

Abstract
The cell interior is a complex and demanding environment. An incredible variety of molecules jockey to identify the correct position–the specific interactions that promote biology that are hidden among countless unproductive options. Ensuring that the business of the cell is successful requires sophisticated mechanisms to impose temporal and spatial specificity–both on transient interactions and their eventual outcomes. Two strategies employed to regulate macromolecular interactions in a cellular context are co-localization and compartmentalization. Macromolecular interactions can be promoted and specified by localizing the partners within the same subcellular compartment, or by holding them in proximity through covalent or non-covalent interactions with proteins, lipids, or DNA– themes that are familiar to any biologist. The net result of these strategies is an increase in effective molarity: the local concentration of a reactive molecule near its reaction partners. We will focus on this general mechanism, employed by Nature and adapted in the lab, which allows delicate control in complex environments: the power of proximity to accelerate, guide, or otherwise influence the reactivity of signaling proteins and the information that they encode.
Keywords: cell signaling, effective molarity, chemically induced dimerization, templated catalysis
1. Introduction
The interior of a cell is not unlike Grand Central Station at 5 pm. Hordes of people (or molecules) in all shapes and sizes, each one (well, almost) guided by a defined destination and departure time. Ensuring that everyone reaches their destination requires sophisticated mechanisms to impose temporal and spatial specificity on both transient interactions and eventual outcomes. Two strategies employed to regulate macromolecular interactions in a cellular context are co-localization and compartmentalization. Macromolecular interactions can be promoted and specified by localizing the partners within the same subcellular compartment or by holding them in proximity through covalent or non-covalent interactions with proteins, lipids, or DNA (Figure 1). The net result ov both strategies is an increase in effective molarity between signaling partners. In this review, we chose specific examples to illustrate one mechanism employed by nature to ensure the faithful passage of information: the power of proximity to accelerate, guide, or otherwise influence the reactivity of signaling proteins and the information that they encode.
Figure 1.

A slow bimolecular reaction characterized by the rate constant kintermolecular, (units of M-1 •sec-1) can be accelerated when the two reactants are held in close proximity. In this case, a unimolecular reaction ensues that is characterized by the rate constant kintramolecular (units of sec-1). The effective molarity of one substrate relative to the other in the latter case is defined by the ratio of the two rate constants and possesses units of M.
2. Increasing effective molarity using natural protein domains
Many applications of effective molarity, especially those that employ natural protein domains, fall within the burgeoning field of synthetic biology, where researchers aim to gain insight into the molecular logic of signaling systems by engineering novel pathways and studying their behavior. This sort of molecular engineering demands both a toolkit of modular parts as well as an understanding of how these elements can be combined reliably into functional units.[1] Once a detailed understanding of each part has been obtained, perturbations can be introduced that illuminate the influence of factors like architecture, affinity, dynamics, and allostery on pathway output. Beyond understanding how signaling pathways are controlled, engineered pathways may have direct applications to improve human health[2] and the production of commercial chemicals such as biofuel.[3]
2.1: Early work
Early progress in this area made extensive use of naturally modular protein domains to rewire signaling pathways.[1] Simply tagging a catalytic domain (often a kinase) with a novel targeting domain (an FKBP, SH3, or PDZ domain, for example) is often sufficient to redirect catalytic activity, effectively rewiring the signaling pathway. An early materialization of this idea was reported by Howard et al ., who engineered a novel adaptor protein to redirect signaling through the epidermal growth factor receptor (EGFR) (Figure 2).[4] Normally, activated EGFR recruits the adaptor protein Grb2 through an interaction between the SH2 domain of Grb2 and phosphotyrosine 1068 on the EGFR C-terminal tail.[5] Grb2 then recruits downstream signaling proteins via its SH3 domains to induce proliferation. To redirect the response to EGFR activation, Howard et al. expressed a chimeric adaptor in which the SH2 domain of Grb2 was linked to the death effector domain (DED) of the Fadd adaptor in place of both SH3 domains of Grb2. The Fadd receptor normally serves to link activation of the Fas receptor to downstream apoptotic proteins. Indeed, expression of this chimeric adaptor protein in mammalian cells rewired the cell signaling pathways so that activation of EGFR led to apoptosis rather than proliferation. Notably, this strategy did not require modification of the endogenous signaling protein, EGFR.
Figure 2.

Howard et al reenigineered EGFR signaling by creating a chimeric adaptor protein. (A) In both EGFR and Fas signaling, a modular adaptor protein links activated receptor proteins to downstream signaling events. (B) A chimeric adaptor protein was created that linked EGFR activation to apoptosis.[4]
2.2: Effective molarity as a tool in metabolic engineering
Harnessing key concepts found in nature, scaffold proteins have been engineered to increase the production of desirable metabolites in microorganisms. Several natural enzymes employ substrate channeling to increase metabolite production, including tryptophan synthase, carbomoy| phosphate synthase and polyketide synthases.[6] Colocalizing the necessary enzymes on one scaffold increases the effective concentration of metabolic intermediates while also preventing their accumulation to toxic levels. In a landmark example, Dueber and Keasling engineered E. coli to express a scaffold that organizes the enzymes necessary for the production of mevalonate, a isoprenoid precursor. The chimeric scaffold possessed three domains: SH3, PDZ and a GTPase binding domain, and the peptide ligands for these domains were fused to three kinetically relevant enzymes in the mevalonate biosynthetic pathway. E. coli expressing the optimized scaffold and reaction components produced 77-fold more mevalonate than those lacking the scaffold. This early success established a strong foundation for the design and implementation of synthetic systems capable of metabolite production.
