

Abstract
BACKGROUND
Toll-like receptor 2 (TLR2) contributes to sepsis pathogenesis such as deleterious systemic inflammation, cardiac dysfunction, and high mortality in animal studies. Mitochondrial dysfunction is a key molecular event that is associated with organ injury in sepsis. The role of TLR2 in sepsis-induced mitochondrial dysfunction remains unclear.
METHODS
Intracellular hydrogen peroxide (H2O2) and mitochondrial superoxide (O2−), mitochondrial membrane potential (ΔΨm) and intracellular adenosine triphosphate (ATP) were measured in peritoneal leukocytes. A mouse model of polymicrobial sepsis was generated by cecum ligation and puncture (CLP). Wild-type and TLR2-deficient (TLR2−/−) mice were subjected to sham or CLP. Mitochondrial functions including reactive oxygen species (ROS), ΔΨm, intracellular ATP, and complex III activity were measured.
RESULTS
TLR2/1 activation by Pam3Cys enhanced intracellular H2O2 and mitochondrial O2- production in leukocytes, but had no effect on mitochondrial ΔΨm and ATP production. The effect was specific for TLR2/1 as TLR3 or TLR9 ligands did not induce ROS production. Polymicrobial sepsis induced mitochondrial dysfunction in leukocytes, as demonstrated by increased H2O2 and mitochondrial O2− production (CLP vs. sham; H2O2: 3,173 ± 498, n = 5 vs. 557 ± 38, n = 4; O2−: 707 ± 66, n = 35 vs. 485 ± 35, n = 17, mean fluorescence intensity, mean ± SEM), attenuated complex III activity (13 ± 2, n = 16 vs. 30 ± 3, n = 7, milli-optical densities per minute, mOD/min), loss of mitochondrial ΔΨm, and depletion of intracellular ATP (33 ± 6, n = 11 vs. 296 ± 29, n = 4, nmol/mg protein). In comparison, there was significant improvement in mitochondrial function in septic TLR2−/− mice as evidenced by attenuated mitochondrial ROS production, better- maintained mitochondrial ΔΨm and higher cellular ATP production.
CONCLUSIONS
TLR2 signaling plays a critical role in mediating mitochondrial dysfunction in peritoneal leukocytes during polymicrobial sepsis.
INTRODUNCTION
Sepsis is defined as the systemic inflammatory response syndrome that occurs during infection 1. It has an estimated incidence of 751,000 cases each year 2. Both the incidence of sepsis and the overall sepsis-related mortality have increased significantly between 1993 and 2003 3. Similarly, the rate of severe postoperative sepsis in surgical patients has more than doubled between 2001 and 2006 4. Sepsis is the 10th leading cause of death in the United States 5.
A major cause of death in patients with severe sepsis is multiple organ failure, but the underlying pathogenesis of which is not fully understood. Mitochondrial damage and dysfunction has been recognized as an important molecular pathology in sepsis 6-9 and linked to the severity of organ dysfunction and possibly outcome of sepsis10,11. The increased production of cellular reactive oxygen species (ROS) of mitochondrial origin during sepsis can cause significant oxidative stress to cells 12 and may severely inhibit oxidative phosphorylation and adenosine triphosphate (ATP) generation 13, which can potentially cause multi-organ failure14-16.
While the host innate immune response is necessary to eradicate invading pathogens, excessive inflammatory responses during sepsis is harmful and may lead to tissue injury, in part, by damaging mitochondrial structure and function 17,18. Several molecular mechanisms have been proposed responsible for mitochondrial dysfunction 19. These include attenuated activity of mitochondrial electron transport chain enzyme complexes, inhibitory effects of reactive nitrogen and oxygen species on oxidative phosphorylation and ATP production, increased expression of mitochondrial uncoupling proteins, and the formation of the mitochondrial permeability transition pore. However, the upstream signaling that mediates these molecular events leading to mitochondrial dysfunction in sepsis is poorly understood.
Toll-like receptors (TLRs) play an essential role in the host immune and inflammatory responses during sepsis as well as certain non-infectious tissue injury 20-22. TLRs may also play a role in regulating mitochondrial function. Djafarzadeh and colleagues have shown that TLR3 activation attenuates maximal mitochondrial respiration in cultured human hepatocytes 23. West et al. demonstrate that TLR1/2/4 signaling augments macrophage bactericidal activity through mitochondrial ROS production 24. Yet, others have suggested a dual role for TLR4 signaling in modulating mitochondrial function. TLR4 activation not only triggers endotoxin-induced oxidative stress and mitochondrial DNA (mtDNA) damage, but also mediates mitochondrial biogenesis by up-regulation of mitochondrial complex IV and mitochondrial transcription factors 25,26. These data suggest that TLR signaling may have a significant impact on mitochondrial function during bacterial sepsis.
TLR2 forms a heterodimer with either TLR1 or TLR6. The resulting TLR2/TLR1 and TLR2/TLR6 complexes recognize distinct ligands triacyl and diacyl lipoproteins, respectively. We have previously demonstrated the significant contribution of TLR2 signaling to the pathogenesis of polymicrobial sepsis 27-29. TLR2 activation by bacterial wall components induces cardiomyocyte inflammatory response and dysfunction in vitro 27. TLR2 mediates intracellular hydrogen peroxide (H2O2) production29 and contributes to cardiac dysfunction and mortality 28 in septic animals. The survival benefit of TLR2 deficiency was also confirmed recently 30 and in Pseudomonas aeruginosa sepsis model 31. In the current study, we tested the hypothesis that TLR2 mediates mitochondrial dysfunction during polymicrobial sepsis. Specifically, we tested the effect of TLR activation on mitochondrial function in isolated leukocytes in vitro and determined the impact of TLR2 deletion on mitochondrial dysfunction in a mouse model of peritoneal polymicrobial sepsis.
MATERIALS AND METHODS
Animals
Eight to 12 week-old gender- and age-matched mice were used for the studies. Wild-type (WT) (C57BL/6J) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and housed in a animal facility at Massachusetts General Hospital for at least 1 week before experiments. TLR2−/− mice were generated by Takeuchi et al 32. All animals were housed in pathogen-free, temperature-controlled, and air-conditioned facilities with 12 h/12 h light/dark cycles and fed with the same bacteria-free diet. Animal care and procedures were performed according to the protocols approved by the Massachusetts General Hospital Subcommittee on Research Animal Care and were in compliance with the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health. Simple randomization method was used to assign animals to various experimental conditions.
Reagents
Pam3Cys, Poly (I:C), CpG were purchased from Enzo Life Science (Farmingdale, NY). Lipopolysaccharide (Escheridhia coli 0111:B4) and lipoteichoic acid (LTA) were from Sigma- Aldrich (St Louis, MO). Dichlorodihydrofluorescein diacetate (H2-DCF-DA), MitoSOX red reagent, and 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) were purchased from Invitrogen-Molecular Probes (Eugene, OR). Antimycin A and tetramethylrhodamine ethyl ester perchlorate (TMRE) were purchased from Sigma-Aldrich. ATP bioluminescence assay kit CLS II was purchased from Roche Molecular Biochemicals (Indianapolis, IN). MitoTox™ OXPHOS Complex III Activity Kit was from Abcam (Cambridge, MA).
