

Abstract
Modulation of mRNA translatability either by trans-acting factors (proteins or sRNAs) or by in cis-acting riboregulators is widespread in bacteria and controls relevant phenotypic traits. Unfortunately, global identification of post-transcriptionally regulated genes is complicated by poor structural and functional conservation of regulatory elements and by the limitations of proteomic approaches in protein quantification. We devised a genetic system for the identification of post-transcriptionally regulated genes and we applied this system to search for Pseudomonas aeruginosa RNA thermometers, a class of regulatory RNA that modulates gene translation in response to temperature changes. As P. aeruginosa is able to thrive in a broad range of environmental conditions, genes differentially expressed at 37°C versus lower temperatures may be involved in infection and survival in the human host. We prepared a plasmid vector library with translational fusions of P. aeruginosa DNA fragments (PaDNA) inserted upstream of TIP2, a short peptide able to inactivate the Tet repressor (TetR) upon expression. The library was assayed in a streptomycin-resistant merodiploid rpsL+/rpsL31 Escherichia coli strain in which the dominant rpsL+ allele, which confers streptomycin sensitivity, was repressed by TetR. PaDNA fragments conferring thermosensitive streptomycin resistance (i.e., expressing PaDNA–TIP2 fusions at 37°C, but not at 28°C) were sequenced. We identified four new putative thermosensors. Two of them were validated with conventional reporter systems in E. coli and P. aeruginosa. Interestingly, one regulates the expression of ptxS, a gene implicated in P. aeruginosa pathogenesis.
Keywords: bacterial riboregulators, Pseudomonas aeruginosa, Escherichia coli, PA5194, ptxS, lpxT
INTRODUCTION
Bacteria modulate gene expression in response to a variety of chemical and physical signals in order to cope with the challenges posed by a changing environment. Temperature is one of the main physical parameters influencing bacterial growth because it affects both enzymatic reaction rate and macromolecules state. Thus, it is not surprising that bacteria have evolved complex regulatory networks to face sub-lethal temperature changes. In Escherichia coli, for instance, both a heat-shock and a cold-shock response involving changes in the expression of tens of genes have been described (Phadtare 2004; Guisbert et al. 2008). On the other hand, a modest temperature increase, within the range of permissive growth temperatures for mesophilic bacteria, is used by some bacterial pathogens as a signal of warm-blooded host invasion to trigger the expression of virulence genes (for review, see Konkel and Tilly 2000).
To either avoid irreversible cell damage or establish a successful host infection, the speediness of the response to a sudden temperature variation is critical, thus posing the problem of fast and precise thermosensing. Bacteria exploit different macromolecules as molecular thermosensors (Klinkert and Narberhaus 2009); in particular, RNA thermometers (RNATs) have been found to control the expression of a variety of heat shock and virulence genes in Gram-negative and Gram-positive bacteria (for review, see Waldminghaus et al. 2005; Kortmann and Narberhaus 2012). An RNAT can be described as a thermolabile secondary structure that sequesters the translation initiation region (TIR) of the mRNA at low temperature. Local denaturation due to temperature increase allows ribosome binding and mRNA translation. On this very general theme, which applies to most RNATs described so far, variations of length and localization relative to the AUG of the regions involved in the thermometer structure have been found. For instance, the E. coli rpoH thermosensor, for which the definition of RNA thermometer was originally proposed, encompasses RNA regions well within the coding sequence, whereas a widely disseminated class of RNATs like ROSE (Repression Of heat Shock genes Expression) elements form complex secondary structure within the 5′ UTR of the mRNA (Narberhaus et al. 1998; Morita et al. 1999a,b; Nocker et al. 2001). Moreover, some RNATs, such as those controlling the expression of bacteriophage λ cIII or E. coli cspA, rely on the temperature-dependent formation of alternative secondary structures, instead of the simple melting of an unstable one (Altuvia et al. 1989; Giuliodori et al. 2010).
Only two classes of RNATs with common structural themes have been defined so far, ROSE and FourU elements; in both cases, sequence conservation is limited within each class to very short stretches of 4–5 bases in the proximity of the Shine–Dalgarno (SD) region (Waldminghaus et al. 2005, 2007b). Poor sequence conservation hampers the bioinformatic identification of RNATs, which should mainly rely on structural properties (Waldminghaus et al. 2007a); however, the available bioinformatic tools usually overlook non-Watson–Crick interactions that seem to play a relevant role in temperature sensing by some RNATs (Chowdhury et al. 2006). Moreover, unique RNATs, completely unrelated to both ROSE and FourU elements, have been found in different groups of bacteria (Cimdins et al. 2014; for review, see Kortmann and Narberhaus 2012) making it virtually impossible, at the moment, to perform an exhaustive prediction of RNATs by bioinformatic search for conserved structures. Recently, the RNAtips (temperature-induced perturbation of structure) web server was presented. It predicts clusters of nucleotides with temperature-sensitive pairing within an RNA sequence (Chursov et al. 2013).
We present here a genetic approach, henceforth called Tet-Trap, to identify RNATs. We have applied Tet-Trap to Pseudomonas aeruginosa, for which only a ROSE-like RNAT controlling the expression of the small heat shock protein IbpA has been reported so far (Krajewski et al. 2013). Pseudomonas aeruginosa is a Gram-negative, mesophilic bacterium, endowed with a remarkable metabolic versatility reflected by a large genome (Stover et al. 2000). It can infect hosts as diverse as worms, flies, and mammals. In humans, it behaves as an opportunistic pathogen and it is responsible for a variety of serious nosocomial infections (Driscoll et al. 2007). As P. aeruginosa is a facultative pathogen, it is conceivable that it can exploit body temperature as a signal for activating the expression of virulence genes specifically required during infection of the warm-blooded host. In fact, a recent transcriptomic survey by RNA-seq of P. aeruginosa grown at 28°C and 37°C detected genes preferentially expressed at the body temperature of the mammalian host, among which virulence genes were significantly enriched (Wurtzel et al. 2012). Using Tet-Trap we identified four genes post-transcriptionally regulated by a temperature upshift from 28°C to 37°C. Interestingly, one of them, namely ptxS, encodes a protein previously implicated in P. aeruginosa virulence (Colmer and Hamood 1998).
RESULTS
The Tet-Trap genetic tool
Tet-Trap is a genetic tool aimed at identifying post-transcriptionally regulated genes, based on the assumption that the cis-acting determinant of regulation is located within the 5′ UTR and the 5′ translated region of the regulated ORF (5′-UTR-TR). In principle, it is made up of three components: a signaling, a sensor, and a reporter system (Fig. 1A). In the signaling system, a sequence encoding a small peptide (TIP2, see below) is fused in frame with the 5′-UTR-TR region of an ORF. Thus, expression of TIP2 as the C-terminus of a fusion protein depends on the ORF translation regulation mechanism. The sensor system exploits the Tn10 tetA gene regulatory circuitry to control the expression of the reporter system. In the absence of tetracycline, tetA transcription from the tetAp promoter is prevented by TetR repressor, which binds to the operator tetO; tetracycline acts as the inducer of tetA by binding TetR and causing its dissociation from the cognate operator. TIP2 is a gratuitous inducer that mimics the tetracycline effect on TetR, as it leads to repressor dissociation from tetO. TIP2 retains this property in end-fused chimeric polypeptides (Goeke et al. 2012). For the reporter system, various reporter genes may be cloned under tetO–tetAp so that reporter expression depends on and correlates with TIP2 translation.
FIGURE 1.

Escherichia coli reporter strains of Tet-Trap system. (A) Outline of Tet-Trap system and regulatory elements. Repression by TetR (the sensor) is relieved by TIP2 expression (the signal) and this triggers the transcription of the reporter gene controlled by tetAp. The reporter cassette is integrated between the tonB and yciA loci (nucleotides 1309870–1309872); the tetR cassette between attB and bioB (806594–808521). Details about strain construction are reported in Materials and Methods. Construct elements are represented by an arbitrary scale. Empty boxes, open reading frames; bent arrows, promoters; triangles, intrinsic bidirectional transcription terminators. (B) Reporter gene expression regulation in the Tet-Trap system. The reporter strains carry genes conferring either spectinomycin resistance (aadA:GFP gene; upper part, strain C-5907) or streptomycin sensitivity (rpsL+; lower part, strain C-5920) downstream from the tetAp promoter. In C-5920, the endogenous rpsL allele carries the rpsL31 (streptomycin-resistance) recessive mutation. In both strains, the tetR gene is constitutively expressed from Pcat, and transcription from tetAp is repressed unless TIP2 is expressed. (Gray ovals) TetR; (dark gray bar) TIP2 peptide; (thick arrows) mRNAs. Other symbols are as in A.
We constructed two E. coli strains with different reporter genes to either select for or counter-select against cells expressing TIP2-tagged polypeptides (Fig. 1B). In these strains, a reporter cassette constituted by the Tn10 tet regulatory region (tetRp–tetO–tetAp) (Bertrand et al. 1983) controlling transcription of the reporter gene (under tetAp promoter) was inserted in the chromosome. Because the gene for tetR is also integrated in the chromosome and constitutively expressed by the Pcat promoter in these reporter strains, transcription of the reporter genes is normally switched off. Repression by TetR can be relieved by TIP2, which leads to reporter gene expression. For positive selection of TIP2-expressing cells (strain C-5907), we used the aadA:GFP gene as a reporter. It encodes a chimeric protein conferring spectinomycin resistance and green fluorescence (Rizzi et al. 2008). The strain for negative selection against TIP2 expression (C-5920) harbours the streptomycin-resistant rpsL31 recessive allele and, as reporter cassette, the rpsL+ allele, which confers streptomycin sensitivity to the otherwise resistant strain (Lederberg 1951). Expression of TIP2-tagged polypeptides from control plasmids renders the strains C-5907 and C-5920 spectinomycin-resistant and streptomycin-sensitive, respectively (data not shown and Fig. 2).
FIGURE 2.