Scaffolds can also be generated to assemble hydrolytic enzyme complexes capable of processing cellulose.[7] In nature, the enzymes responsible for cellulose hydrolysis are organized into the cellulosome, a multienzyme complex located on the bacterial cell surface.[8] The cellulosome is organized around scaffoldin, a scaffold protein composed of a cellulose-binding module and several cohesin domains. The cohesin domains bind to the dockerin domains of cellulases via a high-affinity (> 10-9M) interaction.[7a, 7b] When compared to non-complexed cellulases, hydrolytic enzymes organized onto this scaffold increase the rate of cellulose hydrolysis, particularly in the case of recalcitrant cellulose.[7c] The modular architecture of the cellulosome has enabled the engineering of synthetic cellulosomes that hydrolyze cellulose, with the eventual goal of producing biofuels.[7d, 7e] In one example, yeast that express scaffolds for two copies each of endoglucanase and β- glucosidase exhibited a 4.2-fold enhancement in cellulose hydrolysis when compared to yeast lacking he scaffold. Furthermore, cells displaying the engineered cellulosome exhibited a 2-fold increase in ethanol production compared to cells displaying only single copies of endoglucanase and β-glucosidase. The use of adaptor scaffoldins is a flexible strategy to control the number of enzymes present on the cell surface that could be generally applicable toward other reactions. Engineering systems based around the dockerin-cohesin interaction has been used to template the formation of other cascade enzymes, including those involved in glycolysis and gluconeogenesis.[7f]
2.3 Directing bacterial two-component systems through novel adaptors
Bacterial two-component systems differ from eukaryotic signaling pathways in that they generally do not rely upon adaptor and scaffold proteins to relay signals. Bacterial two- component systems are organized around two proteins: a histidine kinase and a response regulator. The histidine kinase senses an environmental signal, undergoes auto-phosphorylation, and then phosphorylates the response regulator, which usually responds by altering transcriptional activity.[9] Recently Whitaker et al. described an adaptor protein that exploited induced proximity to re-direct a histidine kinase to particular response regulators (Figure 3). The Taz histidine kinase was tagged with an SH3 domain and the response regulator CpxR was tagged with a mutually orthogonal leucine zipper segment. A series of a daptor proteins were designed to associate with both the SH3 domain and the orthogonal leucine zipper, effectively recruiting non-native response regulators to the histidine kinase. In bacteria lacking native two-component pathways, the authors observed a 17-fold increase in gene activation over that observed with an adaptor lacking the SH3 ligand. The authors extended their preliminary results to create an autoinhibited histidine kinase that is functionalized with both an SH3 domain for binding the adaptor and an SH3 ligand that binds the SH3 domain in the absence of the adaptor. Association of the adaptor with the autoinhibited histidine kinase releases the autoinhibitory interaction, allowing phosphotransfer to the response regulator. The success of this design demonstrates that colocalizing the two-component signaling proteins is sufficient to control phosphotransfer specificity. Future experiments could focus on engineering two-component systems to sense and respond to non-natural signals, such as toxins.
Figure 3.

Bacterial two-component signaling can be directed with adaptor proteins designed to promote the interaction between a histidine kinase and an unnatural response regulator. Ternary complex formation mediated by the adaptor promotes phosphotransfer from the histidine kinase to the response regulator, leading to transcriptional activation.[10]
2.4: Creating novel signaling proteins through domain recombination
As an alternative to creating novel adaptor and scaffolding proteins to control protein- protein interactions, signaling enzymes themselves can be altered to rewire signaling.[11] In a landmark example, Dueber et al. engineered the actin regulatory switch neuronal Wiskott-Aldrich syndrome protein (N-WASP) so that it was controlled by novel factors. Normally, N-WASP is held in an inactive conformation by two intra-molecular interactions that both must be displaced to induce activity. To change the signaling properties of N-WASP, the endogenous autoinhibitory interactions were replaced with a PDZ domain and its peptide ligand and an SH3 domain and its ligand. The engineered N-WASP was activated in the presence of both a PDZ peptide ligand and an SH3 peptide ligand, recapitulating the AND gate that exists naturally. The authors varied the affinities between the peptide ligands and domains as well as the linker lengths between domains to gain insight into the features necessary for repression and activation.
Similar concepts were applied to reprogram cell morphology by engineering synthetic guanine nucleotide exchange factors (GEFs) that respond to unnatural signals, such as protein kinase A (PKA) phosphorylation.[11b] GEFs promote the exchange of bound GDP to GTP to activate Rho family members that are essential in maintaining the actin cytoskeleton. GEFs possess a modular structure in which the catalytic domain is adjacent to an autoinhibitory domain. Yeh et a. took advantage of this modularity by flanking the GEF catalytic domain by both a PDZ domain and a PDZ peptide ligand. In the absence of phosphorylation by PKA, the catalytic motif was held in an autoinhibited form by the intra- molecular interaction between the PDZ domain and its ligand. Phosphorylation of the PDZ ligand by PKA releases the intra- molecular interaction, activating the catalytic domain. Both of these studies take advantage of the natural modularity of proteins and signaling pathways to engineer cellular signaling proteins capable of responding to unnatural inputs. Engineering signaling proteins by domain recombination has been particularly useful in understanding enzyme mechanisms and how enzymes function in signaling pathways.