Peritoneal cell isolation after thioglycollate injection
Peritoneal cells were elicited chemically by intra-peritoneal injection of 4% thioglycollate. Twelve to 16 hours later, 6 ml of Dulbecco's phosphate-buffered saline (DPBS) without calcium and magnesium was injected into the peritoneal space and mixed thoroughly by gentle massage. The peritoneal lavage fluid was collected and centrifuged at 1,500 rpm for 5 min. The supernatants were discarded and the cell pellets were suspended in RPMI 1640 containing 0.05% bovine serum albumin. We have previously shown that more than 85% of the peritoneal cells are Gr-1+ neutrophils 33.
Peritoneal cell collection following surgery
Twenty-four hours after sham or cecum ligation and puncture (CLP) surgery, 6 ml of ice-cold DPBS without calcium and magnesium was injected into the peritoneal space and mixed thoroughly by gentle massage. Five ml of the peritoneal lavage were collected and centrifuged at 1,500 rpm for 5 min. The supernatants were discarded and the cell pellets were suspended in RPMI 1640. We have previously shown that more than 90% of the peritoneal cells from the CLP mice are Gr-1+ neutrophils 34.
ROS
Total intracellular H2O2 was measured with dichlorodihydrofluorescein diacetate (H2-DCF-DA, Cat. D399, Invitrogen), whereas mitochondrial superoxide (O2−) was assayed with MitoSOX (Cat. M36008, Invitrogen). Specifically, peritoneal neutrophils were harvested, plated in 96-well plate, and treated with antimycin A (Cat. A8674, Sigma) or TLR ligands as indicated. At the end of treatment, cells were incubated with freshly prepared H2-DCF-DA or MitoSOX at 37 °C in the dark for 30 min. Unstained controls were handled similarly except that treatments and dyes were omitted. Dye-loaded cells were resuspended in cold DPBS containing 1% FBS and analyzed immediately by flow cytometry at fluorescein isothiocyanate or R-phycoerythrin channel. Ten thousand cells were routinely counted by flow cytometry, and data expressed as the median fluorescence intensity in arbitrary units from at least three separate experiments. In some experiments, MitoSOX-stained cells attached to pre-coated plates (with 5 µg/ml of fibronectin and 20 µg/ml of gelatin) were analyzed for ROS production under fluorescence microscope (Texas Red channel).
Mitochondrial membrane potential
Two methods were employed to measure mitochondrial membrane potential (ΔΨm). First, we used TMRE (Cat. 87917, Sigma) to measure levels of ΔΨm. TMRE is a cationic dye that is rapidly and reversibly accumulated by healthy mitochondria. Decrease in the levels of TMRE indicates reduction in mitochondrial membrane potential levels. Experimentally, a fraction of cells (5 × 105) from the peritoneal lavage was labeled with freshly prepared TMRE at 37 °C in the dark for 30 minutes. Unstained controls were treated similarly, except that ligand treatment and dyes were omitted. Dye-loaded cells were immediately re-suspended in cold DPBS containing 1% FBS and analyzed immediately by flow cytometry at the R-phycoerythrin channel. Ten thousand cells were routinely collected, and data were expressed as the mean fluorescence intensity in arbitrary units from the average of at least three separate experiments. Second, we measured mitochondrial ΔΨm using JC-1 dye (Invitrogen, MP 03168). Specifically, peritoneal leukocytes were treated with antimycin A or stimulated with TLR ligands as indicated. At the end of treatments, cells were incubated with 2 μM JC-1 at 37°C for 30 min and washed twice with DPBS. Finally, fluorescence was read at red fluorescence (excitation 535 nm, emission 590 nm) and green fluorescence (excitation 485 nm, emission 530 nm) using a fluorescence plate reader. The level of ΔΨm was calculated by ratio of red fluorescence to green fluorescence.
ATP assay
Intracellular ATP level was measured by a luciferase-based assay using the ATP Bioluminescence Assay Kit CLS II (Roche Molecular Biochemicals). In brief, intracellular ATP was released using a boiling method. Specifically, peritoneal neutrophils were treated with antimycin A or stimulated with TLR ligands as indicated. At the end of treatments, cells were then harvested, washed twice with ice-cold DPBS, drained and resuspended in boiling buffer (100 mM Tris and 4 mM EDTA, pH 7.75). The suspensions were pipetted, vortexed, and snap frozen in liquid nitrogen. Frozen cells were boiled for 3 min in a water bath, placed on ice for 5 min, and then centrifuged at 14,000 rpm for 10 min at 4 °C. The supernatant was transferred to a fresh tube and kept on ice until measurement. Finally, 35 μl of luciferase reagent was added to 35 μl of the sample or standard. Experiments were performed in triplicates, and data were standardized to the protein concentration using the Bradford protein assay.
Mouse model of polymicrobial sepsis
A mouse model of polymicrobial sepsis was generated by CLP as described previously 28. In brief, the cecum was ligated 1.0 cm from the tip. A through and through puncture was made with an 18-gauge needle and a small amount (droplet) of feces was extruded to ensure the patency of the puncture site before returned it back to the abdominal cavity. The sham-operated mice underwent laparotomy but without CLP. The abdominal wall incision was closed in layers. After surgery, pre-warmed normal saline (50 ml/kg) was administered subcutaneously. Postoperative pain control was managed with subcutaneous injection of bupivacaine (3 mg/kg) and buprenorphine (0.1 mg/kg). Of note, this model of polymicrobial peritonitis in C57BL/6 mice leads to severe sepsis as evidenced by multi-organ dysfunction such as cardiac dysfunction and acute kidney injury with 60-90% of mortality 28,34.
Mitochondrial complex III activity assay
Mitochondrial complex III activity was measured using MitoTox™ OXPHOS Complex III Activity Kit (Cat. ab109905, Abcam) according to the manufacturer's protocol with some modifications. Briefly, cells were lysed by sonication and mitochondrial fractions were re- suspended in ice-cold DPBS. Complex III activity was then measured in a mixture (1:1 ratio) of cell suspension and assay solution containing succinate, rotenone, potassium cyanide, cytochrome c by monitoring complex III-sensitive cytochrome c reduction (λ=550nm). Data were collected every 20 s for 5 min after initiation of the reaction.
Mitochondrial gene expression
Mitochondrial transcript factor A (Tfam) and cytochrome c oxidase subunit II (COX2), both coded by mtDNA, were quantified by real-time qRT-PCR.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA). The distributions of the continuous variables were expressed as the mean ± SEM. Data were analyzed by one-way ANOVA with Tukey or two-way ANOVA with Bonferroni post hoc tests for statistic significance. Of note, the sample sizes were based on our prior experiences rather than a formal statistical power calculation. The null hypothesis was rejected for P < 0.05 with the two-tailed test.
