

Abstract
Hepatitis C virus (HCV) infections represent a major global health problem. End-stage liver disease caused by chronic HCV infection is a major indication for liver transplantation. However, after transplantation the engrafted liver inevitably becomes infected by the circulating virus. Direct acting antivirals are not yet approved for use in liver transplant patients, and limited efficacy and severe side effects hamper the use of pegylated interferon combined with ribavirin in a post-transplant setting. Therefore, alternative therapeutic options need to be explored. Viral entry represents an attractive target for such therapeutic intervention. Understanding the mechanisms of viral entry is essential to define the viral and cellular factors involved. The HCV life cycle is dependent of and associated with lipoprotein physiology and the presence of lipoproteins has been correlated with altered antiviral efficacy of entry inhibitors. In this review, we summarise the current knowledge on how lipoprotein physiology influences the HCV life cycle. We focus especially on the influence of lipoproteins on antibodies that target HCV envelope proteins or antibodies that target the cellular receptors of the virus. This information can be particularly relevant for the prevention of HCV re-infection after liver transplantation.
Keywords: Hepatitis, Lipids, Neutralisation, Antiviral therapy, Animal models
Core tip: We reviewed the influence of lipids and lipoproteins on the hepatitis C virus life cycle and their impact on viral neutralization by antibodies that target the viral envelope proteins or that target the receptors used by the virus to infect the hepatocyte.
INTRODUCTION
Approximately 3% of the world’s population is chronically infected with the hepatitis C virus. Depending on the genotype of the infecting virus, 50% to 80% of chronically infected patients clear the virus upon treatment with pegylated interferon and ribavirin[1]. Addition of one of the recently approved protease inhibitors: telaprevir, boceprevir or simeprevir; or the nucleotide analogue polymerase inhibitor sofosbuvir, significantly increases the response rate in patients infected with HCV genotype 1[2-4]. However, side effects and drug-drug interactions severely complicate the use of first wave protease inhibitors in triple therapy in chronically infected patients in need for liver transplantation[5,6]. Therefore, cocktails of other direct acting antivirals (DAAs) without interferon are needed to treat this expanding patient population. Very recently, sofosbuvir has been approved in combination with ribavirin for the treatment of chronic HCV patients with hepatocellular carcinoma awaiting liver transplantation in order to prevent post-transplant HCV recurrence. No drug interactions with immunosuppressive therapy were observed and post-transplantation sustained virologic response (SVR) was achieved in more than half of the patients[7]. Although promising results have been presented of studies investigating the effect of DAA-based regimens in patients suffering from recurrent HCV[8-10], today no therapy exists that protects the liver graft from being infected in viremic pre-transplant patients.
The viral entry process offers multiple targets for the development of alternative strategies for the prevention of HCV recurrence[11]. The viral envelope glycoproteins, E1 and E2, are major components of the viral particle and play a pivotal role in the entry process. The envelope protein E2, and especially its hypervariable region 1 (HVR1), is the main target for neutralizing antibodies that are generated by the humoral immune response. Presumably, broadly cross-neutralizing monoclonal antibodies (mAbs) directed against HCV E2 could represent a promising tool for effective passive immunotherapy. However, due to the high propensity of the virus to continually mutate, and the high variability of this region in particular, neutralizing anti-HVR1 antibodies have only limited cross-genotype neutralization potency[12]. Nevertheless, neutralizing antibodies play a role in viral protection and disease outcome[13,14]. Therefore, the applicability of HCV neutralizing antibodies as a means of preventing HCV re-infection of liver allografts has been explored. Although proof of concept pre-clinical studies showed promising results[15-18], no or only transient effects on HCV RNA levels and HCV recurrence after liver transplantation were observed in clinical studies[19-22]. This lack of effectiveness could partially be explained by the observation that human serum, and HDL in particular, lower the neutralization efficacy of anti-HCV neutralizing antibodies[23-25].
HCV relies on multiple host cell membrane molecules to establish initial cell contact and other more specific interactions to initiate the ensuing entry into the hepatocyte. Glycosaminoglycans and the LDL-receptor have been proposed to as initial attachment factors for HCV to the surface of the hepatocyte[26,27]. For actual endocytosis, the virus first needs to interact with SR-BI and CD81 before being directed to the tight junction proteins claudin-1 (CLDN1) and occludin[28-31]. Blockade of one or more of these host cell factors represent an attractive strategy for antiviral intervention.
During the HCV entry process and different other steps in the viral life cycle, lipids and lipoproteins have been reported to intervene. For example, essential roles for HCV entry have been dedicated to lipid receptors, free lipoproteins and apolipoproteins (Apo) associated with the viral particle, whereas lipid droplets and ApoE expression are important during assembly and release of the viral particle respectively. In this review, we focus on the interaction of lipid physiology with HCV virology and to what extent these interactions influence the antiviral efficacy of entry inhibitors, more specifically neutralizing antibodies targeting either the viral envelope or the cellular (co-)receptors used by HCV for its entry.
LIPIDS AND LIPOPROTEIN METABOLISM
Blood plasma and interstitial fluid represent an aqueous environment wherein hydrophilic interactions predominate. To transport hydrophobic lipids throughout the body these are incorporated into water-soluble aggregates of protein and lipid, known as lipoproteins. Lipoproteins are defined by their apolipoprotein, cholesterol, triglyceride and phospholipid content and subdivided by their buoyant densities into high, low, intermediate and very low density lipoproteins (HDL, LDL, IDL and VLDL respectively) and chylomicrons[32]. A schematic representation of lipoprotein metabolism can be found in Figure 1.
Figure 1.
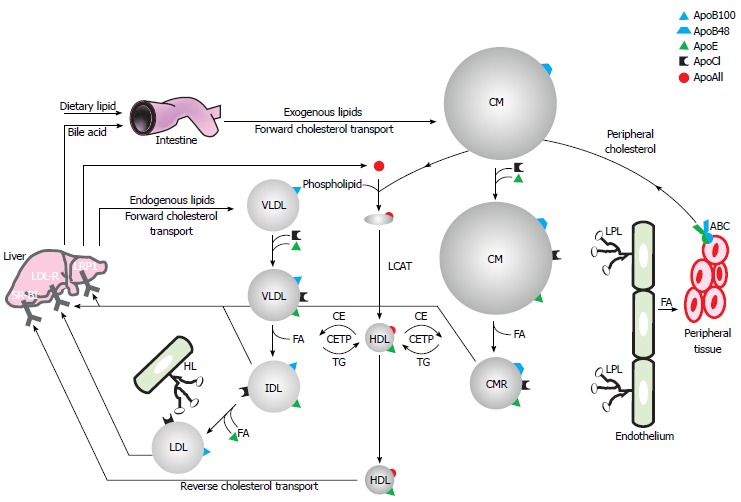
Schematic representation of lipoprotein metabolism. Refer to text for further details. CM: Chylomicron; CMR: Chylomicron remnant; Apo: Apolipoprotein; VLDL: Very low density lipoprotein; IDL: Intermediate density lipoprotein; LDL: Low density lipoprotein; HL: Hepatic lipase; LPL: Lipoprotein lipase; SR-BI: Scavenger receptor class B type I; LDL-R: Low density lipoprotein receptor; LRP1: Low density lipoprotein receptor-related protein 1; CETP: Cholesteryl ester transfer protein; TG: Triglycerides.
In the enterocyte, absorbed dietary lipids are re-esterified to triglycerides (TG), cholesteryl esters and phospholipids that, together with fat-soluble vitamins, are assembled and secreted into the bloodstream as TG- and ApoB48-rich chylomicrons. In contrast, endogenously synthesized lipids are secreted by the liver in VLDL particles containing ApoB100. Both these ApoB-containing lipoproteins are involved in the lipid delivery pathway and therefore circulate in the blood in order to distribute lipids to peripheral and specific target tissues. This process is controlled by the apolipoproteins present in these particles that act as receptor ligands or enzyme cofactors. ApoCII for example acts as a cofactor for lipoprotein lipases (LPL), which catalyzes the hydrolysis of TG into 2-monoacylglycerol and free or non-esterified fatty acids for tissue utilization as an energy source, for energy storage and thermoregulation in specialized tissues such as muscles, white and brown adipose tissues respectively. The physiological site of action of the lipase is located at the luminal surface of the capillary endothelial cells, because TG-rich lipoprotein particles are too large to cross the capillary endothelium in most tissues. The enzyme is attached to the endothelium via highly charged, membrane-bound chains of heparan sulphate-proteoglycans (HSPG). ApoB-containing lipoproteins acquire ApoCII and ApoE in circulation, immediately after secretion or due to protein exchange with HDL. Chylomicron TG can then be hydrolyzed into free fatty acids by LPL, leading to the formation of smaller chylomicron remnants, which are taken up by the liver via ApoE interaction with the LDL-R or the low density lipoprotein receptor-related protein 1. In addition, LPL converts VLDL into ApoE- and cholesterol-rich IDL that can also be removed by these receptors. Assisted by hepatic lipase (HL), LPL can further metabolise IDL to LDL, upon which it loses most of its ApoE and can be recognized and internalized by the hepatic LDL-R via its ApoB moiety. The lipid-proteoglycan bridging capacity of these lipases facilitates clearance of lipolytic remnant particles by presentation to hepatic surface proteoglycans before receptor-mediated endocytosis. Although mainly recycled to the liver, LDL can also be taken up by peripheral cells by the LDL-R. Importantly, excess LDL and chylomicron remnants can invade the arterial wall, become oxidized and be taken up by the scavenger receptor on arterial wall macrophages that are hence transformed into foam cells, a process leading to atherosclerosis[33,34].
Besides TG, also cholesterol is transported through the bloodstream via lipoprotein particles. Cholesterol is an essential component of the plasma membrane by maintaining the barrier function between intra- and extracellular environment, modulating its fluidity, and creating “rafts” that concentrate signalling molecules. Cholesterol is transported back to the liver in a process called reverse cholesterol transport that implicates HDL. Nascent HDL is generated by the transfer of phospholipids and cholesterol from peripheral tissues, intestine and liver onto ApoA-1. This process is catalyzed by the ATP-binding cassette A1 transporter. The cholesterol contained in this nascent HDL is then esterified by lysolecithin cholesterol acyltransferase thereby forming more spherical mature HDL. Additional cholesterol can be loaded onto mature HDL by another ABC transporter, ABCG1. HDL can further capture free cholesterol from membrane pools via interactions with SR-BI, lipid rafts and caveolae. These processes are important in preventing atherosclerotic vessel disease by allowing macrophages to efflux artery wall cholesterol. During their passage through the circulation the ApoE content of HDL increases due to protein exchange with VLDL. In addition, the cholesteryl ester transfer protein can transfer cholesteryl ester from HDL to chylomicrons, VLDL and their remnants in exchange for TG. HDL-cholesteryl-esters can be utilized by the liver through the SR-BI receptor. After hydrolysis, free cholesterol can be metabolized to bile acids that are excreted into the digestive tract via biliary secretion. Extrahepatically, SR-BI supports HDL-cholesteryl-esters consumption as a precursor for the manufacture of all steroid hormones[35,36].