Maximizing precision
Researchers in the burgeoning field of synthetic biology have made good use of natural protein domains to exploit effective molarity in the creation of novel synthetic systems. As these natural domains are encodable, they can be evolved to display new or improved signaling properties. Because the interactions between natural protein domains and their ligands are often well studied, implementing these protein-ligand pairs in synthetic biology contexts is straightforward and predictable. However, because the parts they contain are natural, their specificities are in general, not unique, leading to interactions with many other signaling proteins in side the cell. These ‘off-target’ interactions can result in undesirable outcomes because of interaction of protein modules with endogenous cellular proteins.[12] Furthermore, this method requires the modification of the protein of interest. As stated previously, the substitution of endogenous protein domains with other natural protein domains presents the possibility for unintended cross- talk.[12-13] One strategy for minimizing cross-talk makes use of modular protein domains in organisms that naturally lack these domains. Other strategies, discussed in the sections that follow, make use of small molecules, nucleic acid templates, and wholly unnatural protein domains.
3. Inducing specificity with small molecules
3.1. Chemical Inducers of Dimerization
One strategy for introducing greater or alternative specificity into an engineered signaling pathway exploits small molecules–natural or designed–that bind two protein partners simultaneously. In classic work that ushered in an era of chemical biology research, Schreiber and Crabtree developed this idea in a cellular context using molecules referred to as ‘chemical inducers of dimerization’ (abbreviated as CID). In the initial report, the synthetic ligand FK1012, containing two copies of the FKBP-binding moiety FK506, effectively dimerized and thereby activated T antigen cell surface receptors fused to FKBP modules. on the surface of living cells[14] More recent developments based on the interaction of rapamycin or its analogs with FKBP and FRB increased versatility by expanding the scope to include heterodimeric interactions. Myriad chemical inducers of dimerization have been used to illuminate the role of proximity in biology and to engineer a diverse array of unnatural interactions and processes.[15] More recently, CID has be used to localize a protein of interest to a specific organelle (Figure 4).[16] Chemical inducers of dimerization are attractive scaffolds to increase effective molarity because they are cell permeable, bind tightly to target proteins, and can diffuse more rapidly than larger biomolecules making it easier to control cellular availability. However, like scaffolds based on natural proteins, some small molecule dimerizers can engage in crosstalk and off-target effects; one example are the inhibitory interactions of rapamycin with endogenous mTOR.[17] Use of a mutant FRB fusion protein and a rapalog that selectively binds to the mutant FRB over mTOR can mimimize cross-talk.[18] Alternatively, the use of orthogonal fusion proteins, such as bacterial DHFR, or ligands that lack endogenous binding partners can help.
Figure 4.

Chemical inducers of dimerization. A, The interaction of rapamycin with FKBP and FRB brings ‘protein of interest 1’ (PO1) into close proximity with ‘protein of interest 2’ (PO2) to initiate a cellular response. B, Application of CID to localize a protein of interest to a specific organelle.[16]
3.1.1. Initiating transcription
In a related way, the yeast three-hybrid system exploits a designed, bifunctional small molecule to colocalize the DNA-binding and activation regions of a transcription factor and thereby turn on transcription. The DNA-binding domain is fused to one of the small molecule partners while the activation domain is fused to the other. Addition of the small molecule guides the activation domain in proximity of the DNA binding domain, bound to DNA, promoting transcription. In its first incarnation, three hybrid system exploited an FK506-dexamethasone hybrid small molecule to identify FKBP12 from within a Jurkat cDNA library.[19] Since that time, the yeast three hybrid system has been applied in many contexts, including the identification of novel enzyme catalysts [20] and as a screen to identify protein-ligand-protein interactions useful for small molecule induced dimerization.[18a,21]
3.1.2. Probing histone modifications
A recent application of chemical inducers of dimerization in the context of epigenetics focused on the posttranslational trimethylation of histone H3K9 (H3K9me3). To control H3K9me3, the chromatin shadow domain of HP1α (csHP1α), a domain that recruits H3K to specific histone methylases, was fused to FRB while a DNA binding domain was fused to FKB. This arrangement insures that csHP1α is only recruited to chromatin when rapamycin is present. Using this system, it was discovered that recruitment of csHP1α represses gene expression and increases production of H3K9me3. The CID also facilitated analysis of how removing rapamycin and therefore stopping the recruitment of csHP1α affected repression of genes across generations of cells.[22]
3.2 Facilitating complex biology
Chemically induced dimerization has also been used to generate orthogonal Boolean logic gates in living cells. In recent work, this goal was accomplished using two orthogonal chemical dimerizers, rapamycin and a gibberellin analog. Gibberellin works as a dimerizer by binding to the protein gibberellin insensitive dwarf1 (GID1) and causing a change in conformation that allows GID1 to bind a second protein, gibberellin insensitive (GAI). Two ‘proteins of interest’ can be fused to either GAI or GID1; dimerization is induced upon addition of gibberellin or an appropriate analog. The gibberellin analog used (GA3-AM) binds GID1 only after the acetoxymethyl group is cleaved off by an endogenous esterase. OR gates were created by expressing both GAI-effector and FRB-effector fusion proteins; in this way, the cellular reponse can be controlled by addition of rapamycin or GA3-AM. AND gates were created by fusing GAI to a localization domain, fusing FKBP to GID1, and fusing FRB to an effector protein; in this way, both rapamycin and GA3-AM are required to localize the effector protein to the membrane and produce a cellular response.[23]
Induced proximity also provides the basis for a small-molecule strategy to guide the proteosome to specific—presumably undesirable—protein substrates. In early work, ‘proteolysis-targeting chimeric molecules’ were designed to approximate a protein of interest and a ubiquitin ligase (an enzyme that mediates the ubiquitination of a target protein), bringing about protein ubiquitination and subsequent degradation.[24] These bifunctional small molecules are composed of a target protein binding ligand and an E3 ubiquitin ligase ligand. In theory, these molecules effectively reprogram the substrate specificity of the E3 ubiquitin ligase to any target for which a small molecule ligand exists.
