

Abstract
DNA glycosylases remove damaged or enzymatically modified nucleobases from DNA, thereby initiating the base excision repair (BER) pathway, which is found in all forms of life. These ubiquitous enzymes promote genomic integrity by initiating repair of mutagenic and/or cytotoxic lesions that arise continuously due to alkylation, deamination, or oxidation of the normal bases in DNA. Glycosylases also perform essential roles in epigenetic regulation of gene expression, by targeting enzymatically-modified forms of the canonical DNA bases. Monofunctional DNA glycosylases hydrolyze the N-glycosidic bond to liberate the target base, while bifunctional glycosylases mediate glycosyl transfer using an amine group of the enzyme, generating a Schiff base intermediate that facilitates their second activity, cleavage of the DNA backbone. Here we review recent advances in understanding the chemical mechanism of monofunctional DNA glycosylases, with an emphasis on how the reactions are influenced by properties of the nucleobase leaving-group, the moiety that varies across the vast range of substrates targeted by these enzymes.
Introduction
The nucleobases in DNA are reactive and subject to continuous modification by endogenous and exogenous agents, producing a broad spectrum of damage that can pose a severe threat to genomic integrity and contribute to aging and diseases including cancer.1 Three major types of damage occur to the canonical bases in DNA, including alkylation by endogenous and exogenous electrophiles, hydrolytic deamination of the exocyclic amino groups (not relevant for thymine), and oxidation by a slew of reactive oxygen species (ROS). The resulting lesions are corrected by DNA base excision repair (BER), a pathway that is conserved in all forms of life and is initiated by a DNA glycosylase.2 These enzymes use a nucleotide-flipping mechanism to recognize chemically modified or mismatched bases in DNA, and remove them by cleaving the carbon-nitrogen bond that connects the target base to the anomeric carbon of the 2’-deoxyribose sugar.3
DNA glycosylases are found in two basic types, monofunctional and bifunctional.4 Monofunctional glycosylases catalyze hydrolysis of the N-glycosidic bond, yielding an apurinic/apyrimidinic (AP) or abasic site in the DNA (scheme 1). Bifunctional glycosylases cleave the N-glycosidic bond using an amine nucleophile of the enzyme, giving a Schiff base (imine) intermediate that facilitates a second enzymatic activity, cleavage of the phosphodiester backbone on the 3’ side of the lesion (β-elimination). Some bifunctional glycosylases also cleave the DNA on the 5’ side of the lesion (δ-elimination). The various reaction products of monofunctional and bifunctional glycosylases can be highly mutagenic and cytotoxic, and they are recognized and processed by follow-on BER enzymes to restore the original DNA sequence.
Scheme 1.

N-glycosidic bond hydrolysis in DNA by monofunctional and bifunctional DNA glycosylases
Our focus here is largely on the mechanism of N-glycosidic bond hydrolysis by monofunctional glycosylases. We also consider the relevant non-enzymatic reactions, hydrolysis of 2’-deoxynucleosides, which inform the enzymatic mechanisms. The emphasis is on developments since the most recent reviews on this topic by Berti and McCann (2006)5 and Stivers and Jiang (2003),4 and in particular on the role of the leaving group base, the moiety which varies across the sea of substrates targeted by these enzymes. We generally do not discuss aspects of the deoxynucleoside sugar or nucleophile activation or positioning, and would direct the reader to the reviews noted above.4, 5
Shown in Fig. 1 are the four canonical nucleobases found in DNA, four modified forms of cytosine that are enzymatically generated in the DNA of vertebrates, and four of examples from the vast range of damaged bases that are recognized and excised by DNA glycosylases. While some of these enzymes act predominantly on a single type of lesion, others excise a variety of modified bases, in keeping with findings that the number of different glycosylases in any given organism is typically far smaller than the number of different lesions that must be excised to initiate repair. Some glycosylases excise one of the four canonical bases, if that base resides in a mismatched pair. For example, MutY excises adenine that is mispaired with 8-oxoguanine,6 and other glycosylases (TDG, MBD4) selectively excise thymine bases that are mismatched with guanine.7
Fig. 1.

The four canonical bases in DNA (top), four enzymatically-generated forms of cytosine found in vertebrates (middle), and four examples of damaged bases (bottom) from among the many dozens that arise in DNA.
In addition to removing damaged bases, some glycosylases excise modified bases that are enzymatically derived from one of the four normal DNA bases. For example, active cytosine deaminases convert cytosine to uracil (Fig. 1), which is then excised by uracil DNA glycosylase (UDG), a process that is important for maturation of antibody affinity and the innate defense against retroviruses and retroelements.8, 9 In addition, methyltransferases generate bases including 5-methylcytosine (mC), a modification that is important for maintaining genomic stability and transcriptional regulation.10 In mammals, TET (ten-eleven translocation) enzymes catalyze oxidation of mC, in a stepwise manner, to give 5-hydroxymethyl-C (hmC),11, 12 5-formyl-C (fC), and 5-carboxyl-C (caC).13–15 Glycosylase-mediated excision of mC (in plants)16–18 or of fC or caC (vertebrates)14, 19 is an essential step in active DNA demethylation, a process that converts mC back to C and plays a central role in epigenetic regulation of gene expression.10
Transition-state analysis of reaction mechanisms
The detailed mechanism of many N-glycoside hydrolysis reactions has been studied by a variety of approaches, including structure-activity relationships and transition-state analysis. The latter involves the determination of kinetic isotope effects (KIEs) for reactants harboring an isotopic label at multiple positions and computational analysis to identify the TS structure(s) that best match the observed KIEs.5, 20 Transition-state analysis has been completed for many enzymatic and non-enzymatic ribonucleotide N-glycoside hydrolysis reactions, but only for a limited number of deoxyribonucleoside reactions.
The two limiting mechanisms for hydrolysis of the N-glycosidic bond are shown in Figure 2.4, 5, 21 In a stepwise DN*AN (or SN1) mechanism, departure of the nucleobase leaving group (DN) gives rise to a discrete but highly unstable oxacarbenium ion intermediate (*) that exits for a finite (albeit short) lifetime before addition of the nucleophile (AN). Alternatively, in a concerted biomolecular ANDN (or SN2) mechanism, nucleophile addition (AN) begins prior to departure of the leaving group (DN), such that bond order to both groups exists in a single transition state (Fig. 2). For N-glycoside hydrolyses, the ANDN reactions are typically highly dissociative, with a single oxacarbenium-ion-like transition state in which leaving group departure is greatly advanced over nucleophile approach.5
Fig. 2.

Two limiting mechanisms for N-glycosidic bond hydrolysis in (deoxy)ribonucleosides. The stepwise reaction (top) has two transition states, for departure of the leaving group (DN) and nucleophile addition (AN).
The stepwise reactions can be further resolved, where the rate limiting step, indicated by the double dagger (‡), is either departure of the leaving-group (DN‡*AN) or addition of the nucleophile (DN*AN‡).22 Notably, in the latter mechanism, cleavage of the glycosidic bond is reversible. The asterisk in DN*AN indicates that the intermediate is too short-lived for the leaving group to diffuse into solution prior to nucleophile attack. By contrast, for a DN + AN mechanism, the intermediate lifetime allows for diffusional equilibration of the leaving group with solvent.