INTERPLAY BETWEEN PATIENT LIPID METABOLISM, CHRONIC HCV AND ANTI-HCV THERAPY EFFICACY
Chronic HCV infection has been linked to various lipid metabolism disorders. HCV perturbs lipid homeostasis while supporting its own survival but thereby causing liver disease. These HCV-induced lipid homeostasis alterations affect serum lipid profiles that lead to hepatic steatosis, the accumulation of hepatocellular lipid droplets[37]. Especially genotype 3 HCV infections are associated with reduced levels of total and LDL cholesterol and with the development of hepatic steatosis[38]. In these patients, steatosis and hypocholesterolemia are associated with high viral load[39].
It has been observed that HCV infection in humanized mice mediates changes in the hepatic expression of genes that regulate lipid metabolism[40]. Also during the early stages of HCV infection in chimpanzees that permanently or transiently cleared the virus upon IFN-γ induction, host genes involved in lipid metabolism were shown to be differentially regulated[41]. These observations suggest that lipid metabolism is essential for the HCV life cycle or viral clearance[40] and that changes in lipid metabolism can influence the efficacy of anti-HCV treatment. Indeed, pre-treatment serum LDL and cholesterol levels in HCV infected patients were found to directly correlate with response to interferon-based therapy[42], while liver steatosis was associated with a lower sustained response rate to interferon-based therapy[39]. Furthermore, cholesterol-lowering statins possess anti-HCV capacities[43,44]. However it should be mentioned that their anti-HCV property is not considered to depend on their inhibitory effects on cholesterol biosynthesis, but rather on their capacity to inhibit geranylgeranyl pyrophosphate synthesis, which is important for HCV RNA replication. Although clinical proof of anti-HCV activity of statin monotherapy is lacking or contradictory[45-48], its combination with interferon-based therapy may improve virologic response rates[49-52]. Another inhibitor of cholesterol synthesis, bezafibrate, only slightly reduced HCV viremia in chronic hepatitis C patients with concomitant hyperlipidaemia[53]. Finally, HCV viremia has been shown to inversely correlate with serum triglyceride levels[54]. In fact, high triglyceridemia during acute HCV infection may facilitate viral clearance since triglyceridemia was increased in patients that cleared HCV infection[55]. Chronic HCV infection is associated with the development of insulin resistance which further promotes the progression of steatosis and fibrosis[56]. Since insulin resistance induces interferon resistance, the management of insulin resistance by means of insulin sensitizers such as metformin[57] or pioglitazone[58] has been proposed to improve interferon-based HCV treatment outcomes, however insulin therapy reduced SVR[52].
VIRAL LIFE CYCLE IS TIGHTLY ASSOCIATED WITH HEPATIC LIPID METABOLISM PATHWAYS
HCV circulates in the bloodstream as a very heterogeneous population, not only genetically (quasispecies) but also physicochemically. The latter is the result of the association of viral particles with immunoglobulins and lipoproteins. HCV particles vary in density between 1.03 and 1.34 g/mL and the very low-density population appears to be particularly infectious[59]. This population is known as lipoviroparticles, i.e. lipoprotein-like structures composed of triglyceride-rich lipoproteins containing at least ApoB and E, viral RNA and HCV core protein[60-62].
Effects on HCV entry
HCV hepatocyte entry involves lipid receptors: Scavenger receptor class B type I . The scavenger receptor class B type I (SR-BI) is a highly glycosylated 509 amino acid long glycoprotein with a large extracellular domain anchored to the plasma membrane at both the amino- and carboxy-termini. It is concentrated in microdomains that correspond to cholesterol and sphingolipid-enriched plasma membrane microdomains (lipid rafts) called caveolae[63]. SR-BI is the principal HDL receptor but it also binds VLDL, LDL and oxidized lipoproteins. SR-BI predominantly mediates selective uptake of HDL-cholesteryl esters (CE) in a two-step process that first involves HDL binding with subsequent incorporation of CE to the plasma membrane pool, followed by hydrolysis to free cholesterol by a neutral CE hydrolase and storage in an intracellular cholesterol pool. This process occurs without catabolism of the HDL particle. In addition, an alternative lipid exchange pathway exists that is thought to gain importance in case of disturbed lipid metabolism and which comprises concomitant endocytosis of SR-BI and whole HDL particles followed by particle re-secretion. HDL re-secretion could be linked to cholesterol efflux, since re-secreted HDL particles are enriched in cellular cholesterol. SR-BI is also involved in bidirectional free cholesterol flux. As mentioned before, it is expressed predominantly in the liver and steroidogenic tissues. In hepatocytes, SR-BI transports cholesteryl esters for membrane function and synthesis of bile acids, thereby participating in reverse cholesterol transport and cholesterol homeostasis. In steroidogenic tissues, such as the adrenal cortex, it delivers cholesteryl esters for the synthesis of glucocorticoids[63-65].
Attachment through ApoE: SR-BI was initially discovered as an HCV (co-)receptor based on its ability to interact with soluble E2 (sE2). Deletion of HVR1 impaired the binding of sE2 to SR-BI suggesting that this region is particularly involved in this interaction[29]. However, more recent data of Dao Thi et al[66] revealed that the HCV-associated lipoproteins are even more important for the interaction of the virus with SR-BI. Indeed, intermediate density HCV particles (1.10-1.16 g/mL) bound efficiently to human SR-BI expressed on BRL3A cells that are devoid of endogenous SR-BI. Surprisingly, a mutation in HVR1 (L339R) that abrogates sE2 binding to SR-BI, did not alter the ability of cell culture-derived HCV (HCVcc) to bind human SR-BI. In addition, BRL3A cells expressing mouse SR-BI, which is unable to bind sE2, also supported HCVcc binding. Furthermore, despite the observation that Blocker of Lipid Transport-4 (BLT-4) and anti-E2 antibodies both block sE2-SR-BI binding, they did not abrogate HCV-SR-BI binding. In contrast, HCVcc did not attach to a mouse SR-BI variant that is unable to bind lipoproteins. These findings indicate that lipid components of HCV rather than E2 are mainly responsible for HCV-SR-BI binding. This idea was further strengthened by the observation that purified VLDL and ApoE inhibited HCV-SR-BI binding[66]. Earlier indications that HCV-associated lipoproteins are involved in SR-BI attachment, rather than E2 itself, were reported by Maillard et al[67]. They observed that SR-BI-binding of HCV from sera of infected patients was insensitive to anti-E2-HVR1 antibodies, whereas this interaction was inhibited by ApoE and VLDL.
Lipid transfer function of SR-BI is involved during post-attachment HCV entry: SR-BI has also been demonstrated to mediate post-binding events during HCV entry[68]. Besides its primary HCV attachment function through HCV-associated ApoE, SR-BI is involved in HCV entry by means of its lipid transfer ability. Antibodies that inhibit HDL binding and SR-BI-mediated lipid transfer potently inhibit HCV infection of different genotypes both in cell culture and in humanized uPA-SCID mice[69,70]. In addition to SR-BI-specific antibodies, small-molecule inhibitors of SR-BI-mediated cholesteryl ester lipid uptake (ITX-5061, ITX-7650, Rimcazole and BLT (block lipid transport)-2, 3 and 4) also have proven anti-HCV activity[23,71-76]. However, it should be noted that besides their ability to block SR-BI-mediated lipid transfer, these molecules all abrogate the sE2-SR-BI interaction as well.
Increasing evidence for the role SR-BI’s lipid transfer function plays during HCV entry was provided by Dao Thi et al[66]. These investigators showed that the inhibition of HCV pseudoparticle (HCVpp) entry in murine SR-BI expressing cells by BLT-4 correlated with the level of lipid transfer abrogation. Due to the inability of HCV E2 to bind murine SR-BI, this finding indicates the essential involvement of SR-BI’s lipid transfer function in mediating HCV entry in a process that is completely independent of E2’s attachment to and/or interaction with SR-BI. Additional proof of this concept was delivered by the observation that an HCVpp E2 mutant (L399R), unable to bind SR-BI, was still dependent on the presence of SR-BI for its infectivity. This mutant therefore used SR-BI for cell entry by other means than its E2 binding capacity. Furthermore, mouse SR-BI mutants, unable to bind E2 and impaired in their lipid transfer, reduced HCVpp infection proportional to their impairment of lipid transfer ability[66]. In addition, anti-SR-BI mAbs have been reported that interfere specifically with post-binding steps during the HCV entry process linked to SR-BI’s lipid transfer function without affecting the SR-BI-E2 or SR-BI-HDL interactions[77].
Post-attachment SR-BI-E2 interaction: Although it appears increasingly unlikely that E2 is directly involved in SR-BI mediated primary attachment of HCV to the host cell, a functional role for E2 during the interaction of HCV with SR-BI should not be ignored. Indeed, Catanese et al[78] identified residues in SR-BI that are involved in sE2 binding (AA 70-87 and residue E210), but these are distinct from the HDL binding site (C323[79]). Mutations in this region that confer SR-BI defective for sE2 binding (however retaining its oligomerization, HDL binding and cholesterol efflux activity) impaired the ability of SR-BI to mediate HCV infection. This indicates that the E2 binding ability of SR-BI remains important for HCV infection[78]. These observations convince us to believe that the ability of E2 to interact with SR-BI is not predominantly involved in primary HCV attachment but rather becomes important during a post-attachment process. Accordingly, the interaction of E2 with SR-BI is essential for the infection enhancement-process mediated by HDL (cfr. infra)[66].
In conclusion, currently available data reveal three important SR-BI functions involved in HCV entry: (1) SR-BI-ApoE attachment; (2) SR-BI lipid transfer; and (3) SR-BI-E2 interaction. Altogether, it can be suggested that SR-BI functions as a primary HCV receptor that interacts with virus-associated lipoproteins before subsequent direct interactions occur with the HCV glycoprotein E2. Through its lipid transfer function, SR-BI may crucially facilitate the accessibility or the recruitment of the HCV receptor complex by membrane cholesterol enrichment. Finally, the ability of SR-BI to simultaneously interact with HDL and E2 might be essential for the support or even enhancement of HCV infection.
LDL-receptor. This receptor transfers LDL particles into cells through clathrin-coated pits and vesicles before lysosomal degradation. Within the cell, LDL-derived cholesterol elicits several regulatory functions in cholesterol metabolism and homeostasis, including feedback inhibition of cholesterol synthesis[80]. VLDL is not a ligand for LDL-R whereas its lipolytic remnants IDL and LDL, respectively resulting from sequential triglyceride hydrolysis catalyzed by LPL and HL, are[33].
