

Abstract
Background
Protein kinase C (PKC) is a major regulator of platelet function and secretion. The underlying molecular pathway from PKC to secretion, however, is poorly understood. By a proteomics screen we identified the guanine nucleotide exchange factor cytohesin-2 as a candidate PKC substrate.
Objectives
We aimed to validate cytohesin-2 as a PKC substrate in platelets and to determine its role in granule secretion and other platelet responses.
Methods and results
Immunoprecipitation was performed with a phosphoserine PKC substrate antibody followed by mass spectrometry, leading to the identification of cytohesin-2. By western blotting we showed that different agonists induced cytohesin-2 phosphorylation by PKC. Protein function was investigated using a pharmacological approach. The cytohesin inhibitor SecinH3 significantly enhanced platelet dense granule secretion and aggregation, as measured by lumi-aggregometry. Flow cytometry data indicate that α-granule release and integrin αIIbβ3 activation were not affected by cytohesin-2 inhibition. Lysosome secretion was assessed by a colorimetric assay and was also unchanged. As shown by western blotting, ARF6 interacted with cytohesin-2 and was present in an active GTP-bound form under basal conditions. Upon platelet stimulation, this interaction was largely lost and ARF6 activation decreased, both of which could be rescued by PKC inhibition.
Conclusions
Cytohesin-2 constitutively suppresses platelet dense granule secretion and aggregation by keeping ARF6 in a GTP-bound state. PKC-mediated phosphorylation of cytohesin-2 relieves this inhibitory effect, thereby promoting platelet secretion and aggregation.
Keywords: ADP-ribosylation factor 6, cytohesin-2, platelets, protein kinase C, secretion
Introduction
Platelet function and secretion are critically regulated by protein kinase C (PKC), which is activated downstream of a multitude of cell surface receptors 1. The conventional isoforms PKCα and PKCβ are the major positive regulators of platelet function in human and mouse platelets 2,3. PKCα knockout mice show reduced thrombus formation and platelet secretion, suggesting PKCα to be critically involved in these processes 4. A similar role has been reported for the highly related PKCβ 5. The mechanism by which PKC regulates platelet secretion is unclear. When activated, conventional PKC isozymes preferentially phosphorylate substrates containing serine or threonine within a defined consensus sequence, with arginine or lysine at the −3, −2 and +2 positions, and hydrophobic amino acids at position +1 6. A few PKC substrates in platelets are well characterized, such as pleckstrin 7 and MARCKS 8. More recently, we showed that SHP-1 is phosphorylated by PKC 9 and that GSK3 and PDE3A are regulated in a PKC-dependent manner 10,11. However, the majority of proteins phosphorylated by conventional PKC isoforms, and their relationship to platelet secretion, are presently undefined.
Using an anti-phosphopeptide antibody-based approach followed by a proteomics screen we identified cytohesin-2, also known as ARF nucleotide-binding site opener (ARNO), as a candidate conventional PKC substrate in platelets. Cytohesin-2 is a guanine-nucleotide exchange factor (GEF) for the small GTPase called ADP-ribosylation factor 6 (ARF6). The conserved SEC7 domain catalyses GDP release from, and GTP binding to, ARF6, resulting in its activation 12. ARF6 regulates vesicle trafficking 13 and has been shown to be located at the membrane and to regulate exocytosis in chromaffin cells 14, adipocytes 15 and neuroendocrine cells 16,17. More importantly, ARF6 has been reported to be involved in collagen-induced platelet aggregation and spreading, and ARF6-GTP levels decrease upon stimulation of platelet activation in a PKC-dependent manner 18,19. In cell lines, PKC phosphorylates Ser392 of cytohesin-2, causing its translocation to the cytosol 20. Anti-cytohesin-2 antibodies were reported to inhibit catecholamine secretion in chromaffin cells 21 and cytohesin-2 co-localized with proteins involved in exocytosis in neuroendocrine cells 22. The exact mechanism of cytohesin-2/ARF6 regulation in platelets, however, has not been established.
The aims of our study were to elucidate how PKC regulates the cytohesin-2/ARF6 pathway and to examine the role of this pathway in granule secretion and function. Here we show that cytohesin-2 has a negative regulatory role in resting platelets, by keeping ARF6 in an activated GTP-bound state, which suppresses granule secretion and aggregation. Upon platelet activation, phosphorylation of cytohesin-2 by PKC causes it to dissociate from ARF6, which is then converted into its inactive GDP-bound form. This results in the relief of platelet inhibition, allowing granule secretion and aggregation to occur.
Materials and methods
Materials
The phospho-(Ser) PKC substrate (pSer PKC substrate) antibody was from Cell Signaling Technology (Danvers, MA, USA). The cytohesin-2 western blot antibody, ARF6 antibody and GAPDH antibody were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The cytohesin-2 immunoprecipitation antibody was from Thermo Scientific (Loughborough, Leicestershire, UK). Horseradish peroxidase (HRP)-conjugated donkey anti-rabbit, anti-mouse and anti-goat secondary antibodies were from Jackson ImmunoResearch Laboratories (Newmarket, UK). The PKC inhibitor bisindolylmaleimide (BIM) IX (Ro 31-8220) and the inactive analogue BIM V were from Calbiochem (Nottingham, UK). The PKC inhibitors BIM I (GF 109203X), Go 6983 and ruboxistaurin (LY 333531) and the cytohesin-2 inhibitor SecinH3 were from Tocris (Bristol, UK). The GST-GGA3 construct was a generous gift from Professor Sidney Whiteheart (University of Kentucky, USA). Cross-linked collagen-related peptide (CRP) was synthesized by Professor Richard Farndale (University of Cambridge, UK). PE-P-selectin and FITC-PAC1 antibodies were from Emfret Analytics (Eibelstadt, Germany). Luciferin-luciferase was from Chronolog (Labmedics, Stockport, UK). NuPAGE LDS sample buffer was from Invitrogen (Carlsbad, CA, USA). ECL reagent was from GE Healthcare (Amersham, UK). All other reagents were from Sigma-Aldrich (Poole, UK).