In an ambitious manifestation of proximity-induced reactions, Spiegel and Barbas, building on early precedent of Schultz,[25] made use of proximity to specify a sophisticated immune response as opposed to degradation of a single protein,[26] Using antibody recruiting molecules (ARMs) possessing an antibody binding motif and a target cell binding motif, it has been possible to target HIV positive cells, [27] prostate cancer cells, [28] and metastatic cancer cells.[29] This approach goes beyond simply rewiring signaling enzyme specificity, but it utilizes the same concepts, such as ternary complex formation to bring biomolecules in proximity to change biological output. An analytical treatment of three component equilibria that should prove useful for optimizing occupancy of the ternary complex has also recently been reported.
4.DNA/RNA templated reactions
Nucleic acids can also be used as scaffolds to increase the effective molarity of chemical and biochemical reactions in solution (Figure 5). Indeed, the natural processes of transcription, translation, replication, recombination, splicing, and DNA repair all rely on reactions promoted by nucleic acid templates. In the context of unnatural processes, substrates and reagents can be co-localized to specific nucleic acid regions either by hybridization or specific protein-or small molecule-nucleic acid interfaces. Because engineering these interactions is often straightforward and predictable, utilization of DNA and RNA to bring two molecules in to proximity in a programmable manner is particularly versatile.
Figure 5.

Three strategies by which substrates and reagents can be co-localized to an oligomeric nucleic acid scaffold. B, DNA templates can template reactions and lead to the discovery of new reactions[36] and, C, complex RNA architectures can organize more efficient metabolic pathways.[51]
4.1: Novel reactions
Early examples of unnatural reactions template by sequence-specific nucleic acid interactions phosphodiester bond formation[31], include duplex- and triplex-mediated alkylation[32], and DNA cleavage[33] reactions. More recently, the increased effective molarity provided by co-localized DNA hybridization has been exploited to facilitate difficult chemical reactions, discover novel reactivity, and template the assembly of unnatural biopolymers[34] For example, Liu and coworkers have harnessed DNA hybridization to promote entropically unfavorable reactions, such as macrocyclization, which are impeded by undesirable intermolecular side reactions.[15, 35] These previously detrimental synthetic obstacles are overcome by low solution reactant concentrations (nM-μM), in which only the templated reaction proceeds to any significant extent because of the high local concentration induced by the nucleic acid. This technology has also been applied in a discovery mode to identify novel carbon-carbon bond forming reactions, such as in the case of a palladium catalyzed reaction between an alkyne and alkene that occurs only because of the increased effective molarity between reactants.[36]
4.2: Complex molecule and library syntheses
For many DNA-templated reactions, base pairing–not chemistry–is rate limiting. In these cases the rate of the templated reaction is independent of the distance (along the DNA strand) connecting the binding sites of the two reactants,[37] allowing multiple synthetic steps to be template on a single oligomer. This versatility provides for the construction of molecules with significant synthetic complexity as well as diverse libraries.[38] Complex molecules can also be synthesized by templating each step of the reaction separately[39] Because the reactions do not proceed unless templated, this process can take place in a single reaction vessel, greatly simplifying the synthesis single.[39] In the most recent manifestation of this idea, Liu and coworkers have used DNA templated synthesis to couple β-peptide building blocks in a sequence-programmed manner.[34] It should be noted, however, that product inhibition is a genuine challenge for all DNA- templated reactions (as well as all proximity-aided reactions). While methods have been developed to disrupt the product- template interaction[40] product inhibition remains a genuine limitation and highlights that specificity, not turnover, is the true advantage of these templated reactions.
4.3: Templated reactions inside the cell
The utility of DNA and RNA for inducing proximity of ligands both large and small is not restricted to reactions performed in the test tube. Ma and Taylor have developed a technology that utilizes a DNA templated reaction to potentially target a drug to cancerous cells.[41] In an early example, a DNA template programmed as an endogenous oncogene templates the co-localization to a prodrug-DNA conjugate and a uniquely functionalized oligonucleotide–one modified with an imidazole group capable of catalyzing release of the drug[41] Although much effort has been put into the development of DNA-templated reactions for drug release, problems of biocompatibility and delivery of oligonucleotide- conjugated drugs have thwarted progress.[42] conjugated drugs Recently, Winssinger and coworkers have overcome some of these challenges by utilizing an azide reduction reaction that potentially could be used to release any functionalized small molecule (Figure 6).[43]
Figure 6.

