

Abstract
Severe sepsis and septic shock are leading causes of morbidity and mortality worldwide. Infection-associated inflammation promotes the development and progression of adverse outcomes in sepsis. The effects of heterodimeric Interleukin-27 (IL-27; p28/EBI3) have been implicated in the natural course of sepsis, while the molecular mechanisms underlying regulation of gene expression and release of IL-27 in sepsis are poorly understood. In this report we studied the events regulating the p28 subunit of IL-27 in endotoxic shock and polymicrobial sepsis following cecal ligation and puncture. Neutralizing antibodies to IL-27(p28) improved survival rates, confined cytokine release and reduced bacterial burden in C57BL/6 mice during sepsis. Genetic disruption of IL-27 signaling enhanced the respiratory burst of macrophages. Experiments using splenectomized mice or treatment with clodronate liposomes suggested macrophages in the spleen may be a significant source of IL-27(p28) during sepsis. In cultures of TLR4-activated macrophages, the frequency of F4/80+CD11b+IL-27(p28)+ cells was reduced with addition of IL-10. IL-10 antagonized both MyD88-dependent and TRIF-dependent release of IL-27(p28). Genetic deletion of STAT3 in Tie2-Cre/STAT3flox macrophages completely interrupted the inhibition of IL-27(p28) by IL-10 after TLR4-activation. In contrast, IL-10 remained fully active to suppress IL-27(p28) with deletion of SOCS3 in Tie2-Cre/SOCS3flox macrophages. Blockade of the IL-10 receptor by antibody or genetic deficiency of IL-10 resulted in 3-5-fold higher concentrations of IL-27(p28) in endotoxic shock and polymicrobial sepsis. Our studies identify IL-10 as a critical suppressing factor for IL-27(p28) production during infection-associated inflammation. These findings may be helpful for a beneficial manipulation of adverse IL-27(p28) release during sepsis.
Keywords: Inflammation, macrophages, cecal ligation and puncture, shock, lipopolysaccharide
Introduction
Sepsis-associated mortality is a major burden on public health. It is estimated that approximately 2,800,000 annual cases of sepsis occur in high-income countries leading to mortality rates of 20-50% (1, 2). The molecular events of sepsis remain incompletely understood. There is a wide consensus that unbalanced inflammatory responses during bacterial, fungal or viral infections contribute to the catastrophic progression of sepsis by the release of immune mediators and reactive oxygen species (3, 4).
Interleukin-27 (IL-27) is a novel mediator formed by association of the subunit proteins, IL-27(p28) and EBI3 (5, 6). IL-27 belongs to the IL-6/IL-12 family of cytokines and binds to the IL-27RA (WSX-1) and gp130 receptor complex (7). IL-27(p28) (also termed IL-30) and EBI3 are not always expressed in the same cells and have additional binding partners, suggesting that each subunit may have unique functions (7-9). EBI3 can also associate with the p35 subunit of IL-12 to form IL-35(10).
At present there is an ongoing controversy about the classification of IL-27 as a mediator that will either promote or dampen inflammation. In a model of chronic colitis, interruption of IL-27 signaling resulted in reduced inflammation and higher numbers of Foxp3+ regulatory T cells (11). Blockade of heterodimeric IL-27 by administration of IL-27RA-Fc fusion soluble receptor improved the outcome of peritonitis-induced sepsis (12). Furthermore, IL-27 signaling was associated with increased susceptibility during experimental arthritis (13). These studies suggest IL-27 is proinflammatory.
In contrast, IL-27 also has potent anti-inflammatory effects. For example, IL-27RA-/- mice developed a lethal T-cell mediated inflammatory disease and excessive cytokine production (IL-6, TNFα) in protozoan infections (14, 15). In experimental autoimmune encephalitis, both heterodimeric IL-27 as well as purified IL-27(p28) suppressed Th17 cells (16, 17). A major mechanism for IL-27 limiting chronic inflammatory responses is its capacity to induce IL-10 production from Th1, Th2, Th17, Treg and CD8+ cells in a STAT1/STAT3 dependent manner (18, 19).
IL-10 mediates its immunosuppressive effects and macrophage M2 polarization via the IL-10Rα/IL-10Rβ receptor complex (20, 21). Upon binding of IL-10, IL-10Rα associates with Jak1, IL-10Rβ and tyrosine kinase 2 (Tyk2). Phosphorylation of Jak1 and Tyk2 facilitates direct interaction with STAT3 and subsequent IL-10 induced gene activation (20, 22).
While the role of IL-27 for induction of IL-10 by lymphocytes has been well characterized, little is known about IL-10 reciprocally controlling the release of IL-27(p28) from antigen-presenting cells, which according to in vitro studies appear to be a predominant cellular source of IL-27.The aim of our current study was to assess the regulatory networks associated with IL-27(p28) production during experimental sepsis. We uncover that IL-27(p28) release was powerfully antagonized by IL-10 via STAT3 without requirement of SOCS3. The increased susceptibility of IL-10-/- mice during sepsis was partially explained by an uncontrolled release of detrimental IL-27(p28).
Materials and Methods
Animals
All procedures with animals were performed in accordance with the U.S. National Institutes of Health guidelines, the University Committee on Use and Care of Animals (UCUCA) of the University of Michigan, the animal protection act of Germany, the State Investigation Office of Rhineland-Palatinate and directive 2010/63/EU of the European Parliament and of the Council of the European Union. TRIF-/- mice were bred at the of Michigan. STAT3 deficient mice (Tie2-Cre/STAT3f1/f1) and SOCS3 deficient mice (Tie2-Cre/SOCS3f1/f1) were generated by breeding Tie2-Cre mice with floxed STAT3 mice or floxed SOCS3 mice at St. Jude Children's Research Hospital (23). STAT3 deficient mice were phenotyped by Western blotting demonstrating missing phospho-STAT3 in IL-6-activated macrophages. Male mice of the strains C57BL/6J, IL-10-/- (B6.129P2-Il10tm1Cgn), MyD88 (B6.129P2(SJL)-Myd88tm1.1Defr/J) and TLR4-/- (B6.B10ScN-Tlr4lps-del/JthJ) were from the Jackson Laboratories (Bar Harbor, USA). IL-27RA-/- mice (approximate C57BL6/NJ background) were obtained from the Jackson Laboratories, backcrossed to C57BL/6J mice for one generation at the University Medical Center Mainz and used together with wild type littermate controls. All animals were housed under specific pathogen free conditions.
In Vivo Experiments
Splenectomy was performed in male C57BL/6J mice. A left-sided infracostal incision (1-1.5 cm) was made, the peritoneum opened, the splenic mesenterium ligated with 3-0 silk suture and the spleen removed before wound closure with 6-0 silk suture. For sham surgery the peritoneum was opened in a similar manner without ligation and removal of the spleen. After the surgery animals received 1 mg Piperacillin/Tazobactam in 1 ml NaCl 0.9% s.c.
For endotoxemia randomized and age/sex matched groups were used and bodyweight of each individual animal was measured directly before injection with LPS (10 mg/kg BW i.p., E. coli 0111:B4, Sigma-Aldrich).
Cecal ligation and puncture was performed as described before (24). The surgeon was blinded to the nature of the randomized and age/sex matched groups of mice. A through-and-through puncture was performed with an 18G (for ‘high-grade CLP’) or 21G (for ‘mid-grade CLP’) needle and feces extruded to ensure patency. Sham mice underwent anesthesia, laparotomy and wound closure. After surgery animals received 1 ml NaCl 0.9% s.c for fluid resuscitation. Body surface temperature was measured with an YSI4600/YSI427 thermometer. Blood plasma was collected using EDTA (5-10 mM). During survival studies mice were monitored every 12 h for at least 10 days.