Human platelet preparation
Human blood was drawn from healthy volunteers, under local ethics committee agreement and after fully informed consent, and washed platelets were prepared as described previously 23. In brief, blood anticoagulated with 0.4% (v/v) trisodium citrate and acidified with 16% (v/v) acidic citrate dextrose (85 mm trisodium citrate, 71 mm citric acid, 111 mm glucose) was centrifuged at 180 ×g for 17 min. The platelet-rich plasma was subsequently centrifuged at 650 ×g for 10 min in the presence of 10 μm indomethacin and 0.02 U mL−1 apyrase. Platelets were resuspended to the required density in HEPES-Tyrode's buffer pH 7.2 (10 mm HEPES, 145 mm NaCl, 3 mm KCl, 0.5 mm Na2HPO4, 1 mm MgSO4), modified with 0.1% (w/v) glucose, 10 μm indomethacin and 0.02 U mL−1 apyrase. Platelets for use in immunoprecipitation (IP) studies were double washed.
Mouse platelet preparation
A colony of PKCα knockout (PKCα−/−) mice was kindly provided by Professor J. Molkentin (Cincinnati Children's Hospital, USA). Littermate PKCα wild-type (WT) mice were used as controls. Animals were sacrificed by CO2 asphyxiation and blood was drawn by cardiac puncture under terminal anesthesia into 0.4% trisodium citrate. Blood was acidified with 20% ACD, diluted with 500 μL of modified HEPES-Tyrode's buffer pH 7.2, and centrifuged at 180 ×g for 8 min. PRP was removed and platelets were isolated by centrifugation at 520 ×g for 10 min in the presence of 10 μm indomethacin and 0.02 U mL−1 apyrase. Pelleted platelets were resuspended to the required density in modified HEPES-Tyrode's buffer pH 7.2.
Platelet stimulation and lysis
Washed human platelets (4 × 108 mL−1) or mouse platelets (2 × 108 mL−1) were incubated for 15 min with the indicated inhibitor or 0.2% dimethylsulfoxide (DMSO) vehicle. Next, platelets were stimulated at 30 °C under non-stirring conditions. For IP, co-IP and ARF-GTP pull down studies, platelets were lysed with an equal volume of ice-cold 2× RIPA buffer pH 7.4 (25 mm HEPES, 200 mm NaCl, 1 mm EDTA, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 20 mm sodium β-glycerol phosphate, 10 mm sodium pyrophosphate, 1 mm benzamidine), NP40 buffer pH 7.5 (25 mm HEPES, 120 mm NaCl, 1 mm EDTA, 1% NP40, 20 mm sodium β-glycerol phosphate, 10 mm sodium pyrophosphate, 1 mm benzamidine) or ARF buffer pH 7.5 (50 mm Tris, 150 mm NaCl, 1% Triton x-100, 0.5% sodium deoxycholate, 0.1% SDS, 10 mm MgCl2), respectively, to which protease inhibitors were added. Cell extracts were centrifuged at 10 000 ×g at 4 °C, and the supernatant was taken for subsequent analysis. Alternatively, for western blotting (whole cell lysate), platelets were lysed in 4× NuPAGE LDS sample buffer, which was supplemented with 50 mm dithiothreitol (DTT).
IP and ARF-GTP pull down
Protein A and G sepharose beads were used for IP studies with rabbit and mouse antibodies, respectively. The ARF activation was assessed as described previously 18. In brief, GST-GGA3 fusion proteins, which specifically bind ARF-GTP, coupled to glutathione-agarose beads were prepared by E. Aitken in our laboratory. 250 μL platelet lysate was incubated overnight under constant rotation at 4 °C with 10 μL beads and, in the case of IP, 10 μL antibody. Beads were washed three times in 1× lysis buffer and bound proteins were eluted in 2× NuPAGE LDS sample buffer, which was supplemented with 50 mm DTT at 70 °C for 10 min.
Electrophoresis and immunoblotting
Samples were separated by SDS-PAGE on 10% polyacrylamide gels. Proteins were transferred at 100 V for 1 h to PVDF membranes in transfer buffer (22.5 mm Tris, 172.5 mm glycine, 20% methanol). The membranes were blocked using 1× Sigma blocking buffer or, in the case of ARF6 blotting, 1% milk in Tris-buffered saline with Tween (20 mm Tris pH 7.6, 137 mm NaCl, 0.1% Tween). Blots were probed with primary and horseradish peroxidase-conjugated secondary antibodies. Proteins were detected using ECL reagents. Membranes were stripped in stripping buffer pH 6.8 (62.5 mm Tris, 2% SDS, 100 mm 2-mercaptoethanol) and reprobed as appropriate.
Platelet aggregation and ATP secretion assay
Platelet aggregation and ATP secretion from dense granules were monitored simultaneously at 37 °C under constant stirring. Platelets (2 × 108 mL−1) were treated for 15 min with vehicle (0.2% DMSO) or 15 μm SecinH3. Before stimulation, platelets were incubated with 5 μL luciferase-luciferin reagent at RT. Light transmission and luminescence were recorded in a Chronolog 590-2A aggregometer.
Flow cytometry
Platelets (2 × 107 mL−1) were treated with vehicle or 15 μm SecinH3 for 15 min before stimulation in the presence 1 : 12 PE-conjugated anti-P-selectin antibody and 1 : 6 FITC-conjugated PAC-1 antibody, to assess α-granule secretion and αIIbβ3 integrin activation, respectively. Platelets were stimulated for 10 min at RT prior to fixation in 1% paraformaldehyde for 30 min. Fluorescent analysis was conducted by flow cytometry on a FACSCalibur flow cytometer.