While HCV envelope glycoprotein E2 did not bind to LDL-R[81], it was proposed to function as an HCV attachment/entry factor through interaction with HCV associated ApoE and ApoB[60]. Indeed, LDL has been shown to compete with serum-derived HCV for LDL-R[82]. In addition, Molina and co-workers demonstrated the involvement of LDL-R in HCV infection of primary hepatocytes[83]. The virus is thought to hijack the ability of LDL-R to capture lipoproteins from the circulation for uptake of cholesterol into cells. This mechanism is not exclusive to Flaviviridae but may be a general feature among viruses that are associated with lipoproteins[27]. However, LDL-R implication in the HCV life cycle has been controversial. Although it has been suggested that HCV infectivity is facilitated by interactions between HCV’s ApoE moiety and LDL-R[84-86], recent data indicate that this is not necessarily the case. Prentoe et al[87] observed that LDL-R antibodies interfering with ApoE- and ApoB-dependent LDL-R association blocked HCV infectivity less efficiently compared to LDL-R antibodies with lower interference of lipoprotein association. Furthermore, addition of exogenous LPL increased HCV binding to LDL-R, which increased HCV RNA internalization but reduced HCV infectivity[88]. These data encourage us to speculate that HCV can indeed interact with LDL-R but this event does not necessarily lead to a productive infection. Since LDL-R blocking studies clearly assigned a supporting role for LDL-R in the life cycle of HCV, we assume that this function must be distinct from its lipoprotein binding capacities. Interestingly, recent work from Albecka et al[88] confirms a role for LDL-R in the life cycle of HCV but indicates that it is mainly involved in viral replication rather than supporting productive HCV entry. In summary, the function of LDL-R during the HCV life cycle seems double-edged: (1) inhibition of infection through HCV-associated lipoprotein recognition, which supposedly paves the way to non-productive HCV entry; and (2) support of infection by enhancing replication.
NPC1L1. The most recently described host cell protein that modulates HCV cell entry is the Niemann-Pick C1-like 1 cholesterol uptake receptor (NPC1L1)[89]. NPC1L1 is a 13-transmembrane-domain cell surface cholesterol-sensing receptor expressed on the apical surface of intestinal enterocytes and the bile canalicular membrane of human hepatocytes[90]. It is critical for intestinal cholesterol absorption[91] and regulation of biliary cholesterol concentration[92]. Probably, the cholesterol uptake activity of NPC1L1 is involved in HCV entry. Antibody-mediated receptor blockade of NPC1L1 large extracellular loop 1 (LEL1), but not LEL 2 or LEL3, and NPC1L1 silencing impairs HCVcc infection initiation. Ezetimibe, a 2-azetidinone-class NPC1L1 antagonist that has been approved by the FDA as a cholesterol-lowering medicine[93] inhibits HCV uptake of all major genotypes in vitro at a post-binding step and delays genotype 1b infection in humanized mice[89].
HSPG, LPL and hepatic triglyceride lipase. Although LPL exerts its physiological action at the luminal surface of for example cardiac, muscle and adipose tissue endothelial cells, it is found to be synthesized by parenchymal cells and thought to be translocated to its site of action afterwards. LPL is a secretory water-soluble protein that belongs to the triglyceride lipase family and has triacylglycerol hydrolase activity that targets lipoproteins such as chylomicrons and VLDL through their ApoCII moiety. By hydrolyzing the triacylglycerol component, LPL provides fatty acids to peripheral tissues and therefore plays a central role in overall lipid metabolism. In addition, LPL has a non-catalytic function based on its heparin- and lipoprotein-binding domains, which enables it to form a bridge between lipoproteins and cell surface HSPG, especially with strong ligand affinity to hepatocyte HSPG (syndecan-1). Since HCV particles are tightly associated to lipoproteins, hepatocyte HSPG has been proposed as a possible attachment receptor for HCV[94-96]. In addition, ApoE has been implicated in virus attachment to the host cell by an interaction that involves cell surface HSPG, in particular syndecan-1[97,98].
Although extrahepatically expressed, peripheral LPL was shown to be an important component of lipoproteins capable of mediating their binding to hepatic receptors and, thereby targeting lipoproteins to the liver for internalization and degradation in hepatocytes[34,99,100]. As it is involved in hepatic clearance of lipoproteins from the circulation, LPL’s role has been investigated in HCV cell entry. It was observed that exogenous LPL indeed mediates cellular binding of HCV through HCV-associated lipoproteins in an HSPG-dependent manner. These findings suggest an indirect interaction of HCV with HSPG, through LPL. As mentioned however, while LPL enhances HCVcc binding to hepatoma cells, it actually decreases HCV infectivity[88,101,102].
Different mechanisms can be proposed to explain this abortive LPL-dependent infection process. LPL could mediate hepatic lipoviral clearance either by means of its lipoprotein bridging abilities to liver HSPG (syndecan-1), that leads to direct endocytosis through the HSPG-metabolic pathway and degradation of HCV[101], or alternatively by its lipolytic activity, or by a combination of both. The enzymatic activity of LPL could transform HCV-associated lipoproteins into lipoprotein remnants that facilitate virus binding to LDL-R, also a non-productive HCV entry pathway[88,103]. Finally, using electron microscopy Maillard et al[102] provided evidence for infectivity inhibition by retention of the viral particle at the cell surface, rather than non-productive cellular uptake, that depended on both LPL-mediated lipolytic modification of the viral lipoprotein content and its strong bridging effect.
As mentioned, LPL is a non-hepatically expressed triglyceride lipase, mainly involved in converting VLDL to IDL and LDL. Another enzyme catalyzing IDL to LDL conversion is the hepatically expressed hepatic triglyceride lipase (HTGL). Modification of HCVcc-associated lipoproteins exerted by these lipases, resulted in a decreased amount of HCV-associated ApoE and HCV of higher density, which caused loss of HCV infectivity. HTGL blockade partially neutralized LPL-mediated infection inhibition and its knockdown even increased HCV infectivity[103]. Altogether, these observations tempt us to hypothesize that interactions with lipases can target the lipoviroparticle to an abortive entry pathway that involves LDL-R and/or HSPG.
Lipid receptor ligands influence HCV entry: Several SR-BI receptor ligands have been reported to alter HCV infectivity; HDL enhances HCV entry (see next paragraph) whereas VLDL, LDL, oxidized LDL and amyloid-α have inhibitory effects. OxLDL presumably inhibits HCV infection by two different mechanisms. First, it affects the biophysical properties of the viral particle; and second it perturbs the interaction between HCV and SR-BI in a non-competitive manner[104,105]. Serum amyloid A, an acute phase protein and SR-BI receptor ligand, was also shown to possess anti-HCV activity by interacting with the viral particle[106,107].
Both LDL and VLDL reduce infectivity of HCVpp[23,108]. VLDL inhibits HCVcc probably by abrogating the binding between virus associated ApoE and SR-BI[66]. To some degree, LDL also inhibits SR-BI interaction with HCV from serum of infected patients but less efficiently than VLDL[67]. Accordingly, LDL and VLDL outcompete serum-derived lipoviroparticle binding to hepatocyte cell lines[60]. LDL has also been shown to compete with serum-derived HCV for LDL-R[82].
HDL enhances HCV infection: Different groups reported that infectivity of HCVcc and HCVpp in cell culture is enhanced by the presence of human serum, in which the HDL component was identified as the responsible agent[23-25,69,72,73,108,109]. It was discovered that the HVR1 region of E2 is particularly important for this process, since HVR1 deletion or mutations in this region greatly hamper the infection enhancement ability of HDL. Accordingly, we observed that HVR1 deletion or other mutations in the envelope glycoproteins that alter HCV’s SR-BI-dependency for viral entry render these viruses unable to benefit from HDL-mediated infection enhancement[110]. SR-BI is indeed the host cell factor supporting this phenomenon. This is endorsed by the observations that HDL-mediated infection enhancement can be abolished by both small-molecule inhibitors of SR-BI’s lipid transfer[23,72,73] and mutations in SR-BI that reduce its lipid transfer function[109]. However, besides lipid transfer abrogation, these receptor mutations and small molecule inhibitors also decreased the binding potential of sE2 to SR-BI, making it impossible to discriminate the involvement of these two receptor functions in the enhancing process. Dao Thi et al[66] recently confirmed the essential role of the E2 binding function of SR-BI in this context. They observed that mouse SR-BI, which has functional lipid transfer activity but lacks E2 binding properties, did not allow HDL to enhance HCV infectivity. In addition, HDL did not enhance cell entry of HCVpp harboring SR-BI binding defective E2.
Since, to our knowledge, it has never been shown that HDL infection enhancement solely relies on the ability of SR-BI to interact with E2, independent of SR-BI’s lipid transfer, we propose a concerted action of both receptor functions in this process. We postulate that the critical E2-SR-BI interaction positions the HCV viral particle in such a way that it significantly benefits from SR-BI’s lipid transfer, leading to infection enhancement. By promoting the SR-BI mediated lipid transfer, HDL could either change cholesterol contents of the lipoviroparticle itself or initiate cholesterol enrichment of the host cell plasma membrane resulting in facilitation of post-attachment entry events. It has indeed been reported that HCV entry is dependent on the cholesterol content of the host cell membrane[111]. Possibly, cholesterol-mediated elevated plasma membrane fluidity assists and accelerates essential events in the HCV entry process such as receptor recruitment and interactions, particle internalization and fusion. Of note, HDL does not stimulate HCVpp or sE2 binding to CD81 or SR-BI, and no indication for direct interactions between HDL and HCVpp have been observed[73,112].
Increased receptor recruitment and interactions. E2-specific antibodies do not abrogate HCV binding to SR-BI, yet they neutralized infectivity of HCV particles of all densities[66]. Although E2 is therefore not involved in primary HCV-SR-BI attachment, it does mediate HCV cell entry by enabling HDL to enhance its infectivity and through interaction with more downstream entry factors. It is observed that the HVR1 region of E2 masks the viral binding site to CD81[113], the central regulator of HCV entry[114]. Possibly by inducing a conformational change to the viral envelope, HCV-E2-SR-BI interaction activates the virus for CD81 receptor engagement. A kinetic analysis of HCVcc entry illustrated that SR-BI acts at an early step in the infection process closely linked to the interactions of the virus with CD81 and CLDN1 suggesting that HCV entry may be mediated through the formation of co-receptor complexes[68,115]. Additional evidence for co-receptor complex formation between CD81 and CLDN1 was demonstrated using FRET analysis[116].
SR-BI localizes to cholesterol-enriched plasma membrane microdomains[117] where also tetraspanins such as CD81 associate with each other, with other tetraspanin and with non tetraspanin proteins. These associations are modulated by cholesterol[118,119]. Therefore, SR-BI/HDL mediated cholesterol enrichment of the plasma membrane could increase the efficiency of co-receptor complex formation. For this reason, the SR-BI-HDL-HCV interaction should act in concert with downstream HCV receptors. CD81 is indeed required for SR-BI/HDL-mediated HCV entry enhancement since silencing of CD81 expression abolished this process[68]. In addition, CD81 and SR-BI cooperatively mediate HCV infection, and this is dependent on membrane cholesterol content[111]. Cellular cholesterol depletion also decreased HCV infectivity by reducing CLDN1 localization, CLDN1-CD81 and CD81-CD81 association at the plasma membrane[120]. Furthermore, SR-BI and CD81 mutants defective for receptor palmitoylation, which is essential for their partitioning in cholesterol-rich microdomains, reduced HCV entry[109,121]. Hydrolysis of sphingomyelin, another important plasma membrane lipid constituent, reduced HCV entry by decreasing CD81 cell surface levels[122]. Altogether, these observations emphasize that the function of HCV cell entry factors strongly depends on specific membrane lipid contents. Therefore, it can be assumed that these HCV receptor associations benefit from SR-BI/HDL-mediated cholesterol delivery.