DNA-templated activation of a pro-drug. In this work, an oncogenic DNA sequence serves to co-localize a phosphine reducing agent and a pro-drug in order to potentially deliver a small molecule therapeutic to a diseased cell.[43]
4.4: More efficient metabolic pathways
Nucleic acid scaffolds can also promote multi-step transformations and improve the flux through metabolic pathways. The broad toolkit of programmable oligomeric structures makes DNA and RNA invaluable scaffolds.[44] Colocalization of enzymes in a metabolic pathway can improve reaction efficiency by increasing the effective molarity of intermediate (see section 2, above) and, if the proteins are close enough, intermediate transfer is even more efficient as diffusion is dimensionally restricted along the protein hydration shells.[45]
Reaction optimization on these scaffolds is idiosyncratic: the properties of the system are dependent on a large number of difficult-to-predict factors, including the details of scaffold structure and secondary scaffold-enzyme interactions. For example, the activity of some enzymes that are colocalized to a scaffold have been observed to change,[46] while others have found the enzymatic activity to remain constant.[45] Therefore, careful examination is necessary to confirm the system behaves as engineered. While this technology has been successfully applied, in vitro modeling of the diffusion of intermediates in a scaffolded enzyme pair has revealed that the DNA scaffold persists only on the timescale of minutes before a sufficient concentration of intermediate has been built up.[48] While this may be true for in vitro experiments in simple buffered solutions, one could imagine a greater benefit of scaffolds in the complex milieu of a cell, and further investigations into the benefits of such template reactions in vivo must be conducted.
This technology has also been successfully implemented in live cells. However, the chemical attachment of nucleotides to the proteins of interest is not feasible for cellular experiments, therefore, encodable methods must be utilized such as RNA aptamer- protein[49] or zinc finger-DNA [50] interactions. Aldaye and coworkers were able to colocalize proteins involved in hydrogen production in an organelle-like area inside the cell by engineering aptamer domains into the scaffold and fusing aptamer-binding proteins to the proteins of interest. [51] This scaffold complexity, which protein scaffolds lack, significantly improved hydrogen production in cells in comparison to RNA scaffolds that were more dimensionally simple.[51] Additionally, other labs have used DNA templates and zinc finger protein fusion to engineer metabolic pathways.[52] A major setback to the use of oligomer scaffolds to alter cellular pathways is the requirement that the protein of interest be genetically tagged, thus not allowing for the reorganization of endogenous signaling pathways. In summary, the precision of base pairing, the diversity of oligonucleotide nanostructures and the ease of design has made DNA and RNA templates a helpful tool in scaffolding reactions from the molecular scale up to the organization of complex metabolic pathways, regardless of the obstacles associated with biocompatibility.
5. Unnatural domains
A final potential solution to guide macromolecular associations in a crowded environment and decrease cellular cross-talk exploits adaptors or scaffolds composed of wholly unnatural domains with prescribed (and presumably optimized) recognition properties. More than fifty different protein domains possessing unique and orthogonal structures and binding properties have been developed over the past 15 years. Examples include include affibodies,[9] monobodies,[5] designed ankyrin repeat proteins (DARPins),[2] tetratricopeptide repeats (TPRs,),[53] as well as the pancreatic-fold based miniature proteins that we have studied.[54] One advantage of adaptors composed of wholly unnatural recognition domains is that they can be evolved on the basis of one function only–specificity–and thus have the potential for fewer off-target interactions than a natural prote in domain whose evolution has been guided by multiple, often antagonistic ideals. Another advantage of a synthetic adaptor approach is that their function does not require the modification of signaling proteins in the pathway being manipulated.
5.1: A toolkit of coiled coil domains
One benefit of twenty-five years of research on coiled coil interactions is a clear understanding of the rules governing strand orientation, stoichiometry, and preference for homo-or hetero-dimeric interactions. Woolfson and coworkers have now made use of this information to develop a toolkit of mutually interacting protein parts based on de novo designed coiled-coil peptides.[32] A set of parts (peptides) were reported, whose lengths span 21, 24, or 28 residues, and which interact in a hetero-dimeric manner with equilibrium dissociation constants in the micromolar to sub-nanomolar range (Figure 7). The designs incorporate a buried asparagine residue whose hydrogen bonding preferences specify uniqueness in both stoichiometry and axial orientation. Notably, despite the sophistication of the design, the sequences of the peptide parts are not fully defined and can be further modified at a number of residues (including those that are solvent-exposed). This level of flexibility is particularly desirable for applications other than just simple oligomerization: for instance, in more complex association/dissociation processes involving complex dynamics and subunit exchange.
Figure 7.
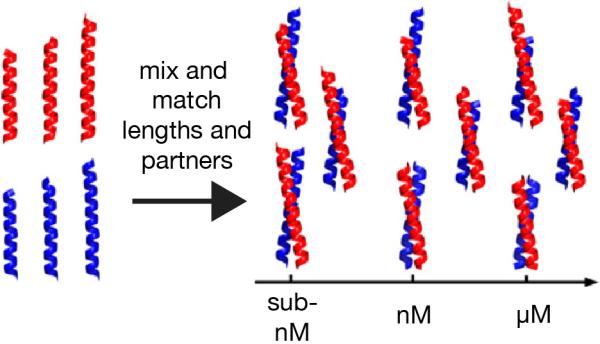
A toolkit of mutually interacting protein parts based on de novo designed coiled-coil peptides.[32]
5.2: Wholly unnatural adaptor proteins
We recently reported a wholly unnatural adaptor protein that acts as a catalyst to redirect the Src family kinase Hck to phosphorylate hDM2, a negative regulator of the p53 tumor suppressor and a naturally poor Hck substrate (Figure 8). The design of this adaptor began with two previously reported miniature proteins, YY2 and 3.3. Miniature protein YY2 uses residues within its PPII helix to interact with the SH3 domains of certain Src family kinases, such as Hck. This interaction disrupts the intra-molecular SH3 domain interaction that down-regulates kinase activity and results in kinase activation. Miniature protein 3.3 uses residues within its α-helix to bind to hDM2, thereby inhibiting the association of hDM2 with p53. A series of adaptor proteins varying in the linker length were constructed; one was shown to exploit templated catalysis to redirect the Src family kinase Hck to phosphorylate hDM2. Phosphorylation occurs with multiple turnover and at a single site targeted by the non-Src family kinasec-Abl kinase in the cell. Notably, miniature proteins are encodable and evolvable and facilitate induced proximity of endogenous (unmodified) cellular proteins using orthogonal domains with less potential for unintended cross-talk.