RESULTS
TLR2 activation leads to intracellular and mitochondrial ROS production in peritoneal leukocytes
To establish a system that is reliable and sufficiently sensitive to detect cellular ROS production, we first tested the effect of antimycin A on intracellular H2O and mitochondrial O2− production in the peritoneal leukocytes. Antimycin A is a potent inhibitor of the mitochondrial respiratory chain enzyme complex III and known for its ability to induce mitochondrial O2− production as demonstrated in figure 1A. As illustrated in figure 1B-C, antimycin A treatment led to both intracellular H2O2 and mitochondrial O2− production as measured by flow cytometry and fluorescent microscopy. To determine whether or not TLR signaling induces ROS production, we next stimulated leukocytes with various TLR ligands. Similar to antimycin A, Pam3Cys (a TLR1/2 ligand, 20 μg/ml) induced a significant increase in both intracellular H2O2 and mitochondrial O2−2 levels as demonstrated by flow cytometry (H2O2: con vs. Pam3, 531 ± 57 vs. 2426 ± 89; O2−: con vs. Pam3, 848 ± 38 vs. 1,621 ± 91, mean fluorescence intensity [MFI]) (fig. 2A-D) and fluorescent microscopy (fig. 1). In contrast, at the same concentration, LTA (a TLR2/6 ligand), Poly (I:C) (a TLR3 ligand) or CpG (a TLR9 ligand) had no effect on intracellular or mitochondrial ROS production. Lipopolysaccharide (a TLR4 ligand) only induced a modest increase in mitochondrial O2− level (fig. 2A-D). The effect of Pam3Cys was dose-dependent and partially mediated via TLR2 as Pam3Cys-induced mitochondrial O2− production was significantly attenuated in TLR2-deficient leukocytes (WT vs. knockout [KO], 2766±259 vs. 2,044 ± 57, MFI) (fig. 2E-G).
Figure 1. Antimycin A and Pam3Cys induce intracellular H2O2 and mitochondrial O2− production in peritoneal leukocytes.

A, Antimycin A leads to a dose-dependent mitochondrial O2− production. Thioglycollate-elicited peritoneal leukocytes were treated with antimycin A for 1 h and analyzed for mitochondrial O2− production with flow cytometry. n=3 in each group. * P < 0.05, ** P < 0.01 versus the untreated controls. Each error bar represents mean ± SEM. MFI, mean fluorescence intensity. The experiments were performed twice with similar results. B-C, Antimycin A- or Pam3Cys-induced intracellular or mitochondrial ROS production. Representative histograms of flow cytometry (B) and fluorescent images (C) are presented. Peritoneal leukocytes were treated with antimycin A (10 μg/ml) or Pam3Cys (20 μg/ml) for 1 h, incubated with 10 μM DCF or 2.5μM MitoSOX, and then analyzed for cellular H2O2 or mitochondrial O2− production, respectively, with flow cytometry (B) or fluorescent microscope (C). DCF = dichlorodihydrofluorescein diacetate; MitoSOX fluo = MitoSOX fluorescence; Mito O2− = mitochondrial superoxide; H2O2 = hydrogen peroxide.
Figure 2. Effect of various TLR ligands on intracellular H2O2 and mitochondrial O2− production in peritoneal leukocytes.

A-B, Effect of TLR ligands on intracellular H2O2 generation. Thioglycollate-elicited peritoneal leukocytes were treated with TLR ligands as indicated: Pam3Cys, LTA or LPS (20 μg/ml) for 1 h, incubated with 10 μM of DCF and analyzed for intracellular H2O2 with flow cytometry. Representative histograms are presented in A and combined MFI data in B. n = 3 in each group. *** P < 0.001 versus control. The experiments were performed twice with the similar results. C- D, Effect of TLR ligands on mitochondrial O-2production. Cells were treated with TLR ligands as indicated: Pam3Cys, LTA, LPS, Poly(I:C) or CpG (20 μg/ml) for 1 h, incubated with 2.5μM of MitoSOX and analyzed for mitochondrial O2− production. Representative histograms are presented in C and combined MFI data in D. n = 3 in each group. * P < 0.05, *** P < 0.001 versus control. The experiments were performed four times. E, TLR2 activation induces a dose- dependent mitochondrial O2− production. N = 3 in each group. * P < 0.05, *** P < 0.001 versus untreated control. The experiments were performed twice. F-G, TLR2 mediates Pam3Cys- induced mitochondrial O2− production. Peritoneal leukocytes harvested from WT or TLR2−/− mice were treated with Pam3Cys (20 μg/ml) for 1 h and analyzed for mitochondrial O2− production with flow cytometry. Representative histograms are presented in F and combined MFI data in G. n = 3 in each group. *** P < 0.001 versus control. ## P < 0.01 versus WT. Each error bar represents mean ± SEM. MFI = mean fluorescence intensity; DCF = dichlorodihydrofluorescein diacetate; Mito O2− = mitochondrial superoxide; WT = wild- type; TLR2KO = TLR2 knockout; LTA - lipoteichoic acid; LPS = lipopolysaccharides; H2O2 = hydrogen peroxide. T he experiments were performed twice with similar results.
TLR2 activation has no impact on mitochondrial membrane potential and intracellular ATP production
The mitochondrial membrane potential (ΔΨm) is generated by protons transport across the mitochondrial inner membrane. This process is catalyzed by the enzyme complexes I, III, and IV of the electron transport chain and produces the proton motive force to generate ATP. Previous studies have shown that a positive correlation exists between ΔΨm reduction and ROS production 35-37 and that ATP depletion represents a hallmark of mitochondrial dysfunction 38. We therefore analyzed ΔΨm response to TLR ligands using two mitochondrial membrane potential-sensitive fluorescent probes, namely TMRE and JC-1. As illustrated in figure 3A-B, while antimycin A, a complex III inhibitor, induced a dose-dependent reduction in the mitochondrial ΔΨm, most TLR ligands tested, i.e., Pam3Cys, LTA, lipopolysaccharide and CpG, had no effect on ΔΨm. poly (I:C) led to a higher ΔΨm. Consequently, antimycin A led to marked reduction in ATP production in leukocytes (fig. 3C-D). Moreover, absence of glucose in culture media markedly reduced ATP production in the untreated cells (Control) and further abolished ATP production in the antimycin A-treated leukocytes (fig. 3C vs. 3D). Similar to ΔΨm data, Pam3cys and lipopolysaccharide did not reduce cellular ATP production in leukocytes (fig. 3C-D). These data suggest that unlike the complex III blocker antimycin A, TLR activation is not sufficient to induce mitochondrial dysfunction.
Figure 3. TLR2 activation has no impact on mitochondrial ΔΨm and intracellular ATP production.