Accelerated particle internalization. HDL-mediated HCVpp and HCVcc entry acceleration has been observed[24,73]. HDL accelerates HCV endocytosis by reducing the time that viral particles are cell-bound before being internalized[73]. This pre-internalization phase may reflect the time needed to form HCV receptor complexes, a process that likely benefits from SR-BI/HDL-mediated cholesterol delivery (cfr supra). This is in agreement with the rate-limiting role fulfilled by SR-BI in HCV entry[123,124].
Increased fusion. After formation of the HCV-receptor complex, HCV utilizes clathrin-dependent receptor mediated endocytosis to enter cells. Of note, the function of these specialized cholesterol-enriched plasma membrane domains, clathrin-coated pits, is also dependent on membrane cholesterol levels[125]. The internalization step of endocytosis involves the budding of vesicles from the plasma membrane, which are then targeted to and fused with an early endosomal compartment. In the early endosome, acidification probably induces a conformational change in the E1E2 glycoproteins, after which the viral envelope fuses with the endosomal membrane, thereby releasing the viral nucleocapsid into the cytoplasm[126,127]. This fusion process is significantly facilitated by the presence of cholesterol and sphingomyelin in the target membrane[128,129] and ApoCI, an exchangeable apolipoprotein that predominantly resides in HDL, on the viral membrane[112]. Since HDL did not directly interact or exchange ApoCI with HCV, the authors propose a mechanism for ApoCI exchange mediated by a close interaction of HDL with HCV at their common receptor, SR-BI[112]. We hypothesize that this exchange depends on a correct approximation of HCV with HDL, mediated by an intact SR-BI-E2 interaction that is essential for HDL-mediated infection enhancement.
Effects on viral RNA replication
Genomic HCV RNA replication[130] has also been linked to lipid metabolism[131]. Changes in hepatocyte lipid content could therefore affect the replication of the HCV genome. Indeed, an anti-LDL-R mAb decreased HCV RNA replication by altering intracellular lipid content[88]. For example decreased free/esterified cholesterol and phosphatidylcholine/phospatidylethanolamine ratios were observed[88]. Of particular interest is the latter ratio of membrane phospholipids which, when decreased, has been linked to steatohepatitis. A decrease of this phospholipid ratio affects endoplasmic reticulum membrane integrity where HCV replication takes place[132]. In addition, the HCV polymerase, NS5B, contains a sphingolipid binding motif and a sphingolipid biosynthesis inhibitor was found to block HCV replication[133]. Finally, the lipid kinase phosphatidylinositol 4-kinaseIIIα (PI4KIIIα) was shown to be a host cofactor essential for efficient HCV replication by supporting the formation of the membranous web, an altered membrane structure specialized in HCV RNA replication[134,135].
Effects on assembly and secretion
Lipid droplets establish an HCV production microenvironment: The lipid droplet (LD) is an organelle used for the storage of neutral lipids and the maintenance of intracellular lipid homeostasis. LDs consist of a core of triglycerides and cholesteryl esters, surrounded by a phospholipid monolayer and associated proteins. It moves through the cytoplasm where it can interact with the endoplasmic reticulum, thereby facilitating lipid and protein transport to other organelles. In addition, cell signaling and trafficking events have been linked to LDs[136].
Moradpour et al[137] found that HCV core protein is located on the endoplasmic reticulum membrane and on the surface of cytoplasmic LDs[138]. Moreover, it was shown that LD-association of HCV proteins is essential for production of infectious HCV particles. HCV core proteins on the LDs seem to induce endoplasmic reticulum derived LD-associated membrane rearrangements. These rearrangements might establish an HCV production microenvironment recruiting HCV RNA and non-structural proteins around LDs[139]. In addition, the HCV core protein induces LD overproduction which might be linked to steatosis and abnormal lipid metabolism caused by chronic HCV infection[140]. Besides lipid storage, the LD is involved in the lipoprotein secretory pathway[141], which is found to intersect with the HCV assembly process[142].
HCV secretion intersects with the VLDL secretory pathway: The first step in the VLDL assembly process involves co-translational lipidation of ApoB-100, the main protein component of VLDL. Thereto, microsomal triglyceride transfer protein (MTP) interacts with ApoB100 to deliver lipids from LDs into the endoplasmic reticulum lumen. Additional lipidation results in a triglyceride-poor form of VLDL. After transportation to the Golgi apparatus, depending on the triglyceride level, it can either be secreted as such or further lipidated, and associated to other apolipoproteins such as ApoE, into mature triglyceride-rich VLDL by fusing with bulk lipids from luminal LDs[141].
HCV and VLDL are both uniquely secreted by hepatocytes and circulate in blood associated to each other[60,61,143]. This complexation likely occurs during viral particle assembly and release, processes found to intersect with VLDL assembly and secretion pathways. On isolated membrane vesicles containing the HCV replication complex, Huang et al[142] observed enrichment of proteins involved in VLDL assembly, such as ApoB, ApoE and MTP. In addition, HCV production was reduced by VLDL assembly blockade using ApoB silencing and MTP inhibition[142,144,145]. These findings, together with the observation that infectious intracellular precursors of HCV have a higher buoyant density compared to secreted HCV[146,147] argues for a lipidation of HCV during a maturation process that parallels VLDL formation. Additional evidence for the overlap of HCV and VLDL assembly processes is provided by the work of Icard et al[148]. They detected HCV glycoproteins in ApoB containing triglyceride rich lipoproteins when expressed in Caco-2 and HepG2 cells, but not in Huh7 hepatoma cells, an event that was prevented by MTP inhibition. Moreover, HCV is thought to divert cellular processes towards its own generation, which is detrimental to the hepatocyte’s VLDL production. An HCV-induced deficiency in arylacetamide deacetylase, an enzyme involved in VLDL production by lipolysis of triacylglycerol from LDs, was identified to cause VLDL production impairment during the early peak of HCV infection in culture[149].
Besides MTP and ApoB, ApoE is essential during VLDL and HCV production. Fazio et al[150] demonstrated that ApoE is not exclusively important for lipoprotein uptake alone, but it can be recycled to be involved in lipoprotein assembly as well. Interestingly, its knockdown hampers the production of infectious HCV particles[151,152]. Production of secreted infectious HCV was very poorly restored after ectopic expression of an ApoE mutant that is unable to be secreted; infectious HCV particles accumulated intracellularly instead[86]. It should be noted that Jiang et al[152], emphasize the importance of ApoE during HCV production but show that ApoB inhibition does not affect this process. They attribute the effect of MTP inhibitors on HCV production suppression to its inhibitory consequences on ApoE expression rather than its blockade of ApoB lipidation. Accordingly, production of HCV from non-liver cells depends on ectopic expression of ApoE[153] and not ApoB or MTP[154]. ApoE interacts with HCV nonstructural protein 5A (NS5A) that is involved in HCV production. Hence assembly-defective HCV mutants had strongly impaired ApoE-NS5A binding[152,155]. ApoE is suggested to be involved in an assembly step that acts between capsid envelopment and secretion of infectious HCV[154].
Apolipoproteins and lipids are HCV particle constituents: While the requirement of ApoB for HCV assembly is controversial, ApoB-containing lipoviroparticles have been identified in patients[60]. Although VLDL and both anti-ApoB and ApoE block serum derived lipoviroparticle binding to hepatocyte cell lines[60], only VLDL and anti-ApoE can efficiently compete them out for interaction with SR-BI[67]. The presence of ApoE on the viral particle is confirmed in different reports and is assigned a very important function in HCV entry, especially since ApoE specific antibodies neutralize HCV infectivity[84,151,156]. As mentioned, ApoE, rather than HCV glycoprotein E2, is involved in primary attachment of HCV to SR-BI[66,67]. In addition, ApoE is involved in direct cell-to-cell transmission[154] and the loss of HCV associated ApoE, after LPL treatment, resulted in decreased HCV infectivity[103].
ApoCI, an exchangeable apolipoprotein that predominantly resides in HDL, has been identified as a component of the HCV viral particle[157]. While ApoCI is not a minimal requirement for HCV assembly[154], its association occurs within the cell prior to virus release[157]. Like HDL, pre-incubation of HCVpp with ApoCI resulted in a genotype-independent enhancement of infection. The observation that anti-ApoCI suppressed the HDL-mediated infection enhancement further suggests that ApoCI is a key mediator of this process[108,112,157]. As mentioned, ApoCI enhances HCV infectivity by specifically promoting HCV fusion. It should be noted that higher concentrations of ApoCI inhibited HCVpp infectivity by specifically disrupting the viral membrane[112].
In support of the tight association with lipoproteins, it was observed that the lipid composition of HCV particles is strikingly different from cell membranes, but its increased ratio of cholesteryl esters-to-phospholipids rather resembles that of LDL and VLDL[156]. Furthermore, the association of cholesterol and sphingolipids with HCV is essential for cell internalization, virus maturation and infectivity[158,159]. In addition, HCV-associated cholesterol might contribute to the virion’s interaction with ApoE, since cholesterol-extracted HCVcc showed dramatically reduced reactivity with anti-ApoE[159].
EFFECT OF LIPOPROTEINS ON ANTIVIRAL EFFICACY OF ANTI-HCV AND ANTI-RECEPTOR ANTIBODIES
Effect of lipoproteins on escape from anti-HCV neutralizing Abs
HCV-associated lipoproteins: Lipoproteins associated to virus particles could mask neutralizing epitopes, thereby facilitating viral escape from the humoral immune response. Besides viral components, cell entry of HCV is initiated and mediated by endogenous proteins providing a mechanism of escape from the humoral immune response. Indeed, HCV uses associated host lipoprotein components to attach to lipid receptors on the hepatocyte. This explains, at least partially, the inefficiency of anti-envelope antibodies to prevent primary hepatocyte attachment of HCV and to neutralize HCV infectivity[66,67]. Indeed, it has been postulated that virus-lipoprotein interactions play a role in immune evasion[60]. Some evidence has been obtained to support this hypothesis. It was observed that HCV particles of lower density are more resistant to neutralizing antibodies, a feature that might however be specific for gt2a virus only[160-162]. In addition, serum-derived HCV particles have been observed that were seemingly totally bound to beta-lipoprotein, a condition that coincided with the absence of HCV-IgG complexes, suggesting that virus associated lipoproteins may restrict the access of anti-envelope antibodies[163].
Previous studies have indicated a role for HVR1 and E2 residue 451 in covering the CD81 binding site, shielding neutralizing epitopes and infectivity of low density viral particles[113,160]. In addition, Tao and coworkers described a mutant (I414T) with increased susceptibility to E2 neutralizing Abs. Although prior to release, no difference in neutralization behavior was observed between mutant and wild type virus, the latter became more resistant to neutralization upon cell secretion. This observation was attributed to increased ApoE and ApoCI abundance on the viral particle[147]. Accordingly, HCV production in ApoE knockdown cells generates viral particles with increased sensitivity to neutralization[164]. In addition, a correlation was observed between ApoE association and the ability of a viral isolate to escape from neutralizing antibodies and infect the liver graft after liver transplantation[164]. This might explain viral quasispecies evolution during liver transplantation. Clinical virus isolates selected during the liver transplantation process were indeed characterized by enhanced viral entry and escape from neutralizing antibodies compared to non-selected variants[165].