While most ribonucleotides are hydrolyzed through a highly dissociative ANDN mechanism with some rare examples of DN*AN, all studies to date find that hydrolysis of deoxyribonucleotides proceeds through a DN*AN mechanism.23–26 As indicted in Fig. 2, these reactions are nearly always irreversible, though one DNA glycosylase can catalyze the synthesis of an N-glycosidic bond, using abasic DNA and one particular nucleobase.27
Non-enzymatic N-glycoside hydrolysis of dAMP
Transition-state analysis of non-enzymatic N-glycoside hydrolysis has been reported for only one deoxynucleoside, that being 2’-deoxyadenosine monophosphate (dAMP).26, 28 The acid-catalyzed reaction, collected in 0.1 M HCl, proceeds through a stepwise mechanism in which the glycosidic bond breaks and reforms repeatedly, giving an equilibrium between the reactant and the intermediate, an adenine·oxacarbenium contact ion pair complex (CPIC).26 Expulsion of the adenine leaving-group is catalyzed by protonation at two endocyclic nitrogens, N1 and N7, such that it departs as a monocation. The first irreversible step is either nucelophile (water) attack on the oxacarbenium in the CIPC, 80% of the time, or dissociation of the CPIC followed by nucleophile addition, 20% of the time. These conclusions were based in part on methanolysis reactions, collected in 50% MeOH/0.1 M HCl, which also indicated that the oxacarbenium-ion intermediate has a lifetime of 10−11-10−10 s. Thus, the mechanism is 80% DN*AN‡ and 20% DN + AN (more specifically, DN*P‡ + AN, where “P” indicates diffusional separation). The results suggest that dAMP hydrolysis at neutral pH follows a DN*AN‡ mechanism.
Differing substantially from dAMP, acid-catalyzed hydrolysis of AMP exhibits a concerted ANDN transition-state, with simultaneous approach of the nucleophile and departure of the leaving group.29 The vastly different mechanisms for hydrolysis of AMP and dAMP may reflect the ~4 kcal/mol lower stability of a ribocation relative to a 2′-deoxyribocation.30
Ricin-catalyzed N-glycoside hydrolysis of dA in DNA
Ricin, a ribosome-inactivating enzyme that normally excises adenine from RNA, was employed to study the mechanism of enzymatic N-glycoside hydrolysis of dA nucleotides in DNA.24 The KIEs indicated a stepwise mechanism but did not distinguish clearly between a DN‡*AN or a DN*AN‡ mechanism. Subsequent computational studies suggest that the KIEs support a DN‡*AN mechanism, with rate-limiting and irreversible rupture of the N-glycosidic bond.26 An important component of the ricin-catalyzed reaction is activation (protonation) of the adenine leaving group, as discussed below.
MutY-catalyzed N-glycoside hydrolysis of dA in DNA
MutY is a DNA repair enzyme that excises adenine bases that are mispaired with 8-oxo-G. Transition-state analysis using multiple KIEs shows that MutY-catalyzed N-glycosidic bond hydrolysis follows a stepwise DN*AN‡ mechanism, where the C-N bond breaks and reforms repeatedly prior to irreversible addition of the water nucleophile (AN).25 The results also demonstrate that the adenine base is protonated (at N7) prior to nucleophile addition, probably before C-N bond cleavage. MutY imparts ground-state distortion to favor a sugar ring conformation that resembles the oxacarbenium ion intermediate. Findings that the commitment to catalysis is negligible and that C-N bond cleavage is reversible lead to the remarkable conclusion that once the oxacarbenium ion intermediate arises in the active site, the most likely outcone is that the glycosidic bond will be reformed and the intact DNA substrate will dissociate from the enzyme. The results also indicate that, following bond cleavage, the adenine leaving group remains in a position that allows for reformation of the C-N bond, at least until the AN transition state. Thus, the MutY reaction and acid-catalyzed dAMP hydrolysis both follow a DN*AN‡ mechanism, while ricin-catalyzed dA hydrolysis involves a DN‡*AN mechanism.
UNG-catalyzed N-glycoside hydrolysis of dU in DNA
Uracil DNA glycosylase (UNG) catalyzes hydrolysis of 2’-deoxyuridine (dU) residues in DNA, and transition-state analysis revealed a stepwise DN*AN mechanism, with an oxacarbenium ion uracil anion intermediate (Fig. 3A).23 This conclusion was supported by computational studies of the enzymatic reaction using hybrid quantum-mechanical/molecular-mechanical (QM/MM) methods.31 The oxacarbenium ion intermediate adopts a slightly 3′-exo sugar pucker that differs from the 2′-endo conformation of B-type DNA but resembles that observed in the enzyme-substrate complex.32 This suggests that UNG imparts ground-state strain to impose a sugar conformation that favors a highly dissociative oxacarbenium-ion-like transition-state on path to the oxacarbenium ion intermediate.23 The KIEs and other findings indicate that the first step, C-N bond cleavage, is rate-limiting, that is, a DN‡*AN mechanism.23, 26, 31
Fig. 3.

UNG-catalyzed excision of uracil from DNA. (A) Transition-state analysis and computational studies indicate that UNG follows a stepwise mechanism with irreversible C-N bond cleavage. NMR studies show that the uracil anion is stabilized in the ternary product complex. (B) The uracil anion and a conserved Asp side chain interact with the 1-aza-2'-deoxyribose cation, suggesting similar stabilization of the oxacarbenium ion in the chemical transition state(s).
Observation of a rate-limiting DN step typically suggests that the leaving group is either (i) activated via protonation, (ii) electrostatically stabilized after departure, or (iii) sequestered in order to suppress reformation of the C-N bond. Consistent with the second mechanism, NMR studies show that UNG dramatically stabilizes the uracil anion in the ternary (UNG-DNA-uracil) product complex at neutral pH (Fig. 3A).33–35 Uracil is also anionic in a ternary complex with UNG and DNA containing a cationic mimic of the oxacarbenium ion intermediate (Fig. 3B).36 These findings, which were not predicted by crystal structures, suggest a similar mechanism for stabilization of the uracil anion in the chemical transition state(s), as discussed below. Together, the uracil anion and a conserved Asp side chain stabilize the cationic oxacarbenium-ion via an “electrostatic sandwich” (Fig. 3B).34, 36
A crystal structure of UDG bound to DNA containing a cationic 1-aza-2′-deoxyribose and the uracil anion provides a mimic of the second (AN) transition state.37 Remarkably, the structure indicates an electrophile migration mechanism, whereby electrostatic forces pull the electrophile (oxacarbenium ion) towards the bound water nucleophile (Fig. 3A).
While transition state analysis of non-enzymatic glycosidic bond hydrolysis has not been reported for dU or any other pyrimidine deoxynucleotide, other studies are informative. Non-enzymatic hydrolysis of dU, dT, and 5-halogen-substituted dU proceeds with a strong dependence on leaving group quality (see below) and a low and positive entropy of activation (TΔS), suggesting a highly dissociative ANDN mechanism or perhaps a stepwise (DN*AN) mechanism.38, 39 Computational studies of dU hydrolysis using QM/MM methods indicate the former.31
Phosphorylase-catalyzed hydrolysis of dT in DNA
Thymidine phosphorylase (TP) catalyzes reversible phosphorolysis of the N-glycosidic bond in thymidine (dT), and in the absence of phosphate it can use water as the nucleophile to hydrolyze dT. Transition state analysis shows that the hydrolytic reaction proceeds through a stepwise DN*AN‡ mechanism, with reversible C-N bond cleavage followed by irreversible and rate-limiting capture of the water nucleophile by the deoxyribocation intermediate.40 Thus, the intermediate is in equilibrium with the reactant (dT) prior to nucleophile capture, as observed for acid-catalyzed dAMP hydrolysis and the MutY reaction. The equilibrium isotope effect (EIE) between free dT and N1-protonated thymine indicates that a departing thymine anion is protonated at N1 prior to nucleophile capture of the deoxyribocation. The near-unity 15N KIE indicates little or no rehybridization of N1, hence no participation of the leaving group, at the rate-determining transition state for nucleophile addition. The steep reaction barrier for AN is thought to reflect improper alignment of water or its weak nucleophilic nature relative to the preferred phosphorolytic reaction of TP. The results suggest general base activation of the water nucleophile, potentially by a His side chain. In remarkable contrast, the phosphorolytic reaction follows a nearly synchronous ANDN transition state with no cationic character.41
Role of the Leaving Group
The various nucleotides in DNA, be they canonical, enzymatically modified, or damaged, differ in the chemical nature of the nucleobase. As such, it is of interest to consider how N-glycosidic bond hydrolysis may be influenced by the base, given its key role as the leaving group in these reactions. Findings that a stepwise (DN*AN) pathway predominates in glycosidic bond hydrolysis for 2’-deoxynucleosides indicate that properties of the leaving-group can have a large impact on the reaction mechanism and rate.