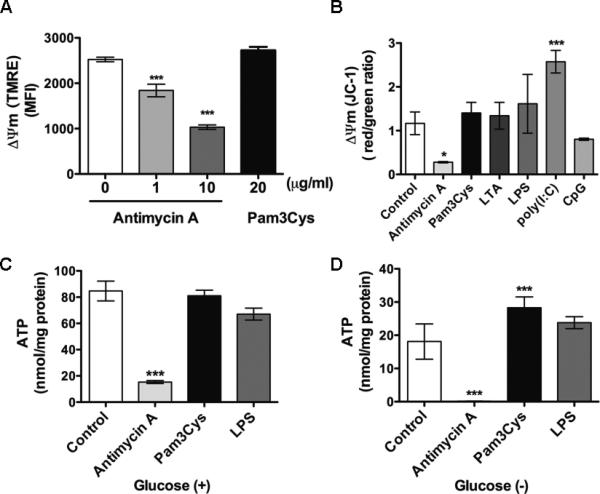
A-B, Mitochondrial ΔΨm measurements. Mitochondrial ΔΨm was detected with TMRE (A) or JC-1 (B) dye. A, Peritoneal leukocytes were treated with the indicated concentrations of antimycin A or Pam3Cys for 1 h and analyzed for ΔΨm with flow cytometry. *** P < 0.001 versus control. The numbers of samples in each group: 0 μg/ml Antimycin A, n=7; 1 μg/ml Antimycin A, n = 5; 10 μg/ml Antimycin A, n = 3; 20 μg/ml Pam3cys, n = 4. The experiments were performed twice. B, Cells were treated with antimycin A or TLR ligands as indicated: Pam3Cys, LTA, LPS, Poly (I:C) or CpG, all at 20 μg/ml, for 1 h and analyzed for ΔΨm with fluorescence ratio detection. n=3 in each group. * P < 0.05, *** P < 0.001 versus control. The experiments were performed twice. C-D, ATP production in the presence or absence of glucose. C, Cells were treated with antimycin A, Pam3Cys or LPS (all at 20 μg/ml) in glucose containing medium for 4 h and analyzed for ATP production with a ATP bioluminescence assay kit. n=3 in each group. The experiments were performed three times. D, Cells were treated with antimycin A, Pam3Cys or LPS (all at 20 μg/ml) in glucose-free medium for 1 h and analyzed for intracellular ATP level, *** P < 0.001 versus control. Each error bar represents mean ± SEM. The numbers of samples in each group: Control, n = 5; Antimycin A, n = 5; Pam3cys, n = 5; LPS, n = 5. MFI = mean fluorescence intensity; ΔΨm = membrane potential; TMRE = tetramethylrhodamine ethyl ester perchlorate; ATP = adenosine triphosphate; LTA = lipoteichoic acid; LPS = lipopolysaccharides.
TLR2 mediates mitochondrial ROS production in leukocytes during polymicrobial sepsis
Next, we tested whether or not TLR2 plays a role in leukocyte mitochondrial ROS production in sepsis. We subjected WT and TLR2−/− mice to sham or CLP procedure, a clinically relevant animal model of peritoneal polymicrobial sepsis. Twenty-four hours after the procedures, the peritoneal cells were harvested and the intracellular H2O2 and mitochondrial O2− were measured using flow cytometry. As indicated in figure 4, there was a basal level of ROS signal in the peritoneal leukocytes isolated from sham mice. However, in leukocytes harvested from WT septic mice, there was a significant increase in cellular H2O and mitochondrial O2− levels. In comparison, both intracellular H2O2 and mitochondrial O2− were markedly reduced in TLR2−/− septic mice (intracellular H2O2: 3,173 ± 498 vs. 1,628 ± 324; mito O2−: 707 ± 66 vs. 451 ± 37, WT-CLP vs. TLR2 KO-CLP, MFI) (fig. 4). These data clearly suggest that TLR2 signaling plays an important role in mediating cellular H2O2 and mitochondrial O -2 production in the peritoneal leukocytes during polymicrobial sepsis.
Figure 4. Absence of TLR2 attenuates leukocyte cellular H2O2 and mitochondrial O2− production during polymicrobial sepsis.

WT and TLR2−/− mice were subjected to sham or CLP procedures. Twenty-four hours later, peritoneal leukocytes were harvested, stained with either 10 μM of DCF or 2.5 μM of MitoSOX, and analyzed with flow cytometry for intracellular H2O2 (A-B) or mitochondrial O2− (C-D) production. The numbers of samples in panel B: WT-Sham, n = 4; WT-CLP, n = 5; TLR2KO- Sham, n = 5; TLR2KO-CLP, n = 5. The numbers of samples in panel D: WT-Sham, n = 17; WT- CLP, n = 35; TLR2KO-Sham, n = 15; TLR2KO-CLP, n = 23. * P < 0.05, *** P < 0.001 versus sham. # P < 0.05, ## P < 0.01 versus WT. Each error bar represents mean ± SEM. MFI = mean fluorescence intensity; DCF = dichlorodihydrofluorescein diacetate; WT = wild type; KO = knockout; CLP = cecum ligation and puncture; Mito O2− = mitochondrial superoxide; H2O2 = hydrogen peroxide.
TLR2 signaling contributes to mitochondrial dysfunction during polymicrobial sepsis
Given the role of mitochondrial ΔΨm and ATP production in mitochondrial ROS generation, we tested the mitochondrial ΔΨm and intracellular ATP concentration in peritoneal leukocytes of animals with polymicrobial peritonitis. We found that there was marked reduction in the mitochondrial ΔΨm and ATP generation in septic WT mice as compared with the sham-operated controls (ΔΨm: 4,455 ± 400 vs. 1,694 ± 352, MFI; ATP: 296 ± 29 vs. 33 ± 6 nmol/mg protein; sham vs. CLP in WT, MFI) (fig. 5). TLR2 deletion significantly improved the mitochondrial ΔΨm and ATP production (ΔΨm: 1,694 ± 352 vs. 2,866 ± 167, MFI; ATP: 33 ± 6 vs. 71 ± 8, nmol/mg protein; WT-CLP vs. TLR2KO-CLP) (fig. 5). These results suggest that TLR2 signaling contributes to the leukocyte mitochondrial dysfunction during polymicrobial sepsis.
Figure 5. TLR2−/− mice have improved leukocyte mitochondrial ΔΨm and intracellular ATP production during severe sepsis.

A-B, Mitochondrial ΔΨm. WT and TLR2−/− mice were subjected to sham or CLP surgical procedures. Twenty-four hours later, the peritoneal cells were harvested, stained with TMRE and analyzed for ΔΨm. A, Representative flow cytometry histograms; B, Combined MFI. The numbers of samples in each group: WT-Sham, n = 5; WT-CLP, n = 12; TLR2KO-Sham, n = 5; TLR2KO-CLP, n = 12. *** P < 0.001 versus sham. ## P < 0.01 versus WT. C, Cellular ATP. Mice were subjected to sham or CLP and 24 h later the peritoneal cells were harvested and analyzed for intracellular ATP level by ATP bioluminescence assay. The numbers of samples in each group: WT-Sham, n = 4, WT-CLP: n = 11, TLR2KO-Sham, n = 5, TLR2KO-CLP, n = 12. *** P < 0.001 versus sham. ## P < 0.01 versus WT. Each error bar represents mean ± SEM. MFI = mean fluorescence intensity. ΔΨm = membrane potential. TMRE = tetramethylrhodamine ethyl ester perchlorate. WT = wild type; TLR2KO = TLR2 knockout; CLP = cecum ligation and puncture; ATP = adenosine triphosphate;
Polymicrobial sepsis inhibit mitochondrial complex III activity in peritoneal via a TLR2- independent mechanism
Studies have demonstrated that complex III is one of the principal sites responsible for mitochondrial ROS generation 39. We next examined the complex III activities in leukocytes isolated from sham and septic animals and tested the impact of TLR2 deficiency on their activities during polymicrobial sepsis. As illustrated in figure 6, there was a marked reduction in the complex III activity in WT CLP mice as compared to the sham control mice (30 ± 3 vs. 13 ± 2, sham vs. CLP in WT, mOD/min). However, TLR2-deficient mice did not have improved complex III function as compared with WT mice following CLP. This data clearly suggest that TLR2 signaling mediates ROS production and mitochondrial dysfunction during polymicrobial sepsis via a complex III-independent mechanism.