We hypothesize that the interaction of HCV-associated ApoE with the host cell receptor SR-BI initiates changes to the viral particle by altering its lipid content, mediated by SR-BI’s lipid transfer activity. Alternatively, the ApoE-SR-BI interaction could precede and direct E2 presentation to SR-BI, which in turn initiates a conformational change in E2. These modifications might result in exposing CD81 binding sites and neutralizing epitopes in E2. This gradual epitope exposure could be a system used by HCV to preclude neutralizing antibody recognition and might be needed for E2 to interact with the downstream receptor CD81. It has indeed been suggested that CD81 binding epitopes are not yet exposed on circulating particles[81,166,167]. This implies that anti-E2 antibodies interfering with E2-CD81 interactions may only exert their neutralizing activity from the moment of epitope exposure after SR-BI docking until the interaction with the downstream coreceptor complex. Importantly, HDL reduces this time window presumably by facilitating receptor complex formation[73].
Virus-free lipoproteins: As mentioned before, HDL is able to enhance HCV infectivity via the SR-BI lipid transfer and E2 binding function. It was observed that HDL reduces the lag between HCV-host cell binding and its actual internalization. This might reflect a reduction in the time needed to form a functional receptor complex and the time needed for viral glycoprotein E2 to engage with CD81. Therefore, HDL can reduce the antiviral efficiency of molecules blocking these interactions by shortening the time window during which such molecules are active. In fact, the virus might use this entry acceleration to escape humoral immune responses that target this virus-host factor interaction[73]. This was confirmed by a kinetic analysis revealing that HCVpp entry enhancement by HDL is initiated earlier than the activity of neutralizing antibodies[24]. Accordingly, by enhancing HCV entry, HDL lowers the neutralization efficacy of such anti-HCV antibodies. This neutralization attenuation effect was specifically observed for antibodies interfering with the E2-CD81 interaction (such as AP33, 3/11 and H48) and was not observed for polyclonal anti-E1 or an antibody with slower neutralization kinetics that interfered with E2-SR-BI (designated 9/27)[24,73]. In addition, antibodies isolated from HCV-infected patients also suffer from reduced neutralization efficacy in the presence of human serum or HDL[23-25]. Inhibition of SR-BI’s lipid transfer restores neutralization by anti-HCV antibodies in the presence of human serum[23,24,73]; resistance to neutralization by an anti-E2 neutralizing Ab (C1) correlated with increased SR-BI expression levels[123]; and, the neutralizing activity of patient sera increased with lower SR-BI levels[24]. These observations indicate that SR-BI is the responsible factor, activated by HDL, for neutralization attenuation of anti-HCV antibodies. In conclusion, although antibodies that bind to the viral envelope, and thereby interfere with receptor interactions, are able to neutralize HCV infection, SR-BI/HDL interactions accelerate the entry of HCV particles that are not yet neutralized. This process therefore might help the virus to persist in patients. Indeed, persisting viral strains were more efficiently facilitated by serum than those that were controlled during the acute phase[25].
Effect of lipoproteins on escape from anti-receptor Abs
Anti-CD81 antibodies: Another class of HCV entry inhibitors that interfere with CD81 and E2 interactions are CD81-receptor blockers. If this receptor blocker is not present before the virus initiates its entry, HDL could again reduce its antiviral efficacy by shortening the window of effective antiviral activity. Accordingly, HCV infection inhibition using an anti-CD81 mAb, designated JS81, was strongly reduced when added during infection in the presence of human serum[73]. Once more, the attenuation of anti-CD81 antiviral efficacy was alleviated by blocking SR-BI, indicating that compounds that block E2-CD81 interaction are less potent when SR-BI is activated. The same antibody was effective in preventing HCV infection in a human-liver chimeric mouse model, but was unable to resolve infection when administered shortly (six hours to three days) after viral challenge[70,168]. This again highlights the importance of CD81 receptor blocker availability before exposure to the virus.
Anti-SR-BI antibodies: In contrast to CD81 receptor blockade, the timing of SR-BI inhibition seems less delicate since administration of anti-SR-BI mAb therapy three days after HCV inoculation still successfully protects humanized mice from infection[70,169]. Accordingly, we have shown that this therapy effectively inhibits direct cell-to-cell transmission, hence indicating that it exerts its antiviral activity even after the primary infection is established. Therefore, compared to anti-CD81 therapy, it appears that the time frame after infection during which the anti-SR-BI therapy remains effective is significantly larger. As mentioned, HDL reduces the antiviral efficacy of molecules interfering with CD81-E2 interactions by shortening the time window during which such molecules are active[73]. In contrast, it was shown that HDL did not negatively affect anti-SR-BI’s antiviral efficacy[69]. Moreover, we observed that human HDL positively influences the ability of an anti-SR-BI specific antibody, mAb1671, to inhibit HCV infectivity[110]. Addition of in vivo-like concentrations of human HDL to anti-SR-BI mAb1671 in the HCV cell culture system significantly increases its antiviral efficacy, enabling almost complete infection inhibition. This observation might explain why the antibody reaches higher protection levels in humanized mice than it does in the HCV cell culture system; and why it has a larger therapeutic window compared to anti-CD81 in vivo. Of note, anti-SR-BI small molecule ITX-5061’s antiviral efficacy was not increased by the addition of in vivo-like concentrations of human HDL in the HCV cell culture system.
CONCLUSION
As mentioned, today no anti-HCV therapy is approved for prevention of re-infection after liver transplantation. Interestingly, anti-SR-BI therapy was strongly protective in humanized mice after exposure to a serum-derived viral variant that escaped the control of the adaptive immune response and became dominant after liver transplantation[165,169]. These variants have an increased entry phenotype and escape more easily from envelope neutralizing antibodies, possibly due to increased HCV-ApoE associations[164]. These data suggest that SR-BI receptor blockade may be a novel therapeutic approach to prevent graft reinfection in liver transplant patients. Furthermore, since active SR-BI was shown to be the responsible factor for HDL-mediated infection enhancement and reduced efficacy of anti-CD81 and anti-E2 neutralizing Abs, SR-BI inhibitors might increase their antiviral efficacy in vivo. Although HCV variants have been described that are more resistant to anti-SR-BI therapy in culture, these variants remain fully responsive to anti-SR-BI therapy in humanized mice[110]. These observations encourage us to speculate that combinations of anti-SR-BI with anti-E2 antibodies might be worth the considering in the context of liver transplantation.
ACKNOWLEDGMENTS
We also want to thank Ms. Julie Vercauteren for graphical assistance.
Footnotes
Supported by Ghent University, Grants No. 01G00507 and No. 01G01712; Research Foundation - Flanders, Projects No. 1500910N, No. G0212.10N and No. G052112N (FWO-Vlaanderen); Belgian Federal Government, No. IUAP P6/36-HEPRO and No. P7/47-HEPRO-2; and European Union No. FP6 HEPACIVAC; FP7, HepaMab; Mesalam AA is a recipient of a PhD Fellowship provided by the Egyptian government
P- Reviewer: Shimizu Y S- Editor: Ma YJ L- Editor: A E- Editor: Liu XM
References
- 1.Zeuzem S. Interferon-based therapy for chronic hepatitis C: current and future perspectives. Nat Clin Pract Gastroenterol Hepatol. 2008;5:610–622. doi: 10.1038/ncpgasthep1274. [DOI] [PubMed] [Google Scholar]
- 2.Sarrazin C, Hézode C, Zeuzem S, Pawlotsky JM. Antiviral strategies in hepatitis C virus infection. J Hepatol. 2012;56 Suppl 1:S88–S100. doi: 10.1016/S0168-8278(12)60010-5. [DOI] [PubMed] [Google Scholar]
- 3.Manns M, Marcellin P, Poordad FPF, de Araujo ESA, Buti M, Horsmans Y, Janczewska EJE, Villamil F, Peeters M, Lenz O, et al. Simeprevir (Tmc435) with Peginterferon/Ribavirin for Treatment of Chronic Hcv Genotype-1 Infection in Treatment-Naive Patients: Results from Quest-2, a Phase Iii Trial. J Hepatol. 2013;58:S568–S568. [Google Scholar]
- 4.Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368:1878–1887. doi: 10.1056/NEJMoa1214853. [DOI] [PubMed] [Google Scholar]
- 5.Peveling-Oberhag J, Zeuzem S, Hofmann WP. Antiviral therapy of chronic hepatitis C in patients with advanced liver disease and after liver transplantation. Med Microbiol Immunol. 2010;199:1–10. doi: 10.1007/s00430-009-0131-8. [DOI] [PubMed] [Google Scholar]
- 6.Charlton M. Telaprevir, boceprevir, cytochrome P450 and immunosuppressive agents--a potentially lethal cocktail. Hepatology. 2011;54:3–5. doi: 10.1002/hep.24470. [DOI] [PubMed] [Google Scholar]
- 7.Curry MP, Forns X, Chung RT, Terrault N, Brown RS, Fenkel JM, Gordon FD, O’Leary JG, Kuo A, Schiano TD, et al. Pretransplant Sofosbuvir and Ribavirin to Prevent Recurrence of HCV Infection after Liver Transplantation. Hepatology. 2013;58:314a–315a. doi: 10.1053/j.gastro.2014.09.023. [DOI] [PubMed] [Google Scholar]
- 8.Charlton MR, Gane EJ, Manns MP, Brown RS, Curry MP, Kwo PY, Fontana RJ, Gilroy R, Teperman LW, Muir AJ, et al. Sofosbuvir and Ribavirin for the Treatment of Established Recurrent Hepatitis C Infection After Liver Transplantation: Preliminary Results of a Prospective, Multicenter Study. Hepatology. 2013;58:1378a–1378a. [Google Scholar]
- 9.Forns X, Fontana RJ, Moonka D, McHutchison JG, Symonds WT, Denning JM, McNair L, Chang P, Kivett VA, Shiffman ML, et al. Initial Evaluation of the Sofosbuvir Compassionate Use Program for Patients with Severe Recurrent HCV Following Liver Transplantation. Hepatology. 2013;58:732a–733a. [Google Scholar]