The leaving-group quality of a nucleobase depends on its capacity to accommodate the increased electron density generated by heterolytic cleavage of the glycosidic bond (Fig. 4A). In the absence of catalysis, a neutral base departs as an anion, which is usually unstable in solution and a relatively good nucleophile, and typically a poor leaving group. By contrast, a cationic base departs as a neutral species, which is generally a better leaving group than the anion. As such, leaving-group quality of a neutral base can be enhanced by protonation (acid catalysis), typically prior to C-N bond cleavage, and/or electrostatic stabilization of the departing anion. Alkylation, if it imparts a positive charge, improves leaving group quality because the base departs as a neutral species.
Fig. 4.

Leaving group quality of the nucleobase depends on the acidity (pKa) of its glycosidic nitrogen. (A) In the stepwise reactions for glycosidic bond hydrolysis, a neutral base departs as an anion and a cationic base departs as a neutral species. (B) Acidity of the glycosidic nitrogen for a given base depends on the stability of the N-deprotonated species (conjugate base). The glycosidic nitrogen is N1 for pyrimidines, N9 for purines. Acidity (pKa) is shown for U, 5FU, A, and 3-methyl-A.
A key indicator of leaving-group quality is the acidity of the glycosidic nitrogen (N1 for pyrimidines, N9 for purines). Greater acidity, or a lower pKa, reflects a more stable N-deprotonated species (conjugate base) and a weaker nucleophile, hence a better leaving group for deoxynucleotide hydrolysis reactions (Fig. 4B). For instance, N1 is more acidic for 5FU relative to U because the N1 anion is more stable for 5FU (F substituent stabilizes the anion), and the leaving-group quality is better for 5FU relative to U. The lower N9 pKa for 3-methyl-A relative to A reflects greater stability of the N9-deprotonated species for the former, which is a better leaving group (Fig. 4B).
Below, we consider mechanisms for catalyzing leaving-group departure in glycosidic bond hydrolysis reactions, first for purines and then for pyrimidines.
Catalysis of purine departure
Hydrolysis of purine deoxynucleosides (dA, dG) is strongly acid catalyzed via protonation of nitrogen(s) in the nucleobase, which can dramatically improve leaving-group quality, as noted above.5, 42, 43 The mono-protonated base departs as a neutral species, a much better leaving group than the anion, and the di-protonated base departs as a monocation, an even better leaving group.
The pH profiles for hydrolysis of purine nucleosides (dA, dG) and for DNA depurination show acid-catalysis at pH 7.4, even as the pKa values for the most basic ring nitrogens are well below 7.4. The depurination rates are very slow at physiological pH, reflecting the very low but significant population of the protonated species. For hydrolysis of dA at pH 7.4, acid catalysis involves N1 protonation (pKaN1 3.7) and gives an estimated rate enhancement of 103.6-fold.5, 42 An additional rate enhancement of about 105.3-fold is realized by protonation at a second site, N7 (pKaN7 −1.3) to give diprotonated dA. Notably, transition-state analysis gives direct evidence for protonation at the second site (N7) in acid-catalyzed hydrolysis of dAMP (performed at pH 1).26 Similarly, pH profiles indicate that acid catalyzed dG hydrolysis at pH 7.4 involves N7 protonation (pKaN7 2.4) and gives an estimated rate enhancement of 105-fold.5, 42 An additional rate enhancement of roughly 104.7-fold is realized by ionization at a second site, N3 (pKaN3 −2.4) to give di-protonated dG.5, 42
As noted previously,5 the pKa of −1.3 for N7 of dA is a macroscopic value, reflecting the interconversion of mono- and di-protonated species. However, protonation of neutral dA at N7 occurs with a microscopic pKa of about 2.4, thus acidity is not dramatically greater for N7 relative to N1. Similar reasoning likely applies to N3, the most acidic purine nitrogen. This is relevant to acid catalysis by enzymes, which do not always protonate the most basic nitrogen of the purine leaving-group.
Enzymatic catalysis of adenine departure
Ricin-catalyzed hydrolysis of dA in DNA has been proposed to involve mono- or di-protonation of adenine to activate it for departure, but the site(s) of nitrogen protonation are not yet established. Transition state analysis suggests that N7 is protonated in ricin-catalyzed hydrolysis of adenosine in RNA.30 It was proposed that N7 and possibly N1 are protonated in ricin-mediated hydrolysis of dA in DNA.24 The enzyme has no side chain that can directly protonate N7 via general acid catalysis, but it does provide backbone oxygen contacts that could favor N7 protonation, with protons derived from solvent.44 Structural and mutagenesis studies suggest that a conserved Arg side chain could protonate adenine at N3.44–46 Thus, evidence to date favors N3 and N7 as sites of protonation for ricin-catalyzed dA hydrolysis.
Transition-state analysis demonstrates that MutY-mediated hydrolysis of dA in DNA involves protonation of the adenine base at N7, prior to glycosidic bond cleavage.25 N7-protonation increases the calculated acidity of adenine (N9-H) by over 100 kcal/mol in the gas-phase.47 A conserved Asp side chain, which is essential for base excision, contacts adenine N7, suggesting a role in general acid catalysis of adenine expulsion.48–50 MutY activity is reduced at least 104.4-fold when N7 of adenine is replaced by carbon; it cannot excise 7-deaza-A.47, 51 Moreover, activity is reduced 40-fold for 1-deaza-A, 100-fold for 3-deaza-A,47, 52 and 6000-fold for an adenine analogue lacking both N1 and N3,47, 53 suggesting that electrostatic contacts to N1 and N3 stabilize the departing adenine in the transition-state. An Arg side chain contacts N1, and an ordered water molecule contacts N3 in the enzyme-substrate (ES) complex.49 While N1 is more basic than N3 and N7, it is not the site of protonation and appears to be the least important endocyclic nitrogen in the enzymatic reaction.
Enzymatic excision of damaged purines
Mammalian alkyladenine DNA glycosylase (AAG) protects against DNA damage caused by alkylation and oxidative deamination of purine bases in DNA. Acid catalysis is observed for AAG excision of two damaged forms of purine, hypoxanthine (Hx, Fig. 2) and 1,N6-ethenoadenine (εA, Fig. 5).54 The pH profiles suggest an essential protonated group in the ES complex, with pKa values of 6.4 and 7.3 for Hx and εA, respectively. Because mutational analysis revealed no general acid for AAG,54 the data could potentially reflect ionization of the nucleobase in the ES complex, with a proton derived from solvent. This could be facilitated by an ordered water molecule, and one is proximal to N7 of εA in the ES complex.5, 55 Evidence that acid catalysis for Hx involves N7-protonation is indicated by findings that activity is reduced by over 107-fold when N7 is replaced by carbon; AAG cannot excise 7-deaza-Hx (Fig. 5).54 This idea is supported by the absence of acid catalysis for 7-methylguanine (7meG, Fig. 5).54 7meG is cationic (pre-activated for departure), and N7-methylation mimics the effect of N7-protonation. Indeed, the glycosidic nitrogen (N9) is much more acidic for 7meG relative to Hx and εA, indicting 7meG is an inherently good leaving group.56 Catalysis for εA could involve protonation at N10, pKa 4.05,57 which has greater proton affinity than N7.58 Notably, it was also proposed that AAG excises some lesions without acid catalysis, such that the base departs as an anion; the glycosidic nitrogen (N9) is more acidic for Hx and εA relative to A and G in the gas phase, and this trend may hold in a hydrophobic active site, which could make Hx and εA more susceptible to enzymatic excision.56, 58, 59
Fig. 5.