Determination of Colony forming units (CFU)
Blood and peritoneal lavage fluids were serially diluted with sterile PBS, plated on 5% sheep blood agar (Remel, Lenexa, USA) and incubated at 37°C for 24 h under aerobic conditions. CFU were then counted and numbers multiplied by the dilution factor.
Cell Cultures of Macrophages
For bone marrow derived macrophages (BMDM) femurs and tibias were flushed with PBS through 40 μm filters. Cells were incubated in RPMI 1640 (25 mM HEPES, 100 units/ml penicillin-streptomycin, 20% FCS, 30% L-cell conditioned media) for 7 days. BMDM were plated at 5×105 cells/ml in RPMI 1640 (25 mM HEPES, 100 units/ml penicillin-streptomycin, 0.1% BSA) and incubated at 37°C, 5% CO2. Peritoneal elicited macrophages (PEM) were collected 4 days after i.p. injection with 1.5 ml thioglycollate 2.4% (w/v) (Becton Dickinson). For experiments with splenocytes, spleens were removed from C57BL/6J mice, processed through 40 μm filters and washed with PBS before counting. RAW 264.7 and MH-S cells were maintained in RPMI 1640 (25 mM HEPES, 100 units/ml penicillin, 10% FCS).
Quantification of Proteins by ELISA and Bead-based Immunoassay
ELISA kits for mouse IL-27(p28) and IL-10 were from R&D Systems. The IL-27(p28) ELISA has <5% cross-reactivity with rmIL-27(EBI3/p28) and a detection limit of ∼10 pg/ml. Bead-based immunoassays were used for detections of multiple mediators (Bioplex Pro™ mouse cytokine bead-based immunoassay, BioRad, USA) in plasma or phosphorylated signaling proteins (Akt (Ser473), c-Jun (Ser63), CREB (Ser133), ERK1/2 (Thr202/Tyr204, Thr185/Tyr187), JNK (Thr183/Tyr185), MEK1 (Ser217/Ser221), NFκB (Ser536), p38 MAPK (Thr180/Tyr182), STAT3 (Ser727), all from BioRad). Quantification was performed on the Luminex xMAP™/Bioplex-200 System with Bioplex Manager™ Software 5.0.
Isolation of mRNA and Real Time PCR
Total RNA was obtained by either the Trizol method or RNeasy Kit (Qiagen). The cDNA was generated with TaqMan® Reverse Transcription Reagents (Applied Biosystems) in a GeneAmp® PCR System 9700. Amplification was performed with SYBR® Green Mastermix in the 7500 Real Time PCR System (Applied Biosystems). Results were analyzed by the 2−ddCt relative quantification method and normalized to GAPDH. For primer sequences see Supplementary Table 1.
Flow Cytometry
For intracellular cytokine detection, cells were incubated with Monensin (2 μM, Sigma-Aldrich) and stained using the Cytofix/Cytoperm Kit (BD Biosciences) and BD Fc-Block. For phosphoprotein detection, cells were permeabilized with Perm-Buffer III (BD Biosciences). 50,000 events were acquired on a BD LSR II flow cytometer or BD FACSCanto II (BD Biosciences) and data analyzed with WinList for Win32 3.0 Software (Verity Software). As a gating strategy we used FSC and SSC to exclude cell aggregates and debris from analysis. All antibodies used were anti-mouse together with matched fluorochrome labeled isotype controls. From BD Pharmingen: PE p-STAT3 (pY705) (clone 4/P-STAT3). From eBioscience: PE IL-27(p28) (clone MM27-7B1), APC F4/80 (clone BM8), efluor450 CD11b (clone M1/70).
Quantification of Oxidative Burst Activity
Peritoneal elicited cells (PECs) were harvested 20 h after i.p. injection with 1.5 ml thioglycollate 2.4% (w/v). 1×106 cells/sample were incubated with 2-4×107 opsonized E. coli for 10 min at 37°C (Phagoburst kit; Orpegen Pharma, Heidelberg, Germany). Dihydrorhodamine (DHR) 123 (10 min incubation) and antibodies were added before analysis by flow cytometry as described above.
Fluorescence Microscopy
Cryosections of spleens were fixed with 4% formaldehyde solution (Thermo Scientific). Macrophages were grown in Lab-Tek chamber slides (Thermo Scientific) and stimulated in the co-presence of Monensin. Blocking (7% BSA, 10% normal mouse serum) was followed by overnight incubation with primary antibodies. After incubation with secondary antibodies the slides were mounted with prolong gold with DAPI (Invitrogen). Antibodies used (from eBioscience) were rat anti-mouse F4/80 (clone BM8), rat IgG2aκ isotype control, mouse isotype IgG2aκ control, mouse anti-mouse IL-27(p28) (clone MM27-7B1, a gift from Dr. P. Just, eBioscience) and (from Invitrogen) rabbit anti-mouse AF594 IgG, donkey anti-rat AF594 IgG. Microscopic images were acquired using an Olympus BX-51 microscope (40×/0.9, 60×/1.4 oil, 100×/1.4 oil) with Olympus DP-70 camera and DP Controller Software.
Antibodies and Reagents
LPS was from E. coli (0111:B4, Sigma-Aldrich). Neutralizing polyclonal goat anti-mouse IL-27(p28) IgG, neutralizing polyclonal goat anti-mouse IL-10, total goat IgG, rmIL-10 were all from R&D Systems. Neutralizing anti-mouse IL-10R (clone 1B1.3a) was affinity purified using protein G–Sepharose (GE Healthcare, Munich, Germany) according to a standard protocol and used together with purified rat IgG1κ isotype antibody (BioLegend). Multilamellar liposomes were prepared from phosphatidylcholine and cholesterol (from Sigma) and filled with clodronate (dichloromethylene-bisphosphonate, from Roche Diagnostics) or PBS (25).
Statistical Analysis
The GraphPad Prism Version 5.04 software was used for statistical analysis. In vitro experiments were performed independently two or three times and in vivo data were generated with the numbers of mice as indicated in the figure legends. All values are expressed as mean and error bars represent s.e.m. Data sets were analyzed by one-way ANOVA, two-tailed Student's t-test and survival curves by the log-rank (Mantel-Cox) test. We considered differences significant when P < 0.05.