- 10.Kwo P, Mantry P, Coakley E, Te H, Vargas H, Brown R, Gordon F, Levitsky J, Terrault N, Burton J, et al. Results of the phase 2 study M12-999: Interferon-free regimen of ABT-450/r/ABT-267 ABT-333 ribavirin in liver transplant recipients with recurrent HCV genotype 1 infection. J Hepatol. 2014;60:s47–s48. [Google Scholar]
- 11.Vercauteren K, Leroux-Roels G, Meuleman P. Blocking HCV entry as potential antiviral therapy. Future Virol. 2012;7:547–561. [Google Scholar]
- 12.Keck ZY, Machida K, Lai MM, Ball JK, Patel AH, Foung SK. Therapeutic control of hepatitis C virus: the role of neutralizing monoclonal antibodies. Curr Top Microbiol Immunol. 2008;317:1–38. doi: 10.1007/978-3-540-72146-8_1. [DOI] [PubMed] [Google Scholar]
- 13.Chapel HM, Christie JM, Peach V, Chapman RW. Five-year follow-up of patients with primary antibody deficiencies following an outbreak of acute hepatitis C. Clin Immunol. 2001;99:320–324. doi: 10.1006/clim.2001.5036. [DOI] [PubMed] [Google Scholar]
- 14.Pestka JM, Zeisel MB, Bläser E, Schürmann P, Bartosch B, Cosset FL, Patel AH, Meisel H, Baumert J, Viazov S, et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci USA. 2007;104:6025–6030. doi: 10.1073/pnas.0607026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farci P, Shimoda A, Wong D, Cabezon T, De Gioannis D, Strazzera A, Shimizu Y, Shapiro M, Alter HJ, Purcell RH. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc Natl Acad Sci USA. 1996;93:15394–15399. doi: 10.1073/pnas.93.26.15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanwolleghem T, Bukh J, Meuleman P, Desombere I, Meunier JC, Alter H, Purcell RH, Leroux-Roels G. Polyclonal immunoglobulins from a chronic hepatitis C virus patient protect human liver-chimeric mice from infection with a homologous hepatitis C virus strain. Hepatology. 2008;47:1846–1855. doi: 10.1002/hep.22244. [DOI] [PubMed] [Google Scholar]
- 17.Meuleman P, Bukh J, Verhoye L, Farhoudi A, Vanwolleghem T, Wang RY, Desombere I, Alter H, Purcell RH, Leroux-Roels G. In vivo evaluation of the cross-genotype neutralizing activity of polyclonal antibodies against hepatitis C virus. Hepatology. 2011;53:755–762. doi: 10.1002/hep.24171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morin TJ, Broering TJ, Leav BA, Blair BM, Rowley KJ, Boucher EN, Wang Y, Cheslock PS, Knauber M, Olsen DB, et al. Human monoclonal antibody HCV1 effectively prevents and treats HCV infection in chimpanzees. PLoS Pathog. 2012;8:e1002895. doi: 10.1371/journal.ppat.1002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willems B, Ede M, Marotta P, Wall W, Greig P, Lilly L, Kneteman N, Wong WN, Roy A, Marleau D, et al. Anti-HCV human immunoglobulins for the prevention of graft infection in HCV-related liver transplantation, a pilot study. J Hepatol. 2002;36:32–32. [Google Scholar]
- 20.Davis GL, Nelson DR, Terrault N, Pruett TL, Schiano TD, Fletcher CV, Sapan CV, Riser LN, Li Y, Whitley RJ, et al. A randomized, open-label study to evaluate the safety and pharmacokinetics of human hepatitis C immune globulin (Civacir) in liver transplant recipients. Liver Transpl. 2005;11:941–949. doi: 10.1002/lt.20405. [DOI] [PubMed] [Google Scholar]
- 21.Schiano TD, Charlton M, Younossi Z, Galun E, Pruett T, Tur-Kaspa R, Eren R, Dagan S, Graham N, Williams PV, et al. Monoclonal antibody HCV-AbXTL68 in patients undergoing liver transplantation for HCV: results of a phase 2 randomized study. Liver Transpl. 2006;12:1381–1389. doi: 10.1002/lt.20876. [DOI] [PubMed] [Google Scholar]
- 22.Chung RT, Gordon FD, Curry MP, Schiano TD, Emre S, Corey K, Markmann JF, Hertl M, Pomposelli JJ, Pomfret EA, et al. Human monoclonal antibody MBL-HCV1 delays HCV viral rebound following liver transplantation: a randomized controlled study. Am J Transplant. 2013;13:1047–1054. doi: 10.1111/ajt.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartosch B, Verney G, Dreux M, Donot P, Morice Y, Penin F, Pawlotsky JM, Lavillette D, Cosset FL. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J Virol. 2005;79:8217–8229. doi: 10.1128/JVI.79.13.8217-8229.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voisset C, Op de Beeck A, Horellou P, Dreux M, Gustot T, Duverlie G, Cosset FL, Vu-Dac N, Dubuisson J. High-density lipoproteins reduce the neutralizing effect of hepatitis C virus (HCV)-infected patient antibodies by promoting HCV entry. J Gen Virol. 2006;87:2577–2581. doi: 10.1099/vir.0.81932-0. [DOI] [PubMed] [Google Scholar]
- 25.Lavillette D, Morice Y, Germanidis G, Donot P, Soulier A, Pagkalos E, Sakellariou G, Intrator L, Bartosch B, Pawlotsky JM, et al. Human serum facilitates hepatitis C virus infection, and neutralizing responses inversely correlate with viral replication kinetics at the acute phase of hepatitis C virus infection. J Virol. 2005;79:6023–6034. doi: 10.1128/JVI.79.10.6023-6034.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barth H, Schafer C, Adah MI, Zhang F, Linhardt RJ, Toyoda H, Kinoshita-Toyoda A, Toida T, Van Kuppevelt TH, Depla E, et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J Biol Chem. 2003;278:41003–41012. doi: 10.1074/jbc.M302267200. [DOI] [PubMed] [Google Scholar]
- 27.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci USA. 1999;96:12766–12771. doi: 10.1073/pnas.96.22.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 29.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 31.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffin BA. Lipid metabolism. Surgery (Oxford) 2013;31:267–272. [Google Scholar]
- 33.Chappell DA, Medh JD. Receptor-mediated mechanisms of lipoprotein remnant catabolism. Prog Lipid Res. 1998;37:393–422. doi: 10.1016/s0163-7827(98)00017-4. [DOI] [PubMed] [Google Scholar]
- 34.Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: structure, function, regulation, and role in disease. J Mol Med (Berl) 2002;80:753–769. doi: 10.1007/s00109-002-0384-9. [DOI] [PubMed] [Google Scholar]
- 35.Rosenson RS, Brewer HB, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown WV. High-density lipoprotein and transport of cholesterol and triglyceride in blood. J Clin Lipidol. 2007;1:7–19. doi: 10.1016/j.jacl.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Serfaty L, Andreani T, Giral P, Carbonell N, Chazouillères O, Poupon R. Hepatitis C virus induced hypobetalipoproteinemia: a possible mechanism for steatosis in chronic hepatitis C. J Hepatol. 2001;34:428–434. doi: 10.1016/s0168-8278(00)00036-2. [DOI] [PubMed] [Google Scholar]
- 38.Siagris D, Christofidou M, Theocharis GJ, Pagoni N, Papadimitriou C, Lekkou A, Thomopoulos K, Starakis I, Tsamandas AC, Labropoulou-Karatza C. Serum lipid pattern in chronic hepatitis C: histological and virological correlations. J Viral Hepat. 2006;13:56–61. doi: 10.1111/j.1365-2893.2005.00655.x. [DOI] [PubMed] [Google Scholar]
- 39.Poynard T, Ratziu V, McHutchison J, Manns M, Goodman Z, Zeuzem S, Younossi Z, Albrecht J. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology. 2003;38:75–85. doi: 10.1053/jhep.2003.50267. [DOI] [PubMed] [Google Scholar]
- 40.Walters KA, Joyce MA, Thompson JC, Smith MW, Yeh MM, Proll S, Zhu LF, Gao TJ, Kneteman NM, Tyrrell DL, et al. Host-specific response to HCV infection in the chimeric SCID-beige/Alb-uPA mouse model: role of the innate antiviral immune response. PLoS Pathog. 2006;2:e59. doi: 10.1371/journal.ppat.0020059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci USA. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gopal K, Johnson TC, Gopal S, Walfish A, Bang CT, Suwandhi P, Pena-Sahdala HN, Clain DJ, Bodenheimer HC, Min AD. Correlation between beta-lipoprotein levels and outcome of hepatitis C treatment. Hepatology. 2006;44:335–340. doi: 10.1002/hep.21261. [DOI] [PubMed] [Google Scholar]
- 43.Ikeda M, Abe K, Yamada M, Dansako H, Naka K, Kato N. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology. 2006;44:117–125. doi: 10.1002/hep.21232. [DOI] [PubMed] [Google Scholar]
- 44.Delang L, Paeshuyse J, Vliegen I, Leyssen P, Obeid S, Durantel D, Zoulim F, Op de Beeck A, Neyts J. Statins potentiate the in vitro anti-hepatitis C virus activity of selective hepatitis C virus inhibitors and delay or prevent resistance development. Hepatology. 2009;50:6–16. doi: 10.1002/hep.22916. [DOI] [PubMed] [Google Scholar]
- 45.Milazzo L, Meroni L, Galazzi M, Cesari M, Caramma I, Marchetti G, Galli M, Antinori S. Does fluvastatin favour HCV replication in vivo? A pilot study on HIV-HCV coinfected patients. J Viral Hepat. 2009;16:479–484. doi: 10.1111/j.1365-2893.2009.01104.x. [DOI] [PubMed] [Google Scholar]
- 46.O’Leary JG, Chan JL, McMahon CM, Chung RT. Atorvastatin does not exhibit antiviral activity against HCV at conventional doses: a pilot clinical trial. Hepatology. 2007;45:895–898. doi: 10.1002/hep.21554. [DOI] [PubMed] [Google Scholar]
- 47.Bader T, Fazili J, Madhoun M, Aston C, Hughes D, Rizvi S, Seres K, Hasan M. Fluvastatin inhibits hepatitis C replication in humans. Am J Gastroenterol. 2008;103:1383–1389. doi: 10.1111/j.1572-0241.2008.01876.x. [DOI] [PubMed] [Google Scholar]
- 48.Forde KA, Law C, O’Flynn R, Kaplan DE. Do statins reduce hepatitis C RNA titers during routine clinical use? World J Gastroenterol. 2009;15:5020–5027. doi: 10.3748/wjg.15.5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milazzo L, Caramma I, Mazzali C, Cesari M, Olivetti M, Galli M, Antinori S. Fluvastatin as an adjuvant to pegylated interferon and ribavirin in HIV/hepatitis C virus genotype 1 co-infected patients: an open-label randomized controlled study. J Antimicrob Chemother. 2010;65:735–740. doi: 10.1093/jac/dkq002. [DOI] [PubMed] [Google Scholar]
- 50.Atsukawa M, Tsubota A, Kondo C, Itokawa N, Narahara Y, Nakatsuka K, Hashimoto S, Fukuda T, Matsushita Y, Kidokoro H, et al. Combination of fluvastatin with pegylated interferon/ribavirin therapy reduces viral relapse in chronic hepatitis C infected with HCV genotype 1b. J Gastroenterol Hepatol. 2013;28:51–56. doi: 10.1111/j.1440-1746.2012.07267.x. [DOI] [PubMed] [Google Scholar]
- 51.Harrison SA, Rossaro L, Hu KQ, Patel K, Tillmann H, Dhaliwal S, Torres DM, Koury K, Goteti VS, Noviello S, et al. Serum cholesterol and statin use predict virological response to peginterferon and ribavirin therapy. Hepatology. 2010;52:864–874. doi: 10.1002/hep.23787. [DOI] [PubMed] [Google Scholar]