Plating efficiency at different temperatures of clones carrying putative RNATs in TIP2 fusions. (A) Map of the plasmid constructs encoding ST-TIP2. ST-TIP2 is composed by a flexible (SG4)5 linker at the N-terminus, followed by an E. coli TrxA-Tip2 fusion that effectively induces TetR (Goeke et al. 2012). pGM956 and pGM957 differ by the presence in the former of an in-frame TIR driving translation of ST-TIP2, which is absent in pGM957. Details about plasmid construction and coordinates of the cloned regions are reported in Materials and Methods. (Dotted lines) vector sequence; (bent arrows) IPTG-inducible promoter Ptac; (open box) ST-TIP2 ORF; (star) SmaI restriction site exploited for P. aeruginosa DNA cloning in pGM957. (B) Thermosensitive-streptomycin resistance upon induction of TIP2-tagged protein expression. Serial 10-fold dilutions of C-5920 overnight cultures carrying putative P. aeruginosa RNATs cloned upstream of ST-TIP2 in pGM957 were replicated on LD-chloramphenicol plates in the presence or absence of streptomycin (Str) and IPTG and incubated for 16–20 h at the indicated temperatures. C-5920 carrying pGM956 and pGM957 were used as positive and negative controls, respectively, of ST-TIP2 dependent rpsL+ expression.
Construction of a P. aeruginosa 5′-UTR-enriched genomic library
As first step toward the identification of P. aeruginosa RNATs, we constructed a P. aeruginosa genomic library enriched for 5′-UTR-TRs, where regions controlling translation initiation usually map in bacteria. Random fragments of PAO1 and PA14 genomic DNA ranging in length between 300 and 800 bp were pooled and cloned in pGM957 (Fig. 2A). This plasmid carries an artificial gene encoding the (SG4)5:TrxA:TIP2 chimeric polypeptide (hereafter indicated as ST-TIP2) downstream from Ptac. Pseudomonas aeruginosa DNA fragments were inserted between Ptac and ST-TIP2. To select for cells expressing TIP2-tagged peptides, the aadA:GFP reporter strain (C-5907) was transformed with the pooled PAO1 and PA14 genomic library, and spectinomycin-resistant transformants were selected in the presence of IPTG. Transformants were also plated in permissive conditions (without spectinomycin or IPTG) to estimate the coverage of the genomic library. As reported in Table 1, the library reached >99% coverage of the combined PAO1 and PA14 genomes. About 1.7% of all transformants (∼48,000 clones) were SpcR; these clones were expected to express ST-TIP2 and thus constituted the translational fusion library.
TABLE 1.
Features of the P. aeruginosa genomic library

To estimate the coverage of the enriched library, we pooled all SpcR clones, extracted their plasmid DNA, and amplified the genomic inserts by PCR for 454-pyrosequencing. The reads generated by pyrosequencing were mapped on the PAO1 and PA14 genomes. We found that ∼60% of the P. aeruginosa annotated coding genes (61.5% and 58.1% for PAO1 and PA14, respectively) were represented in the translational fusion library. For 2389 coding genes (69.7% of all genes represented in the library), the cloned region encompassed the 5′ UTR and the beginning of the annotated ORF.
Given the high genomic coverage of the library, we expect that missing genes may belong essentially to the following categories: (1) genes physiologically poorly translated; (2) genes translationally coupled to other genes; (3) genes whose translation requires positive regulators absent in E. coli; and (4) genes that are positively regulated by small molecules absent in the experimental conditions of the screening. Moreover, constructs encoding toxic fusion proteins would also have been lost.
Application of Tet-Trap to the identification of putative P. aeruginosa RNA thermometers
In the rpsL+ reporter strain (C-5920), the presence of an RNAT in the DNA fragment cloned in frame with ST-TIP2 should result in thermosensitive streptomycin resistance. Thus, no growth should be observed upon transformation of such strain (C-5920) with the translation fusion library on streptomycin plates at 37°C. Conversely, by plating the transformants in the presence of the antibiotic at 28°C, one should expect to select for clones carrying P. aeruginosa inserts translationally silent under these conditions and, thus, enrich for putative RNATs. However, upon transformation with the library, we observed a high background of StrR clones at both 28°C and 37°C, as the plating efficiency on streptomycin was only ∼250-fold lower than in the absence of selection. To identify clones carrying putative thermosensors, we screened 1152 StrR transformants grown at 28°C by replica plating on streptomycin and IPTG at 28°C and 37°C. We isolated 16 clones exhibiting thermosensitive, IPTG-dependent streptomycin resistance. The clones carried 12 different P. aeruginosa DNA inserts, as determined by sequencing (Table 2 and data not shown). Four clones contained the 5′ UTR and 5′ end of ORFs in frame with ST-TIP2. In particular, codons 1–53 of ptxS (clone #2), 1–49 of PA5194 (#3), and 1–34 of dsbA (#4) were fused with ST-TIP2. In clone #1, the cloned ORF is annotated as an intergenic region in the Pseudomonas database, but as it is in frame with the downstream PA1031, it could actually represent the 5′ end of this gene. Concerning the other fusions, three carried internal fragments of annotated ORFs in frame with ST-TIP2, and in the other five clones the ORFs in frame with ST-TIP2 overlapped with annotated ORFs in a different frame or in antisense orientation (data not shown). It is possible that regulatory sites may actually lie within the internal portion of an ORF. However, as RNATs usually map in the 5′ UTR and the first part of the coding region of the genes, we focused on clones carrying such regions fused with ST-TIP2.
TABLE 2.
Pseudomonas aeruginosa regions carrying putative thermosensors
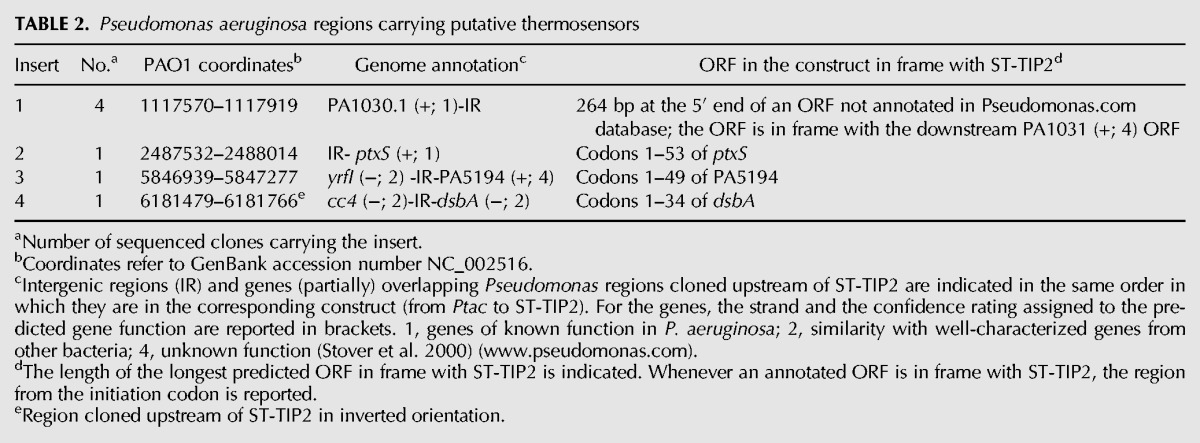
We estimated the efficiency of plating (e.o.p.) at 37°C versus 28°C of cultures carrying the constructs 1–4 (Fig. 2B). The ptxS fusion showed the strongest effect, with a ≥105 e.o.p. reduction at 37°C on plates supplemented with streptomycin and IPTG; for PA1031 and dsbA, the e.o.p. was reduced ∼100-fold. Finally, for PA5194, the e.o.p. was reduced ∼10-fold and the colonies were very small at 37°C. All strains showed comparable e.o.p. at the two temperatures in the absence of either streptomycin or IPTG. This indicates (1) that the constructs do confer thermosensitive streptomycin resistance and not a generic thermosensitive phenotype, and (2) that the increase in ST-TIP2 expression was not due to temperature-dependent activation of promoters in the cloned regions, as it depends on Ptac induction. This was confirmed by Northern blotting analysis of the transcription pattern of the four constructs at 28°C and 37°C. In all four cases we did not observe any relevant difference in the transcription pattern at the two temperatures (data not shown). On the whole, this analysis confirmed that the expression of the TIP2-tagged peptides 1–4 was post-transcriptionally activated by the temperature upshift.
Validation of the PA5194 and ptxS RNA thermosensors in E. coli and P. aeruginosa
To validate the results of the Tet-Trap, we analyzed translational fusions of two putative thermosensors, i.e., those identified in constructs #2 (ptxS) and #3 (PA5194), with conventional reporter genes. For both ptxS and PA5194 two constructs each were generated, one carrying the entire P. aeruginosa regions originally found in the #2 and #3 ST-TIP2 plasmids, respectively, and the other with the same 3′ end of the #2 and #3 inserts, but starting with the first nucleotide transcribed from the endogenous PA5194 or ptxS promoters in P. aeruginosa (Fig. 3A; Dotsch et al. 2012). As the 5′ end of the PA5194 mRNA was not known, we first mapped it by primer extension at 25°C and 37°C and found a single signal of comparable intensity at the two temperatures at position 5847081, 50 nt upstream of the ORF start codon (Fig. 3B).
FIGURE 3.

Constructs used for validation of putative RNATs and transcriptional analysis. (A) Map of plasmids encoding PtxS-BgaB and PA5194-sfGFP. Control plasmids carrying the 5′ end of the E. coli recA gene in frame with the reporter genes are also represented. Details about plasmid construction and coordinates of the cloned regions are reported in Materials and Methods. Pseudomonas aeruginosa and E. coli 5′ UTRs and ORFs are drawn to scale. (Dotted lines) vector sequences; (bent arrows) araBp promoter; (empty boxes) P. aeruginosa and E. coli DNA; (gray boxes) reporter genes. (B) Mapping the 5′ end of the PA5194 mRNA by primer extension. Primer extension was performed with the radiolabeled oligo 3003 on 30 µg of RNA extracted from exponential cultures of PAO1 grown at 25°C and 37°C. The same oligonucleotide was used for Sanger-sequencing of construct 3. The coordinate of the identified 5′ end and the sequence context (in bold the 5′ end T) are reported beside the panel. (C) Northern blot analysis of ptxS-bgaB and PA5194-sfGFP fusions in P. aeruginosa. Cultures of the PA01 strain carrying the plasmids indicated above the lanes were grown at 25°C up to OD600 = 0.5. The cultures were induced with 0.1% arabinose, split and further incubated at the temperature indicated above the lanes for 45 min. Northern blotting was performed as described in Materials and Methods after electrophoresis in a 1.5% denaturing agarose gel. The filter was hybridized with oligonucleotides specific for bgaB or sfGFP, as indicated on top of the panels. To check gel loading, the filters were either hybridized with the 16S rRNA-specific 1396 oligonucleotide (bgaB panels) or stained with methylene blue before the hybridization (sfGFP panels; the bands corresponding to 16S rRNA are shown). The transcript size was roughly estimated by comparison with rRNA migration as follows: pGM981 and pGM989, 2.5 kb; pGM2013, 1.2 kb; pGM2016, 1.1 kb.