Results
Blockade of IL-27(p28) improves the outcome of sepsis
To confirm the biological relevance of IL-27 during sepsis, we used neutralizing polyclonal antibodies directed against the p28 subunit in models of endotoxic shock and polymicrobial sepsis following cecal ligation and puncture (CLP). Treatment with anti-mouse IL-27(p28) antibody as compared to normal IgG dramatically improved survival during endotoxic shock (25% vs. 80%; Fig. 1A). Similarly, in polymicrobial sepsis following CLP, neutralization of IL-27(p28) with anti-IL-27(p28) antibody significantly improved mortality rates (Fig. 1B). Time course studies revealed that maximum concentrations of IL-27(p28) in plasma were detectable by ELISA at 6 h after LPS injections in vivo (Fig. 1C). Blocking of IL-27(p28) during endotoxic shock attenuated the systemic “cytokine storm”, defined by reduced concentrations of mediators such as IFNγ, IL-17, IL-1α, IL1β and MCP-1 in plasma (Fig. 1D). For IFNγ and IL-1β it has been established that these mediators contribute to the detrimental pathophysiology of sepsis (3). Furthermore, blocking of IL-27(p28) during CLP improved bacterial clearance in blood and the peritoneal compartment (Fig. 1E, 1F). Counts of CFUs in blood and peritoneal lavage fluids were significantly reduced at 20 h following CLP, while effects at 10 h were less consistent for peritoneal lavage fluids (Fi.g 1 E, 1F). When plasma mediators such as IL-1β, IL-17 and IFNγ were quantified at 10 h and 20 h following CLP, these mediators were reduced by anti-IL-27(p28) antibody treatment at the 20 h time point as compared to mediator concentrations in the control IgG group at 20 h (Fig. 1G). Differences between the two groups were less prominent at the 10 h time point (Fig. 1G), which may be related to the fact that the release of IL-27(p28) requires several hours to occur (Fig. 1C). Furthermore, blockade of IL-27(p28) during endotoxic shock or polymicrobial sepsis following CLP did not significantly reduce the appearance of IL-10 at all time points studied (Fig. 1D, Fig. 1G).
Figure 1. Neutralization of IL-27(p28) is protective during polymicrobial sepsis and endotoxic shock.

A. Survival of Wt mice during endotoxic shock (LPS 10 mg/kg body weight i.p.) following treatment with neutralizing anti-IL-27(p28) antibody (40 μg/mouse i.p., n=10) or control IgG (40 μg/mouse i.p., n=12). B. Survival of Wt mice after CLP (‘high-grade’) with treatment of either control IgG or neutralizing anti-IL-27(p28) antibody (40 μg/mouse i.p., n=10 for both groups). C. Time course for appearance of IL-27(p28) in plasma of Wt mice during endotoxic shock, n=4-6 mice for each time point, ELISA. D. Reduction of plasma mediators during endotoxic shock in Wt mice using control IgG or anti-IL-27(p28) antibody (n=5/group), 12 h, bead-based assay. E. Quantification of colony forming units (CFU) in peritoneal lavage fluids of Wt mice at different time points after CLP with application of control IgG or blocking anti-IL-27(p28) antibody (40 μg/mouse i.p., n=10 for both groups). F. Determination of bacteremia (CFU) in blood from the same experiment described under E. G. Detection of plasma mediators after 10 h and 20 h following CLP in Wt mice treated with neutralizing anti-IL-27(p28) antibody or control IgG (n≥7/group). Antibodies were injected 1 h before CLP or LPS in all experiments. All experiments were performed with the numbers of mice per group as indicated. * P < 0.05, ** P < 0.01, *** P < 0.001, n.s. denotes not significant.
Next, we sought to study the effects of IL-27 on production of reactive oxygen species (ROS). F4/80+CD11b+ peritoneal elicited macrophages from IL-27RA-/- mice displayed an increased respiratory burst following activation by opsonized E. coli as compared to C57BL/6(Wt) macrophages (Fig. 2A, 2B). This enhanced production of ROS may explain a higher bactericidal activity and improved pathogen clearance following blockade of IL-27 during sepsis. Our findings are in accordance with another report on targeting of heterodimeric IL-27 with IL-27RA-Fc fusion proteinor using EBI3-/- mice in sepsis (12).
Figure 2. Enhanced oxidative burst in macrophages with genetic disruption of IL-27 signaling.
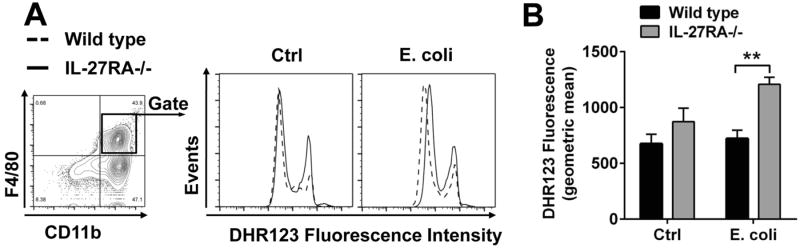
A. Peritoneal elicited cells from IL-27RA-/- mice and littermate Wt mice were incubated with opsonized E. coli (2-4×107) for 10 min before addition of green fluorescent DHR123 for additional 10 min as an indicator of reactive oxygen production (ROS). Cells were then stained with antibodies and flow cytometry gating was used to identify F4/80+CD11b+ macrophages. Following addition of opsonized E. coli F4/80+CD11b+ macrophages derived from IL-27RA-/- mice showed increased DHR123 fluorescence as compared to Wt mice. B. Geometric mean and statistical analysis of DHR123 fluorescence from the same experiment as shown in A. Experiments were performed with n=4 mice/group. ** P < 0.01.
Macrophages in the spleen are a major source of IL-27(p28) during sepsis
To define the cellular sources of IL-27(p28) during sepsis, various organs were collected from C57BL/6J (Wt) mice following endotoxic shock and analyzed for IL-27(p28) mRNA expression (normalized to GAPDH). Highest increases of IL-27(p28) mRNA were detected in spleens (73-fold) and lungs (9-fold), with lowest levels in heart (1-fold) (Fig. 3A). The spleen is known to participate in the inflammatory reflex and splenectomy protects against sepsis-associated lethality (26, 27). When splenectomized mice were subjected to endotoxemia, plasma concentrations of IL-27(p28) were greatly reduced (Fig. 3B). We were not able to visualize IL-27(p28) by immunohistofluorescence in spleen (data not shown), which may be explained by rapid in vivo secretion of IL-27(p28) in the absence of golgi transport inhibitors. Next, we depleted F4/80+ macrophages in spleen (and liver) by i.p. injection of clodronate liposomes and used PBS containing liposomes as negative controls (Fig. 3C). Following treatment with clodronate liposomes, mice during endotoxic shock were deficient in IL-27(p28) production (-86%) (Fig. 3D). Other mediators including IFNγ (-31%), MCP-1 (-50%), RANTES (-24%) and IL-1α (-50%) were less affected by macrophage depletion followed by endotoxic shock (data not shown). As reported before, treatment with clodronate liposomes did neither induce cytokine responses nor activated macrophages (28-30). Collectively, our data suggested that splenic macrophages are a cellular source of IL-27(p28) during sepsis.
Figure 3. Macrophages localized in the spleen are a major cellular source of IL-27(p28) in vivo.
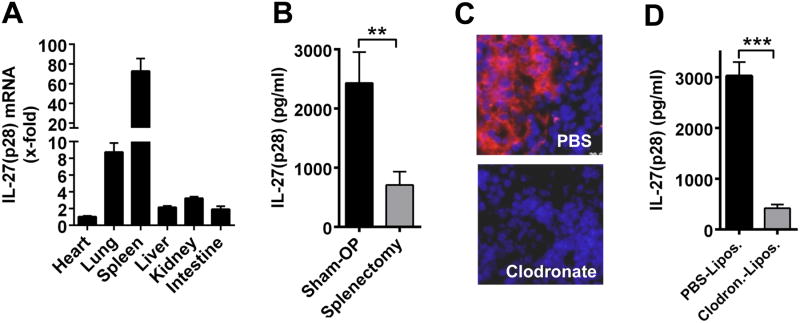
A. RT-PCR of mRNA for IL-27(p28) in organ homogenates from Wt mice 6 h after endotoxic shock (LPS 10 mg/kg body weight i.p., n=5). Relative IL-27(p28) expression levels in the heart were used as 1-fold, normalization using GAPDH. B. IL-27(p28) in plasma 6 h after endotoxic shock. Wt mice underwent either sham surgery (n=10) or splenectomy (n=10) 7 days earlier, ELISA. C. Depletion of F4/80+ macrophages (stained in red) in spleens (nuclei stained in blue) following treatment with PBS liposomes (controls) or clodronate liposomes (300 μl/-72 h and 150 μl/-24 h i.p. before LPS); n=4/group; Olympus BX-51 microscope [100×/1.4 oil] with Olympus DP-70 camera. D. IL-27(p28) in plasma 6 h after endotoxic shock following pretreatment with clodronate liposomes (n=10) or PBS liposomes (n=10) as described above. All experiments were performed with the numbers of mice per group as indicated. ** P < 0.01, *** P < 0.001.