Figure 6. TLR2 deletion has no effect on leukocyte mitochondrial complex II/III enzyme activity during polymicrobial sepsis.
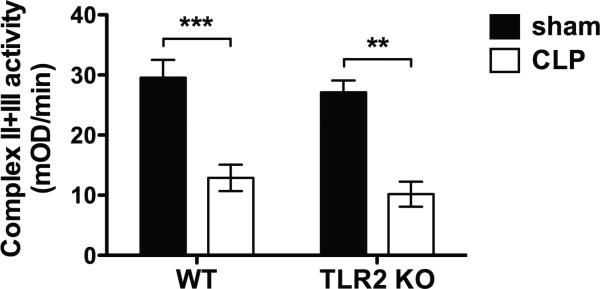
WT and TLR2−/− mice were subjected to sham or CLP surgical procedures. Twenty-four hours later, the peritoneal cells were harvested and analyzed for mitochondrial complex II/III activity by MitoTox™ OXPHOS Complex III Activity Kit. The numbers of animals in each group: WT- Sham, n = 7; WT-CLP, n = 16; TLR2KO-Sham, n = 3; TLR2KO-CLP, n = 7. ** P < 0.01, *** P < 0.001 versus sham. Each error bar represents mean ± SEM. WT = wild type; TLR2KO = TLR2 knockout; CLP = cecum ligation and puncture.
Polymicrobial sepsis induces mitochondrial Tfam and COX 2 depletion
Mitochondrial oxidative stress can lead to mtDNA damage and depletion. The mtDNA is reported more susceptible to oxidative stress than nuclear DNA 40. Studies have demonstrated that lipopolysaccharide induces mitochondrial oxidative stress and mtDNA depletion 41. We examined the effect of polymicrobial sepsis on the expression of the two mitochondrial molecules, namely mitochondrial transcript factor A (Tfam) and cytochrome c oxidase subunit II (COX 2), both coded by mtDNA. As shown in figure 7A, compared with sham mice, CLP led to significantly lower Tfam and COX 2 gene expression in the liver. This effect seemed more prominent in the liver as CLP did not significantly impact on Tfam and COX 2 expression in the heart or peritoneal leukocytes within the same period of time (24 h) (fig. 7B-C). Similar to mitochondrial complex III activity shown in figure 6, TLR2 deficiency did not reverse the reduced Tfam and COX 2 gene expression in the septic liver (fig. 7A).
Figure 7. Mitochondrial gene expression in polymicrobial sepsis.

WT and TLR2−/− mice were subjected to Sham or CLP procedure. Twenty-four h after the surgery, liver, heart and peritoneal cells were harvested. Total RNA was extracted and mitochondrial gene expression was measured by qRT-PCR and normalized to GAPDH levels. * P < 0.05, ** P < 0.01, *** P < 0.001. The numbers of animals in each group: A, n = 6 mice in each group, B, n = 6 in each group, C, Tfam, n = 11 in WT-sham, n = 9 in TLR2KO sham, n = 6 in CLP groups; COX 2: n = 9 in WT-sham, n = 9 in TLR2KO sham, n = 5 in WT CLP group, n = 6 in TLR2KO CLP group. Each error bar represents mean ± SEM. WT= wild type; TLR2KO = TLR2 knockout; CLP = cecum ligation and puncture; Tfam = mitochondrial transcript factor A; COX2 = cytochrome c oxidase subunit II; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; gRT-PCR = ?????.
DISCUSSION
The current study demonstrates a pivotal role of TLR2 signaling in mediating mitochondrial ROS production as well as mitochondrial dysfunction in a clinically relevant mouse model of severe polymicrobial sepsis. First, we found that activation of TLR1/2, but not TLR2/6, TLR3, TLR4, or TLR9, was capable of inducing a robust intracellular and mitochondrial ROS production in leukocytes. We also found that while the inhibition of mitochondrial respiratory complex III reliably caused mitochondrial dysfunction as evidenced by reduced mitochondrial ΔΨm and cellular ATP production, TLR1/2 activation appeared insufficient to induce mitochondrial dysfunction in isolated leukocytes. Second, we found that polymicrobial peritonitis sepsis led to a marked mitochondrial dysfunction in peritoneal leukocytes with increased intracellular and mitochondrial ROS, decreased mitochondrial ΔΨm, reduced intracellular ATP, and markedly inhibited mitochondrial complex III activity. In comparison, mice deficient of TLR2 had significantly improved mitochondrial function with markedly reduced intracellular and mitochondrial ROS production, and significantly improved mitochondrial ΔΨm, and intracellular ATP production. However, TLR2 deficiency had no impact on mitochondrial complex III activity in both sham and sepsis animals. Finally, we found that polymicrobial sepsis in mice led to depletion of mitochondrial Tfam and COX 2 gene expression in the liver and this process seems independent of TLR2
We have observed that TLR1/2 activation by Pam3cys leads to a robust production of both cellular H2O2 and mitochondrial O2−. Cellular ROS (including O2− and H2O2) is generated not only via NADPH oxidase-dependent respiratory burst, but also through mitochondrial oxidative phosphorylation process. Mitochondria are a major site for ROS production 42. During the normal respiration process, ROS is produced as a by-product when high-energy electrons escape before they reach the final acceptor oxygen. The first ROS produced in mitochondria is the highly reactive superoxide anion (O2−), which can mediate oxidative damage to cells. Superoxide dismutase, an intrinsic antioxidant defense system, converts O2− into a much more stable ROS, H2O 39,43. O2− has very limited membrane permeability, but H2O2 can diffuse across membranes 44,45 and leave mitochondrion to cytosol 46. Therefore, it is very much likely that the increased mitochondrial ROS production contributes to a portion of the increased intracellular ROS in the leukocytes following TLR1/2 stimulation or during polymicrobial sepsis.