Measurement of β-hexosaminidase release
Platelets (2 × 108 mL−1) were treated for 15 min with vehicle or 15 μm SecinH3 prior to stimulation for 15 min and centrifuged at 650 ×g for 5 min. Supernatants were taken and incubated with 20 μL of substrate (5 mm nitrophenyl-acetyl-glucosaminide) in citrate phosphate buffer pH 4.2 (0.2 m Na2HPO4, 0.1 m citric acid) in a 96-well plate. The reaction was stopped by the addition of 200 μL of 0.1 m NaOH. β-hexosaminidase release from lysosomes was assessed by measuring samples at 405 nm using a microplate spectrophotometer.
Statistics
Data were analyzed using GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA, USA). All data are presented as the mean ± SEM of the indicated number of independent observations and P-values were calculated using Student's t-test. Differences between concentration-response curves were determined by F test, where the null hypothesis states that both datasets can be fitted using the same parameters.
Results
PKC phosphorylates cytohesin-2 in platelets
We used the commercially available phospho-(Ser) PKC substrate (pSer PKC substrate) antibody, raised against a set of peptides that correspond to conventional PKC consensus phosphorylation sites, to pull down potential non-isoform-specific conventional PKC substrates in thrombin-stimulated platelets. One of the proteins identified by mass spectrometry was cytohesin-2, a GEF for ARF6. Interestingly, Serine 392, which is located at the C-terminus of cytohesin-2, lies perfectly within the consensus phospho-motif of the pSer PKC substrate antibody (Fig.1A). First, we validated that PKC phosphorylates cytohesin-2 in platelets upon cellular activation by immunoblotting pSer PKC substrate antibody immunoprecipitations for cytohesin-2. Cytohesin-2 was phosphorylated in platelets after stimulation with thrombin (Fig.1B), collagen-related peptide (CRP) (Fig.1D) and the thromboxane A2 mimetic U46619 (Fig.1F), reaching a maximum at 2 min. A low agonist concentration was also able to induce cytohesin-2 phosphorylation (Figure S1). To address whether this phosphorylation event was dependent on PKC, platelets were pre-treated with the broad-spectrum PKC inhibitor BIM IX. PKC inhibition blocks platelet dense granule secretion, and thereby also the release of ADP, which mediates autocrine P2Y12 signaling. To avoid any potential side-effects resulting from the blockage of PKC-mediated autocrine-produced ADP, platelets were stimulated in the presence of ADP. Incubation of platelets with ADP alone did not cause cytohesin-2 phosphorylation (Figure S1). Phosphorylation of cytohesin-2 induced by thrombin (Fig.1C), CRP (Fig.1E) and U46619 (Fig.1G) was completely abolished by PKC inhibition.
Figure 1.

PKC phosphorylates cytohesin-2 upon platelet activation. The C-terminus of cytohesin-2 (CYH2) contains the phospho-motif recognized by the pSer PKC substrate antibody (A). X, any amino acid. Hyd, any hydrophobic amino acid. Washed platelets (4 × 108 mL−1) were stimulated with 0.2 U mL−1 α-thrombin (αT) (B), 5 μg mL−1 CRP (D) or 10 μm U46619 (F) and lysed at the indicated time-points. Alternatively, platelets were treated for 15 min with 0.2% DMSO vehicle (Veh) or the PKC inhibitor BIM IX (2 μm) prior to stimulation with the same agonists in the presence of 10 μm ADP (C, E, G). Clarified whole cell lysates (input) and immunoprecipitations (IP) using the pSer PKC substrate antibody were immunoblotted for CYH2. The arrows (➤) indicate the 47 kDa CYH2 band and the asterisk (*) indicates a non-specific band. Results (B–G) are representative of at least three independent experiments.
In addition to BIM IX, other pharmacological inhibitors of PKC were used to assess phosphorylation of cytohesin-2. The broad-spectrum PKC inhibitors BIM I and Go 6983, as well as the PKCα/β selective inhibitor ruboxistaurin 24, blocked phosphorylation of cytohesin-2 upon stimulation (Fig.2A). These results suggest that cytohesin-2 phosphorylation is mediated through the conventional PKC isoform PKCα/β. Moreover, the inactive analogue (BIM V) did not affect cytohesin-2 phosphorylation, thereby serving as a control for non-specific effects. In addition, to further explore which PKC isoform is responsible for the phosphorylation of cytohesin-2, we performed similar experiments using PKCα knockout (PKCα−/−) mouse platelets (Fig.2B). Cytohesin-2 phosphorylation was, however, not affected in PKCα−/− platelets, but was blocked by ruboxistaurin, suggesting that PKCβ may be principally responsible for the phosphorylation of cytohesin-2.
Figure 2.