Damaged forms of adenine and guanine. AAG can remove Hx, εA, and 7meG but not 7-deaza-Hx.
The E. coli 3-methyladenine DNA glycosylase I (TAG) excises 3-methyladenine (3meA) and 3-methylguanine (3meG). These cationic bases are expelled as neutral species after C-N bond cleavage. The effect of N3-methylation on leaving group quality is reflected by the much greater acidity of the glycosidic nitrogen (N9) for 3meA and 3meG relative to A.56 Structures of TAG and structure-activity relationships for binding 3meA analogues indicate that the hydrophobic enzyme active site stabilizes the neutral form of 3meA, i.e., the leaving group, relative to the cationic ground-state.60–62 The results suggest TAG imposes ground-state electrostatic strain on the cationic nucleotide substrate and selectively stabilizes the departing neutral base in the transition state. Specificity against neutral substrates may involve tight ground-state binding and poor stabilization of a departing anion in the transition state.
In contrast to AAG and TAG, the E. coli 3-methyladenine DNA glycosylase II (AlkA) does not distinguish between methylated and undamaged bases.63, 64 Rather, it excises a broad variety of alkylated purines and pyrimidines and has some activity for undamaged DNA bases. The remarkably indiscriminant active site of AlkA exhibits a relatively uniform rate enhancement of ~10−5-fold for excision of alkylated and normal bases. As such, specificity depends largely on the reactivity of the N-glycosidic bond.64
The Fpg-MutM enzymes target oxidized purines such as formamidopyrimidines and 8-oxoguanine (oxoG, Fig. 1).65 The reaction mechanism is unknown but could be stepwise (DN*AN), where an oxocarbenium intermediate captures a nucleophile provided by the enzyme, the α-nitrogen of an N-terminal Pro residue.66 Structures suggest these enzymes could catalyze departure of the oxoG anion via electrostatic stabilization.66 The glycosidic nitrogen (N9) is more acidic for oxoG relative to G,67 due perhaps to resonance stabilization of the oxoG anion (Fig. 6), such that leaving group quality is likely better for oxoG relative to G. Remarkably, Fpg provides four hydrogen bonds (backbone N-H) to the O6 oxygen of oxoG, which could stabilize an oxoG anion.66 Findings that acid-catalyzed C-N hydrolysis is slower for 8-oxo-dG versus dG68 likely reflect the fact that acid catalysis involves N7-protonation for dG (see above), which cannot occur for 8-oxo-dG. Rather, acid-catalyzed (nonenzymatic) hydrolysis of 8-oxo-dG could involve N3-protonation; N3 has a calculated pKa of 0.2.67 While acid catalysis at N3 cannot be excluded for the Fpg-MutM reaction, the site is rather acidic and the enzymes have no obvious group to perform such a role.
Fig. 6.
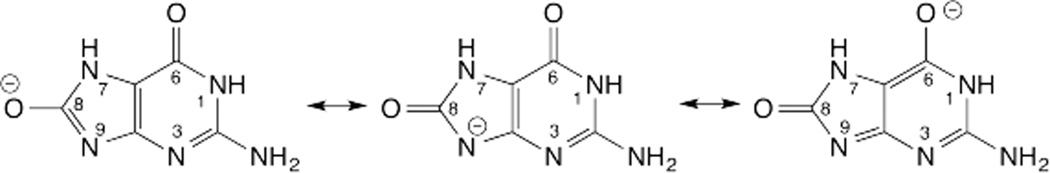
Resonance structures of the 8-oxoguanine anion.
OGG1 (8-oxoguanine DNA glycosylase) enzymes also excise oxoG,69 and the nucleophile can be either an enzyme group (Lys in this case) or a water molecule.70 OGG1 provides no contacts to O8 and few contacts to other regions of oxoG.71 Contacts to O6 involve only two water molecules for OGG1, compared to four backbone N-H contacts for Fpg. Despite the absence of contacts to O8, structure-activity relationships with oxoG analogues indicate that a carbonyl oxygen at C8 is important for OGG1 activity; 8-methoxy-G is not a substrate.72 While this finding was interpreted as evidence that OGG protonates O8 to activate oxoG,72 it is also consistent with departure of an oxoG anion, stabilized by charge delocalization to O8 (Fig. 6). Interestingly, it was proposed that an anionic oxoG leaving group participates in the lyase activity of OGG1, by deprotonating the Lys nucleophile to generate the Schiff base intermediate (scheme 1).73
Some lesions resulting from purine deamination are excised by enzymes in the UDG superfamily, including TDG-MUG, SMUG1, UDGa/b, and HDG.74–78 Yeast TDG (thymine DNA glycosylase) excises Hx and xanthine (X, Fig. 7).75 Remarkably, E. coli MUG (mismatch uracil glycosylase) has greater activity for X than it does for U.77 As discussed below, enzymes of the UDG superfamily do not use acid catalysis to activate uracil departure, and this might also be true for their excision of purine lesions. The glycosidic nitrogen of Hx is more acidic (pKaN9 8.9) than that of U (pKaN1 9.8), suggesting that the Hx anion could be a good leaving group.39, 56 The glycosidic nitrogen of X is even more acidic (pKaN9 7.3, calculated), thus the X monoanion is predicted to be an excellent leaving group (Fig. 7).56, 79 However, N3-H is also relatively acidic, with a pKaN3 of 5.7 for the xanthosine nucleoside,80 such that 2’-deoxyxanthosine (dX) is predominantly anionic at pH 7.4 (Fig. 7). Non-enzymatic hydrolysis of dX in ssDNA is pH-independent for pH >7 and acid-catalyzed for pH 3–7, with an apparent pKa of ~5 that likely reflects N3-protonation of the dX anion (though the results were suggested to reflect N7-protonation of neutral dX).81 The predominance of the dX anion in DNA at neutral pH likely explains its resistance to depurination,82 which was not initially expected.1 Because the dianion of X is likely to be a very poor leaving group, excision of the X monoanion seems unlikely. Thus, enzymatic hydrolysis of dX in DNA could potentially be acid-catalyzed via N3-protonation, such that X departs as a monoanion, which is predicted to be a good leaving group (Fig. 7).
Fig. 7.

2’-deoxyxanthosine (dX) ionizes at N3 with a pKa of about 5.7 (based on xanthosine) and is predominantly anionic at pH 7.4. Hydrolysis of neutral dX results in departure of the xanthine (X) monoanion, which should be a good leaving group, given that N9 is acidic for X relative to other purines, due perhaps to charge delocalization (N7, O6).