IL-10 mediates suppression of IL-27(p28) release
To characterize how production of IL-27(p28) is regulated during inflammation, we used cultures of bone marrow derived macrophages (BMDM). BMDM were phenotyped by flow cytometry as >95% F4/80+CD11b+ double-positive and CD11clow (Supplementary Fig. 1A). LPS in a dose range of 1 ng/ml – 1000 ng/ml was effective to induce IL-27(p28) from BMDM (Supplementary Fig. 1B).
Since IL-27 signaling is known to induce IL-10 secretion by T cells, we sought to investigate if IL-10 would mediate a negative feedback on production of IL-27(p28) in macrophages. Indeed, recombinant IL-10 caused long-lasting suppression of IL-27(p28) release in supernatants from LPS-activated BMDM derived from Wt mice (Fig. 4A). Induction of mRNA for IL-27(p28) preceded the appearance of secreted protein since mRNA for IL-27(p28) already peaked after 3 h following TLR4-activation by LPS in BMDM (Fig. 3B). The levels of mRNA for IL-27(p28) were also greatly antagonized by recombinant IL-10 (Fig. 4B). IL-10 potently suppressed production of IL-27(p28) in a dose-dependent manner (IC50: 200-400 pg/ml) in macrophages (Fig. 4C).
Figure 4. Suppression of IL-27(p28) production by IL-10 in macrophages.

A. Time course of IL-27(p28) release from BMDM (Wt) after LPS (1 μg/ml) +/- IL-10 (10 ng/ml), ELISA. B. RT-PCR of IL-27(p28) mRNA levels in BMDM (Wt) with LPS or LPS plus IL-10 (10 ng/ml). C. Dose response studies of IL-27(p28) release from LPS-activated BMDM (Wt) with several IL-10 concentrations, 10 h. Ctrl indicates resting BMDM. D. Release of IL-27(p28) by LPS-activated BMDM from Wt or IL-10-/- mice, 10 h. E. Release of IL-27(p28) by BMDM (Wt) after LPS with addition of control IgG or neutralizing anti-IL-10 antibody (10 μg/ml), 24 h. F. Flow cytometry of BMDM (Wt) after LPS +/- IL-10, 10 h. G. Immuno-cytofluorescence of BMDM (Wt) with red staining for IL-27(p28) (Olympus BX-51 microscope [100×/1.4 oil] with Olympus DP-70 camera), 14 h. H. Relative inhibition of IL-27(p28) by IL-10 in BMDM, peritoneal elicited macrophages (PEM), RAW 264.7 macrophages, MH-S macrophages (SV40 transformed mouse alveolar macrophage cell line) and splenocytes from C57BL/6J mice, 10 h. Data are representative of three independent experiments each performed in duplicates. * P < 0.05, ** P < 0.01, *** P < 0.001.
In our cultures of BMDM the addition of LPS triggered a release of endogenous IL-10 in a range of 100-300 pg/ml after 24 h (data not shown). When BMDM derived from IL-10-/- mice were compared to BMDM from Wt mice, the IL-10-/- BMDM showed nearly 4-fold elevated levels of IL-27(p28) secretion following TLR4-activation (Fig. 4D). In addition, blocking of endogenous IL-10 with neutralizing antibody in cultures of Wt BMDM enhanced the release of IL-27(p28) (Fig. 4E). This suggested that in cultures of macrophages low levels of endogenous IL-10 are sufficient to limit the production of IL-27(p28) in an autocrine and/or paracrine fashion. Variations in absolute amounts of IL-27(p28) from individual experiments with Wt BMDM (e.g. in Fig. 4D and Fig. 4E) are explained by different incubation times and day-to-day variations. For the experiments shown in Fig. 4E, we used longer incubation time points (24 h) as compared to Fig. 4D, since 10 h incubation periods did not reliably result in increased IL-27(p28) concentrations. This was most likely explained by the limited efficacy of IL-10 blocking antibodies and the delayed release of IL-10 during in vitro cultures of LPS-activated macrophages (data not shown). Next, we used flow cytometry to assess intracellular IL-27(p28) finding profound reductions in frequencies of F4/80+CD11b+IL-27(p28)+ BMDM in the presence of exogenous IL-10, when added to LPS-activated BMDM as compared to treatment with LPS alone (Fig. 4F). Optimal staining for intracellular IL-27(p28) required a methodology including Monensin (golgi transport inhibitor), while only few BMDM stained positive with the matched isotype control antibody (Supplementary Fig. 1C). The suppression of IL-27(p28) synthesis by IL-10 was visualized using immuno-cytofluorescence with staining for intra-cellular IL-27(p28) following stimulation of BMDM with LPS and IL-10 alone or in combination (Fig. 4G).
Finally, we wanted to study if the curtailment of IL-27(p28) by IL-10 is a phenomenon restricted to BMDM. All tested cell types of the monocytic-macrophage cell lineage including peritoneal elicited macrophages (PEM), RAW 264.7 macrophages and MH-S macrophages (SV40 transformed mouse alveolar macrophage cell line) produced abundant IL-27(p28) in response to LPS. Recombinant IL-10 antagonized I 27(p28) levels in all monocytic-macrophage cell types tested as observed for PEM, RAW 264.7 and MH-S macrophages (Fig. 4H). LPS-induced IL-27(p28) release was also suppressed by IL-10 in cultures of splenocytes (Fig. 4H). When C57BL/6 mice were treated with clodronate liposomes before isolation of splenocytes, the amounts of IL-27(p28) released were greatly reduced, suggesting that macrophages/phagocytic cells are the source of IL-27(p28) in cultures of splenocytes (data not shown).
In summary, these data (obtained using multiple methodologies) suggest that IL-10 may negatively feedback on the synthesis of the IL-10-inducing cytokine IL-27 in cultures of phagocytic cells.
IL-10 inhibition of MyD88-/TRIF-dependent IL-27(p28) is mediated via STAT3 but not SOCS3
We sought to characterize the intracellular mechanisms underlying regulation of IL-27(p28) gene expression by IL-10. LPS ligation with TLR4 recruits both MyD88 and TRIF pathways to induce gene expression of IL-27(31, 32). We confirmed these previous findings by intracellular staining for IL-27(p28) using macrophages from Wt, MyD88-/- and TRIF-/- mice. After 10 h of incubation with LPS, 45.6% of Wt macrophages had converted to an F4/80+IL-27(p28)+ phenotype, while only 8.6% of MyD88-/- macrophages and virtually no TRIF-/- macrophages were F4/80+IL-27(p28)+ (Fig. 5A). We quantified the reduction of IL-27(p28) concentrations with the genetic absence of MyD88 (∼65-75%) or TRIF (∼90%) by ELISA (Fig. 5B). More importantly, IL-10 suppressed the residual IL-27(p28) release in both MyD88-/- and TRIF-/- BMDM (Fig. 5B). This suggests that IL-10 non-selectively inhibits both MyD88 and TRIF dependent pathways during IL-27(p28) production.