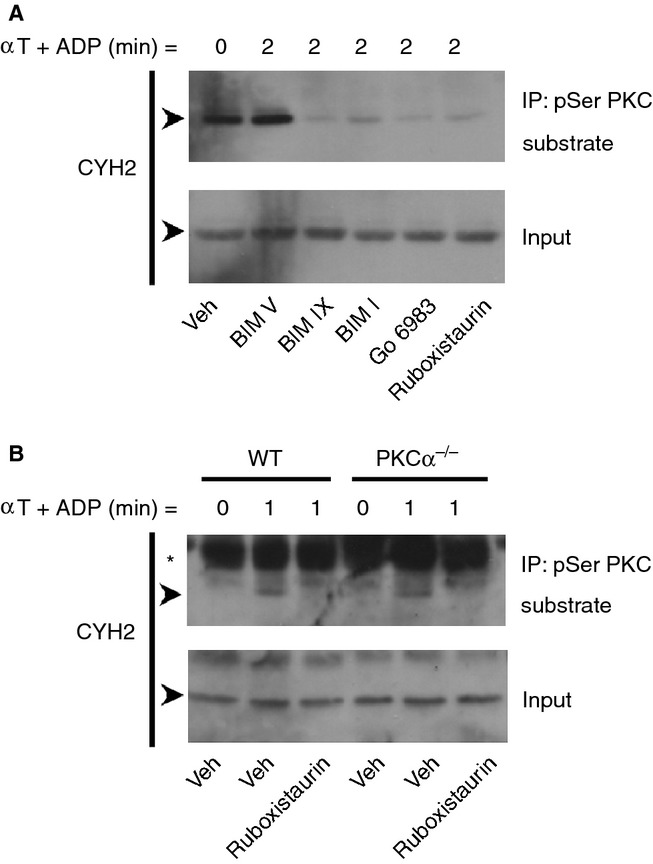
Conventional PKC isoforms mediate the phosphorylation of cytohesin-2. Washed human platelets (4 × 108 mL−1) were treated for 15 min with 0.2% DMSO vehicle (Veh), the control BIM compound BIM V (10 μm), the broad-spectrum inhibitors PKC inhibitor BIM IX (2 μm), BIM I (5 μm) and Go 6983 (10 μm), or the PKCα/β selective inhibitor ruboxistaurin (10 μm) (A). Alternatively, mouse wild-type (WT) and PKCα knockout (PKCα−/−) mouse platelets (2 × 108 mL−1) were used (B). Platelets were stimulated with 0.2 U mL−1 α-thrombin (αT) in the presence of 10 μm ADP and lysed at the indicated time-points. Clarified whole cell lysates and immunoprecipitations using the pSer PKC substrate antibody were immunoblotted for cytohesin-2 (CYH2). The arrows (➤) indicate the 47 kDa CYH2 band and the asterisk (*) indicates a non-specific band. Results are representative of at least three independent experiments.
Cytohesin-2 inhibition enhances dense granule secretion and aggregation
To investigate the role of cytohesin-2 in platelet function, we used the pharmacological ARF-GEF inhibitor SecinH3, which displays selectivity for the cytohesin family, mainly cytohesin-2 25. SecinH3 was shown to potently inhibit receptor-mediated ARF6 activation in multiple cells, including platelets 26–29.
We incubated platelets with SecinH3 prior to stimulation with thrombin and CRP to analyze the function of the cytohesin-2/ARF6 pathway. Platelet luminometry and optical aggregometry were performed to assess ATP secretion from dense granules and aggregation responses, respectively. Platelets were stimulated with a low agonist concentration to be able to detect both negative and positive functional changes. SecinH3 did not stimulate ATP secretion or aggregation by itself (data not shown). ATP secretion induced by 0.05 U mL−1 (Fig.3Ai,ii), 0.075 U mL−1 (Fig.3Ai,iii) and 0.2 U mL−1 (Fig.3Ai,iv) thrombin was significantly enhanced by SecinH3 (Figure3A). When platelets were stimulated with 0.3 μg mL−1 (Fig.3Ci-ii) and 1 μg mL−1 (Fig.3Ci,iii) CRP, but not 5 μg mL−1 CRP (Fig.3Ci,iv), similar results were seen. Platelet aggregation in response to low and intermediate concentrations of both agonists was also significantly enhanced by SecinH3 (Fig.3B,D). Using a higher concentration of SecinH3 did not result in a greater effect on ATP secretion or aggregation (Figure S2).
Figure 3.
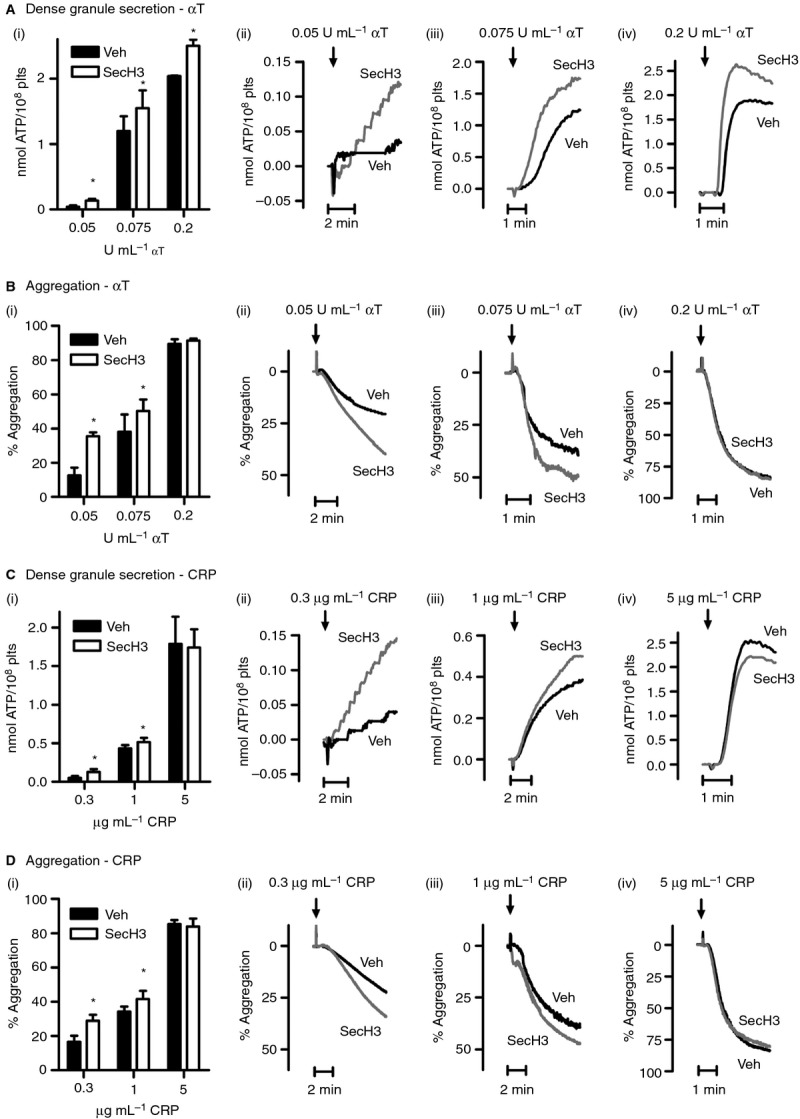
Cytohesin-2 inhibition by SecinH3 enhances dense granule secretion and platelet aggregation. Washed platelets (2 × 108 mL−1) were treated for 15 min with 0.2% DMSO vehicle (Veh) or 15 μm SecinH3 (SecH3). ATP release from dense granules (A, C) was assessed by luminometry, simultaneously with platelet aggregation (B, D). Platelets were stimulated with the indicated concentrations of α-thrombin (αT) (A, B) or CRP (C, D). The bar graphs (i, mean ± SEM, n ≥ 3) show ATP release (nmol ATP/108 plts, A, C) and aggregation (% of maximal aggregation, B, D), whereas ii–iv (A–D) show representative traces. *P < 0.05 (Student's paired t-test).