Two different reporter genes were used in the constructs. In particular, for the ptxS fusions we exploited the bgaB gene, which encodes a thermostable β-galactosidase (Hirata et al. 1984) and has been already used for the analysis of RNATs (Waldminghaus et al. 2007a; Klinkert et al. 2012). To validate PA5194, which is predicted to encode a membrane protein with a signal peptide at the N-terminus, we cloned the PA5194 fragments in frame with sfGFP, a GFP variant which, unlike β-galactosidase (Oliver and Beckwith 1981), is known to be functional in the periplasm upon translocation across the membrane (Dinh and Bernhardt 2011). β-Galactosidase activity or fluorescence of the chimeric proteins were then monitored at different temperatures. As control, fusions of the 5′ UTR and the first nine codons of the E. coli recA gene with bgaB or sfGFP were assayed. All constructs were cloned in the E. coli–P. aeruginosa shuttle vector pGM931 under the araBp promoter. The expression of the reporter genes was monitored in E. coli DH10B cultures grown at 28°C and 42°C upon transcription induction with arabinose (Tables 3 and 4). β-gal expression in the presence of the ptxS-bgaB fusions was strongly temperature-dependent in E. coli, as cultures carrying the long (pGM980) or the short (pGM981) construct showed >20- and 10-fold increases, respectively, in enzymatic activity at 42°C. Conversely, BgaB activity in the recA control plasmid (pGM989) increased only twofold. It should be noted that β-galactosidase expression by the long pGM980 construct was much lower at both temperatures than by the short construct pGM981 (Table 3). Reduced reporter expression by the longer fusion could be due to decreased efficiency of translation and/or stability of the mRNA transcribed from araBp, which bears at the 5′ end a sequence absent in the P. aeruginosa ptxS mRNA. We then assayed the expression of the ptxS-bgaB fusion by pGM981 in the P. aeruginosa strain PAO1 at 25°C and 37°C. We performed the experiments at lower temperatures than in E. coli to mimic the temperature upshift due to infection of a mammalian host. We found that ptxS-bgaB expression was also clearly thermoregulated in P. aeruginosa, as it showed a more than fivefold higher activity at 37°C, whereas recA-bgaB expression by the pGM989 control plasmid was not significantly affected by the temperature upshift (Table 3).
TABLE 3.
ptxS RNAT validation
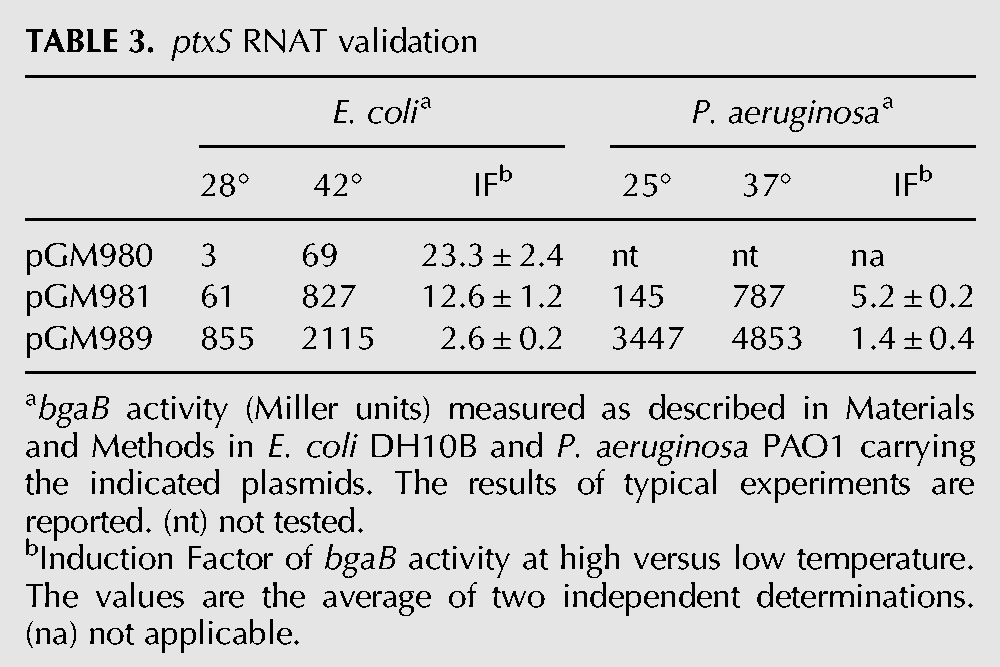
TABLE 4.
PA5194 RNAT validation
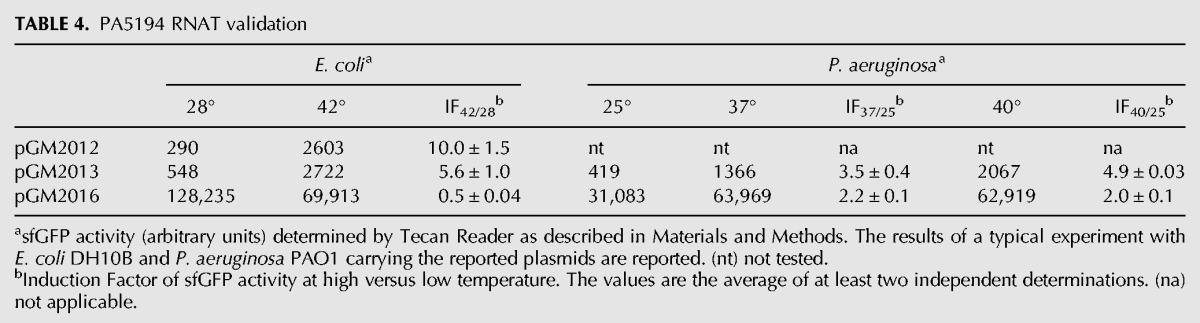
For PA5194-sfGFP, we found a sharp increase in the fluorescence of the fusion proteins by both the long and the short constructs at 42°C in E. coli, whereas the RecA-sfGFP fluorescence intensity did not increase with temperature (Table 4). In P. aeruginosa, a 3.5-fold increase in signal strength was observed at 37°C versus 25°C. An upshift of the temperature to 40°C further increased PA5194-sfGFP expression, whereas RecA-sfGFP expression increased twofold at both 37°C and 40°C (Table 4). Thermoregulated expression of PA5194-sfGFP was confirmed by direct visualization of the cells by fluorescence microscopy (Fig. 4). On the whole, these data show that the ptxS and PA5194 regions encoded by the Tet-Trap constructs confer thermoregulated expression to the reporter genes, thus indicating that the determinants of this regulation lie in the cloned portion of the two P. aeruginosa genes. Moreover, since the levels of the transcripts of the reporter constructs did not change with temperature, as assessed by Northern blotting (Fig. 3C), translation initiation is the most likely target of this regulation.
FIGURE 4.
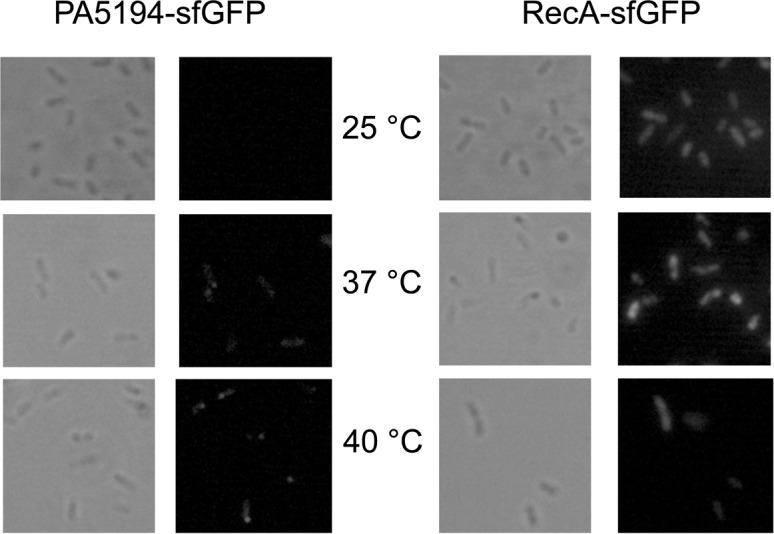
Temperature-dependent expression of PA5194-sfGFP by P. aeruginosa cells. Images of PAO1 cells carrying plasmids pGM2013 (PA5194-sfGFP) or pGM2016 (RecA-sfGFP) were acquired on a Leica fluorescence microscope. The cells were sampled from cultures grown at the temperatures indicated between the panels as described in Materials and Methods. The same viewfield was captured in contrast phase and fluorescence imaging mode.
To check whether the results obtained with the translational fusions applied also to the full-length ptxS and PA5194 proteins, we analyzed the expression in P. aeruginosa of variants of the two genes with an HA tag inserted immediately before the stop codon. In agreement with the data obtained with the reporter constructs, the expression of the full-length PtxS and PA5194 proteins was barely detectable at 25°C and strongly induced at higher temperatures (Fig. 5A). Conversely, ptxS transcript was expressed at 25°C and increased two- to threefold at temperature ≥37°C, whereas PA5194 mRNA was equally expressed at all the tested temperatures (Fig. 5B). Similar expression profiles were shown also by the endogenous PA5194 and ptxS mRNAs (i.e., transcribed from the P. aeruginosa chromosome) (Fig. 5C).
FIGURE 5.