Non-enzymatic hydrolysis of dU and its analogs
While glycosidic bond hydrolysis is acid catalyzed at neutral pH for dA, dG, and dC (below), hydrolysis of dU (and dT) is pH-independent for pH 2–8 and acid catalyzed for pH <2.5, 39 For these pyrimidines, N3 is protonated at neutral pH (pKaN3 > 9) and the two carbonyl oxygens (O2, O4) are highly acidic, with pKa < −3. Accordingly, for non-enzymatic hydrolysis of dU and dT at neutral pH, the base is not protonated prior to C-N bond cleavage and it departs as an anion (Fig. 8A). While acid-catalysis of base departure has been proposed for many glycosylases that excise U or T, such a mechanism is unlikely due to the acidity of O2 and O4.33, 34 Moreover, acid catalysis is not required, because charge delocalization to the two carbonyl oxygens (O2, O4) stabilizes the departing anion and thereby improves its leaving-group quality (Fig. 8B).
Fig. 8.

(A) Hydrolysis of dU, dT, and dU analogs is not acid catalyzed at neutral pH, for enzymatic or non-enzymatic reactions, because the carbonyl oxygens are highly acidic (pKa < −3), so the base departs as an anion. (B) Resonance stabilization of the uracil anion involves the two carbonyl oxygens (O2, O4).
Given that a stepwise (DN*AN) mechanism is observed for glycosidic bond hydrolysis reactions in deoxynucelotides (see above), the rate can be expected to depend on the leaving-group quality of departing base, at least for reactions in which the DN step is rate limiting. One indicator of leaving-group quality is the acidity of the glycosidic nitrogen (N1 for pyrimidines), where greater acidity (lower pKa) indicates a more stable anion, a weaker nucleophile, and thus a better leaving group.
Supporting this idea, a Brønsted-type linear free energy relationship (LFER) is observed for the dependence of reaction rate (log kobs) on leaving-group acidity (N1 pKa) for non-enzymatic hydrolysis of 5-substituted deoxyuridines, including dU, dT, 5-F-dU, 5-Cl-dU, and 5-Br-dU (Fig. 9).38, 39, 83 The electron-withdrawing (σm > 0) halogen substituents stabilize the uracil anion, enhancing its leaving-group quality and increasing the reaction rate.
Fig. 9.

Brønsted-type linear free energy relationship (LFER) for non-enzymatic hydrolysis of 5-substituted deoxyuridines.83 The dependence of log kobs on N1 pKa (acidity of the leaving-group nitrogen) for hydrolysis of dT, dU, 5-F-dU, 5-Br-dU, and 5-Cl-dU gives a slope of βlg = −0.87 ± 0.03. The kobs values were extrapolated to 22 °C using values at higher temperatures,39, 87 as described.83 For 5-Br-dU, a corrected N1 pKa of 8.24 was used83 rather than the previously reported 8.49.39
Enzymatic hydrolysis of dU and its analogs
We next consider how enzymes facilitate leaving group departure during glycosidic bond hydrolysis of dU, dT, and related nucleotides in DNA. As noted above, UNG catalyzes the hydrolytic excision of uracil from DNA. In the stepwise (DN‡*AN) UNG reaction,23, 31 uracil departs as an anion, stabilized by hydrogen bonds to O2 and O4 from backbone and side-chain groups. Most notable among these is the imidazole group of a His residue that is strictly conserved in UNG enzymes and some other members of the UDG superfamily.32, 84 The catalytic imidazole is neutral through all steps of the UNG reaction, as demonstrated by NMR, and it provides a strong hydrogen bond to uracil O2, stabilizing the anion by ~5 kcal/mol (Fig. 10A).33–35, 85 Remarkably, UNG binds uracil as an anion in a ternary product complex with abasic DNA at neutral pH (Fig. 10A).35 Thus, UNG stabilizes the uracil anion by about 5 kcal/mol, reducing its N1 pKa from 9.8 in solution to 6.4 in the enzyme-product (EP) complex,35 due largely to the strong hydrogen bond noted above.85 The uracil anion is even further stabilized (pKaN1 <4.5) when bound in an EP complex that includes a cationic mimic of the oxacarbenium ion (Fig. 10B).36 These observations suggest that the catalytic His residue provides a strong hydrogen bond to stabilize the departing uracil anion in the chemical transition state(s) of the UNG reaction. Notably, the catalytic His residue is also conserved in SMUG1 enzymes,86 which belong to the UDG superfamily, though its role in these enzymes has not been rigorously examined.
Fig. 10.

Enzymatic stabilization of anionic leaving group. UNG provides a strong hydrogen bond to stabilize the uracil anion in the product complex, as indicated by a reduced N1 pKa of 6.4 relative to that of 9.8 for uracil in solution. (B) The uracil N1 pKa is even further reduced in a ternary complex with UNG and DNA that contains a mimic of the cationic oxacarbenium ion.
The UNG superfamily includes the TDG-MUG family of enzymes, which can excise many pyrimidines from DNA, but do not have the catalytic His residue that facilitates leaving-group departure in UNG enzymes. The findings discussed above for glycosidic bond hydrolysis in deoxynucleotides suggest that the TDG-MUG enzymes follow a stepwise (DN*AN), or perhaps a highly dissociative (ANDN) mechanism. As such, excision of U, T, or U analogs by TDG-MUG enzymes likely involves expulsion of an anionic leaving group. Consistent with this idea, the activity of TDG (kobs) depends on the N1 acidity of the excised pyrimidine base, as demonstrated by a Brønsted-type LFER for the dependence of log kobs on pKaN1 for uracil and cytosine bases harboring various C5-substituents (Fig. 11).83 Moreover, differences in leaving-group quality are enhanced in the hydrophobic TDG active site compared to aqueous solution, as indicated by the steeper slope of the LFER for TDG (βlg = −1.6) relative to non-enzymatic reactions (βlg = −0.87).83 As noted previously, a steeper (more negative) βlg can be expected for reactions in environments that are less-polar than aqueous solution, because charge development in the transition state of the latter is stabilized to a greater extent by solvent.88, 89
Fig. 11.

Brønsted-type LFER for enzymatic (TDG) hydrolysis of 5-substituted dU and dC nucleotides in DNA as reported by Bennett et al.83 The dependence of log kobs on N1 pKa for the base gives βlg = −1.6 ± 0.2; included in the fitting are U, T, FU, ClU, hoU, hmU, FC, hoC, and C (O). Data for BrU, IU, and BrC (□) were not included in the fitting, because kmax values suggested limited access of these bases to the TDG active site.83
Hydrolysis of deoxycytidine and its analogues
As noted above, protonation of the departing base is a common mechanism for catalyzing glycoside hydrolysis, because it enhances leaving-group quality. Non-enzymatic hydrolysis of deoxycytidine (dC) is acid catalyzed via protonation at N3, which ionizes with a pKa of 4.3.38, 90, 91 Leaving-group quality is greatly improved for N3-protonated relative to neutral dC, but the rate of non-enzymatic dC hydrolysis is still very slow at physiological pH because the N3-protonated species is only sparsely populated.
Non-enzymatic hydrolysis of 5-Br-dC is also acid catalyzed by N3 protonation, but the electron-withdrawing halogen substituent enhances acidity at both N1 and N3, with counteracting effects on the reaction rate. While the 5-Br substituent enhances leaving-group quality by increasing N1 acidity (ΔpKaN1 = −2), it also increases N3 acidity (ΔpKaN3 = 1.6) such that such that acid catalysis is less effective.92, 93 Thus, leaving-group quality is better for 5-Br-C versus C, but dC is hydrolyzed as rapidly as 5-Br-dC (95 °C, pH 5) because acid catalysis is more effective for dC than 5-Br-dC. The effect of electron-withdrawing substituents on acidity (N1, N3) and glycosidic bond hydrolysis for dC analogues is an important factor when considering the enzymatic removal of the oxidized forms of 5-methylcytosine found in mammalian DNA, as discussed below.