Our previous study shows that activation of TLR1/2, but not TLR3, TLR4 or TLR9, in induces a marked intracellular H2O2 production in rat cardiomyocytes and mouse bone marrow- derived neutrophils 29. Consistent with this, the current study demonstrates a highly selective and robust effect for TLR1/2 in its ability to induce mitochondrial O2− production in neutrophils. A similar finding has been reported in macrophages, where TLR1/2 activation induces mitochondrial ROS production via a mechanism involving TRAF-6 mitochondrial translocation and interaction with a complex I-associated protein ECSIT (evolutionarily conserved signalling intermediate in Toll pathways) 24. Interestingly, under the same conditions and unlike antimycin A (a complex III inhibitor), TLR1/2 activation by Pam3cys seems incapable of causing mitochondrial dysfunction. Pam3cys treatment has no effect on mitochondrial ΔΨm and intracellular ATP production, which has been linked to mitochondrial O2− production. Importantly, while mitochondria may produce more ROS at higher membrane potential 35,36, lower ΔΨm and decreased activity of the respiratory chain during mitochondrial dysfunction is associated with a simultaneous increase in ROS production 37 as we have demonstrated in antimycin A-treated leukocytes. These data suggest that TLR1/2 activation alone does not induce depolarization of mitochondrial ΔΨm and subsequent impairment of oxidative phosphorylation and thus is insufficient to impair mitochondrial function. Mitochondrial ROS generation has been linked with several key cellular processes, such as cell death, cellular oxidative stress, inflammatory cytokine production 47 and macrophage bactericidal activity 24. We have shown that TLR2 activation leads to several pro-inflammatory cytokine production 29. Thus, it is possible that TLR2-induced ROS production may serves as an intracellular signal transducing molecules in cytokine production, rather than a sign of mitochondrial dysfunction and oxidative stress in normal peritoneal leukocytes. Interestingly, in our study, TLR4 activation by lipopolysaccharide fails to induce intracellular H2O2 production and only induces a very modest increase in mitochondrial O2− level in neutrophils. However, in macrophages, lipopolysaccharide reportedly induces marked ROS production including mitochondrial ROS 24,48 and results in mitochondrial dysfunction and biogenesis in the heart and liver 49-52.
We demonstrate that polymicrobial sepsis leads to a robust increase in intracellular and mitochondrial ROS production in leukocytes isolated from the infectious peritonitis site. Moreover, TLR2 deficiency markedly reduces ROS production in the peritoneal leukocytes compared to WT mice, suggesting that TLR2 signaling may contribute to leukocyte ROS production during polymicrobial sepsis. To further probe the underlying mechanisms, we tested the effect of TLR2 on mitochondrial function and identified that polymicrobial infection led to marked mitochondrial dysfunction in leukocytes with significantly reduced mitochondrial ΔΨm and intracellular ATP production. In comparison, mice deficient of TLR2 had preserved mitochondrial ΔΨm and significantly improved intracellular ATP production. These data suggest that TLR2 signaling may play a contributory role in mitochondrial dysfunction and subsequent mitochondrial ROS production during polymicrobial sepsis. As demonstrated before, TLR2- deficient mice have markedly improved neutrophil migratory and phagocytic function, enhanced blood bacterial clearance and reduced systemic cytokine productions compared with WT mice during polymicrobial sepsis 28,29,53. Collectively, these studies demonstrate that TLR2 signaling plays a central role in regulating mitochondrial function, cellular ROS production, leukocyte migration, and phagocytosis during polymicrobial sepsis.
Antimycin A is a specific inhibitor of mitochondrial complex III. It inhibits succinate and NADPH oxidase, and mitochondrial electron transport between cytochromes b and c. The inhibition of electron transport causes the production of ROS and results in a collapse of the proton gradient across the mitochondrial inner membrane, thereby breaking down the mitochondrial ΔΨm and reducing intracellular ATP generation 54-58. Distinctly different from antimycin A, TLR 2 activation exhibits no effect on mitochondrial ΔΨm and ATP production even it leads to increased ROS production. This implies that TLR2-mediated mitochondrial ROS production is not associated with mitochondrial dysfunction including that of complex III activity. Similarly, in vivo, septic mice exhibit marked reduction in mitochondrial complex III activity and reduced gene expression of Tfam and COX2. However, TLR2 deficiency does not protect against complex III activity inhibition or mtDNA depletion during polymicrobial sepsis although it does improve mitochondrial ΔΨm and intracellular ATP production. Further investigation will be needed to understand the molecular mechanisms by which TLR2 signaling mediates mitochondrial ROS generation in healthy condition and then contributes to mitochondrial dysfunction and ROS production during severe polymicrobial sepsis.
A significant amount of work has been done in determining the role of oxidative stress and mitochondrial dysfunction in sepsis-induced organ injury 59. Lowes and colleagues found that mitochondria-targeted antioxidant Mito Q reduces ROS production in lipopolysaccharide - treated endothelial cells, arguments mitochondrial membrane potential in major organs, and reduces acute liver and kidney dysfunction after lipopolysaccharide -peptidoglycan administration 60. Moreover, in vivo administration of superoxide dismutase, a free-radical scavenger, prevents endotoxin-induced cardiac dysfunction 61. These studies appear to suggest that cell oxidative stress and mitochondrial dysfunction during endotoxemia can lead to organ dysfunction. However, the role of mitochondrial ROS in organ dysfunction during polymicrobial sepsis is less clear. While we have demonstrated the importance of TLR2 in mitochondrial dysfunction as well as cardiac dysfunction in polymicrobial sepsis 28,29, whether mitochondrial dysfunction and oxidative stress induce cardiac functional impairment remains to be investigated.
Different animal models of sepsis have been created and categorized as three classes: 1) bacterial infusion models, 2) endotoxin models, and 3) polymicrobial peritonitis models. Infusion models utilize bolus or short term infusion of bacteria62. These models do not correlate well with the clinical situations where in most cases, there is a focus of infection providing continuous dissemination of bacteria. Endotoxin model simulates the clinical situation of hyperinflammation and septic shock 63. Endotoxin models are highly reproducible and can provide great insight into inflammatory processes 64. However, these models lack an infectious focus and do not closely mimic the pathophysiology observed in septic patients. Bacterial peritonitis models closely resemble the clinical condition of sepsis following bowel perforation. The most widely used peritonitis model is CLP. Similar to many clinical cases of sepsis, CLP model induces polymicrobial sepsis, but the model has a wide variability in terms of the host inflammatory and physiological responses, and the degree of bacteremia and mortality rates64,65. Another limitation of the CLP model is the lack of clear information on the specific pathogens and the associated pathogen components in the pathogenesis of sepsis as the models involve a mixture of several types of bacteria including both Gram-positive and Gram-negative organisms 66.
In summary (fig. 8), our data suggest that TLR1/2 activation by Pam3cys is capable of inducing intracellular H2O and mitochondrial O2− production although it seems insufficient to cause mitochondrial dysfunction. In a mouse model of severe polymicrobial sepsis and employing TLR2-deficient mice, we demonstrate that TLR2 signaling contributes to intracellular and mitochondrial ROS production and mitochondrial dysfunction as evidenced by depleted ATP production and loss of mitochondrial membrane potential (ΔΨm) in leukocytes. However, sepsis-induced other mitochondrial dysfunction in leukocytes such as complex III dysfunction in leukocytes and mtDNA reduction in the liver seems to be TLR2-independent. Nevertheless, this study illustrates an important role of TLR2 in mitochondrial dysfunction, which might contribute to the pathogenesis of organ failure during severe sepsis.
Figure 8. Schematic view of the proposed role of TLR2 in mitochondrial dysfunction during polymicrobial sepsis.