Temperature-dependent expression of PA5194 and ptxS in P. aeruginosa. Proteins (A) and RNA (B) were extracted upon induction with 0.1% arabinose, as detailed in Materials and Methods, from cultures of PAO1 carrying either pPA5194-HA (left) or pPtxS-HA (right). Time and temperature of incubation are indicated above the lanes. (A) Proteins were separated by 10% SDS-PAGE, blotted onto an Hybond ECL filter, and hybridized with HA- or S1-specific antibodies, as indicated beside the panels. No bands were detected in the lanes loaded with not induced samples (data not shown). The PA5194-HA (expected MW, 33.2 kDa) and PtxS-HA (expected MW, 39.9 kDa) migrate in this kind of gels slightly slower than the 28 and 36 kDa bands of the PageRuler Plus Prestained Ladder (ThermoScientific), respectively. (B) The RNA was run on a 5% denaturing urea-polyacrylamide gel, blotted onto a nylon Hybond N membrane and hybridized with either the radiolabeled HA oligonucleotide (PA5194) or the PTXS riboprobe. Migration of the 1.0 and 1.5 kb bands of RiboRuler High Range RNA ladder (ThermoScientific) are reported beside the panels. (C) Northern blot analysis of PA5194 and ptxS endogenous mRNAs. Cultures of PAO1 were grown overnight in LD at 37°C and diluted to OD600 = 0.2 in three flasks containing LD. The flasks were incubated at 25°C–37°C–40°C with agitation up to OD600 = 0.8. Ten milliliters of samples were taken for RNA extraction. Twenty micrograms of RNA were run on a 5% denaturing urea-polyacrylamide gel, blotted onto a nylon Hybond N membrane, and hybridized with either the radiolabeled PA5194 or the PTXS riboprobes. Migration of the 1.0- and 0.5-kb band of RiboRuler High Range RNA ladder (ThermoScientific) is reported on the left of the panels. The numbers below the panels represent relative expression (RE) of each mRNA with respect to its expression at 25°C. 5S rRNA signals were used for normalization.
Analysis of ptxS and PA5194 translation initiation regions' secondary structures
The structure of the first 137 and 126 nt of the ptxS and PA5194 mRNAs was probed at different temperatures (25°C–37°C–42°C) by partial digestion with the RNases T1 and A, which cut at the 3′ of unpaired guanines and pyrimidines, respectively, and with lead acetate, which cleaves the RNA within single-stranded regions. The results are shown in Figure 6A.
FIGURE 6.

In vitro and in silico analysis of the secondary structure of the ptxS and PA5194 5′ UTRs. (A) Structural analysis of in vitro transcribed ptxS (left) and PA5194 (right) RNA with RNase T1 (T1), lead acetate (Pb2+) and RNase A (A) at different temperatures (indicated above the lanes in degrees Celsius). (-), ptxS or PA5194 RNA incubated at 37°C in the absence of RNases or lead acetate; (T) ptxS or PA5194 RNA digested with RNase T1 under denaturing conditions; (L) alkaline digestion of the PA5194 transcript. The dotted boxes encircle regions predicted to fold into secondary structures (shown in panel B) that involve the TIRs of the two RNA molecules. (B) Structure models of ptxS and PA5194 with probing results. Cleavage sites are indicated as full gray circles; in particular, positions showing thermo-dependent cleavage are indicated in dark gray. Stem–loops encompassing the SD and start codons of the two genes are enclosed in dotted boxes. The ptxS GUG start codon encompasses bases 78–80, the PA5194 AUG start codon bases 51–53. The three U's preceding the AUG in PA5194 that are mutated in pGM2013AAA and pGM2013CC are boxed. (C) Output of the RNAtips analysis of the two RNAs showing the density plot of positions along the ptxS (upper panel) or PA5194 (lower panel) sequences whose pairing probability is significantly affected by a temperature change from 28°C to 42°C. (D) Structure model of the E. coli lpxT TIR. The secondary structure was predicted with the software RNAfold. The start codon of the ORF is boxed; the black bar flanks the five uridine nucleotides involved in the pairing with the TIR. RNA secondary structures in B and D were drawn with the VARNA applet (Darty et al. 2009).
No cuts were observed in the ptxS RNA region from nucleotides 30 to 40 at any temperature, whereas the downstream region of the probed RNA was more accessible even at 25°C. Mfold (Zuker 2003) analysis of the transcript predicted several alternative secondary structures with comparable free energy (Fig. 6B and data not shown), among which the structure presented in Figure 6B is in good agreement with the experimental data. The structure consists of two stem–loops (SL1, U18-A40, and SL2, G66-U84) separated by an unstructured region. SL1 is predicted to be stable (ΔG −10.40 kcal/mol), whereas SL2, which involves the GUG start codon, should be relatively unstable (ΔG −5.6 kcal/mol). Moreover, the RNAtips program (Chursov et al. 2013) predicted a cluster of nucleotides with thermosensitive pairing in a region overlapping SL2 (Fig. 6C). Indeed, several positions in SL2 (G at positions 66, 67, 73, 74, 78, 84, and 85) were cut by RNase T1 only when the temperature was increased to 37/42°C (Fig. 6A), suggesting that SL2 can be in equilibrium between an open and a folded conformation and that the temperature upshift may promote unfolding.
For PA5194, we found that the temperature upshift had a modest effect on the accessibility of the RNA to RNases and lead acetate (Fig. 6A). Interestingly, upon degradation with lead acetate and RNase A, increased reactivity at several positions was detected at 42°C, but not at 37°C. This observation is in agreement with in vivo data that showed an effect on PA5194-sfGFP expression by increasing the temperature from 25°C to 37°C and again upon a further increase at 40°C (Table 4). Also for this mRNA, the RNAtips analysis suggests that a cluster of residues with thermosensitive pairing overlaps the translation initiation region (Fig. 6C).
The centroid structure calculated at 25°C by RNAfold (Ding et al. 2005) predicts that PA5194 translation initiation region may be involved in the formation of an unstable stem–loop. In particular, three guanidines in the putative Shine–Dalgarno sequence (nucleotides 36–41) should pair with a stretch of uridines (anti-SD) preceding the AUG start codon. The AUG is also predicted to be involved in pairing within the stem (Fig. 6B). Since the results of PA5194 mRNA structural probing were compatible with different predicted secondary structures of the transcript, we decided to test in vivo whether the putative anti-SD could actually play a role in PA5194 regulation. We replaced the three anti-SD uridines (47–49 in Fig. 6B) with either three cytidines (2013CCC mutation) or three adenosines (2013AAA) in the backbone construct pGM2013. The two mutations should either strengthen (2013CCC) or destabilize (2013AAA) the SD-anti-SD pairing and thus have opposite effects on PA5194-sfGFP expression. In particular, the mutated stem–loop in 2013CCC is predicted to have a free energy of −10.38 kcal/mol and could prevent translation also at high temperature (Neupert et al. 2008). In agreement with the bioinformatics prediction, we found that PA5194-sfGFP expression by 2013CCC was repressed in E. coli even at 42°C (Fig. 7). Conversely, the 2013AAA mutation led to an increase in fluorescence at all temperatures. In particular, the fluorescence reached by the wild-type construct at 40°C–42°C was already shown by the mutant at 35°C–37°C. Overall, these data are compatible with a role of the predicted SD–anti-SD interaction in PA5194 regulation.
FIGURE 7.

Effect of point mutations on PA5194-sfGFP thermoregulation. Cultures of E. coli DH10B carrying plasmids pGM2013 (2013) and its mutated derivatives pGM2013CCC (2013CCC) and pGM2013AAA (2013AAA) were grown at the temperatures indicated below the bars and processed as detailed in Materials and Methods. pGM2016 (2016) was analyzed as negative control and showed modest (less than twofold) thermal induction. Fluorescence emission (F485/535) was measured in a Packard FluoroCount microplate reader at 485/530 nm. The fluorescence was expressed in arbitrary units (AU) as the ratio F485/535/OD600. The average results of two independent measurements are reported with standard deviations. Relative fluorescence with respect to pGM2013 at 28°C is indicated by the numbers on top of the bars.
DISCUSSION
In this work we identified two P. aeruginosa genes, namely ptxS and PA5194, whose translation is regulated in response to temperature changes. The 5′ UTRs of the two genes confer thermoregulated expression to reporter genes in such distantly related bacteria as E. coli and P. aeruginosa, thus making it unlikely that specific factors acting in trans may be responsible for this regulation. In fact, no ortholog of the ptxS gene is present in E. coli and no significant sequence similarity exists between the 5′ UTRs of PA5194 and its probable orthologous gene lpxT (see below). We thus propose that two new RNATs may regulate the translation of ptxS and PA5194 in P. aeruginosa.
Transcriptional analysis of full-length ptxS and PA5194 (Fig. 5) supports this view. In fact, in spite of strong repression of the two proteins at low temperature, their mRNAs are expressed at 25°C and do not (PA5194) or modestly (ptxS) increase at higher temperatures. This is unusual for many (most?) transcripts, as efficiently translated mRNAs are generally more stable than untranslated ones (for review, see Dreyfus 2009), but it has already been reported for other RNATs (Johansson et al. 2002; Kortmann et al. 2011) suggesting that, at low temperature, untranslated mRNAs bearing RNATs may be protected from degradation. This would provide the cells with a pool of translationally silent mRNAs that could be rapidly activated in response to a sudden temperature increase.
No structural similarity of the ptxS and PA5194 mRNAs was found with known RNATs after analysis with Mfold (Zuker 2003). However, in vitro probing of the 5′ end of the ptxS mRNA suggested a secondary structure and a possible mechanism for temperature-dependent translation regulation. In fact, the TIR and the 5′ end of the coding sequence may fold into a thermosensitive stem (SL2), which would sequester the TIR at low temperature. As for PA5194, our data support bioinformatic predictions suggesting that the TIR of the gene may fold into an unstable stem–loop with both the SD and the region encompassing the AUG codon involved in base-pairing. However, abolishing the anti-SD–SD pairing in the 2013AAA mutant had only a mild effect on thermal induction. This suggests that this RNAT may primarily act by modulating the start codon accessibility and that the anti-SD–SD interaction could be implicated in the fine tuning of the response. Interestingly, it has recently reported that the cyanobacterial avashort RNAT primarily acts by sequestering the AUG start codon (Cimdins et al. 2014).