α-granule secretion, integrin αIIbβ3 activation and lysosome secretion are not affected by cytohesin-2 inhibition
Next, we investigated whether cytohesin-2 inhibition also affects α-granule secretion. Therefore, we assessed P-selectin exposure, a marker of α-granule release, by flow cytometry. Platelet pre-incubation with SecinH3 did not alter the expression of P-selectin induced by a range of concentrations of thrombin (Fig.4A) and CRP (Fig.4D). Integrin αIIbβ3 activation (Fig.4B,E), which is required for crosslinking of platelets by binding to fibrinogen, was measured simultaneously and was also not affected. Furthermore, we analyzed the secretion of β-hexosaminidase from lysosomes using a colorimetric assay. SecinH3 had no effect on lysosome secretion in response to platelet stimulation with various concentrations of thrombin (Fig.4C) or CRP (Fig.4F), compared with the vehicle control.
Figure 4.

Alpha granule secretion, integrin activation and lysosome release are not affected by SecinH3. Washed platelets (4 × 107 mL−1) were treated with 0.2% DMSO vehicle (Veh) or 15 μm SecinH3 (SecH3) before stimulation for 10 min with a range of concentrations of α-thrombin (αT) (A, B) or CRP (D, E) in the presence of 1 mm CaCl2. P-selectin expression (A, D) as a result of α-granule secretion and integrin αIIbβ3 activation (B, E) were measured by flow cytometry. Data are shown as the percentage of maximal response. Alternatively, washed platelets (2 × 108 mL−1), treated with Veh or 15 μm SecH3, were stimulated for 15 min with αT (C) or CRP (F) in the presence of 1 mm CaCl2 and the release of β-hexosaminidase from lysosomes was measured by a colorimetric assay. Data are expressed as the percentage of total content. Curves (A–F) were fitted by F-test. Mean ± SEM, n ≥ 4.
PKC negatively regulates the cytohesin-2/ARF6 pathway upon platelet stimulation
As cytohesin-2 is a GEF for the small GTPase ARF6, we assessed the kinetics of ARF6 activation upon platelet stimulation with thrombin using a GGA3-GST pull down method 30. Under basal conditions ARF6 was detected in an active GTP-bound state (Fig.5A). Platelet stimulation with thrombin induced a decrease in active ARF6, which was minimal after 2–5 min and largely restored after 20 min. We investigated the role of PKC in this process by treating platelets with BIM IX. PKC inhibition completely rescued the observed decrease in ARF6-GTP levels upon platelet activation (Fig.5B).
Figure 5.
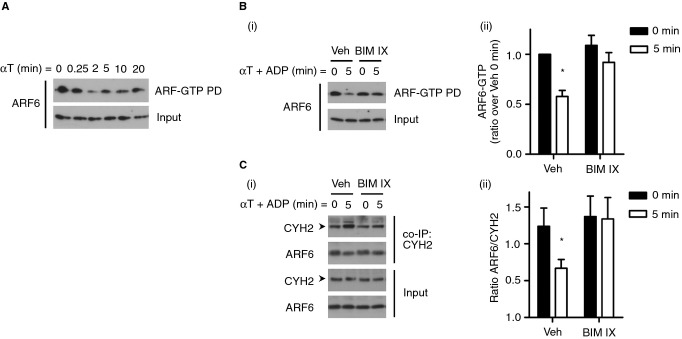
ARF6 activation and interaction with cytohesin-2 decrease upon platelet stimulation. Washed platelets (4 × 108 mL−1) were stimulated with 0.2 U mL−1 αT and lysed at the indicated time-points and ARF6-GTP was pulled down (PD) using GGA3-GST beads (A). Alternatively, platelets were incubated for 15 min with 0.2% DMSO vehicle (Veh) or 2 μm BIM IX prior to stimulation with 0.2 U mL−1 αT and 10 μm ADP, followed by ARF6-GTP pull down (Bi) or cytohesin-2 co-IP (Ci). Samples were immunoblotted for ARF6 and cytohesin-2 (CYH2). Densitometry was performed using ImageJ software and levels of ARF6-GTP (Bii) and ratios of ARF6 over CYH2 were calculated (Cii). *P < 0.05 (Student's paired t-test), mean ± SEM, n ≥ 4. Shown are representative blots. The arrows (➤) indicate the 47 kDa CYH2 band.