Activation of TLR1/2 heterodimer by Pam3cys leads to production of ROS, including cellular H2O2 or mitochondrial O2− in peritoneal leukocytes. Polymicrobial sepsis induces mitochondrial dysfunction as evidenced by mROS production, ATP depletion, loss of mitochondrial membrane potential (ΔΨm), complex III dysfunction in leukocytes and mtDNA reduction in the liver. mROS production, ATP depletion, and ΔΨm reduction are mediated via TLR2-dependent mechanisms. ΔΨm = membrane potential; ATP = adenosine triphosphate; Comp. III = complex III; mtDNA = mitochondrial DNA; TLR2 = toll- like receptor 2; mROS = mitochondrial reactive oxygen species; H2O2 = hydrogen peroxide; O2− = mitochondrial superoxide.
Acknowledgments
Disclosure: This work was supported in part by the National Institutes of Health (Bethesda, Maryland) grant R01-GM080906 and R01-GM097259 (to Dr. Chao) and a mentored research award from International Anesthesia Research Society (San Francisco, California) (to Dr. Zou).
Footnotes
The authors declare no competing interests.
REFERENCES
- 1.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G. SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 2003. 2001;31:1250–6. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: A trend analysis from 1993 to 2003. Crit Care Med. 2007;35:1244–50. doi: 10.1097/01.CCM.0000261890.41311.E9. [DOI] [PubMed] [Google Scholar]
- 4.Bateman BT, Schmidt U, Berman MF, Bittner EA. Temporal trends in the epidemiology of severe postoperative sepsis after elective surgery: A large, nationwide sample. Anesthesiology. 2010;112:917–25. doi: 10.1097/ALN.0b013e3181cea3d0. [DOI] [PubMed] [Google Scholar]
- 5.Minino AM, Heron MP, Smith BL. Deaths: Preliminary data for 2004. Natl Vital Stat Rep. 2006;54:1–49. [PubMed] [Google Scholar]
- 6.Crouser ED. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion. 2004;4:729–41. doi: 10.1016/j.mito.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 7.Exline MC, Crouser ED. Mitochondrial mechanisms of sepsis-induced organ failure. Front Biosci. 2008;13:5030–41. doi: 10.2741/3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azevedo LC. Mitochondrial dysfunction during sepsis. Endocr Metab Immune Disord Drug Targets. 2010;10:214–23. doi: 10.2174/187153010791936946. [DOI] [PubMed] [Google Scholar]
- 9.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–91. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 10.Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–23. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- 11.Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, Smolenski RT, Singer M. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol. 2004;286:R491–7. doi: 10.1152/ajpregu.00432.2003. [DOI] [PubMed] [Google Scholar]
- 12.Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. 2011;107:57–64. doi: 10.1093/bja/aer093. [DOI] [PubMed] [Google Scholar]
- 13.Taylor DE, Ghio AJ, Piantadosi CA. Reactive oxygen species produced by liver mitochondria of rats in sepsis. Arch Biochem Biophys. 1995;316:70–6. doi: 10.1006/abbi.1995.1011. [DOI] [PubMed] [Google Scholar]
- 14.Andrades M, Ritter C, de Oliveira MR, Streck EL, Fonseca Moreira JC, Dal-Pizzol F. Antioxidant treatment reverses organ failure in rat model of sepsis: Role of antioxidant enzymes imbalance, neutrophil infiltration, and oxidative stress. J Surg Res. 2011;167:e307–13. doi: 10.1016/j.jss.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 15.Ritter C, Andrades ME, Reinke A, Menna-Barreto S, Moreira JC, Dal-Pizzol F. Treatment with N-acetylcysteine plus deferoxamine protects rats against oxidative stress and improves survival in sepsis. Crit Care Med. 2004;32:342–9. doi: 10.1097/01.CCM.0000109454.13145.CA. [DOI] [PubMed] [Google Scholar]
- 16.Ritter C, Andrades M, Moreira JC, Dal-Pizzol F, Hussain SN. Superoxide production during sepsis development. Am J Respir Crit Care Med. 2003;167:474. doi: 10.1164/ajrccm.167.3.297e. author reply 474-5. [DOI] [PubMed] [Google Scholar]
- 17.Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence. 2013;5:66–72. doi: 10.4161/viru.26907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. 2011;107:57–64. doi: 10.1093/bja/aer093. [DOI] [PubMed] [Google Scholar]
- 19.Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med. 2007;35:1599–608. doi: 10.1097/01.CCM.0000266683.64081.02. [DOI] [PubMed] [Google Scholar]
- 20.Ishii KJ, Akira S. Toll-like receptors and sepsis. Curr Infect Dis Rep. 2004;6:361–6. doi: 10.1007/s11908-004-0034-1. [DOI] [PubMed] [Google Scholar]
- 21.Feng Y, Chao W. Toll-like receptors and myocardial inflammation. Int J Inflam. 2011;2011:170352. doi: 10.4061/2011/170352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chao W. Toll-like receptor signaling: A critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol. 2009;296:H1–H12. doi: 10.1152/ajpheart.00995.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Djafarzadeh S, Vuda M, Takala J, Ochs M, Jakob SM. Toll-like receptor-3-induced mitochondrial dysfunction in cultured human hepatocytes. Mitochondrion. 2011;11:83–8. doi: 10.1016/j.mito.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 24.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–80. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suliman HB, Welty-Wolf KE, Carraway MS, Schwartz DA, Hollingsworth JW, Piantadosi CA. Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated E. coli. FASEB J. 2005;19:1531–3. doi: 10.1096/fj.04-3500fje. [DOI] [PubMed] [Google Scholar]
- 26.Bauerfeld CP, Rastogi R, Pirockinaite G, Lee I, Huttemann M, Monks B, Birnbaum MJ, Franchi L, Nunez G, Samavati L. TLR4-mediated AKT activation is MyD88/TRIF dependent and critical for induction of oxidative phosphorylation and mitochondrial transcription factor A in murine macrophages. J Immunol. 2012;188:2847–57. doi: 10.4049/jimmunol.1102157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu X, Bagchi A, Zhao H, Kirschning CJ, Hajjar RJ, Chao W, Hellman J, Schmidt U. Toll-like receptor 2 activation by bacterial peptidoglycan-associated lipoprotein activates cardiomyocyte inflammation and contractile dysfunction. Crit Care Med. 2007;35:886–92. doi: 10.1097/01.CCM.0000256723.37586.A2. [DOI] [PubMed] [Google Scholar]
- 28.Zou L, Feng Y, Chen YJ, Si R, Shen S, Zhou Q, Ichinose F, Scherrer-Crosbie M, Chao W. Toll-like receptor 2 plays a critical role in cardiac dysfunction during polymicrobial sepsis. Crit Care Med. 2010;38:1335–42. doi: 10.1097/CCM.0b013e3181d99e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zou L, Feng Y, Zhang M, Li Y, Chao W. Nonhematopoietic toll-like receptor 2 contributes to neutrophil and cardiac function impairment during polymicrobial sepsis. Shock. 2011;36:370–80. doi: 10.1097/SHK.0b013e3182279868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergt S, Wagner NM, Heidrich M, Butschkau A, Noldge-Schomburg GE, Vollmar B, Roesner JP. Hydrocortisone reduces the beneficial effects of toll-like receptor 2 deficiency on survival in a mouse model of polymicrobial sepsis. Shock. 2013;40:414–9. doi: 10.1097/SHK.0000000000000029. [DOI] [PubMed] [Google Scholar]