The physiological role of ptxS thermoregulation is not obvious. The gene encodes a repressor of the LacI family that regulates transcription of its own gene and of operons for gluconate transport and degradation (Swanson et al. 1999; Swanson and Hamood 2000). Gluconate transport is the major pathway of glucose uptake when glucose is abundant, upon its oxidation to gluconate (Whiting et al. 1976). The specific effector of PtxS is 2-ketogluconate, whose binding causes dissociation of PtxS from DNA. Recently, it was shown that PtxS interacts with nanomolar affinity with PtxR, whose gene is adjacent to and transcribed divergently from ptxS, and that 2-ketogluconate abolishes the interaction (Daddaoua et al. 2012). PtxR is a positive regulator of transcription of toxA, which encodes the most toxic virulence factor of P. aeruginosa (Hamood et al. 1996; Daddaoua et al. 2012). It has been proposed that in the absence of 2-ketogluconate, PtxS acts as a negative regulator of toxA by preventing the interaction of PtxR with the RNA polymerase (Daddaoua et al. 2012). Thus, while high glucose concentrations may induce ToxA expression by inhibiting PtxS interaction with PtxR, PtxS thermal induction may help prevent ToxA expression at high temperature but low glucose concentration. Interestingly, although glucose is far less concentrated in the airways surface liquid (ASL) that lines the respiratory tract than in plasma, in cystic fibrosis patients, which are highly susceptible to P. aeruginosa infections, glucose concentration in the ASL is considerably higher than in healthy individuals (Baker et al. 2007; Garnett et al. 2012).
The PA5194 gene is located in a chromosomal region where other functions involved in the response to temperature upshift map. In fact, it is flanked by two divergently transcribed genes, namely yrfI, which encodes the chaperone protein HSP33, and PA5195, which is described in the Pseudomonas Genome Database as a probable heat shock protein. The PA5194 gene product is annotated as a membrane protein with unknown function. However, it has an overall 32% identity and 49% similarity with the E. coli protein LpxT, and the two genes are predicted to be orthologous by Reciprocal Best-Blast analysis (Montague and Hutchison 2000). Interestingly, the 5′ UTR of the lpxT mRNA is predicted by RNAfold to form a structure that resembles a FourU RNAT, suggesting that lpxT translation may also be thermoregulated (Fig. 6D). LpxT catalyzes the phosphorylation of Lipid A at the 1-position using undecaprenyl pyrophosphate as a phosphate donor, thus increasing the negative charge of the bacterial surface. This modification is relevant not only for LPS stability, but also significantly affects its endotoxicity, as Lipid A lacking one or both phosphate groups has proven to be less toxic in several bacteria (Kong et al. 2011; John et al. 2012; Needham and Trent 2013). The effect of temperature on Lipid A decoration has not been extensively explored so far. However, increased Lipid A phosphorylation in response to a modest temperature upshift (from 37°C to 39°C–41°C) has been observed in Porphyromonas gingivalis (Curtis et al. 2011). Moreover, in P. aeruginosa the degree of another modification of Lipid A, i.e., acylation, is modulated by temperature (Ernst et al. 2006); it would be interesting to see whether phosphorylation also responds to this stimulus.
Tet-Trap has allowed the identification of two genes post-transcriptionally regulated in response to temperature changes that would have presumably escaped other experimental or bioinformatic approaches. In fact, ptxS and PA5194 do not show obvious similarity with known RNATs. This also holds true for dsbA and PA1031 5′ UTR (data not shown), which were also identified in the Tet-Trap screening. Moreover, ptxS and PA5194 are comparably expressed at 25°C and 37°C (Fig. 5C) and, in fact, they were not identified among thermoregulated genes in a recent transcriptomic survey of P. aeruginosa (Wurtzel et al. 2012). Finally, as the two genes seem to be inefficiently translated even at high temperature (compare the PtxS and PA5194 fusions with expression of the RecA control fusion in Tables 3 and 4), the study of their regulation through proteomic approaches would probably have been challenging. We expect that other P. aeruginosa RNATs may wait for identification. In fact, we did not fish in our screen the ibpA RNAT (Krajewski et al. 2013), which was represented in the P. aeruginosa ST-TIP2-fusion library (data not shown). Other rounds of Tet-Trap may be run to identify new P. aeruginosa RNATs. Moreover, it would be interesting to apply a Tet-Trap search for RNATs to other Gram-negative bacteria, such as pathogenic strains of E. coli or Salmonella. As TIP-mediated induction of TetR-controlled gene expression has been proven to be functional also in the Gram-positive bacterium Staphylococcus aureus and even in the eukaryote Saccharomyces cerevisiae (Gauger et al. 2012; Stoeckle et al. 2012), the Tet-Trap could potentially be adapted to search for RNATs in these systems.
MATERIALS AND METHODS
Bacterial strains and plasmids
Bacterial strains, plasmids, and oligonucleotides sequences are reported in Supplemental Table S1. Pseudomonas aeruginosa strains used here were PAO1 (Stover et al. 2000) and PA14 (Rahme et al. 1995; Lee et al. 2006) (GenBank accession numbers NC_002516 and NC_008463, respectively). Escherichia coli coordinates throughout this work refer to GenBank accession number U00096.2, whereas P. aeruginosa coordinates refer to PAO1.
Construction of E. coli Tet-Trap reporter strains
The SpcR and StrS reporter cassettes were prepared as follows. A cassette constituted by the Tn10 tet regulatory region (tetRp–tetO–tetAp) (Bertrand et al. 1983) controlling transcription of kanR (under tetAp promoter) and lacZα (under tetRp) was integrated into the C-1a (Sasaki and Bertani 1965) chromosome between the tonB and yciA loci. The cassette was obtained by three-step-PCR as follows. Three partially overlapping fragments corresponding to the Tn10 tet regulatory region, lacZα and kanR were synthesized by PCR. Oligonucleotides 2602–2603 on C-5868 genomic DNA (Carzaniga et al. 2009) were used for Tn10 tet amplification; 2604–2605 on pUC19 (Yanisch-Perron et al. 1985) for lacZα; 2600–2601 on pQE31S1 (Sukhodolets and Garges 2003) for kanR. Then Tn10 tet and lacZα fragments were used as templates in a PCR reaction with oligonucleotides 2603–2604, obtaining the fragment lacZα-tet. Finally, the full-length cassette was obtained by amplification of the lacZα-tet and kanR fragments with primers 2604–2606 and was cloned in the SmaI site of pGM742 (Regonesi et al. 2004), giving plasmid pGM932. The insert was excised by digestion with NotI and XmnI and integrated in C-1a/pKD46 between nucleotides 1309870 and 1309872 by λ Red-mediated homologous recombination (Datsenko and Wanner 2000), obtaining C-5898. To replace the kanR gene with the aadA:GFP fusion gene, which confers spectinomycin resistance and fluorescence, the aadA:GFP ORF was amplified from plasmid pZR80-2 (Rizzi et al. 2008) with the oligonucleotides 2683–2684, harboring 50-nt long tails homologous to the regions flanking the kanR ORF in C-5898. C-5898/pKD46 was transformed with the above-mentioned PCR fragment; the recombinants were selected on spectinomycin plates and their fluorescence evaluated by Versadoc imaging of the plates. The cassette from a highly fluorescent clone was sequenced and revealed a spontaneous A to G transition within the tetA Shine–Dalgarno region (position 916 of Tn10; GenBank accession number AY528506.1). This mutant strain was named C-5899. To replace the aadA:GFP gene with rpsL, the rpsL ORF and the cat gene with two flanking FRT sites were amplified by PCR with the oligonucleotides 2713–2714 on C-1a genomic DNA and the oligonucleotides 2636–2712 on pKD3 (Datsenko and Wanner 2000), respectively. These two partially overlapping amplicons were used as DNA templates for a PCR reaction with oligonucleotides 2638–2602. The resulting DNA fragment was used to transform C-5899/pKD46, obtaining C-5912. The reporter construct was then transduced into C-5708, a C-1a derivative carrying the recessive rpsL31 allele (K42T substitution in S12), which confers streptomycin resistance (Lederberg 1951). The cat cassette was excised from the StrS recombinant strain (C-5916) by FLP-mediated recombination, obtaining C-5918. Pcat-tetR-kanR cassettes were amplified (oligonucleotides 2685–2686) from WH1001 derivatives carrying either the wt or the weaker −10CATTTA mutant of the Pcat promoter upstream of tetR (Georgi et al. 2012). Region 806595–808520 of the BW25113 genome was replaced with the two cassettes by λ Red-mediated homologous recombination, obtaining strains KG264 (Pcat+) and KG265 (Pcat −10CATTTA), respectively. We transduced the Pcat+-tetR-kanR region from KG264 into C-5899 obtaining C-5901; C-5907 is a C-5901 derivative in which the kanR cassette was removed by FLP-mediated recombination (Datsenko and Wanner 2000). C-5920 was obtained by P1 transduction from KG265 into C-5918.
Tet-Trap plasmids
pGM956 and pGM957 carry a chimeric gene composed of an (SG4)5 linker and trxA fused upstream of TIP2. The construct was obtained by three-step PCR. (SG4)5 was obtained by annealing the partially overlapping oligonucleotides 2689–2690 and extension with Pfu polymerase (Stratagene). The trxA:TIP2 sequence was amplified by PCR from pWH2354 (Georgi et al. 2012) with the oligonucleotides 2691–2692. The final PCR was performed on the two above-mentioned fragments with oligonucleotides 2692–2693 obtaining the full-length construct (SG4)5:trxA:TIP2. This was digested with SphI–EcoRI and cloned in pGZ119HE (Lessl et al. 1992) obtaining pGM957. pGM956 is a pGM957 derivative that carries a translation initiation region (TIR) in frame with (SG4)5:trxA:TIP2. The TIR was obtained by annealing the oligonucleotides 2617–2618 and cloning the fragment in pGM957 between HindIII and SphI sites. Both plasmids were checked by sequencing the relevant regions.