To investigate whether ARF6 binds to cytohesin-2, we performed immunoprecipitation studies using a cytohesin-2 antibody. ARF6 bound to cytohesin-2 under basal conditions and this interaction was significantly reduced upon platelet stimulation (Fig.5C). This was most obvious when comparing the ratio of co-immunoprecipitated ARF6 with the levels of immunoprecipitated cytohesin-2, as cytohesin-2 levels were generally slightly enhanced under stimulated conditions (Fig.5Ci). PKC inhibition with BIM IX completely reversed the decrease in interaction of cytohesin-2 with ARF6 observed upon platelet activation (Fig.5C).
Discussion
PKC plays a central role in platelet granule secretion, but the downstream pathways that regulate this process are poorly understood. In this study, we aimed to elucidate the underlying mechanism by which PKC regulates secretion and established a novel mechanism of PKC-regulated dense granule secretion in human platelets (Fig.6).
Figure 6.
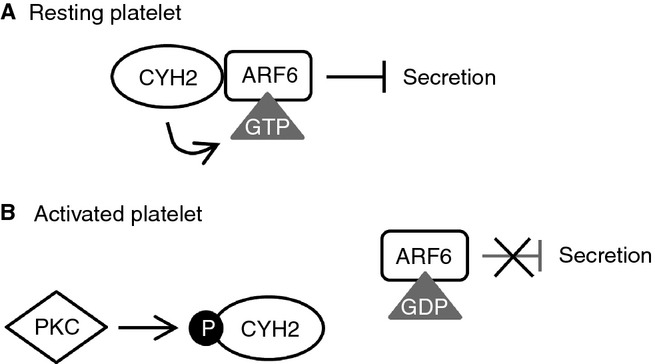
Model of regulation of the cytohesin-2/ARF6 pathway by PKC. (A) In resting platelets, cytohesin-2 (CYH2) is associated with ARF6 and maintains it in an active GTP-bound state, which constitutively suppresses dense granule secretion. (B) Upon platelet stimulation PKC is activated and phosphorylates cytohesin-2. This results in the dissociation of cytohesin-2 from ARF6, which therefore becomes inactive and GDP bound, allowing dense granule secretion to occur.
We used a proteomic approach with an anti-PKC substrate antibody to identify novel PKC substrates that may be involved in platelet activation, and identified cytohesin-2 as a novel conventional PKC substrate. Cytohesin-2 becomes phosphorylated in a PKC-dependent manner upon platelet activation by thrombin, CRP and the TxA2 analogue U46619. Using different PKC inhibitors we confirmed that the conventional PKC isoforms α and β are responsible for the phosphorylation of cytohesin-2. Interestingly, cytohesin-2 phosphorylation was unchanged in PKCα knockout mouse platelets, suggesting that cytohesin-2 phosphorylation may be mediated by PKCβ. However, a redundant role for PKCα cannot be excluded.
As cytohesin-2 is a GEF for the small GTPase ARF6, PKC phosphorylation of cytohesin-2 is likely to be involved in regulating ARF6 activity in human platelets. The regulation and activity of ARF6 in platelets, however, is not firmly established. Choi et al. (2006) found that ARF6-GTP is present in resting platelets and diminishes rapidly upon activation with collagen or thrombin, thereby allowing collagen-induced aggregation, platelet adherence and spreading 18,19. In contrast, Kanamarlapudi et al. (2012) reported that activation of human platelets with ADP promoted a transient but robust increase in ARF6-GTP levels 27, similar to findings in pancreatic β-cells 26. Our findings correspond to the observations of Choi et al. (2006), showing that under basal conditions ARF6-GTP levels are maximal and decrease upon platelet stimulation with thrombin. Importantly, we also demonstrated that ARF6 interacted with cytohesin-2 in resting platelets and that this interaction decreased upon platelet stimulation. Previous publications have described that phosphorylation of cytohesin-2 by PKC diminishes its membrane affinity, causing it to translocate from the membrane 20,31. As ARF6 has been shown to be predominantly membrane-bound, this translocation in turn leads to the dissociation of cytohesin-2 from ARF6, resulting in the inactivation of ARF6. The mechanism in platelets may differ slightly, because although we were able to show that phosphorylation of cytohesin-2 by PKC affects its interaction with ARF6 and the activation state of ARF6, cytohesin-2 did not apparently translocate from membrane to cytosol in these cells (data not shown). A different molecular mechanism of disengaging cytohesin-2 and ARF6 may therefore apply in platelets, mediated by PKC. PKC inhibition completely rescued both the decrease in ARF6-GTP and the dissociation of cytohesin-2 from ARF6 upon platelet stimulation. PKC-mediated cytohesin-2 phosphorylation can thus directly regulate ARF6 activity in human platelets, by decreasing the association of cytohesin-2 with ARF6. At present, we cannot exclude that, in addition to this regulatory mechanism, PKC may also directly regulate the GEF activity of cytohesin-2.
A role for cytohesin-2 in granule secretion has previously been described for chromaffin and β-cells 21,26. The recent development of the ARF-GEF inhibitor SecinH3 25 allowed us to study the role of cytohesin-2 in platelet granule secretion and function. SecinH3 significantly enhanced platelet dense granule secretion induced by thrombin and CRP, suggesting that GTP-bound ARF6 restrains granule secretion. Aggregation was also elevated, which is likely to be due to the enhanced release of autocrine compounds from dense granules, because cytohesin-2 inhibition did not affect other platelet responses such as alpha granule secretion and integrin activation. Despite the considered relation of lysosomes to dense granules 32, lysosome release was unchanged by cytohesin-2 inhibition. Given the different secretion kinetics of these two granule types, this supports a different molecular mechanism of regulation 33. Taken together, our results show that active GTP-bound ARF6 has a negative regulatory role in platelet dense granule secretion and aggregation. In line with this, it has previously been reported that the GTP-bound form of the related Golgi-regulating small GTPase ARF1 inhibits vesicle traffic in normal rat kidney cells 34. The mechanism whereby active ARF negatively regulates vesicle transport has not been established yet, but it is postulated that association of ARF with specific membrane components prevents membrane fusion 35.