Enzymatic excision of cytosine and its analogues from DNA is relatively rare and has not been extensively studied. A variant of UNG has aberrant activity for removing C from normal GC pairs;94, 95 a conserved Asn that contacts the Watson-Crick region of uracil is replaced by Asp, such that favorable contacts can be made with cytosine. Similar to the non-enzymatic reaction, the Asn→Asp UNG variant employs acid catalysis, with the Asp facilitating cytosine N3-protonation.95
Although no glycosylases in animals can excise mC, this activity is possessed by the DEMETER (DME) and ROS1 glycosylases in plants, and these enzymes perform key functions in epigenetic regulation.16–18 While some mechanistic insight has been obtained, their approach for catalyzing leaving group departure is unknown. Given that C and mC exhibit very weak N1 acidity (pKaN1 > 12) and the N1 anions have poor leaving group quality,83, 92 it seems reasonable that DME and ROS1 enzymes may activate mC by general acid catalysis via N3-protonation. Two residues that are conserved in DME and ROS1 enzymes and essential for mC excision activity include an Asp and a His,96 which appear to be positioned on opposite sides of the flipped mC base. It is conceivable that one of these residues could facilitate N3-protonation to activate mC.
Endo III-Nth and Nei enzyme families excise some oxidized forms of dC, including 5-hydroxy-dC and 5,6-dihydroxy-dC, and they remove a broad range of other lesions, but it is not presently clear how they catalyze leaving group departure.5
In contrast to other DNA glycosylases, TDG excises many cytosine analogues,83, 91 some of which are also removed by the related E. coli MUG enzymes.97, 98 Most of these TDG substrates have an electron-withdrawing (σm > 0) substituent at C5, which enhances the otherwise dismal leaving-group quality of cytosine. These substrates include 5-fluoro-C (5FC), 5-bromo-C (5BrC), and 5-hydroxy-C (5hoC).31 TDG also excises fC and caC,45, 46 two recently-discovered components of DNA that arise by Tet-mediated oxidation of mC (Fig. 1).13
As shown in Fig. 12, fC is acidic (N1) relative to the established TDG substrates U and T, indicating it is a much better leaving group than cytosine.91 This can be explained by the electronic effect of the formyl substituent (σm = 0.35)99 and resonance stabilization of the fC anion via charge delocalization to the formyl and O2 oxygens. By contrast, the electron-donating carboxylate group (σm = −0.10)99 decreases the N1 acidity of caC, which exists predominantly as an anion at physiological pH (Fig. 12).91, 100 As such, the caC anion is less acidic (N1) than C or hmC, bases that are not removed by TDG (Fig. 12).91 This is consistent with the idea that a dianionic base would be a very poor leaving group. However, the neutral forms of caC, particularly the N3-protonated forms, are much more acidic than the anion (Fig. 12), indicating they are much better leaving groups. This suggests that enzymatic excision of the caC anion from DNA requires acid catalysis, such that the base departs as a monoanion rather than a dianion.
Fig. 12.

Calculated N1 acidity for pyrimidines are given as the free energy (ΔG, kcal mol−1) of deprotonation in the gas phase and bulk solution (parenthetical values).91 Lower values indicate greater N1 acidity and better leaving-group quality.
Consistent with this idea, TDG excision of caC is acid catalyzed (Fig. 13), though it does not appear to involve a general acid from the enzyme. Rather, caC ionizes with an apparent pKa of 5.8 in the TDG active site, likely at N3, facilitated by a conserved Asn side chain.91 In contrast, no evidence of acid catalysis is observed for TDG excision of fC (Fig. 13)91 5FC, BrC, or hoC.83 However, this is not unexpected. While N3-protonation would likely enhance TDG excision of these cytosine analogues it is unlikely to be observed because the N3 pKa values are far below the pH range for which TDG is active.90, 92, 101 Thus, acid catalysis is not observed for TDG excision of cytosines bearing an electron-withdrawing substituent that enhances leaving-group quality (lowers N1 pKa), but it is needed for excision of caC, which has an electron-donating substituent that weakens leaving-group quality (raises N1 pKa). A remaining question is how TDG avoids N3-protonation and subsequent excision of C, mC, hmC, which have N3 pKa values close to that of caC and should be similarly activated by N3-protonation.91
Fig. 13.

pH dependence of TDG activity for G.fC (Δ) and G.caC (O) substrates.91 The G.caC data is best fitted to a model for ionization of one essential protonated group (pKa1 = 5.75 ± 0.03) and a second group (pKa2 = 8.2 ± 0.7) that is not essential but confers higher activity when deprotonated (solid line). Fitting is poor for a model with a single essential protonated group (dotted line).
Concluding Remarks
It was noted by Berti and McCann in 2006 that 2’-deoxynucleosides are poorly represented in transition-state (TS) analysis of N-glycosidic bond cleavage.5 While subsequent studies have been reported, the situation has not changed dramatically. This is particularly true for non-enzymatic reactions, which include only one purine, dAMP,26 and no pyrimidines. TS analysis of non-enzymatic hydrolysis reactions for additional deoxynucleotides would likely be highly informative, including dTMP, 5-methyl-dCMP and some of its oxidized derivatives (5-formyl-dCMP, 5-ca-dCMP) and for some important lesions such as 8-oxo-dGMP. Likewise, TS analysis of enzymatic N-glycosidic bond cleavage includes only two DNA glycosylases (UNG, MutY), plus the “moonlighting” reactions catalyzed by ricin and TP (which do not normally act on deoxynucleotides in DNA). While TS analyses of related reactions can be informative, much remains to be learned about the chemical mechanism of DNA glycosylases. However, TS analysis is challenging and is pursued by a limited number of groups, so progress will likely take time.
Of course, many other approaches are used to elucidate enzyme mechanisms, including structural biology, NMR and other spectroscopic methods, transient kinetics, equilibrium binding, structure-activity relationships, mutational analysis, and, increasingly, computational methods. These various tools have already provided much information regarding the mechanism of DNA glycosylases, as discussed above, and a multi-disciplinary approach will be needed to rigorously address many of the remaining questions, some of which are noted above. We anticipate substantial future progress toward understating the chemical mechanism of DNA glycosylases.