- 52.Rao GA, Pandya PK. Statin therapy improves sustained virologic response among diabetic patients with chronic hepatitis C. Gastroenterology. 2011;140:144–152. doi: 10.1053/j.gastro.2010.08.055. [DOI] [PubMed] [Google Scholar]
- 53.Fujita N, Kaito M, Kai M, Sugimoto R, Tanaka H, Horiike S, Konishi M, Iwasa M, Watanabe S, Adachi Y. Effects of bezafibrate in patients with chronic hepatitis C virus infection: combination with interferon and ribavirin. J Viral Hepat. 2006;13:441–448. doi: 10.1111/j.1365-2893.2005.00718.x. [DOI] [PubMed] [Google Scholar]
- 54.Dai CY, Chuang WL, Ho CK, Hsieh MY, Huang JF, Lee LP, Hou NJ, Lin ZY, Chen SC, Hsieh MY, et al. Associations between hepatitis C viremia and low serum triglyceride and cholesterol levels: a community-based study. J Hepatol. 2008;49:9–16. doi: 10.1016/j.jhep.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 55.Marzouk D, Sass J, Bakr I, El Hosseiny M, Abdel-Hamid M, Rekacewicz C, Chaturvedi N, Mohamed MK, Fontanet A. Metabolic and cardiovascular risk profiles and hepatitis C virus infection in rural Egypt. Gut. 2007;56:1105–1110. doi: 10.1136/gut.2006.091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Romero-Gómez M. Insulin resistance and hepatitis C. World J Gastroenterol. 2006;12:7075–7080. doi: 10.3748/wjg.v12.i44.7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Romero-Gómez M, Diago M, Andrade RJ, Calleja JL, Salmerón J, Fernández-Rodríguez CM, Solà R, García-Samaniego J, Herrerías JM, De la Mata M, et al. Treatment of insulin resistance with metformin in naïve genotype 1 chronic hepatitis C patients receiving peginterferon alfa-2a plus ribavirin. Hepatology. 2009;50:1702–1708. doi: 10.1002/hep.23206. [DOI] [PubMed] [Google Scholar]
- 58.Khattab M, Emad M, Abdelaleem A, Eslam M, Atef R, Shaker Y, Hamdy L. Pioglitazone improves virological response to peginterferon alpha-2b/ribavirin combination therapy in hepatitis C genotype 4 patients with insulin resistance. Liver Int. 2010;30:447–454. doi: 10.1111/j.1478-3231.2009.02171.x. [DOI] [PubMed] [Google Scholar]
- 59.Bartenschlager R, Penin F, Lohmann V, André P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011;19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 60.André P, Komurian-Pradel F, Deforges S, Perret M, Berland JL, Sodoyer M, Pol S, Bréchot C, Paranhos-Baccalà G, Lotteau V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J Virol. 2002;76:6919–6928. doi: 10.1128/JVI.76.14.6919-6928.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J Virol. 2006;80:2418–2428. doi: 10.1128/JVI.80.5.2418-2428.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Diaz O, Delers F, Maynard M, Demignot S, Zoulim F, Chambaz J, Trépo C, Lotteau V, André P. Preferential association of Hepatitis C virus with apolipoprotein B48-containing lipoproteins. J Gen Virol. 2006;87:2983–2991. doi: 10.1099/vir.0.82033-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rhainds D, Brissette L. The role of scavenger receptor class B type I (SR-BI) in lipid trafficking. defining the rules for lipid traders. Int J Biochem Cell Biol. 2004;36:39–77. doi: 10.1016/s1357-2725(03)00173-0. [DOI] [PubMed] [Google Scholar]
- 64.Rigotti A, Miettinen HE, Krieger M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr Rev. 2003;24:357–387. doi: 10.1210/er.2001-0037. [DOI] [PubMed] [Google Scholar]
- 65.Röhrl C, Stangl H. HDL endocytosis and resecretion. Biochim Biophys Acta. 2013;1831:1626–1633. doi: 10.1016/j.bbalip.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dao Thi VL, Granier C, Zeisel MB, Guérin M, Mancip J, Granio O, Penin F, Lavillette D, Bartenschlager R, Baumert TF, et al. Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps. J Biol Chem. 2012;287:31242–31257. doi: 10.1074/jbc.M112.365924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maillard P, Huby T, Andréo U, Moreau M, Chapman J, Budkowska A. The interaction of natural hepatitis C virus with human scavenger receptor SR-BI/Cla1 is mediated by ApoB-containing lipoproteins. FASEB J. 2006;20:735–737. doi: 10.1096/fj.05-4728fje. [DOI] [PubMed] [Google Scholar]
- 68.Zeisel MB, Koutsoudakis G, Schnober EK, Haberstroh A, Blum HE, Cosset FL, Wakita T, Jaeck D, Doffoel M, Royer C, et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology. 2007;46:1722–1731. doi: 10.1002/hep.21994. [DOI] [PubMed] [Google Scholar]
- 69.Catanese MT, Graziani R, von Hahn T, Moreau M, Huby T, Paonessa G, Santini C, Luzzago A, Rice CM, Cortese R, et al. High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J Virol. 2007;81:8063–8071. doi: 10.1128/JVI.00193-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meuleman P, Catanese MT, Verhoye L, Desombere I, Farhoudi A, Jones CT, Sheahan T, Grzyb K, Cortese R, Rice CM, et al. A human monoclonal antibody targeting scavenger receptor class B type I precludes hepatitis C virus infection and viral spread in vitro and in vivo. Hepatology. 2012;55:364–372. doi: 10.1002/hep.24692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nieland TJ, Penman M, Dori L, Krieger M, Kirchhausen T. Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. Proc Natl Acad Sci USA. 2002;99:15422–15427. doi: 10.1073/pnas.222421399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Voisset C, Callens N, Blanchard E, Op De Beeck A, Dubuisson J, Vu-Dac N. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J Biol Chem. 2005;280:7793–7799. doi: 10.1074/jbc.M411600200. [DOI] [PubMed] [Google Scholar]
- 73.Dreux M, Pietschmann T, Granier C, Voisset C, Ricard-Blum S, Mangeot PE, Keck Z, Foung S, Vu-Dac N, Dubuisson J, et al. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J Biol Chem. 2006;281:18285–18295. doi: 10.1074/jbc.M602706200. [DOI] [PubMed] [Google Scholar]
- 74.Brimacombe CL, Grove J, Meredith LW, Hu K, Syder AJ, Flores MV, Timpe JM, Krieger SE, Baumert TF, Tellinghuisen TL, et al. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J Virol. 2011;85:596–605. doi: 10.1128/JVI.01592-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mittapalli GK, Jackson A, Zhao F, Lee H, Chow S, McKelvy J, Wong-Staal F, Macdonald JE. Discovery of highly potent small molecule Hepatitis C Virus entry inhibitors. Bioorg Med Chem Lett. 2011;21:6852–6855. doi: 10.1016/j.bmcl.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 76.Syder AJ, Lee H, Zeisel MB, Grove J, Soulier E, Macdonald J, Chow S, Chang J, Baumert TF, McKeating JA, et al. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J Hepatol. 2011;54:48–55. doi: 10.1016/j.jhep.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 77.Zahid MN, Turek M, Xiao F, Thi VL, Guérin M, Fofana I, Bachellier P, Thompson J, Delang L, Neyts J, et al. The postbinding activity of scavenger receptor class B type I mediates initiation of hepatitis C virus infection and viral dissemination. Hepatology. 2013;57:492–504. doi: 10.1002/hep.26097. [DOI] [PubMed] [Google Scholar]
- 78.Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, Paonessa G, Rice CM, Cortese R, Vitelli A, et al. Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J Virol. 2010;84:34–43. doi: 10.1128/JVI.02199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo L, Chen M, Song Z, Daugherty A, Li XA. C323 of SR-BI is required for SR-BI-mediated HDL binding and cholesteryl ester uptake. J Lipid Res. 2011;52:2272–2278. doi: 10.1194/jlr.M019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wünschmann S, Medh JD, Klinzmann D, Schmidt WN, Stapleton JT. Characterization of hepatitis C virus (HCV) and HCV E2 interactions with CD81 and the low-density lipoprotein receptor. J Virol. 2000;74:10055–10062. doi: 10.1128/jvi.74.21.10055-10062.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Monazahian M, Böhme I, Bonk S, Koch A, Scholz C, Grethe S, Thomssen R. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J Med Virol. 1999;57:223–229. doi: 10.1002/(sici)1096-9071(199903)57:3<223::aid-jmv2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 83.Molina S, Castet V, Fournier-Wirth C, Pichard-Garcia L, Avner R, Harats D, Roitelman J, Barbaras R, Graber P, Ghersa P, et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J Hepatol. 2007;46:411–419. doi: 10.1016/j.jhep.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 84.Owen DM, Huang H, Ye J, Gale M. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology. 2009;394:99–108. doi: 10.1016/j.virol.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mazumdar B, Banerjee A, Meyer K, Ray R. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology. 2011;54:1149–1156. doi: 10.1002/hep.24523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hishiki T, Shimizu Y, Tobita R, Sugiyama K, Ogawa K, Funami K, Ohsaki Y, Fujimoto T, Takaku H, Wakita T, et al. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J Virol. 2010;84:12048–12057. doi: 10.1128/JVI.01063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prentoe J, Serre SB, Ramirez S, Nicosia A, Gottwein JM, Bukh J. Hypervariable region 1 deletion and required adaptive envelope mutations confer decreased dependency on scavenger receptor class B type I and low-density lipoprotein receptor for hepatitis C virus. J Virol. 2014;88:1725–1739. doi: 10.1128/JVI.02017-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Albecka A, Belouzard S, Op de Beeck A, Descamps V, Goueslain L, Bertrand-Michel J, Tercé F, Duverlie G, Rouillé Y, Dubuisson J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology. 2012;55:998–1007. doi: 10.1002/hep.25501. [DOI] [PubMed] [Google Scholar]
- 89.Sainz B, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med. 2012;18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu L. The structure and function of Niemann-Pick C1-like 1 protein. Curr Opin Lipidol. 2008;19:263–269. doi: 10.1097/MOL.0b013e3282f9b563. [DOI] [PubMed] [Google Scholar]
- 91.Altmann SW, Davis HR, Zhu LJ, Yao X, Hoos LM, Tetzloff G, Iyer SP, Maguire M, Golovko A, Zeng M, et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303:1201–1204. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- 92.Temel RE, Tang W, Ma Y, Rudel LL, Willingham MC, Ioannou YA, Davies JP, Nilsson LM, Yu L. Hepatic Niemann-Pick C1-like 1 regulates biliary cholesterol concentration and is a target of ezetimibe. J Clin Invest. 2007;117:1968–1978. doi: 10.1172/JCI30060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bays HE, Neff D, Tomassini JE, Tershakovec AM. Ezetimibe: cholesterol lowering and beyond. Expert Rev Cardiovasc Ther. 2008;6:447–470. doi: 10.1586/14779072.6.4.447. [DOI] [PubMed] [Google Scholar]