Figure 8.

A miniature-protein based adaptor protein that redirects the Src family kinase Hck to phosphorylate hDM2, a negative regulator of the p53 tumor suppressor and a naturally poor Hck substrate.[55]
6. Summary and outlook
In this review, we have described several methods that chemists and synthetic biologists have used to rewire signaling pathways. These methods all exploit effective molarity as a way to promote the reaction between to signaling partners. Recent work in the field of synthetic biology has drawn on classic concepts that utilize increased effective molarity to promote unnatural reactions, in particular chemically induced dimerization and templated catalysis. The former benefits from temporal control and the confidence that the requisite domains will perform as advertised. The latter benef its from the ability to function with natural, as opposed to modified signaling domains.
Acknowledgements
The authors are grateful to members of the Schepartz laboratory for valuable discussions and to Jonathan Miller (Yale University) for an astute editing of the manuscript. Some of the work discussed in this review was carried out in the authors’ laboratory and was supported by grants from the US National Institutes of Health and the National Foundation for Cancer Research. Allison S. Walker was supported by the National Science Foundation under Grant No. (NSF grant number).
References
- 1.Bhattacharyya RB, Reményi A, Yeh BJ, Lim WA. Annu. Rev. Biochem. 2006;75:655–680. doi: 10.1146/annurev.biochem.75.103004.142710. [DOI] [PubMed] [Google Scholar]
- 2.Wei P, Wong WW, Park JS, Corcoran EE, Peisajovich SG, Onuffer JJ, Weiss A, Lim WA. Nature. 2012;488:384–388. doi: 10.1038/nature11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KLJ, Keasling JD. Nat. Biotech. 2009;27:753–759. doi: 10.1038/nbt.1557. [DOI] [PubMed] [Google Scholar]
- 4.Howard PL, Chia MC, Del Rizzo S, Liu F-F, Pawson T. Proc. Natl. Acad. Sci. USA. 2003;100:11267–11272. doi: 10.1073/pnas.1934711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koide A, Koide S. In: Protein Engineering Protocols. Arndt K, Müller K, editors. Vol. 352. Humana Press; 2007. pp. 95–109. [Google Scholar]
- 6.Miles EW, Rhee S, Davies DR. J. Biol. Chem. 1999;274:12193–12196. doi: 10.1074/jbc.274.18.12193. [DOI] [PubMed] [Google Scholar]
- 7.a Schaeffer F, Matuschek M, Guglielmi G, Miras I, Alzari PM, Béguin P. Biochemistry. 2002;41:2106–2114. doi: 10.1021/bi011853m. [DOI] [PubMed] [Google Scholar]; b Miras I, Schaeffer F, Béguin P, Alzari PM. Biochemistry. 2002;41:2115–2119. doi: 10.1021/bi011854e. [DOI] [PubMed] [Google Scholar]; c You C, Zhang X-Z, Sathitsukanoh N, Lynd LR, Zhang Y-HP. Appl. Environ. Microbiol. 2012;78:1437–1444. doi: 10.1128/AEM.07138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Fan L-H, Zhang Z-J, Yu X-Y, Xue Y-Y, Tan T-W. Proc. Natl. Acad. Sci. USA. 2012;109:13260–13265. doi: 10.1073/pnas.1209856109. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Tsai S-L, DaSilva NA, Chen W. ACS Synth. Biol. 2012 doi: 10.1021/sb300047u. [DOI] [PubMed] [Google Scholar]; f You C, Myung S, Zhang Y-HP. Angew. Chem. Intl. Ed. 2012;51:8787–8790. doi: 10.1002/anie.201202441. [DOI] [PubMed] [Google Scholar]
- 8.a Bayer EA, Belaich J-P, Shoham Y, Lamed R. Annu. Rev. Microbiol. 2004;58:521–554. doi: 10.1146/annurev.micro.57.030502.091022. [DOI] [PubMed] [Google Scholar]; b Fontes CMGA, Gilbert HJ. Annu. Rev. Biochem. 2010;79:655–681. doi: 10.1146/annurev-biochem-091208-085603. [DOI] [PubMed] [Google Scholar]
- 9.Nygren P-Å. FEBS J. 2008;275:2668–2676. doi: 10.1111/j.1742-4658.2008.06438.x. [DOI] [PubMed] [Google Scholar]
- 10.Douglass EF, Miller CJ, Sparer G, Shapiro H, Spiegel DA. J. Am. Chem. Soc. 2013;135:6092–6099. doi: 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitaker WR, Davis SA, Arkin AP, Dueber JE. Proc. Natl. Acad. Sci. USA. 2012;109:18090–18095. doi: 10.1073/pnas.1209230109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Dueber JE, Yeh BJ, Chak K, Lim WA. Science. 2003;301:1904–1908. doi: 10.1126/science.1085945. [DOI] [PubMed] [Google Scholar]; b Yeh BJ, Rutigliano RJ, Deb A, Bar-Sagi D, Lim WA. Nature. 2007;447:596–600. doi: 10.1038/nature05851. [DOI] [PubMed] [Google Scholar]