In summary, we propose a model for regulation of the GEF cytohesin-2, where cytohesin-2 in resting platelets keeps the small GTPase ARF6 in an active state, thereby constitutively suppressing dense granule secretion. Cytohesin-2 phosphorylation by a conventional PKC isoform, upon platelet stimulation, causes cytohesin-2 to dissociate from ARF6, which then becomes inactive, resulting in the relief of the constitutive inhibition of dense granule secretion (Fig.6).
Addendum
M. T. J. van den Bosch designed, performed and analyzed the experiments and wrote the manuscript. A. W. Poole conceived the experiments, supervised the project and edited the manuscript. I. Hers conceived the experiments, supervised the project and wrote the manuscript.
Acknowledgments
We are grateful for the expert assistance of Elizabeth Aitken with technical support for this work, particularly the generation of the GGA3-GST fusion protein beads. We thank the healthy blood donors within the Medical Sciences Building, University of Bristol, for their generous donations. This work was funded by the British Heart Foundation (FS/11/62/28934, RG/10/006/28299).
Disclosure of Conflict of Interests
The authors state that they have no conflict of interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
ADP does not induce cytohesin-2 phosphorylation.
SecinH3 dose-response.
References
- 1.Strehl A, Munnix IC, Kuijpers MJ, van der Meijden PE, Cosemans JM, Feijge MA, Nieswandt B, Heemskerk JW. Dual role of platelet protein kinase C in thrombus formation: stimulation of pro-aggregatory and suppression of procoagulant activity in platelets. J Biol Chem. 2007;282:7046–55. doi: 10.1074/jbc.M611367200. [DOI] [PubMed] [Google Scholar]
- 2.Harper MT, Poole AW. Isoform-specific functions of protein kinase C: the platelet paradigm. Biochem Soc Trans. 2007;35:1005–8. doi: 10.1042/BST0351005. [DOI] [PubMed] [Google Scholar]
- 3.Harper MT, Poole AW. Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J Thromb Haemost. 2010;8:454–62. doi: 10.1111/j.1538-7836.2009.03722.x. [DOI] [PubMed] [Google Scholar]
- 4.Konopatskaya O, Gilio K, Harper MT, Zhao Y, Cosemans JM, Karim ZA, Whiteheart SW, Molkentin JD, Verkade P, Watson SP, Heemskerk JW, Poole AW. PKCalpha regulates platelet granule secretion and thrombus formation in mice. J Clin Invest. 2009;119:399–407. doi: 10.1172/JCI34665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams CM, Feng Y, Martin P, Poole AW. Protein kinase C alpha and beta are positive regulators of thrombus formation in vivo in a zebrafish (Danio rerio) model of thrombosis. J Thromb Haemost. 2011;9:2457–65. doi: 10.1111/j.1538-7836.2011.04520.x. [DOI] [PubMed] [Google Scholar]
- 6.Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–60. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- 7.Tyers M, Rachubinski RA, Stewart MI, Varrichio AM, Shorr RG, Haslam RJ, Harley CB. Molecular cloning and expression of the major protein kinase C substrate of platelets. Nature. 1988;333:470–3. doi: 10.1038/333470a0. [DOI] [PubMed] [Google Scholar]
- 8.Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature. 1992;356:618–22. doi: 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- 9.Jones ML, Craik JD, Gibbins JM, Poole AW. Regulation of SHP-1 tyrosine phosphatase in human platelets by serine phosphorylation at its C terminus. J Biol Chem. 2004;279:40475–83. doi: 10.1074/jbc.M402970200. [DOI] [PubMed] [Google Scholar]
- 10.Moore SF, van den Bosch MT, Hunter RW, Sakamoto K, Poole AW, Hers I. Dual regulation of glycogen synthase kinase 3 (GSK3)alpha/beta by protein kinase C (PKC)alpha and Akt promotes thrombin-mediated integrin alphaIIbbeta3 activation and granule secretion in platelets. J Biol Chem. 2013;288:3918–28. doi: 10.1074/jbc.M112.429936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunter RW, Mackintosh C, Hers I. Protein kinase C-mediated phosphorylation and activation of PDE3A regulate cAMP levels in human platelets. J Biol Chem. 2009;284:12339–48. doi: 10.1074/jbc.M807536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donaldson JG, Jackson CL. ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat Rev Mol Cell Biol. 2011;12:362–75. doi: 10.1038/nrm3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hofmann I, Thompson A, Sanderson CM, Munro S. The Arl4 family of small G proteins can recruit the cytohesin Arf6 exchange factors to the plasma membrane. Curr Biol. 2007;17:711–6. doi: 10.1016/j.cub.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 14.Galas MC, Helms JB, Vitale N, Thierse D, Aunis D, Bader MF. Regulated exocytosis in chromaffin cells. A potential role for a secretory granule-associated ARF6 protein. J Biol Chem. 1997;272:2788–93. doi: 10.1074/jbc.272.5.2788. [DOI] [PubMed] [Google Scholar]
- 15.Yang CZ, Mueckler M. ADP-ribosylation factor 6 (ARF6) defines two insulin-regulated secretory pathways in adipocytes. J Biol Chem. 1999;274:25297–300. doi: 10.1074/jbc.274.36.25297. [DOI] [PubMed] [Google Scholar]