BgaB, sfGFP, and HA reporter plasmids
To construct the shuttle vector pGM931, the HindIII–PstI pGM362 fragment carrying the transcriptional terminator tΩ (Briani et al. 2000) was cloned in pBAD24-Δ1 (Carzaniga et al. 2012), obtaining pGM930. The MluI–HindIII pGM930 fragment carrying araBp-tΩ was cloned in pHERD20T (Qiu et al. 2008), obtaining pGM931. All reporter constructs were assembled in pGM931 and checked by sequencing. bgaB was amplified by PCR from pBAD2_bgaB (Klinkert et al. 2012) with the primers 2846–2847, digested with NcoI and PstI and cloned in pGM931, obtaining pGM978. pGM978 was digested with NcoI and EcoRI and used as a backbone for the translation fusions. The following portions of the ptxS gene were amplified from the P. aeruginosa PAO1 genome and cloned: region 2487531–2488013 (primers 2850–2851; plasmid pGM980); region 2487779–2488013 (primers 2851–2852; plasmid pGM981). The Superfolder GFP (sfGFP) (Pedelacq et al. 2006) gene was amplified from pXG-10SF (Corcoran et al. 2012) with the oligonucleotides 2803–2804, digested with PstI–KpnI and cloned in pGM931, obtaining pGM2011. Translational fusions were obtained by cloning PCR amplified-KpnI-digested PAO1 genomic regions 5846939–5847277 (oligonucleotides 3004–3005) and 5847080–5847277 (oligonucleotides 3004–3006) in KpnI-digested pGM2011, obtaining plasmids pGM2012 and pGM2013, respectively. pGM2013CCC and pGM2013AAA carry the same region as pGM2013 with the substitution of the three thymidines in position 5847127–5847129 with three cytidines or three adenosines, respectively. The constructs were obtained by three-step PCR on PAO1 DNA with external oligonucleotides 3004 and 3006 and partially overlapping primers 3141–3142 (for pGM2013CCC) or 3141–3143 (for pGM2013AAA), digestion with KpnI of the amplicons and cloning in pGM2011. Control plasmids pGM989 and pGM2016 carry the leader region and the first nine codons of E. coli recA in frame with the bgaB and sfGFP genes, respectively. The recA fragments were amplified from MG1655 genomic DNA with the oligonucleotides 2915–2916 (amplicon Rec1) and 2928–2929 (Rec2). Rec1 was digested with NcoI and EcoRI and cloned in pGM978, obtaining pGM989, whereas Rec2 was digested with KpnI and cloned in pGM2011, obtaining pGM2016. pPA5194-HA was constructed by cloning a KpnI-digested amplicon obtained by PCR with oligonucleotides 3006–3144 on PAO1 DNA in pGM931 digested with KpnI and SmaI. pPtxS-HA was similarly obtained by cloning a NcoI-digested DNA fragment amplified by PCR with oligonucleotides 2852–3145 on PAO1 DNA in pGM931 digested with NcoI and SmaI. All pGM931 derivatives were constructed in E. coli and transferred to PAO1 by triparental conjugation (Goldberg and Ohman 1984).
Bacterial cultures were grown in LD (10 g/L Tryptone, 5 g/L Yeast Extract, 5 g/L NaCl) or M9-glucose (0.1% NH4Cl, 1.6% Na2HPO4·12 H2O, 0.3 KH2PO4, 0.5% NaCl, 0.013% MgSO4, 0.001% CaCl2 and trace elements, 0.4% glucose). When needed, media were supplemented as follows: 100 µg/mL ampicillin; 30 µg/mL chloramphenicol; 100 µg/mL spectinomycin; 25 µg/mL streptomycin; 0.5 mM IPTG; 0.1%–0.2% arabinose. Pseudomonas aeruginosa cultures carrying pGM931 derivatives were grown either in LD supplemented or in M9-glucose supplemented with 300 µg/mL (LD) or 150 (M9-glucose) µg/mL carbenicillin.
Library generation
Genomic DNA was extracted from stationary phase cultures of PAO1 and PA14 with the Puregene Kit. One microgram of PAO1 and PA14 genomic DNA was partially digested with AluI, HaeIII, or the two enzymes for 30 min at 37°C. The digestions were loaded on a 1% agarose gel and bands corresponding to DNA ranging from 300 to 800 nt were cut out from the gel. The digested fragments were purified and pooled. The randomly digested DNA fragments of PAO1 and PA14 were cloned in pGM957 linearized with SmaI, which makes a single cut between Ptac and the (SG4)5-trxA-TIP2 chimeric ORF. C-5907 was then transformed with the ligated DNA, and the library was obtained by extracting plasmid DNA from the pool of clones grown in the presence of spectinomycin.
Library sequencing and data analysis
Pseudomonas aeruginosa inserts cloned in the plasmid library described above were amplified by PCR with the oligonucleotides 2739–2740. Ten nanograms of the library DNA were used as template in the amplification reaction. The amplicons were purified by using Agencourt AMPure XP (Beckmann Coulter) in order to remove primer dimers and fragments shorter that 50 bp; the 454 sequencing library was then prepared following the Method Manual for Rapid Library Preparation, GS FLX Titanium (Roche Applied Science). Four hundred fifty-four adaptors with MID indexes were ligated to the ends of the library fragments. The sequencing library was analyzed with Agilent Bioanalizer High Sensitivity assay and quantified with a NanoDrop fluorimeter by using PicoGreen (Invitrogen, Life Technologies). The library was sequenced in replicate in two lanes corresponding to 1/8 of a GS FLX Titanium Pico Titer Plate (PTP). Genome sequences and annotation files were retrieved from Pseudomonas Genome Database (http://www.pseudomonas.com). The 52,116 reads generated by pyrosequencing, which were on average 526 bp long, were mapped to the PAO1 and PA14 genomes (GenBank accession numbers NC_002516 and NC_008463, respectively) using Newbler (Roche, 454). The overlap with annotated genes and the coverage were assessed using BEDTools (Quinlan and Hall 2010). In order to define the P. aeruginosa core and the PAO1–PA14 strain-specific coding genes, the protein sequences of both strains were blasted against each other (Altschul et al. 1990), 5320 proteins showing sequence similarity ≥90% were defined as “core,” 252 were identified as PAO1-specific and 572 as PA14-specific (Lee et al. 2006; Silby et al. 2011). Three thousand sixty-four core coding genes, 168 PAO1-specific and 194 PA14-specific, for a total of 3426 genes were identified by sequencing. More than 78% of these genes were sequenced with coverage above the detection threshold, which was equal to or >3 reads per gene. The translation initiation region of a gene was considered to be represented in the library if at least one consensus resulting from the mapping covered the start codon of that gene.
β-Galactosidase assays
Escherichia coli DH10B (Grant et al. 1990) cultures were grown at 28°C in LD supplemented with ampicillin up to OD600 = 0.5 and induced with 0.1% arabinose. The cultures were split in two and the sub-cultures incubated at 28°C or 42°C. After 30 min, samples were taken to measure OD600 and β-galactosidase activity. BgaB activity was measured in permeabilized cells as described by Miller (1972), except that the assay was performed at 55°C. The assay on P. aeruginosa was performed by growing PAO1 cultures at 25°C in 40 mL of LD supplemented with carbenicillin (Carb) up to OD600 = 0.5. As PAO1 cultures formed macroscopic aggregates under these conditions, the cells were collected by centrifugation and carefully resuspended in 1 mL of LD to eliminate visible aggregates. The cells were then inoculated in 40 mL of LD supplemented with Carb and 0.1% arabinose at 25°C. Twenty milliliters were immediately withdrawn and shifted to 37°C. After 45-min incubation, 15 mL of each sub-culture were centrifuged and resuspended in 0.3 mL of TEDP (0.1 M Tris–HCl, pH 8; 0.001 M EDTA; 0.1 M DTT; 0.1 M PMSF). Crude extracts were obtained by sonication and the protein concentration was evaluated by Bradford assay (Bradford 1976). β-Galactosidase activity at 55°C was assayed by mixing 0.2 mL of crude extract with 0.8 mL of Z buffer (Miller 1972) and 0.2 mL of 4 g/L ONPG in Z buffer. The reaction was stopped by adding 0.5 mL of 1 M Na2CO3.
Fluorescence detection
Fluorescence of cells carrying sfGFP fusions was detected either by means of a Tecan Infinity PRO 200 reader or a Packard Fluorocount microplate reader. For Tecan experiments, P. aeruginosa PAO1 and E. coli DH10B cultures carrying the reporter plasmids were grown 16 h in LD with the appropriate antibiotics and 0.2% arabinose at different temperatures. Cells were collected by centrifugation, washed, and resuspended in PBS at OD600 = 0.1–0.5. Two hundred microliter cell samples were transferred in black polystyrene 96-well microplates with clear flat bottom (Corning). OD595 and Fluorescence Polarization (FP)485/535 were measured. sfGFP activity was expressed in arbitrary units (AU) as the ratio FP485/535/OD595. For Packard Fluorocount experiments, DH10B cultures carrying the reporter plasmids were grown and processed as described previously for Tecan experiments, with the difference that the cultures were grown 24 h and the cells were resuspended in PBS at OD600 = 0.8–1.0. One hundred microliter cell samples were transferred in black polystyrene 96-well microplates, and fluorescence485/535 (F485/535) was measured. The OD600 of the culture samples in the wells was assessed by Biophotometer (Eppendorf) reading. The sfGFP activity was expressed in arbitrary units (AU) as the ratio F485/535/OD600. Pseudomonas aeruginosa cell images were acquired with a Leica fluorescence microscope (Leica AG) equipped with a digital Leica DC150 camera and a 100× oil immersion objective.