- 13.Lim WA. Nat. Rev. Mol. Cell Biol. 2010;11:393–403. doi: 10.1038/nrm2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zarrinpar A, Park S-H, Lim WA. Nature. 2003;426:676–680. doi: 10.1038/nature02178. [DOI] [PubMed] [Google Scholar]
- 15.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Liu DR. Angew. Chem. Intl. Ed. 2004;43:4848–4870. doi: 10.1002/anie.200400656. [DOI] [PubMed] [Google Scholar]
- 17.a Komatsu T, Kukelyansky I, McCaffery JM, Ueno T, Varela LC, Inoue T. Nat. Meth. 2010;7:206–208. doi: 10.1038/nmeth.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kohler JJ, Bertozzi CR. Chem. Biol. 2003;10:1303–1311. doi: 10.1016/j.chembiol.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 18.Ballou LM, Lin RZ. J. Chem. Biol. 2008;1:27–36. doi: 10.1007/s12154-008-0003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a Bayle JH, Grimley JS, Stankunas K, Gestwicki JE, Wandless TJ, Crabtree GR. Chem. Biol. 2006;13:99–107. doi: 10.1016/j.chembiol.2005.10.017. [DOI] [PubMed] [Google Scholar]; b Liberles SD, Diver ST, Austin DJ, Schreiber SL. Proc. Natl. Acad. Sci. USA. 1997;94:7825–7830. doi: 10.1073/pnas.94.15.7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Licitra EJ, Liu JO. Proc. Natl. Acad. Sci. USA. 1996;93:12817–12821. doi: 10.1073/pnas.93.23.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a Baker K, Bleczinski C, Lin H, Salazar-Jimenez G, Sengupta D, Krane S, Cornish VW. Proc. Natl. Acad. Sci. USA. 2002;99:16537–16542. doi: 10.1073/pnas.262420099. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lin H, Tao H, Cornish VW. J. Am. Chem. Soc. 2004;126:15051–15059. doi: 10.1021/ja046238v. [DOI] [PubMed] [Google Scholar]
- 22.Czlapinski JL, Schelle MW, Miller LW, Laughlin ST, Kohler JJ, Cornish VW, Bertozzi CR. J. Am. Chem. Soc. 2008;130:13186–13187. doi: 10.1021/ja8037728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, Crabtree GR. Cell. 2012;149:1447–1460. doi: 10.1016/j.cell.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyamoto T, DeRose R, Suarez A, Ueono T, Chen M, Sun T, Wolfgang MJ, Mukherjee C, Meyers DJ, Inoue T. Nat. Chem. Biol. 2012;8:465–470. doi: 10.1038/nchembio.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneekloth JS, Jr., Fonseca F, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM. J. Am. Chem. Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 26.Shokat KM, Schultz PG. J. Am. Chem. Soc. 1991;113:1861–1862. [Google Scholar]
- 27.Popkov M, Gonzalez B, Sinha SC, Barbas CF. Proc. Natl. Acad. Sci. USA. 2009;106:4378–4383. doi: 10.1073/pnas.0900147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parker CG, Domaoal RA, Anderson KS, Spiegel DA. J. Am. Chem. Soc. 2009;131:16392–16394. doi: 10.1021/ja9057647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a Murelli RP, Zhang AX, Michel J, Jorgensen WL, Spiegel DA. J. Am. Chem. Soc. 2009;131:17090–17092. doi: 10.1021/ja906844e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang AX, Murelli RP, Barinka C, Michel J, Cocleaza A, Jorgensen WL, Lubkowski J, Spiegel DA. J. Am. Chem. Soc. 2010;132:12711–12716. doi: 10.1021/ja104591m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jakobsche CE, McEnaney PJ, Zhang AX, Spiegel DA. ACS Chem. Biol. 2012;7:316–321. doi: 10.1021/cb200374e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long MJC, Gollapalli DR, Hedstrom L. Chem. Biol. 2012;19:629–637. doi: 10.1016/j.chembiol.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanan MW, Rozenman MM, Sakurai K, Snyder TM, Liu DR. Nature. 2004;431:545–549. doi: 10.1038/nature02920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delebecque CJ, Lindner AB, Silver PA, Aldaye FA. Science. 2011;333:470–474. doi: 10.1126/science.1206938. [DOI] [PubMed] [Google Scholar]
- 34.Naylor R, Gilham PT. Biochemistry. 1966;5:2722–2728. doi: 10.1021/bi00872a032. [DOI] [PubMed] [Google Scholar]
- 35.Grant KB, Dervan PB. Biochemistry. 1996;35:12313–12319. doi: 10.1021/bi9608469. [DOI] [PubMed] [Google Scholar]
- 36.Schultz PG, Taylor JS, Dervan PB. J. Am. Chem. Soc. 1982;104:6861–6863. [Google Scholar]
- 37.Niu J, Hili R, Liu DR. Nat Chem. 2013;5:282–292. doi: 10.1038/nchem.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.a Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. Science. 2004;305:1601–1605. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Percivalle C, Bartolo J-F, Ladame S. Org. Biomol. Chem. 2013;11:16–26. doi: 10.1039/c2ob26163d. [DOI] [PubMed] [Google Scholar]