Figure 5. IL-10 inhibition of MyD88-dependent and TRIF-dependent IL-27(p28) production.
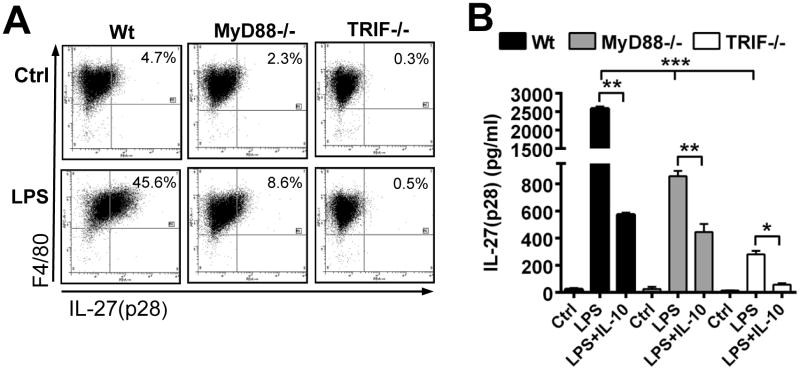
A. Flow cytometry staining of intracellular IL-27(p28) in F4/80+ macrophages derived from Wt mice, MyD88-/- mice or TRIF-/- mice. Cells were left as untreated controls (Ctrl) or activated with LPS (1 μg/ml) for 10 h both in the presence of Monensin (2 μM). B. IL-27(p28) release by BMDM from Wt, MyD88-/- or TRIF-/- mice after stimulation with LPS alone, LPS (1 μg/ml) plus IL-10 (10 ng/ml) or untreated BMDM (Ctrl), 10h, ELISA. Data are representative of two (A) or three (B) independent experiments each performed in duplicates. * P < 0.05, ** P < 0.01, *** P < 0.001.
To further investigate the mechanism of how IL-10 suppresses IL-27(p28), we first studied NFκB and IRF-3, since both factors regulate gene expression of IL-27(p28)(31-33). However, neither NFκB nor IRF-3 was affected by IL-10 in Wt BMDM as studied by DNA-binding immunoassays (data not shown).
The role of STAT3 was also studied. As expected, recombinant IL-10 strongly induced STAT3 phosphorylation in F4/80+ BMDM derived from Wt mice (Fig. 6A). IL-10 was equally effective when used alone or in combination with LPS (Fig. 6A). Time course studies using bead-based immunoassays specific for phosphorylated STAT3 showed maximal phosphorylation already after 20 min following IL-10 treatment in Wt BMDM (Fig. 6B). This is in accordance with previous observations (20). Treatment of BMDM with IL-10 did not affect the patterns of phosphorylation in several other signaling pathways such as Akt, c-Jun, CREB, ERK1/2, JNK, MEK1 or p38MAPK (data not shown).
Figure 6. IL-10 antagonizes secretion of IL-27(p28) via activation of STAT3.

A. Flow cytometry of phospho-STAT3 (pY705) in F4/80+ BMDM (Wt). Macrophages were left untreated (Ctrl) or incubated with IL-10 (10 ng/ml) and LPS (1 μg/ml) alone or in combination for 1 h. B. Time course of phosphorylation of STAT3 in response to IL-10 (10 ng/ml) in BMDM, bead-based assay. C. Suppression of LPS-induced IL-27(p28) by IL-10 in BMDM from Wt or Tie2-Cre/STAT3f1/f1 mice, 24 h. D. Time course of IL-27(p28) release after LPS ± IL-10 using BMDM derived from Wt mice (left panel) or Tie2-Cre/STAT3f1/f1 mice (right panel) Data are representative of at least two independent experiments each in duplicates (A, B) or were done with n=4 mice per group (C, D). ** P < 0.01, *** P < 0.001.
To generate macrophages devoid of STAT3, STAT3f1/f1 mice were crossed with a Tie2-Cre strain, resulting in tissue specific deletion of STAT3 in hematopoietic cells. We used macrophages isolated from neonatal STAT3-deficient mice to bypass the extreme organ inflammation that arises in surviving mice (34). STAT3-deficient mice were phenotyped by Western blotting demonstrating missing phospho-STAT3 in activated macrophages (data not shown). IL-10 completely lost its ability to suppress IL-27(p28) in cultures of BMDM with genetic deficiency of STAT3 (Fig. 6C). Kinetics with BMDM derived from Wt mice and Tie2-Cre/STAT3f1/f1 mice revealed that Tie2-Cre/STAT3f1/f1 BMDM were unresponsive to IL-10 at all the time points studied (Fig. 6D).
The mRNA for SOCS3 was up-regulated by LPS or IL-10 in macrophages, with the combination of LPS and IL-10 being most effective (Fig. 7A). To investigate the relevance of SOCS3 for inhibition of IL-27(p28) by IL-10, we generated macrophages with genetic deficiency of SOCS3 in cells of the hematopoietic lineage by breeding SOCS3f1/f1 mice with the Tie2-Cre strain. As a matter of fact, the relative production of IL-27(p28) by Tie2-Cre/SOCS3f1/f1 macrophages remained equally sensitive to the inhibitory effects of IL-10 as compared with Wt macrophages (Fig. 7B). Indifferent suppression patterns of IL-27(p28) by IL-10 were observed at all studied time points in macrophages from both Wt mice and Tie2-Cre/SOCS3f1/f1 mice (Fig. 7C). The time course of IL-27(p28) release approached a plateau in BMDM from Wt and Tie2-Cre/SOCS3f1/f1 mice, whereas a more linear increase was observed for BMDM from Tie2-Cre/STAT3f1/f1 mice (Fig. 6D, 7C), most likely related to the loss of endogenous IL-10 effects in the Tie2-Cre/STAT3f1/f1 strain. Collectively, these data suggested that IL-10 antagonizes IL-27(p28) production via STAT3, without requirement for SOCS3.
Figure 7. Inhibition of IL-27(p28) production by IL-10 is independent of SOCS3.

A. RT-PCR of C57BL/6J (Wt) macrophages for mRNA of SOCS3 after 6 h incubation with LPS (1 μg/ml) or IL-10 (10 ng/ml) alone or in combination. B. Relative inhibition of LPS-induced IL-27(p28) by IL-10 in macrophages derived from Wt mice or Tie2-Cre/SOCS3f1/f1 mice, 24h. C. Time course of the appearance of IL-27(p28) in cell culture supernatants of LPS-activated macrophages from Wt mice or Tie2-Cre/SOCS3f1/f1 mice in the absence or presence of IL-10. Data are representative of at least two independent experiments each in triplicates (A) or were done with n=4 mice per group (B, C). * P < 0.05, *** P < 0.001.