REFERENCES
- 1.Lindahl T. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Hitomi K, Iwai S, Tainer JA. DNA Repair (Amst) 2007;6:410–428. doi: 10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Brooks SC, Adhikary S, Rubinson EH, Eichman BF. Biochim. Biophys. Acta. 2013;1834:247–271. doi: 10.1016/j.bbapap.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stivers JT, Jiang YL. Chem. Rev. 2003;103:2729–2759. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 5.Berti PJ, McCann JA. Chem. Rev. 2006;106:506–555. doi: 10.1021/cr040461t. [DOI] [PubMed] [Google Scholar]
- 6.David SS, O’Shea VL, Kundu S. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cortazar D, Kunz C, Saito Y, Steinacher R, Schar P. DNA Repair (Amst) 2007;6:489–504. doi: 10.1016/j.dnarep.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 8.Nabel CS, Manning SA, Kohli RM. ACS Chem. Biol. 2012;7:20–30. doi: 10.1021/cb2002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pastor WA, Aravind L, Rao A. Nat. Rev. Mol. Cell Biol. 2013;14:341–356. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohli RM, Zhang Y. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfaffeneder T, Hackner B, Truss M, Munzel M, Muller M, Deiml CA, Hagemeier C, Carell T. Angew Chem Int Ed Engl. 2011;50:7008–7012. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 16.Gong Z, Morales-Ruiz T, Ariza RR, Roldan-Arjona T, David L, Zhu JK. Cell. 2002;111:803–814. doi: 10.1016/s0092-8674(02)01133-9. [DOI] [PubMed] [Google Scholar]
- 17.Choi Y, Gehring M, Johnson L, Hannon M, Harada JJ, Goldberg RB, Jacobsen SE, Fischer RL. Cell. 2002;110:33–42. doi: 10.1016/s0092-8674(02)00807-3. [DOI] [PubMed] [Google Scholar]
- 18.Morales-Ruiz T, Ortega-Galisteo AP, Ponferrada-Marin MI, Martinez-Macias MI, Ariza RR, Roldan-Arjona T. Proc Natl Acad Sci USA. 2006;103:6853–6858. doi: 10.1073/pnas.0601109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maiti A, Drohat AC. J Biol Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schramm VL. Accounts Chem. Res. 2003;36:588–596. doi: 10.1021/ar0200495. [DOI] [PubMed] [Google Scholar]
- 21.Guthrie RD, Jencks WP. Accounts Chem. Res. 1989;22:343–349. [Google Scholar]
- 22.Berti PJ, Tanaka KSE. Adv. Phys. Org. Chem. 2002;37:239–314. [Google Scholar]
- 23.Werner RM, Stivers JT. Biochemistry. 2000;39:14054–14064. doi: 10.1021/bi0018178. [DOI] [PubMed] [Google Scholar]
- 24.Chen XY, Berti PJ, Schramm VL. J. Am. Chem. Soc. 2000;122:6527–6534. [Google Scholar]
- 25.McCann JAB, Berti PJ. J Am Chem Soc. 2008;130:5789–5797. doi: 10.1021/ja711363s. [DOI] [PubMed] [Google Scholar]
- 26.McCann JAB, Berti PJ. J. Am. Chem. Soc. 2007;129:7055–7064. doi: 10.1021/ja067371l. [DOI] [PubMed] [Google Scholar]
- 27.Admiraal SJ, O'Brien PJ. Biochemistry. 2010;49:9024–9026. doi: 10.1021/bi101380d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parkin DW, Leung HB, Schramm VL. J Biol Chem. 1984;259:9411–9417. [PubMed] [Google Scholar]
- 29.Mentch F, Parkin DW, Schramm VL. Biochemistry. 1987;26:921–930. doi: 10.1021/bi00377a037. [DOI] [PubMed] [Google Scholar]
- 30.Chen XY, Berti PJ, Schramm VL. J Am Chem Soc. 2000;122:1609–1617. [Google Scholar]
- 31.Dinner AR, Blackburn GM, Karplus M. Nature. 2001;413:752–755. doi: 10.1038/35099587. [DOI] [PubMed] [Google Scholar]
- 32.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drohat AC, Xiao G, Tordova M, Jagadeesh J, Pankiewicz KW, Watanabe KA, Gilliland GL, Stivers JT. Biochemistry. 1999;38:11876–11886. doi: 10.1021/bi9910880. [DOI] [PubMed] [Google Scholar]
- 34.Drohat AC, Jagadeesh J, Ferguson E, Stivers JT. Biochemistry. 1999;38:11866–11875. doi: 10.1021/bi9910878. [DOI] [PubMed] [Google Scholar]
- 35.Drohat AC, Stivers JT. J. Am. Chem. Soc. 2000;122:1840–1841. [Google Scholar]
- 36.Jiang YL, Drohat AC, Ichikawa Y, Stivers JT. J. Biol. Chem. 2002;277:15385–15392. doi: 10.1074/jbc.M200634200. [DOI] [PubMed] [Google Scholar]
- 37.Bianchet MA, Seiple LA, Jiang YL, Ichikawa Y, Amzel LM, Stivers JT. Biochemistry. 2003;42:12455–12460. doi: 10.1021/bi035372+. [DOI] [PubMed] [Google Scholar]
- 38.Shapiro R, Danzig M. Biochemistry. 1972;11:23–29. doi: 10.1021/bi00751a005. [DOI] [PubMed] [Google Scholar]
- 39.Shapiro R, Kang S. Biochemistry. 1969;8:1806–1810. doi: 10.1021/bi00833a004. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz PA, Vetticatt MJ, Schramm VL. J Am Chem Soc. 2010;132:13425–13433. doi: 10.1021/ja105041j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birck MR, Schramm VL. J Am Chem Soc. 2004;126:2447–2453. doi: 10.1021/ja039260h. [DOI] [PubMed] [Google Scholar]
- 42.Zoltewicz JA, Clark DF, Sharpless TW, Grahe G. J Am Chem Soc. 1970;92:1741–1749. doi: 10.1021/ja00709a055. [DOI] [PubMed] [Google Scholar]
- 43.Sharma S, Lee JK. J. Org. Chem. 2002;67:8360–8365. doi: 10.1021/jo0204303. [DOI] [PubMed] [Google Scholar]
- 44.Ho MC, Sturm MB, Almo SC, Schramm VL. Proc Natl Acad Sci USA. 2009;106:20276–20281. doi: 10.1073/pnas.0911606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim Y, Robertus JD. Protein Eng. 1992;5:775–779. doi: 10.1093/protein/5.8.775. [DOI] [PubMed] [Google Scholar]
- 46.Monzingo AF, Robertus JD. J Mol Biol. 1992;227:1136–1145. doi: 10.1016/0022-2836(92)90526-p. [DOI] [PubMed] [Google Scholar]
- 47.Michelson AZ, Rozenberg A, Tian Y, Sun X, Davis J, Francis AW, O'Shea VL, Halasyam M, Manlove AH, David SS, Lee JK. J Am Chem Soc. 2012;134:19839–19850. doi: 10.1021/ja309082k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA. Nat Struct Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 49.Lee S, Verdine GL. Proc Natl Acad Sci USA. 2009;106:18497–18502. doi: 10.1073/pnas.0902908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brinkmeyer MK, Pope MA, David SS. Chem Biol. 2012;19:276–286. doi: 10.1016/j.chembiol.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Porello SL, Williams SD, Kuhn H, Michaels ML, David SS. J Am Chem Soc. 1996;118:10684–10692. [Google Scholar]
- 52.Livingston AL, O'Shea VL, Kim T, Kool ET, David SS. Nat Chem Biol. 2008;4:51–58. doi: 10.1038/nchembio.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Francis AW, Helquist SA, Kool ET, David SS. J Am Chem Soc. 2003;125:16235–16242. doi: 10.1021/ja0374426. [DOI] [PubMed] [Google Scholar]
- 54.O'Brien PJ, Ellenberger T. Biochemistry. 2003;42:12418–12429. doi: 10.1021/bi035177v. [DOI] [PubMed] [Google Scholar]
