Desbois AP, Coote PJ. Utility of greater wax moth larva (Galleria mellonella) for evaluating the toxicity and efficacy of new antimicrobial agents. Adv Appl Microbiol. 2012;78:25–53. doi: 10.1016/B978-0-12-394805-2.00002-6. [DOI] [PubMed] [Google Scholar]
- 20.Sonnenburg JL, Fischbach MA. Community health care: therapeutic opportunities in the human microbiome. Sci Transl Med. 2011;3:78ps12. doi: 10.1126/scitranslmed.3001626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paddon CJ, et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature. 2013;496:528–532. doi: 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- 22.Duan F, March JC. Engineered bacterial communication prevents Vibrio cholerae virulence in an infant mouse model. Proc Natl Acad Sci USA. 2010;107:11260–11264. doi: 10.1073/pnas.1001294107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu TK, Collins JJ. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA. 2007;104:11197–11202. doi: 10.1073/pnas.0704624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu TK, Collins JJ. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci USA. 2009;106:4629–4634. doi: 10.1073/pnas.0800442106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar R, Friedman N, Molshanski-Mor S, Qimron U. Reversing bacterial resistance to antibiotics by phage-mediated delivery of dominant sensitive genes. Appl Environ Microbiol. 2012;78:744–751. doi: 10.1128/AEM.05741-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seed KD, Lazinski DW, Calderwood SB, Camilli A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature. 2013;494:489–491. doi: 10.1038/nature11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vercoe RB, et al. Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet. 2013;9:e1003454. doi: 10.1371/journal.pgen.1003454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams JJ, Hergenrother PJ. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012;20:291–298. doi: 10.1016/j.tim.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esvelt KM, Smidler AL, Catteruccia F, Church GM. Concerning RNA-guided gene drives for the alteration of wild populations. Elife. 2014:e03401. doi: 10.7554/eLife.03401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Datta S, Costantino N, Court DL. A set of recombineering plasmids for Gram-negative bacteria. Gene. 2006;379:109–115. doi: 10.1016/j.gene.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 32.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 33.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dwyer DJ, Kohanski MA, Hayete B, Collins JJ. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol Syst Biol. 2007;3:91. doi: 10.1038/msb4100135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard. 7. Clinical and Laboratory Standards Institute; Wayne, Pennsylvania, USA: 2006. [Google Scholar]
- 36.Chung CT, Niemela SL, Miller RH. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci USA. 1989;86:2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chasteen L, Ayriss J, Pavlik P, Bradbury ARM. Eliminating helper phage from phage display. Nucleic Acids Res. 2006;34:e145. doi: 10.1093/nar/gkl772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Westwater C, et al. Use of genetically engineered phage to deliver antimicrobial agents to bacteria: an alternative therapy for treatment of bacterial infections. Antimicrob Agents Chemother. 2003;47:1301–1307. doi: 10.1128/AAC.47.4.1301-1307.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong D, Sutaria S, Hwangbo JY, Chen P. A simple and rapid method to isolate purer M13 phage by isoelectric precipitation. Appl Microbiol Biotechnol. 2013;97:8023–8029. doi: 10.1007/s00253-013-5049-9. [DOI] [PubMed] [Google Scholar]
- 40.Ramarao N, Nielsen-Leroux C, Lereclus D. The insect Galleria mellonella as a powerful infection model to investigate bacterial pathogenesis. J Vis Exp. 2012;4392 doi: 10.3791/4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.