IL-10-mediated suppression of IL-27(p28) during sepsis
We determined if the mechanism of IL-10-mediated reduction of IL-27(p28) production was functional in two models of sepsis. Time course studies of plasma concentrations of IL-10 during endotoxic shock revealed a very early surge of IL-10 (maximum concentrations after 1 h, Fig. 8A), which preceded the appearance of IL-27(p28) (maximum concentrations after 6 h, Fig. 1C). Next, C57BL/6J wild type (Wt) mice were treated with neutralizing anti-IL-10R antibody to interrupt IL-10 signaling during endotoxic shock (Fig. 8B). IL-27(p28) plasma concentrations were 5–fold elevated in mice receiving anti-IL-10R (Fig. 8B). Likewise, in IL-10-/- mice (≤8 weeks old) the amounts of IL-27(p28) were 6.5-fold elevated during endotoxic shock (Fig. 8C). In the setting of polymicrobial sepsis following CLP, the amounts of IL-27(p28) in plasma were much higher in Wt mice pre-treated with anti-IL-10R antibody (Fig. 8D). Likewise, more IL-27(p28) was released in IL-10-/- mice as compared to Wt mice during polymicrobial sepsis (Fig. 8E). This is in accordance with findings that the genetic absence of IL-10 is associated with hyperproduction of many proinflammatory mediators (20). As expected for IL-10's broad anti-inflammatory properties, IL-10-/- mice, compared to Wt mice, had lower survival rates (0% vs. 58%, Fig. 8F), even when CLP was performed with less severity (“mid-grade”)(24). Moreover, neutralization of IL-27(p28) was partly effective in rescuing IL-10-/- mice from CLP-associated mortality (Fig. 8F). These findings demonstrated that IL-10 acted as an early natural antagonist of IL-27(p28) in vivo, thereby conferring protection against the adverse outcomes of sepsis.
Figure 8. IL-10 limits IL-27(p28) production during polymicrobial sepsis and endotoxic shock.

A. Time course of IL-10 in plasma of Wt mice during endotoxic shock (LPS 10 mg/kg body weight i.p.), n=4-6/time point, ELISA. B. Wt mice were injected with neutralizing anti-IL-10R antibody (200 μg i.p., n=7) or isotype control IgG1κ (200 μg i.p., n=8) followed by endotoxemia and detection of circulating IL-27(p28) after 8 h, ELISA. C. IL-27(p28) in plasma during endotoxic shock in Wt mice (n=9) compared to IL-10-/- mice (n=6), 10 h. D. Detection of IL-27(p28) in plasma of Wt mice injected with neutralizing anti-IL-10R antibody (n=8) or control IgG1κ (n=7) followed by polymicrobial sepsis induced by CLP, 10 h. E. IL-27(p28) in plasma of Wt mice after sham surgery (n=6) or CLP (n=9) as compared to IL-10-/- mice after CLP (n=8), 10 h. F. Survival after CLP (‘mid-grade’) in Wt mice treated with control IgG and IL-10-/- mice treated with control IgG or neutralizing anti-IL-27(p28) antibody (40 μg/mouse i.p., n=12/group). Data were obtained using the numbers of mice per group as indicated. Antibodies were injected 1 h before LPS or CLP in all experiments. * P < 0.05, ** P < 0.01, *** P < 0.001; frame F: * P < 0.05 for IL-10-/-+Control IgG vs. IL-10-/-+anti-IL-27(p28), # P < 0.05 for Wt+Control IgG vs. IL-10-/-+Control IgG.
Discussion
The classification of IL-27 as a negative or positive regulator of inflammation is a controversial issue. The key to understanding the functions of IL-27 as a promoter or silencer of inflammatory responses may be to distinguish between acute and chronic states of inflammation. We present data suggesting that in the early phases of severe sepsis the IL-27(p28) subunit mediates sepsis-associated mortality, cytokine release and regulates production of reactive oxygen species. At the same time IL-27(p28) expression is effectively silenced by anti-inflammatory IL-10-dependent mechanisms. This would be consistent with the idea that IL-27(p28) is not a universal anti-inflammatory mediator. Moreover, this may suggest the existence of a negative regulatory loop with IL-27 mediating the appearance of IL-10 from T cells and IL-10 subsequently preventing further production of IL-27(p28) from macrophages.
IL-10 functions as a non-redundant anti-inflammatory mediator controlling inflammatory responses by targeting multiple proinflammatory cytokines and chemokines. The cells primarily affected by IL-10 are LPS-activated macrophages and dendritic cells (20). Interestingly, IL-10 only suppresses subsets of genes induced by TLRs but does not antagonize all functions elicited by pattern-recognition receptors (20). According to our data, the group of IL-10 regulated cytokines includes IL-27(p28) and IL-10 limits both MyD88- and TRIF-dependent production of IL-27(p28). Other reports have established the requirement of NFκB and IRF-3 for activation of IL-27(p28) transcription (31-33). However, IL-10 neither inhibited NFκB activity, which is consistent with previous findings (35), nor suppressed IRF-3 activation (data not shown). Instead, IL-10 completely lost its ability to antagonize IL-27(p28) in the absence of STAT3. STAT3 activation subsequently induces SOCS3 expression, although we found SOCS3 not to be required for regulation of IL-27(p28) production. It has been shown that SOCS3 predominantly inhibits the gp130 subunit of the IL-6 receptor(36, 37). According to our data the high susceptibility of IL-10-/- mice in polymicrobial sepsis and endotoxemia is in part contributed by IL-27(p28), since blockade of IL-27(p28) resulted in improved survival. In IL-10-/- mice hyperproduction of many proinflammatory cytokines occurs (IL-1β, IL-6, TNFα) in addition to enhanced secretion of IL-27p28, which may all contribute to the high lethality of IL-10-/- mice in models of sepsis. Our data that splenic macrophages are a major source of IL-27(p28) during sepsis are consistent with other reports that macrophages and dendritic cells are the predominant IL-27-producing cell types in other diseases and in vitro (38).
Several reports have described IL-27 in promoting autoimmune diseases including inflammatory bowel disease (11) and lupus nephritis (39). On the other hand, IL-27 limits the severity of experimental autoimmune encephalomyelitis (17), while there are conflicting data on IL-27 effects during rheumatoid arthritis (13). During malaria infection, IL-27RA signaling prevents Th1-mediated tissue destruction independent of parasite clearance (40). The effects of IL-27 on Th1 responses are intricate because IL-27 can both induce IFNγ (the classical Th1 signature cytokine) but also induce IL-10 (known to suppress Th1 cells). IL-27 and IFNγ promote distinct populations of Treg cells during chronic parasitic disease (41).
In this study, we used polyclonal neutralizing antibodies directed against the p28 subunit of IL-27. Earlier studies did not specifically investigated p28, but rather EBI3 or heterodimeric IL-27 (12). One has to keep in mind, that EBI3 is also a subunit of IL-35 and that data obtained in EBI3-/- mice may therefore not exclusively reflect the biology of IL-27. The subunit p28 appears to be much more specific for IL-27, although others binding partners of p28 such as CLF-1 have been suggested (42). The existence of p28 homodimers is still speculative. However, transgenic mice with overexpression of p28 showed that p28 can act as a natural antagonist of IL-27/gp130 signaling (9).
The murine models of endotoxemia and polymicrobial sepsis after CLP are characterized by detrimental acute inflammatory responses similar to clinical diseases such as systemic inflammatory response syndrome (SIRS) and human sepsis. The work presented here focused on the early hyperinflammatory phases of sepsis rather than later occurring immunosuppression of this disease (4, 43, 44). The CLP model is considered by many of having best correlation to human sepsis, although this view has recently been challenged by a gene expression profiling approach (45). Severe sepsis is among the leading causes of death in developed countries. Despite tremendous scientific efforts, specific treatment options targeting the pathophysiologic mechanisms of sepsis beyond the use of antibiotics are not available (3).