- 39.Gartner ZJ, Liu DR. J. Am. Chem. Soc. 2001;123:6961–6963. doi: 10.1021/ja015873n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. Science. 2004;305:1601–1605. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKee ML, Milnes PJ, Bath J, Stulz E, O'Reilly RK, Turberfield AJ. J. Am. Chem. Soc. 2012;134:1446–1449. doi: 10.1021/ja2101196. [DOI] [PubMed] [Google Scholar]
- 42.a Dose C, Ficht S, Seitz O. Angew Chem Int Edit. 2006;45:5369–5373. doi: 10.1002/anie.200600464. [DOI] [PubMed] [Google Scholar]; b He Y, Liu DR. J. Am. Chem. Soc. 2011;133:9972–9975. doi: 10.1021/ja201361t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Michaelis J, Maruyama A, Seitz O. Chem. Commun. 2013:49. doi: 10.1039/c2cc36162k. [DOI] [PubMed] [Google Scholar]
- 43.Ma Z, Taylor JS. Proc. Natl. Acad. Sci. U.S.A. 2000;97:11159–11163. doi: 10.1073/pnas.97.21.11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacobsen MF, Clo E, Mokhir A, Gothelf KV. ChemMedChem. 2007;2:793–799. doi: 10.1002/cmdc.200700013. [DOI] [PubMed] [Google Scholar]
- 45.Gorska K, Manicardi A, Barluenga S, Winssinger N. Chem. Commun. 2011;47:4364–4366. doi: 10.1039/c1cc10222b. [DOI] [PubMed] [Google Scholar]
- 46.a Afonin KA, Bindewald E, Yaghoubian AJ, Voss N, Jacovetty E, Shapiro BA, Jaeger L. Nature Nanotech. 2010;5:676–682. doi: 10.1038/nnano.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fu JL, Liu MH, Liu Y, Yan H. Accounts Chem Res. 2012;45:1215–1226. doi: 10.1021/ar200295q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fu J, Liu M, Liu Y, Woodbury NW, Yan H. J. Am. Chem. Soc. 2012;134:5516–5519. doi: 10.1021/ja300897h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin J.-L. a. W., I ACS Catalysis. 2013;3:5. doi: 10.1021/cs400088e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.a Wilner OI, Weizmann Y, Gill R, Lioubashevski O, Freeman R, Willner I. Nature nanotechnology. 2009;4:249–254. doi: 10.1038/nnano.2009.50. [DOI] [PubMed] [Google Scholar]; b Nojima T, Konno H, Kodera N, Seio K, Taguchi H, Yoshida M. PloS one. 2012;7:e52534. doi: 10.1371/journal.pone.0052534. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Erkelenz M, Kuo CH, Niemeyer CM. J. Am. Chem. Soc. 2011;133:16111–16118. doi: 10.1021/ja204993s. [DOI] [PubMed] [Google Scholar]
- 50.Idan O, Hess H. Nature Nanotech. 2012;7:769–770. doi: 10.1038/nnano.2012.222. [DOI] [PubMed] [Google Scholar]
- 51.Bunka DHJ, Stockley PG. Nat Rev Microbiol. 2006;4:588–596. doi: 10.1038/nrmicro1458. [DOI] [PubMed] [Google Scholar]
- 52.Nakata E, Liew FF, Uwatoko C, Kiyonaka S, Mori Y, Katsuda Y, Endo M, Sugiyama H, Morii T. Angew. Chem. Int. Edit. 2012;51:2421–2424. doi: 10.1002/anie.201108199. [DOI] [PubMed] [Google Scholar]
- 53.a Conrado RJ, Wu GC, Boock JT, Xu H, Chen SY, Lebar T, Turnsek J, Tomsic N, Avbelj M, Gaber R, Koprivnjak T, Mori J, Glavnik V, Vovk I, Bencina M, Hodnik V, Anderluh G, Dueber JE, Jerala R, DeLisa MP. Nucl. Acids Res. 2012;40:1879–1889. doi: 10.1093/nar/gkr888. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lee JH, Jung S-C, Bui LM, Kang KH, Song J-J, Kim SC. App. Environ. Microbiol. 2013;79:9. doi: 10.1128/AEM.02578-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stumpp MT, Binz HK, Amstutz P. Drug Discov. Today. 2008;13:695–701. doi: 10.1016/j.drudis.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 55.Grove TZ, Hands M, Regan L. Protein Eng. Des. Sel. 2010;23:449–455. doi: 10.1093/protein/gzq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.a Zondlo NJ, Schepartz A. J. Am. Chem. Soc. 1999;121:6938–6939. [Google Scholar]; b Chin JW, Grotzfeld RM, Fabian MA, Schepartz A. Bioorganic & Medicinal Chem. Lett. 2001;11:1501–1505. doi: 10.1016/s0960-894x(01)00139-1. [DOI] [PubMed] [Google Scholar]; c Chin JW, Schepartz A. Angew Chem Int Edit. 2001;40:3806. [PubMed] [Google Scholar]; d Chin JW, Schepartz A. J. Am. Chem. Soc. 2001;123:2929–2930. doi: 10.1021/ja0056668. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Montclare JK, Schepartz A. J. Am. Chem. Soc. 2003;125:3416–3417. doi: 10.1021/ja028628s. [DOI] [PubMed] [Google Scholar]
- 57.Hobert EM, Schepartz A. J. Am. Chem. Soc. 2012;134:3976–3978. doi: 10.1021/ja211089v. [DOI] [PMC free article] [PubMed] [Google Scholar]