- 16.Vitale N, Chasserot-Golaz S, Bader MF. Regulated secretion in chromaffin cells: an essential role for ARF6-regulated phospholipase D in the late stages of exocytosis. Ann N Y Acad Sci. 2002;971:193–200. doi: 10.1111/j.1749-6632.2002.tb04463.x. [DOI] [PubMed] [Google Scholar]
- 17.Begle A, Tryoen-Toth P, de Barry J, Bader MF, Vitale N. ARF6 regulates the synthesis of fusogenic lipids for calcium-regulated exocytosis in neuroendocrine cells. J Biol Chem. 2009;284:4836–45. doi: 10.1074/jbc.M806894200. [DOI] [PubMed] [Google Scholar]
- 18.Choi W, Karim ZA, Whiteheart SW. Arf6 plays an early role in platelet activation by collagen and convulxin. Blood. 2006;107:3145–52. doi: 10.1182/blood-2005-09-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karim ZA, Choi W, Whiteheart SW. Primary platelet signaling cascades and integrin-mediated signaling control ADP-ribosylation factor (Arf) 6-GTP levels during platelet activation and aggregation. J Biol Chem. 2008;283:11995–2003. doi: 10.1074/jbc.M800146200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santy LC, Frank SR, Hatfield JC, Casanova JE. Regulation of ARNO nucleotide exchange by a PH domain electrostatic switch. Curr Biol. 1999;9:1173–6. doi: 10.1016/S0960-9822(00)80019-6. [DOI] [PubMed] [Google Scholar]
- 21.Caumont AS, Vitale N, Gensse M, Galas MC, Casanova JE, Bader MF. Identification of a plasma membrane-associated guanine nucleotide exchange factor for ARF6 in chromaffin cells. Possible role in the regulated exocytotic pathway. J Biol Chem. 2000;275:15637–44. doi: 10.1074/jbc.M908347199. [DOI] [PubMed] [Google Scholar]
- 22.Liu L, Liao H, Castle A, Zhang J, Casanova J, Szabo G, Castle D. SCAMP2 interacts with Arf6 and phospholipase D1 and links their function to exocytotic fusion pore formation in PC12 cells. Mol Biol Cell. 2005;16:4463–72. doi: 10.1091/mbc.E05-03-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunter RW, Hers I. Insulin/IGF-1 hybrid receptor expression on human platelets: consequences for the effect of insulin on platelet function. J Thromb Haemost. 2009;7:2123–30. doi: 10.1111/j.1538-7836.2009.03637.x. [DOI] [PubMed] [Google Scholar]
- 24.Ladage D, Tilemann L, Ishikawa K, Correll RN, Kawase Y, Houser SR, Molkentin JD, Hajjar RJ. Inhibition of PKCalpha/beta with ruboxistaurin antagonizes heart failure in pigs after myocardial infarction injury. Circ Res. 2011;109:1396–400. doi: 10.1161/CIRCRESAHA.111.255687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hafner M, Schmitz A, Grune I, Srivatsan SG, Paul B, Kolanus W, Quast T, Kremmer E, Bauer I, Famulok M. Inhibition of cytohesins by SecinH3 leads to hepatic insulin resistance. Nature. 2006;444:941–4. doi: 10.1038/nature05415. [DOI] [PubMed] [Google Scholar]
- 26.Jayaram B, Syed I, Kyathanahalli CN, Rhodes CJ, Kowluru A. Arf nucleotide binding site opener [ARNO] promotes sequential activation of Arf6, Cdc42 and Rac1 and insulin secretion in INS 832/13 beta-cells and rat islets. Biochem Pharmacol. 2011;81:1016–27. doi: 10.1016/j.bcp.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanamarlapudi V, Owens SE, Saha K, Pope RJ, Mundell SJ. ARF6-dependent regulation of P2Y receptor traffic and function in human platelets. PLoS One. 2012;7:e43532. doi: 10.1371/journal.pone.0043532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanamarlapudi V, Thompson A, Kelly E, Lopez Bernal A. ARF6 activated by the LHCG receptor through the cytohesin family of guanine nucleotide exchange factors mediates the receptor internalization and signaling. J Biol Chem. 2012;287:20443–55. doi: 10.1074/jbc.M112.362087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu W, London NR, Gibson CC, Davis CT, Tong Z, Sorensen LK, Shi DS, Guo J, Smith MC, Grossmann AH, Thomas KR, Li DY. Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability. Nature. 2012;492:252–5. doi: 10.1038/nature11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hafner M, Schmitz A, Famulok M. Quantification of ARF-GTP in HepG2 by pulldown with GST-GGA3(1-316) Protocol Exchange. 2006. DOI: 10.1038/nprot.2006.412.
- 31.Macia E, Paris S, Chabre M. Binding of the PH and polybasic C-terminal domains of ARNO to phosphoinositides and to acidic lipids. Biochemistry. 2000;39:5893–901. doi: 10.1021/bi992795w. [DOI] [PubMed] [Google Scholar]
- 32.Dell'Angelica EC, Mullins C, Caplan S, Bonifacino JS. Lysosome-related organelles. FASEB J. 2000;14:1265–78. doi: 10.1096/fj.14.10.1265. [DOI] [PubMed] [Google Scholar]
- 33.Chen D, Lemons PP, Schraw T, Whiteheart SW. Molecular mechanisms of platelet exocytosis: role of SNAP-23 and syntaxin 2 and 4 in lysosome release. Blood. 2000;96:1782–8. [PubMed] [Google Scholar]
- 34.Zhang CJ, Rosenwald AG, Willingham MC, Skuntz S, Clark J, Kahn RA. Expression of a dominant allele of human ARF1 inhibits membrane traffic in vivo. J Cell Biol. 1994;124:289–300. doi: 10.1083/jcb.124.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones AT, Spiro DJ, Kirchhausen T, Melancon P, Wessling-Resnick M. Studies on the inhibition of endosome fusion by GTPgammaS-bound ARF. J Cell Sci. 1999;112(Pt 20):3477–85. doi: 10.1242/jcs.112.20.3477. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ADP does not induce cytohesin-2 phosphorylation.
SecinH3 dose-response.