Our data on the role of IL-27(p28) during sepsis are supported by previous work of Wirtz et al. reporting on the protective effects of a soluble IL-27RA-Fc fusion protein during polymicrobial sepsis (12). We have recently shown that IL-27RA-/- mice have improved survival rates during endotoxic shock (46). IL-27(p28) production during endotoxic shock and polymicrobial sepsis induced by CLP is partly dependent on tyrosine kinase 2 via an autocrine type I interferon loop (46). Our finding that F4/80+CD11b+ macrophages derived from IL-27RA-/- mice displayed increased ROS production after stimulation with opsonized E. coli is consistent with similar findings in LPS-stimulated phagocytes derived from EBI3-/- mice (12). In EBI3-/- mice, the release of cytokines (IL-1, IL-6) is reduced following CLP (12). On the other hand, the production of IL-10 is not significantly impaired in EBI3-/- mice during polymicrobial sepsis (12), which is in line with the data presented here, using neutralizing anti-IL-27(p28) antibodies during endotoxic shock or polymicrobial sepsis (Fig. 1). Treatment with recombinant IL-27 impaired survival following intra-peritoneal challenge with Pseudomonas aeruginosa (47). IL-27RA-/- mice were protected when ‘low-grade’ CLP (=100 % survival) was followed after 24h by intra-tracheal infection with Pseudomonas aeruginosa (47).
In critically ill children the quantification of plasma IL-27 concentrations was useful as a biomarker for discriminating between patients suffering from SIRS (n=101) or sepsis (n=130) (48). On the other hand, in adult patients with sepsis the detection of IL-27 was only helpful for diagnosis, when applied in combination with procalcitonin (49). The concentrations of IL-27 are higher in children as compared to adults, further arguing for the usefulness of IL-27 for pediatric cases of sepsis (50). It appears that in human sepsis, plasma levels of heterodimeric IL-27 may be substantially higher as compared to IL-27(p28) detected in our CLP model using time points <24 hours (47).
Given the data presented here, directly limiting IL-27(p28) activity with either neutralizing antibodies, administration of recombinant IL-10 or indirectly by modulation of STAT3 activity may be considered as a future treatment strategy for sepsis. However, such endeavors will undoubtedly require further experimental studies to provide a more complete understanding of the double-edged role played by IL-27(p28) during acute and chronic settings of unbalanced inflammation.
Supplementary Material
Acknowledgments
We cordially thank Vinay Patel, Mikel Haggadone and Fabien Meta for technical assistance, as well as Beverly Schumann, Sue Scott and Robin Kunkel for excellent staff support.
Financial Disclosure: This work was supported by grants GM-29507 (P.A.W.), GM-61656 (P.A.W.) and CORE grant P30 CA21765 (P.J.M.) from the US National Institutes of Health, along with the Deutsche Forschungsgemeinschaft (BO3482/3-1 to M.B.; Ra988/4-2, STA984/4-1 to M.P.R.), the Federal Ministry of Education and Research (BMBF 01EO1003, M.B.), the B. Braun Foundation (M.B.), the MAIFOR Program of the University Medical Center Mainz (M.B.), a Marie Curie Career Integration Grant of the European Union (project 334486, M.B.), the Boehringer Ingelheim Fonds (N.F.R.), the Hartwell Foundation (P.J.M.), the American Lebanese Syrian Associated Charities (P.J.M.) and the Austrian Science Fund (FWF-F28, B.S. and M.M.). The funding organizations had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Disclosures: The authors have no financial conflicts of interest.
References
- 1.Adhikari NK, Fowler RA, Bhagwanjee S, Rubenfeld GD. Critical care and the global burden of critical illness in adults. Lancet. 2010;376:1339–1346. doi: 10.1016/S0140-6736(10)60446-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vincent JL, Sakr Y, Sprung CL, Ranieri VM, Reinhart K, Gerlach H, Moreno R, Carlet J, Le Gall JR, Payen D. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. 2006;34:344–353. doi: 10.1097/01.ccm.0000194725.48928.3a. [DOI] [PubMed] [Google Scholar]
- 3.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nature Reviews Immunology. 2008;8:776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. 2009;15:496–497. doi: 10.1038/nm0509-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, Hibbert L, Churakova T, Travis M, Vaisberg E, Blumenschein WM, Mattson JD, Wagner JL, To W, Zurawski S, McClanahan TK, Gorman DM, Bazan JF, de Waal Malefyt R, Rennick D, Kastelein RA. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+) T cells. Immunity. 2002;16:779–790. doi: 10.1016/s1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- 6.Wojno ED, Hunter CA. New directions in the basic and translational biology of interleukin-27. Trends Immunol. 2012;33:91–97. doi: 10.1016/j.it.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13:722–728. doi: 10.1038/ni.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maaser C, Egan LJ, Birkenbach MP, Eckmann L, Kagnoff MF. Expression of Epstein-Barr virus-induced gene 3 and other interleukin-12-related molecules by human intestinal epithelium. Immunology. 2004;112:437–445. doi: 10.1111/j.1365-2567.2004.01895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stumhofer JS, Tait ED, Quinn WJ, 3rd, Hosken N, Spudy B, Goenka R, Fielding CA, O'Hara AC, Chen Y, Jones ML, Saris CJ, Rose-John S, Cua DJ, Jones SA, Elloso MM, Grotzinger J, Cancro MP, Levin SD, Hunter CA. A role for IL-27p28 as an antagonist of gp130-mediated signaling. Nat Immunol. 2010;11:1119–1126. doi: 10.1038/ni.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 11.Cox JH, Kljavin NM, Ramamoorthi N, Diehl L, Batten M, Ghilardi N. IL-27 promotes T cell-dependent colitis through multiple mechanisms. Journal of Experimental Medicine. 2011;208:115–123. doi: 10.1084/jem.20100410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wirtz S, Tubbe I, Galle PR, Schild HJ, Birkenbach M, Blumberg RS, Neurath MF. Protection from lethal septic peritonitis by neutralizing the biological function of interleukin 27. Journal of Experimental Medicine. 2006;203:1875–1881. doi: 10.1084/jem.20060471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao Y, Doodes PD, Glant TT, Finnegan A. IL-27 induces a Th1 immune response and susceptibility to experimental arthritis. J Immunol. 2008;180:922–930. doi: 10.4049/jimmunol.180.2.922. [DOI] [PubMed] [Google Scholar]
- 14.Villarino A, Hibbert L, Lieberman L, Wilson E, Mak T, Yoshida H, Kastelein RA, Saris C, Hunter CA. The IL-27R (WSX-1) is required to suppress T cell hyperactivity during infection. Immunity. 2003;19:645–655. doi: 10.1016/s1074-7613(03)00300-5. [DOI] [PubMed] [Google Scholar]
- 15.Hamano S, Himeno K, Miyazaki Y, Ishii K, Yamanaka A, Takeda A, Zhang M, Hisaeda H, Mak TW, Yoshimura A, Yoshida H. WSX-1 is required for resistance to Trypanosoma cruzi infection by regulation of proinflammatory cytokine production. Immunity. 2003;19:657–667. doi: 10.1016/s1074-7613(03)00298-x. [DOI] [PubMed] [Google Scholar]
- 16.Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, Villarino AV, Huang Q, Yoshimura A, Sehy D, Saris CJ, O'Shea JJ, Hennighausen L, Ernst M, Hunter CA. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 17.Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nature Immunology. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 18.Batten M, Kljavin NM, Li J, Walter MJ, de Sauvage FJ, Ghilardi N. Cutting edge: IL-27 is a potent inducer of IL-10 but not FoxP3 in murine T cells. J Immunol. 2008;180:2752–2756. doi: 10.4049/jimmunol.180.5.2752. [DOI] [PubMed] [Google Scholar]