Analysis of the expression of HA-tagged ptxS and PA5194 variants
Cultures of PAO1 expressing PA5194-HA or PtxS-HA from pGM931 derivatives were grown overnight in LD with carbenicillin at 28°C and diluted to OD600 = 0.2 in three flasks containing M9 medium supplemented with carbenicillin, 0.4% glucose, and 0.2% arabinose. The flasks were incubated at 25°C–37°C–40°C. Twenty-five and 3 mL samples were taken after 1 and 2 h for protein and RNA extraction, respectively. RNA was extracted by the RNAsnap procedure (Stead et al. 2012). Protein extraction was performed by resuspending the cell pellet in 2× SDS buffer (100 mM Tris–HCl pH 6.8, 200 mM DTT, 2% SDS, 0.2% bromophenol blue, and 20% glycerol) and boiling the cells for 15 min. The cells were then centrifuged 10 min at 13,000 rpm, the supernatant was recovered, and an aliquot was analyzed by 10% SDS-PAGE. PageRuler Plus Prestained Protein Ladder weight markers (ThermoScientific) were used as size references. For immunological detection of proteins, the gels were blotted onto a Hybond ECL (GE Healthcare Life Sciences) sheet and incubated with monoclonal anti-HA (12CA5; Roche) or polyclonal anti-S1 (kindly provided by U. Bläsi) antibodies (Sambrook et al. 1989).
Northern blotting and primer extension
Procedures for RNA extraction, Northern blot analysis, in vitro transcription with T7 RNA polymerase and 5′ end labeling of oligonucleotides with T4 polynucleotide kinase and [γ32P] ATP were previously described (Dehò et al. 1992; Briani et al. 2007). Extraction of RNA from P. aeruginosa cultures was performed by phenol–chloroform treatment of cell lysates (Dehò et al. 1992) or by means of the RNeasy Mini Kit (Qiagen), according to the manufacturer's instructions. After blotting, the filters were stained by incubating them for 2 min in 0.02% methylene blue and 0.3 M sodium acetate, pH 5.5. Oligonucleotide probes used for Northern blotting were 1396 (16S rRNA); 2865 (bgaB); 2871 (sfGFP); HA (Delvillani et al. 2011). The riboprobes PTXS and PA5194 were obtained by in vitro transcription with T7 RNA polymerase and [α32P] CTP of DNA fragments obtained by PCR amplification of pPtxS-HA (oligonucleotides 2976–2811) and pPA5194-HA (oligonucleotides 3150–3006), respectively. The 5′ end of the PA5194 mRNA was determined by primer extension as previously described (Forti et al. 1995) with the radiolabeled oligonucleotide 3003 on 30 µg of RNA extracted from exponential cultures of PAO1 grown at 25°C and 37°C. The same oligonucleotide was used for Sanger-sequencing of construct 3. Images and densitometric analysis of Northern blots and primer extension gels were obtained by phosphorimaging using ImageQuant software (Molecular Dynamics).
RNA structural probing
The RNAs for structural probing were synthesized by in vitro transcription of proper PCR fragments with T7 RNA polymerase. The DNA templates were obtained by PCR on PAO1 genomic DNA with the oligonucleotides 2909–2910 (ptxS) and 3040–3041 (PA5194). The probes were radiolabeled at the 5′ end by dephosphorylation with alkaline phosphatase followed by phosphorylation with T4 polynucleotide kinase with [γ-32P] ATP as a phosphate donor. After labeling, the probes were run on a denaturing polyacrylamide gel and the bands corresponding to the full-length RNAs were gel-eluted. For structural analysis, the probes were denatured by incubation at 95°C for 5 min; 2 × 104 cpm samples were then preincubated in reaction buffer (20 mM Tris–HCl, 2 mM Mg2Cl, 100 mM NaCl) and 0.66 μg of tRNA mix at 25°C, 37°C, or 42°C for 10 min. One microliter of RNase T1 (final concentration, 66 pg/µL), RNase A (0.04 pg/µL), or lead acetate (0.1 mM) were added to the samples (final volume, 5 µL) and incubation at different temperatures was performed for 5 min before stopping the reactions with 5 μL of RNA loading dye (2 mg/mL xylene cyanol and bromophenol blue, 10 mM EDTA in formamide). A denaturing RNase T1 degradation was performed by incubating each probe (105 cpm) and 0.6 mg/mL tRNA in 1× RNA seq buffer (Ambion) at 50°C for 5 min. RNase T1 (final concentration 0.166 ng/µL) was added and the samples were incubated at 37°C for 5 min before adding 10 μL of RNA loading dye. The RNA ladder was obtained by digesting 2.5 × 105 cpm of probe PA5194 at 95°C for 10 min with 12 μL of Alkaline Buffer (Ambion) and 6 μg of tRNA mix in a final volume of 15 μL. The samples were run on an 8% denaturing polyacrylamide gel. The gel was dried and analyzed by phosphorimaging using ImageQuant software (Molecular Dynamics).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Daniele Daffonchio and Aurora Rizzi (Università degli Studi di Milano) for the pZR80-2 aadA::GFP plasmid, Franz Narberhaus and Stefanie Krajewski (Ruhr-Universität Bochum—Germany) for the pBAD2_bgaB plasmid and for helpful discussions, and Colin P. Corcoran and Jörg Vogel (University of Würzburg, Germany) for the pXG-10SF plasmid. This work was supported by the German Research Council (DFG) through SFB796/C4 to C.B. and by Fondazione Cariplo (grant 2010.0653). B.S. was supported by a fellowship of the Italian Cystic Fibrosis Research Foundation (grant FFC#8/2013 to F.B., sponsored by the FFC Delegation of Montebelluna “La bottega delle donne”).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.044354.114.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Altuvia S, Kornitzer D, Teff D, Oppenheim AB 1989. Alternative mRNA structures of the cIII gene of bacteriophage λ determine the rate of its translation initiation. J Mol Biol 210: 265–280. [DOI] [PubMed] [Google Scholar]
- Baker E, Clark N, Brennan A, Fisher D, Gyi K, Hodson M, Philips B, Baines D, Wood D 2007. Hyperglycemia and cystic fibrosis alter respiratory fluid glucose concentrations estimated by breath condensate analysis. J Appl Physiol (1985) 102: 1969–1975. [DOI] [PubMed] [Google Scholar]
- Bertrand KP, Postle K, Wray LV Jr, Reznikoff WS 1983. Overlapping divergent promoters control expression of Tn10 tetracycline resistance. Gene 23: 149–156. [DOI] [PubMed] [Google Scholar]
- Bradford MM 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Briani F, Ghisotti D, Dehò G 2000. Antisense RNA-dependent transcription termination sites that modulate lysogenic development of satellite phage P4. Mol Microbiol 36: 1124–1134. [DOI] [PubMed] [Google Scholar]
- Briani F, Del Favero M, Capizzuto R, Consonni C, Zangrossi S, Greco C, De Gioia L, Tortora P, Dehò G 2007. Genetic analysis of polynucleotide phosphorylase structure and functions. Biochimie 89: 145–157. [DOI] [PubMed] [Google Scholar]
- Carzaniga T, Briani F, Zangrossi S, Merlino G, Marchi P, Dehò G 2009. Autogenous regulation of Escherichia coli polynucleotide phosphorylase expression revisited. J Bacteriol 191: 1738–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carzaniga T, Antoniani D, Dehò G, Briani F, Landini P 2012. The RNA processing enzyme polynucleotide phosphorylase negatively controls biofilm formation by repressing poly-N-acetylglucosamine (PNAG) production in Escherichia coli C. BMC Microbiol 12: 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Maris C, Allain FH, Narberhaus F 2006. Molecular basis for temperature sensing by an RNA thermometer. EMBO J 25: 2487–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chursov A, Kopetzky S, Bocharov G, Frishman D, Shneider A 2013. RNAtips: analysis of temperature-induced changes of RNA secondary structure. Nucleic Acids Res 41: W486–W491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimdins A, Klinkert B, Aschke-Sonnenborn U, Kaiser FM, Kortmann J, Narberhaus F 2014. Translational control of small heat shock genes in mesophilic and thermophilic cyanobacteria by RNA thermometers. RNA Biol 11: 594–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmer JA, Hamood AN 1998. Characterization of ptxS, a Pseudomonas aeruginosa gene which interferes with the effect of the exotoxin A positive regulatory gene, ptxR. Mol Gen Genet 258: 250–259. [DOI] [PubMed] [Google Scholar]
- Corcoran CP, Podkaminski D, Papenfort K, Urban JH, Hinton JC, Vogel J 2012. Superfolder GFP reporters validate diverse new mRNA targets of the classic porin regulator, MicF RNA. Mol Microbiol 84: 428–445. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Percival RS, Devine D, Darveau RP, Coats SR, Rangarajan M, Tarelli E, Marsh PD 2011. Temperature-dependent modulation of Porphyromonas gingivalis lipid A structure and interaction with the innate host defenses. Infect Immun 79: 1187–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daddaoua A, Fillet S, Fernandez M, Udaondo Z, Krell T, Ramos JL 2012. Genes for carbon metabolism and the ToxA virulence factor in Pseudomonas aeruginosa are regulated through molecular interactions of PtxR and PtxS. PLoS One 7: e39390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darty K, Denise A, Ponty Y 2009. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics 25: 1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci 97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehò G, Zangrossi S, Sabbattini P, Sironi G, Ghisotti D 1992. Bacteriophage P4 immunity controlled by small RNAs via transcription termination. Mol Microbiol 6: 3415–3425. [DOI] [PubMed] [Google Scholar]
- Delvillani F, Papiani G, Dehò G, Briani F 2011. S1 ribosomal protein and the interplay between translation and mRNA decay. Nucleic Acids Res 39: 7702–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Chan CY, Lawrence CE 2005. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA 11: 1157–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh T, Bernhardt TG 2011. Using superfolder green fluorescent protein for periplasmic protein localization studies. J Bacteriol 193: 4984–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotsch A, Eckweiler D, Schniederjans M, Zimmermann A, Jensen V, Scharfe M, Geffers R, Haussler S 2012. The Pseudomonas aeruginosa transcriptome in planktonic cultures and static biofilms using RNA sequencing. PLoS One 7: e31092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus M 2009. Killer and protective ribosomes. Prog Mol Biol Transl Sci 85: 423–466. [DOI] [PubMed] [Google Scholar]
- Driscoll JA, Brody SL, Kollef MH 2007. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67: 351–368. [DOI] [PubMed] [Google Scholar]