- 31.Pene F, Grimaldi D, Zuber B, Sauneuf B, Rousseau C, El Hachem C, Martin C, Belaidouni N, Balloy V, Mira JP, Chiche JD. Toll-like receptor 2 deficiency increases resistance to Pseudomonas aeruginosa pneumonia in the setting of sepsis-induced immune dysfunction. J Infect Dis. 2012;206:932–42. doi: 10.1093/infdis/jis438. [DOI] [PubMed] [Google Scholar]
- 32.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–51. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 33.Feng Y, Zou L, Si R, Nagasaka Y, Chao W. Bone marrow MyD88 signaling modulates neutrophil function and ischemic myocardial injury. Am J Physiol Cell Physiol. 2010;299:C760–9. doi: 10.1152/ajpcell.00155.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou L, Feng Y, Li Y, Zhang M, Chen C, Cai J, Gong Y, Wang L, Thurman JM, Wu X, Atkinson JP, Chao W. Complement factor B is the downstream effector of TLRs and plays an important role in a mouse model of severe sepsis. J Immunol. 2013;191:5625–35. doi: 10.4049/jimmunol.1301903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–8. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 36.Miwa S, Brand MD. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem Soc Trans. 2003;31:1300–1. doi: 10.1042/bst0311300. [DOI] [PubMed] [Google Scholar]
- 37.Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis. 2003;8:115–28. doi: 10.1023/a:1022945107762. [DOI] [PubMed] [Google Scholar]
- 38.Kirkinezos IG, Moraes CT. Reactive oxygen species and mitochondrial diseases. Semin Cell Dev Biol. 2001;12:449–57. doi: 10.1006/scdb.2001.0282. [DOI] [PubMed] [Google Scholar]
- 39.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94:514–9. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choumar A, Tarhuni A, Letteron P, Reyl-Desmars F, Dauhoo N, Damasse J, Vadrot N, Nahon P, Moreau R, Pessayre D, Mansouri A. Lipopolysaccharide-induced mitochondrial DNA depletion. Antioxid Redox Signal. 2011;15:2837–54. doi: 10.1089/ars.2010.3713. [DOI] [PubMed] [Google Scholar]
- 42.Dupre-Crochet S, Erard M, Nubetae O. ROS production in phagocytes: why, when, and where? J Leukoc Biol. 2013;94:657–70. doi: 10.1189/jlb.1012544. [DOI] [PubMed] [Google Scholar]
- 43.Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 44.Salvador A, Sousa J, Pinto RE. Hydroperoxyl, superoxide and pH gradients in the mitochondrial matrix: A theoretical assessment. Free Radic Biol Med. 2001;31:1208–15. doi: 10.1016/s0891-5849(01)00707-9. [DOI] [PubMed] [Google Scholar]
- 45.Mumbengegwi DR, Li Q, Li C, Bear CE, Engelhardt JF. Evidence for a superoxide permeability pathway in endosomal membranes. Mol Cell Biol. 2008;28:3700–12. doi: 10.1128/MCB.02038-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208:417–20. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsu HY, Wen MH. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J Biol Chem. 2002;277:22131–9. doi: 10.1074/jbc.M111883200. [DOI] [PubMed] [Google Scholar]
- 49.Bauerfeld CP, Rastogi R, Pirockinaite G, Lee I, Huttemann M, Monks B, Birnbaum MJ, Franchi L, Nunez G, Samavati L. TLR4-mediated AKT activation is MyD88/TRIF dependent and critical for induction of oxidative phosphorylation and mitochondrial transcription factor A in murine macrophages. J Immunol. 2012;188:2847–57. doi: 10.4049/jimmunol.1102157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suliman HB, Carraway MS, Welty-Wolf KE, Whorton AR, Piantadosi CA. Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. J Biol Chem. 2003;278:41510–8. doi: 10.1074/jbc.M304719200. [DOI] [PubMed] [Google Scholar]
- 51.Suliman HB, Welty-Wolf KE, Carraway M, Tatro L, Piantadosi CA. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc Res. 2004;64:279–88. doi: 10.1016/j.cardiores.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 52.Carchman EH, Whelan S, Loughran P, Mollen K, Stratamirovic S, Shiva S, Rosengart MR, Zuckerbraun BS. Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver. FASEB J. 2013;27:4703–11. doi: 10.1096/fj.13-229476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alves-Filho JC, Freitas A, Souto FO, Spiller F, Paula-Neto H, Silva JS, Gazzinelli RT, Teixeira MM, Ferreira SH, Cunha FQ. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc Natl Acad Sci U S A. 2009;106:4018–23. doi: 10.1073/pnas.0900196106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gille L, Nohl H. The ubiquinol/bc1 redox couple regulates mitochondrial oxygen radical formation. Arch Biochem Biophys. 2001;388:34–8. doi: 10.1006/abbi.2000.2257. [DOI] [PubMed] [Google Scholar]
- 55.Campo ML, Kinnally KW, Tedeschi H. The effect of antimycin A on mouse liver inner mitochondrial membrane channel activity. J Biol Chem. 1992;267:8123–7. [PubMed] [Google Scholar]
- 56.Alexandre A, Lehninger AL. Bypasses of the antimycin a block of mitochondrial electron transport in relation to ubisemiquinone function. Biochim Biophys Acta. 1984;767:120–9. doi: 10.1016/0005-2728(84)90086-0. [DOI] [PubMed] [Google Scholar]
- 57.Pham NA, Robinson BH, Hedley DW. Simultaneous detection of mitochondrial respiratory chain activity and reactive oxygen in digitonin-permeabilized cells using flow cytometry. Cytometry. 2000;41:245–51. doi: 10.1002/1097-0320(20001201)41:4<245::aid-cyto2>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 58.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–31. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 59.Fink MP. Reactive oxygen species as mediators of organ dysfunction caused by sepsis, acute respiratory distress syndrome, or hemorrhagic shock: potential benefits of resuscitation with Ringer's ethyl pyruvate solution. Curr Opin Clin Nutr Metab Care. 2002;5:167–74. doi: 10.1097/00075197-200203000-00009. [DOI] [PubMed] [Google Scholar]
- 60.Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med. 2008;45:1559–65. doi: 10.1016/j.freeradbiomed.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 61.Supinski GS, Callahan LA. Polyethylene glycol-superoxide dismutase prevents endotoxin-induced cardiac dysfunction. Am J Respir Crit Care Med. 2006;173:1240–7. doi: 10.1164/rccm.200410-1346OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deitch EA. Animal models of sepsis and shock: A review and lessons learned. Shock. 1998;9:1–11. doi: 10.1097/00024382-199801000-00001. [DOI] [PubMed] [Google Scholar]
- 63.Fink MP, Heard SO. Laboratory models of sepsis and septic shock. J Surg Res. 1990;49:186–96. doi: 10.1016/0022-4804(90)90260-9. [DOI] [PubMed] [Google Scholar]
- 64.Schultz MJ, van der Poll T. Animal and human models for sepsis. Ann Med. 2002;34:573–81. doi: 10.1080/078538902321117797. [DOI] [PubMed] [Google Scholar]
- 65.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–6. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: Findings in normal and T- and B-cell-deficient mice. Crit Care Med. 1997;25:1298–307. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]