- 19.Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O'Shea JJ, Hunter CA. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol. 2007;8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- 20.Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Current Opinion in Pharmacology. 2006;6:379–386. doi: 10.1016/j.coph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 21.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. The Journal of clinical investigation. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 23.Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, Liu F, Goldsmith MA, Link DC, Murray PJ, Watowich SS. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood. 2006;108:3682–3690. doi: 10.1182/blood-2006-02-003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 26.Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. J Exp Med. 2012;209:1057–1068. doi: 10.1084/jem.20120571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huston JM, Wang H, Ochani M, Ochani K, Rosas-Ballina M, Gallowitsch-Puerta M, Ashok M, Yang L, Tracey KJ, Yang H. Splenectomy protects against sepsis lethality and reduces serum HMGB1 levels. J Immunol. 2008;181:3535–3539. doi: 10.4049/jimmunol.181.5.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Rooijen N, Sanders A. Elimination, blocking, and activation of macrophages: three of a kind? J Leukoc Biol. 1997;62:702–709. doi: 10.1002/jlb.62.6.702. [DOI] [PubMed] [Google Scholar]
- 29.Salkowski CA, Neta R, Wynn TA, Strassmann G, van Rooijen N, Vogel SN. Effect of liposome-mediated macrophage depletion on LPS-induced cytokine gene expression and radioprotection. J Immunol. 1995;155:3168–3179. [PubMed] [Google Scholar]
- 30.Salkowski CA, Detore G, McNally R, van Rooijen N, Vogel SN. Regulation of inducible nitric oxide synthase messenger RNA expression and nitric oxide production by lipopolysaccharide in vivo: the roles of macrophages, endogenous IFN-gamma, and TNF receptor-1-mediated signaling. J Immunol. 1997;158:905–912. [PubMed] [Google Scholar]
- 31.Molle C, Nguyen M, Flamand V, Renneson J, Trottein F, De Wit D, Willems F, Goldman M, Goriely S. IL-27 synthesis induced by TLR ligation critically depends on IFN regulatory factor 3. Journal of Immunology. 2007;178:7607–7615. doi: 10.4049/jimmunol.178.12.7607. [DOI] [PubMed] [Google Scholar]
- 32.Liu JG, Guan XQ, Ma XJ. Regulation of IL-27 p28 gene expression in macrophages through MyD88- and interferon-gamma-mediated pathways. Journal of Experimental Medicine. 2007;204:141–152. doi: 10.1084/jem.20061440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molle C, Goldman M, Goriely S. Critical role of the IFN-stimulated gene factor 3 complex in TLR-mediated IL-27p28 gene expression revealing a two-step activation process. J Immunol. 2010;184:1784–1792. doi: 10.4049/jimmunol.0902005. [DOI] [PubMed] [Google Scholar]
- 34.Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, Kano A, Iwamoto Y, Li E, Craft JE, Bothwell AL, Fikrig E, Koni PA, Flavell RA, Fu XY. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci U S A. 2003;100:1879–1884. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci U S A. 2005;102:8686–8691. doi: 10.1073/pnas.0500419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nature Immunology. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 37.Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- 38.Batten M, Ghilardi N. The biology and therapeutic potential of interleukin 27. Journal of Molecular Medicine. 2007;85:661–672. doi: 10.1007/s00109-007-0164-7. [DOI] [PubMed] [Google Scholar]
- 39.Batten M, Ramamoorthi N, Kljavin NM, Ma CS, Cox JH, Dengler HS, Danilenko DM, Caplazi P, Wong M, Fulcher DA, Cook MC, King C, Tangye SG, de Sauvage FJ, Ghilardi N. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J Exp Med. 2010;207:2895–2906. doi: 10.1084/jem.20100064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Findlay EG, Greig R, Stumhofer JS, Hafalla JC, de Souza JB, Saris CJ, Hunter CA, Riley EM, Couper KN. Essential role for IL-27 receptor signaling in prevention of Th1-mediated immunopathology during malaria infection. J Immunol. 2010;185:2482–2492. doi: 10.4049/jimmunol.0904019. [DOI] [PubMed] [Google Scholar]
- 41.Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, Harris TH, Grainger J, Wojno ED, Wagage S, Roos DS, Scott P, Turka LA, Cherry S, Reiner SL, Cua D, Belkaid Y, Elloso MM, Hunter CA. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity. 2012;37:511–523. doi: 10.1016/j.immuni.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crabe S, Guay-Giroux A, Tormo AJ, Duluc D, Lissilaa R, Guilhot F, Mavoungou-Bigouagou U, Lefouili F, Cognet I, Ferlin W, Elson G, Jeannin P, Gauchat JF. The IL-27 p28 subunit binds cytokine-like factor 1 to form a cytokine regulating NK and T cell activities requiring IL-6R for signaling. J Immunol. 2009;183:7692–7702. doi: 10.4049/jimmunol.0901464. [DOI] [PubMed] [Google Scholar]
- 43.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, 2nd, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss RS. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA: the journal of the American Medical Association. 2011;306:2594–2605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cavassani KA, Carson WFt, Moreira AP, Wen H, Schaller MA, Ishii M, Lindell DM, Dou Y, Lukacs NW, Keshamouni VG, Hogaboam CM, Kunkel SL. The post sepsis-induced expansion and enhanced function of regulatory T cells create an environment to potentiate tumor growth. Blood. 2010;115:4403–4411. doi: 10.1182/blood-2009-09-241083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG Inflammation, and L. S. C. R. P. Host Response to Injury. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bosmann M, Strobl B, Kichler N, Rigler D, Grailer JJ, Pache F, Murray PJ, Muller M, Ward PA. Tyrosine kinase 2 promotes sepsis-associated lethality by facilitating production of interleukin-27. J Leukoc Biol. 2014;96:123–131. doi: 10.1189/jlb.3A1013-541R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao J, Xu F, Lin S, Song Z, Zhang L, Luo P, Xu H, Li D, Zheng K, Ren G, Yin Y. IL-27 controls sepsis-induced impairment of lung antibacterial host defence. Thorax. 2014 doi: 10.1136/thoraxjnl-2014-205777. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 48.Wong HR, Cvijanovich NZ, Hall M, Allen GL, Thomas NJ, Freishtat RJ, Anas N, Meyer K, Checchia PA, Lin R, Bigham MT, Sen A, Nowak J, Quasney M, Henricksen JW, Chopra A, Banschbach S, Beckman E, Harmon K, Lahni P, Shanley TP. Interleukin-27 is a novel candidate diagnostic biomarker for bacterial infection in critically ill children. Critical care. 2012;16:R213. doi: 10.1186/cc11847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong HR, Lindsell CJ, Lahni P, Hart KW, Gibot S. Interleukin 27 as a sepsis diagnostic biomarker in critically ill adults. Shock. 2013;40:382–386. doi: 10.1097/SHK.0b013e3182a67632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wong HR, Liu KD, Kangelaris KN, Lahni P, Calfee CS. Performance of interleukin-27 as a sepsis diagnostic biomarker in critically ill adults. Journal of critical care. 2014;29:718–722. doi: 10.1016/j.jcrc.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.