- Ernst R, Adams K, Moskowitz S, Kraig G, Kawasaki K, Stead C, Trent M, Miller S 2006. The Pseudomonas aeruginosa lipid A deacylase: selection for expression and loss within the cystic fibrosis airway. J Bacteriol 188: 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forti F, Sabbattini P, Sironi G, Zangrossi S, Dehò G, Ghisotti D 1995. Immunity determinant of phage-plasmid P4 is a short processed RNA. J Mol Biol 249: 869–878. [DOI] [PubMed] [Google Scholar]
- Garnett J, Baker E, Baines D 2012. Sweet talk: insights into the nature and importance of glucose transport in lung epithelium. Eur Respir J 40: 1269–1276. [DOI] [PubMed] [Google Scholar]
- Gauger T, Weihs F, Mayer S, Krismer B, Liese J, Kull M, Bertram R 2012. Intracellular monitoring of target protein production in Staphylococcus aureus by peptide tag-induced reporter fluorescence. Microb Biotechnol 5: 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgi C, Buerger J, Hillen W, Berens C 2012. Promoter strength driving TetR determines the regulatory properties of Tet-controlled expression systems. PLoS One 7: e41620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliodori AM, Di Pietro F, Marzi S, Masquida B, Wagner R, Romby P, Gualerzi CO, Pon CL 2010. The cspA mRNA is a thermosensor that modulates translation of the cold-shock protein CspA. Mol Cell 37: 21–33. [DOI] [PubMed] [Google Scholar]
- Goeke D, Kaspar D, Stoeckle C, Grubmuller S, Berens C, Klotzsche M, Hillen W 2012. Short peptides act as inducers, anti-inducers and corepressors of Tet repressor. J Mol Biol 416: 33–45. [DOI] [PubMed] [Google Scholar]
- Goldberg JB, Ohman DE 1984. Cloning and expression in Pseudomonas aeruginosa of a gene involved in the production of alginate. J Bacteriol 158: 1115–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SG, Jessee J, Bloom FR, Hanahan D 1990. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci 87: 4645–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guisbert E, Yura T, Rhodius VA, Gross CA 2008. Convergence of molecular, modeling, and systems approaches for an understanding of the Escherichia coli heat shock response. Microbiol Mol Biol Rev 72: 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamood AN, Colmer JA, Ochsner UA, Vasil ML 1996. Isolation and characterization of a Pseudomonas aeruginosa gene, ptxR, which positively regulates exotoxin A production. Mol Microbiol 21: 97–110. [DOI] [PubMed] [Google Scholar]
- Hirata H, Negoro S, Okada H 1984. Molecular basis of isozyme formation of β-galactosidases in Bacillus stearothermophilus: isolation of two β-galactosidase genes, bgaA and bgaB. J Bacteriol 160: 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson J, Mandin P, Renzoni A, Chiaruttini C, Springer M, Cossart P 2002. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110: 551–561. [DOI] [PubMed] [Google Scholar]
- John CM, Liu M, Phillips NJ, Yang Z, Funk CR, Zimmerman LI, Griffiss JM, Stein DC, Jarvis GA 2012. Lack of lipid A pyrophosphorylation and functional lptA reduces inflammation by Neisseria commensals. Infect Immun 80: 4014–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkert B, Narberhaus F 2009. Microbial thermosensors. Cell Mol Life Sci 66: 2661–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkert B, Cimdins A, Gaubig LC, Rossmanith J, Aschke-Sonnenborn U, Narberhaus F 2012. Thermogenetic tools to monitor temperature-dependent gene expression in bacteria. J Biotechnol 160: 55–63. [DOI] [PubMed] [Google Scholar]
- Kong Q, Six DA, Roland KL, Liu Q, Gu L, Reynolds CM, Wang X, Raetz CR, Curtiss R III 2011. Salmonella synthesizing 1-dephosphorylated [corrected] lipopolysaccharide exhibits low endotoxic activity while retaining its immunogenicity. J Immunol 187: 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel ME, Tilly K 2000. Temperature-regulated expression of bacterial virulence genes. Microbes Infect 2: 157–166. [DOI] [PubMed] [Google Scholar]
- Kortmann J, Narberhaus F 2012. Bacterial RNA thermometers: molecular zippers and switches. Nat Rev Microbiol 10: 255–265. [DOI] [PubMed] [Google Scholar]
- Kortmann J, Sczodrok S, Rinnenthal J, Schwalbe H, Narberhaus F 2011. Translation on demand by a simple RNA-based thermosensor. Nucleic Acids Res 39: 2855–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski SS, Nagel M, Narberhaus F 2013. Short ROSE-like RNA thermometers control IbpA synthesis in Pseudomonas species. PLoS One 8: e65168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederberg J 1951. Streptomycin resistance: a genetically recessive mutation. J Bacteriol 61: 549–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DG, Urbach JM, Wu G, Liberati NT, Feinbaum RL, Miyata S, Diggins LT, He J, Saucier M, Deziel E, et al. 2006. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol 7: R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessl M, Balzer D, Lurz R, Waters VL, Guiney DG, Lanka E 1992. Dissection of IncP conjugative plasmid transfer: definition of the transfer region Tra2 by mobilization of the Tra1 region in trans. J Bacteriol 174: 2493–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- Montague M, Hutchison C 2000. Gene content phylogeny of herpesviruses. Proc Natl Acad Sci 97: 5334–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Kanemori M, Yanagi H, Yura T 1999a. Heat-induced synthesis of σ32 in Escherichia coli: structural and functional dissection of rpoH mRNA secondary structure. J Bacteriol 181: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita MT, Tanaka Y, Kodama TS, Kyogoku Y, Yanagi H, Yura T 1999b. Translational induction of heat shock transcription factor σ32: evidence for a built-in RNA thermosensor. Genes Dev 13: 655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narberhaus F, Kaser R, Nocker A, Hennecke H 1998. A novel DNA element that controls bacterial heat shock gene expression. Mol Microbiol 28: 315–323. [DOI] [PubMed] [Google Scholar]
- Needham BD, Trent MS 2013. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol 11: 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert J, Karcher D, Bock R 2008. Design of simple synthetic RNA thermometers for temperature-controlled gene expression in Escherichia coli. Nucleic Acids Res 36: e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocker A, Hausherr T, Balsiger S, Krstulovic NP, Hennecke H, Narberhaus F 2001. A mRNA-based thermosensor controls expression of rhizobial heat shock genes. Nucleic Acids Res 29: 4800–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver DB, Beckwith J 1981. E. coli mutant pleiotropically defective in the export of secreted proteins. Cell 25: 765–772. [DOI] [PubMed] [Google Scholar]
- Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS 2006. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24: 79–88. [DOI] [PubMed] [Google Scholar]
- Phadtare S 2004. Recent developments in bacterial cold-shock response. Curr Issues Mol Biol 6: 125–136. [PubMed] [Google Scholar]
- Qiu D, Damron FH, Mima T, Schweizer HP, Yu HD 2008. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl Environ Microbiol 74: 7422–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268: 1899–1902. [DOI] [PubMed] [Google Scholar]
- Regonesi ME, Briani F, Ghetta A, Zangrossi S, Ghisotti D, Tortora P, Dehò G 2004. A mutation in polynucleotide phosphorylase from Escherichia coli impairing RNA binding and degradosome stability. Nucleic Acids Res 32: 1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi A, Pontiroli A, Brusetti L, Borin S, Sorlini C, Abruzzese A, Sacchi GA, Vogel TM, Simonet P, Bazzicalupo M, et al. 2008. Strategy for in situ detection of natural transformation-based horizontal gene transfer events. Appl Environ Microbiol 74: 1250–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T 1989. Molecular cloning. A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Sasaki I, Bertani G 1965. Growth abnormalities in Hfr derivatives of Escherichia coli strain C. J Gen Microbiol 40: 365–376. [DOI] [PubMed] [Google Scholar]
- Silby MW, Winstanley C, Godfrey SA, Levy SB, Jackson RW 2011. Pseudomonas genomes: diverse and adaptable. FEMS Microbiol Rev 35: 652–680. [DOI] [PubMed] [Google Scholar]
- Stead MB, Agrawal A, Bowden KE, Nasir R, Mohanty BK, Meagher RB, Kushner SR 2012. RNAsnap: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res 40: e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckle C, Klotzsche M, Hillen W 2012. Protein expression can be monitored in yeast by peptide-mediated induction of TetR-controlled gene expression. J Biotechnol 161: 265–268. [DOI] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, et al. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406: 959–964. [DOI] [PubMed] [Google Scholar]
- Sukhodolets MV, Garges S 2003. Interaction of Escherichia coli RNA polymerase with the ribosomal protein S1 and the Sm-like ATPase Hfq. Biochemistry 42: 8022–8034. [DOI] [PubMed] [Google Scholar]
- Swanson BL, Hamood AN 2000. Autoregulation of the Pseudomonas aeruginosa protein PtxS occurs through a specific operator site within the ptxS upstream region. J Bacteriol 182: 4366–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson BL, Colmer JA, Hamood AN 1999. The Pseudomonas aeruginosa exotoxin A regulatory gene, ptxS: evidence for negative autoregulation. J Bacteriol 181: 4890–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldminghaus T, Fippinger A, Alfsmann J, Narberhaus F 2005. RNA thermometers are common in α- and γ-proteobacteria. Biol Chem 386: 1279–1286. [DOI] [PubMed] [Google Scholar]
- Waldminghaus T, Gaubig LC, Narberhaus F 2007a. Genome-wide bioinformatic prediction and experimental evaluation of potential RNA thermometers. Mol Genet Genomics 278: 555–564. [DOI] [PubMed] [Google Scholar]
- Waldminghaus T, Heidrich N, Brantl S, Narberhaus F 2007b. FourU: a novel type of RNA thermometer in Salmonella. Mol Microbiol 65: 413–424. [DOI] [PubMed] [Google Scholar]
- Whiting P, Midgley M, Dawes E 1976. The regulation of transport of glucose, gluconate and 2-oxogluconate and of glucose catabolism in Pseudomonas aeruginosa. Biochem J 154: 659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtzel O, Yoder-Himes DR, Han K, Dandekar AA, Edelheit S, Greenberg EP, Sorek R, Lory S 2012. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog 8: e1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanisch-Perron C, Vieira J, Messing J 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33: 103–119. [DOI] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.