- 55.Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T. Proc Natl Acad Sci USA. 2000;97:13573–13578. doi: 10.1073/pnas.97.25.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Michelson AZ, Chen M, Wang K, Lee JK. J Am Chem Soc. 2012;134:9622–9633. doi: 10.1021/ja211960r. [DOI] [PubMed] [Google Scholar]
- 57.Scheller KH, Sigel H. J. Am. Chem. Soc. 1983;105:3005–3014. [Google Scholar]
- 58.Liu M, Xu M, Lee JK. J. Org. Chem. 2008;73:5907–5914. doi: 10.1021/jo800891c. [DOI] [PubMed] [Google Scholar]
- 59.Sun XJ, Lee JK. J. Org. Chem. 2007;72:6548–6555. doi: 10.1021/jo070996x. [DOI] [PubMed] [Google Scholar]
- 60.Cao C, Kwon K, Jiang YL, Drohat AC, Stivers JT. J. Biol. Chem. 2003;278:48012–48020. doi: 10.1074/jbc.M307500200. [DOI] [PubMed] [Google Scholar]
- 61.Drohat AC, Kwon K, Krosky DJ, Stivers JT. Nat. Struct. Biol. 2002;9:659–664. doi: 10.1038/nsb829. [DOI] [PubMed] [Google Scholar]
- 62.Metz AH, Hollis T, Eichman BF. EMBO J. 2007;26:2411–2420. doi: 10.1038/sj.emboj.7601649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berdal KG, Johansen RF, Seeberg E. EMBO J. 1998;17:363–367. doi: 10.1093/emboj/17.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O'Brien PJ, Ellenberger T. J. Biol. Chem. 2004;279:9750–9757. doi: 10.1074/jbc.M312232200. [DOI] [PubMed] [Google Scholar]
- 65.Prakash A, Doublie S, Wallace SS. Prog. Mol. Biol. Transl. Sci. 2012;110:71–91. doi: 10.1016/B978-0-12-387665-2.00004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fromme JC, Verdine GL. J. Biol. Chem. 2003;278:51543–51548. doi: 10.1074/jbc.M307768200. [DOI] [PubMed] [Google Scholar]
- 67.Jang YH, Goddard WA, Noyes KT, Sowers LC, Hwang S, Chung DS. Chem. Res. Toxicol. 2002;15:1023–1035. doi: 10.1021/tx010146r. [DOI] [PubMed] [Google Scholar]
- 68.Bialkowski K, Cysewski P, Olinski R. Z. Naturforsch. C. 1996;51:119–122. doi: 10.1515/znc-1996-1-219. [DOI] [PubMed] [Google Scholar]
- 69.van der Kemp PA, Thomas D, Barbey R, de Oliveira R, Boiteux S. Proc Natl Acad Sci USA. 1996;93:5197–5202. doi: 10.1073/pnas.93.11.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nash HM, Lu R, Lane WS, Verdine GL. Chem Biol. 1997;4:693–702. doi: 10.1016/s1074-5521(97)90225-8. [DOI] [PubMed] [Google Scholar]
- 71.Bruner SD, Norman DP, Verdine GL. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 72.Zharkov DO, Rosenquist TA, Gerchman SE, Grollman AP. J Biol Chem. 2000;275:28607–28617. doi: 10.1074/jbc.M002441200. [DOI] [PubMed] [Google Scholar]
- 73.Fromme JC, Bruner SD, Yang W, Karplus M, Verdine GL. Nat Struct Biol. 2003;10:204–211. doi: 10.1038/nsb902. [DOI] [PubMed] [Google Scholar]
- 74.Sartori AA, Fitz-Gibbon S, Yang H, Miller JH, Jiricny J. EMBO J. 2002;21:3182–3191. doi: 10.1093/emboj/cdf309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dong L, Mi R, Glass RA, Barry JN, Cao W. DNA Repair (Amst) 2008;7:1962–1972. doi: 10.1016/j.dnarep.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 76.Mi R, Dong L, Kaulgud T, Hackett KW, Dominy BN, Cao W. J Mol Biol. 2009;385:761–778. doi: 10.1016/j.jmb.2008.09.038. [DOI] [PubMed] [Google Scholar]
- 77.Lee HW, Brice AR, Wright CB, Dominy BN, Cao W. J Biol Chem. 2010;285:41483–41490. doi: 10.1074/jbc.M110.150003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee HW, Dominy BN, Cao W. J Biol Chem. 2011;286:31282–31287. doi: 10.1074/jbc.M111.249524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rogstad KN, Jang YH, Sowers LC, Goddard WA., 3rd Chem Res Toxicol. 2003;16:1455–1462. doi: 10.1021/tx034068e. [DOI] [PubMed] [Google Scholar]
- 80.Roy KB, Miles HT. Nucleosides Nucleotides. 1983;2:231–242. [Google Scholar]
- 81.Vongchampa V, Dong M, Gingipalli L, Dedon P. Nucleic Acids Res. 2003;31:1045–1051. doi: 10.1093/nar/gkg177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wuenschell GE, O'Connor TR, Termini J. Biochemistry. 2003;42:3608–3616. doi: 10.1021/bi0205597. [DOI] [PubMed] [Google Scholar]
- 83.Bennett MT, Rodgers MT, Hebert AS, Ruslander LE, Eisele L, Drohat AC. J Am Chem Soc. 2006;128:12510–12519. doi: 10.1021/ja0634829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Savva R, McAuley-Hecht K, Brown T, Pearl L. Nature. 1995;373:487–493. doi: 10.1038/373487a0. [DOI] [PubMed] [Google Scholar]
- 85.Drohat AC, Stivers JT. Biochemistry. 2000;39:11865–11875. doi: 10.1021/bi000922e. [DOI] [PubMed] [Google Scholar]
- 86.Wibley JE, Waters TR, Haushalter K, Verdine GL, Pearl LH. Mol. Cell. 2003;11:1647–1659. doi: 10.1016/s1097-2765(03)00235-1. [DOI] [PubMed] [Google Scholar]
- 87.Van Schepdael A, Ossembe N, Herdewijn P, Roets E, Hoogmartens J. J. Pharm. Biomed. Anal. 1993;11:345–351. doi: 10.1016/0731-7085(93)80027-x. [DOI] [PubMed] [Google Scholar]
- 88.Jencks WP. Cold Spring. Harb. Symp. Quant. Biol. 1972;36:1–11. doi: 10.1101/sqb.1972.036.01.004. [DOI] [PubMed] [Google Scholar]
- 89.Hollfelder F, Herschlag D. Biochemistry. 1995;34:12255–12264. doi: 10.1021/bi00038a021. [DOI] [PubMed] [Google Scholar]
- 90.La Francois CJ, Jang YH, Cagin T, Goddard WA, Sowers LC. Chem. Res. Toxicol. 2000;13:462–470. doi: 10.1021/tx990209u. [DOI] [PubMed] [Google Scholar]
- 91.Maiti A, Michelson AZ, Armwood CJ, Lee JK, Drohat AC. J Am Chem Soc. 2013;135:15813–15822. doi: 10.1021/ja406444x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wempen I, Fox JJ. J. Med. Chem. 1963;122:688–693. doi: 10.1021/jm00342a013. [DOI] [PubMed] [Google Scholar]
- 93.Wempen I, Fox JJ. J. Am. Chem. Soc. 1964;86:2474–2477. [Google Scholar]
- 94.Kavli B, Slupphaug G, Mol CD, Arvai AS, Peterson SB, Tainer JA, Krokan HE. EMBO J. 1996;15:3442–3447. [PMC free article] [PubMed] [Google Scholar]
- 95.Kwon K, Jiang YL, Stivers JT. Chem. Biol. 2003;10:351–359. doi: 10.1016/s1074-5521(03)00077-2. [DOI] [PubMed] [Google Scholar]
- 96.Brooks SC, Fischer RL, Huh JH, Eichman BF. Biochemistry. 2014;53:2525–2532. doi: 10.1021/bi5002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.O'Neill RJ, Vorob'eva OV, Shahbakhti H, Zmuda E, Bhagwat AS, Baldwin GS. J Biol Chem. 2003;278:20526–20532. doi: 10.1074/jbc.M210860200. [DOI] [PubMed] [Google Scholar]
- 98.Morera S, Grin I, Vigouroux A, Couve S, Henriot V, Saparbaev M, Ishchenko AA. Nucleic Acids Res. 2012;40:9917–9926. doi: 10.1093/nar/gks714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hansch C, Leo A, Taft RW. Chem. Rev. 1991;91:165–195. [Google Scholar]
- 100.Sumino M, Ohkubo A, Taguchi H, Seio K, Sekine M. Bioorg Med Chem Lett. 2008;18:274–277. doi: 10.1016/j.bmcl.2007.10.081. [DOI] [PubMed] [Google Scholar]
- 101.Maiti A, Drohat AC. DNA Repair. 2011;10:545–553. doi: 10.1016/j.dnarep.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]