- 94.Germi R, Crance JM, Garin D, Guimet J, Lortat-Jacob H, Ruigrok RW, Zarski JP, Drouet E. Cellular glycosaminoglycans and low density lipoprotein receptor are involved in hepatitis C virus adsorption. J Med Virol. 2002;68:206–215. doi: 10.1002/jmv.10196. [DOI] [PubMed] [Google Scholar]
- 95.Barth H, Schnober EK, Zhang F, Linhardt RJ, Depla E, Boson B, Cosset FL, Patel AH, Blum HE, Baumert TF. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J Virol. 2006;80:10579–10590. doi: 10.1128/JVI.00941-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol. 2006;80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol. 2012;86:7256–7267. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shi Q, Jiang J, Luo G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J Virol. 2013;87:6866–6875. doi: 10.1128/JVI.03475-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Heeren J, Niemeier A, Merkel M, Beisiegel U. Endothelial-derived lipoprotein lipase is bound to postprandial triglyceride-rich lipoproteins and mediates their hepatic clearance in vivo. J Mol Med (Berl) 2002;80:576–584. doi: 10.1007/s00109-002-0351-5. [DOI] [PubMed] [Google Scholar]
- 100.Zheng C, Murdoch SJ, Brunzell JD, Sacks FM. Lipoprotein lipase bound to apolipoprotein B lipoproteins accelerates clearance of postprandial lipoproteins in humans. Arterioscler Thromb Vasc Biol. 2006;26:891–896. doi: 10.1161/01.ATV.0000203512.01007.3d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Andréo U, Maillard P, Kalinina O, Walic M, Meurs E, Martinot M, Marcellin P, Budkowska A. Lipoprotein lipase mediates hepatitis C virus (HCV) cell entry and inhibits HCV infection. Cell Microbiol. 2007;9:2445–2456. doi: 10.1111/j.1462-5822.2007.00972.x. [DOI] [PubMed] [Google Scholar]
- 102.Maillard P, Walic M, Meuleman P, Roohvand F, Huby T, Le Goff W, Leroux-Roels G, Pécheur EI, Budkowska A. Lipoprotein lipase inhibits hepatitis C virus (HCV) infection by blocking virus cell entry. PLoS One. 2011;6:e26637. doi: 10.1371/journal.pone.0026637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shimizu Y, Hishiki T, Sugiyama K, Ogawa K, Funami K, Kato A, Ohsaki Y, Fujimoto T, Takaku H, Shimotohno K. Lipoprotein lipase and hepatic triglyceride lipase reduce the infectivity of hepatitis C virus (HCV) through their catalytic activities on HCV-associated lipoproteins. Virology. 2010;407:152–159. doi: 10.1016/j.virol.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 104.von Hahn T, Lindenbach BD, Boullier A, Quehenberger O, Paulson M, Rice CM, McKeating JA. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology. 2006;43:932–942. doi: 10.1002/hep.21139. [DOI] [PubMed] [Google Scholar]
- 105.Westhaus S, Bankwitz D, Ernst S, Rohrmann K, Wappler I, Agné C, Luchtefeld M, Schieffer B, Sarrazin C, Manns MP, et al. Characterization of the inhibition of hepatitis C virus entry by in vitro-generated and patient-derived oxidized low-density lipoprotein. Hepatology. 2013;57:1716–1724. doi: 10.1002/hep.26190. [DOI] [PubMed] [Google Scholar]
- 106.Lavie M, Voisset C, Vu-Dac N, Zurawski V, Duverlie G, Wychowski C, Dubuisson J. Serum amyloid A has antiviral activity against hepatitis C virus by inhibiting virus entry in a cell culture system. Hepatology. 2006;44:1626–1634. doi: 10.1002/hep.21406. [DOI] [PubMed] [Google Scholar]
- 107.Cai Z, Cai L, Jiang J, Chang KS, van der Westhuyzen DR, Luo G. Human serum amyloid A protein inhibits hepatitis C virus entry into cells. J Virol. 2007;81:6128–6133. doi: 10.1128/JVI.02627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meunier JC, Engle RE, Faulk K, Zhao M, Bartosch B, Alter H, Emerson SU, Cosset FL, Purcell RH, Bukh J. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc Natl Acad Sci USA. 2005;102:4560–4565. doi: 10.1073/pnas.0501275102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dreux M, Dao Thi VL, Fresquet J, Guérin M, Julia Z, Verney G, Durantel D, Zoulim F, Lavillette D, Cosset FL, et al. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog. 2009;5:e1000310. doi: 10.1371/journal.ppat.1000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vercauteren K, Van Den Eede N, Mesalam AA, Belouzard S, Catanese MT, Bankwitz D, Wong-Staal F, Cortese R, Dubuisson J, Rice CM, et al. Successful anti-scavenger receptor class B type I (SR-BI) monoclonal antibody therapy in humanized mice after challenge with HCV variants with in vitro resistance to SR-BI-targeting agents. Hepatology. 2014;60:1508–1518. doi: 10.1002/hep.27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J Virol. 2007;81:374–383. doi: 10.1128/JVI.01134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dreux M, Boson B, Ricard-Blum S, Molle J, Lavillette D, Bartosch B, Pécheur EI, Cosset FL. The exchangeable apolipoprotein ApoC-I promotes membrane fusion of hepatitis C virus. J Biol Chem. 2007;282:32357–32369. doi: 10.1074/jbc.M705358200. [DOI] [PubMed] [Google Scholar]
- 113.Bankwitz D, Steinmann E, Bitzegeio J, Ciesek S, Friesland M, Herrmann E, Zeisel MB, Baumert TF, Keck ZY, Foung SK, et al. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J Virol. 2010;84:5751–5763. doi: 10.1128/JVI.02200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brazzoli M, Bianchi A, Filippini S, Weiner A, Zhu Q, Pizza M, Crotta S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J Virol. 2008;82:8316–8329. doi: 10.1128/JVI.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Krieger SE, Zeisel MB, Davis C, Thumann C, Harris HJ, Schnober EK, Mee C, Soulier E, Royer C, Lambotin M, et al. Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology. 2010;51:1144–1157. doi: 10.1002/hep.23445. [DOI] [PubMed] [Google Scholar]
- 116.Harris HJ, Farquhar MJ, Mee CJ, Davis C, Reynolds GM, Jennings A, Hu K, Yuan F, Deng H, Hubscher SG, et al. CD81 and claudin 1 coreceptor association: role in hepatitis C virus entry. J Virol. 2008;82:5007–5020. doi: 10.1128/JVI.02286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rhainds D, Bourgeois P, Bourret G, Huard K, Falstrault L, Brissette L. Localization and regulation of SR-BI in membrane rafts of HepG2 cells. J Cell Sci. 2004;117:3095–3105. doi: 10.1242/jcs.01182. [DOI] [PubMed] [Google Scholar]
- 118.Charrin S, Manié S, Thiele C, Billard M, Gerlier D, Boucheix C, Rubinstein E. A physical and functional link between cholesterol and tetraspanins. Eur J Immunol. 2003;33:2479–2489. doi: 10.1002/eji.200323884. [DOI] [PubMed] [Google Scholar]
- 119.Cherukuri A, Shoham T, Sohn HW, Levy S, Brooks S, Carter R, Pierce SK. The tetraspanin CD81 is necessary for partitioning of coligated CD19/CD21-B cell antigen receptor complexes into signaling-active lipid rafts. J Immunol. 2004;172:370–380. doi: 10.4049/jimmunol.172.1.370. [DOI] [PubMed] [Google Scholar]
- 120.Harris HJ, Davis C, Mullins JG, Hu K, Goodall M, Farquhar MJ, Mee CJ, McCaffrey K, Young S, Drummer H, et al. Claudin association with CD81 defines hepatitis C virus entry. J Biol Chem. 2010;285:21092–21102. doi: 10.1074/jbc.M110.104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bertaux C, Dragic T. Different domains of CD81 mediate distinct stages of hepatitis C virus pseudoparticle entry. J Virol. 2006;80:4940–4948. doi: 10.1128/JVI.80.10.4940-4948.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Voisset C, Lavie M, Helle F, Op De Beeck A, Bilheu A, Bertrand-Michel J, Tercé F, Cocquerel L, Wychowski C, Vu-Dac N, et al. Ceramide enrichment of the plasma membrane induces CD81 internalization and inhibits hepatitis C virus entry. Cell Microbiol. 2008;10:606–617. doi: 10.1111/j.1462-5822.2007.01070.x. [DOI] [PubMed] [Google Scholar]
- 123.Schwarz AK, Grove J, Hu K, Mee CJ, Balfe P, McKeating JA. Hepatoma cell density promotes claudin-1 and scavenger receptor BI expression and hepatitis C virus internalization. J Virol. 2009;83:12407–12414. doi: 10.1128/JVI.01552-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Grove J, Huby T, Stamataki Z, Vanwolleghem T, Meuleman P, Farquhar M, Schwarz A, Moreau M, Owen JS, Leroux-Roels G, et al. Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J Virol. 2007;81:3162–3169. doi: 10.1128/JVI.02356-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pichler H, Riezman H. Where sterols are required for endocytosis. Biochim Biophys Acta. 2004;1666:51–61. doi: 10.1016/j.bbamem.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 126.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouillé Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol. 2006;80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lavillette D, Pécheur EI, Donot P, Fresquet J, Molle J, Corbau R, Dreux M, Penin F, Cosset FL. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J Virol. 2007;81:8752–8765. doi: 10.1128/JVI.02642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lavillette D, Bartosch B, Nourrisson D, Verney G, Cosset FL, Penin F, Pécheur EI. Hepatitis C virus glycoproteins mediate low pH-dependent membrane fusion with liposomes. J Biol Chem. 2006;281:3909–3917. doi: 10.1074/jbc.M509747200. [DOI] [PubMed] [Google Scholar]
- 129.Haid S, Pietschmann T, Pécheur EI. Low pH-dependent hepatitis C virus membrane fusion depends on E2 integrity, target lipid composition, and density of virus particles. J Biol Chem. 2009;284:17657–17667. doi: 10.1074/jbc.M109.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 131.Diamond DL, Syder AJ, Jacobs JM, Sorensen CM, Walters KA, Proll SC, McDermott JE, Gritsenko MA, Zhang Q, Zhao R, et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010;6:e1000719. doi: 10.1371/journal.ppat.1000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li Z, Agellon LB, Allen TM, Umeda M, Jewell L, Mason A, Vance DE. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006;3:321–331. doi: 10.1016/j.cmet.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 133.Sakamoto H, Okamoto K, Aoki M, Kato H, Katsume A, Ohta A, Tsukuda T, Shimma N, Aoki Y, Arisawa M, et al. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat Chem Biol. 2005;1:333–337. doi: 10.1038/nchembio742. [DOI] [PubMed] [Google Scholar]
- 134.Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci USA. 2009;106:7577–7582. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012;8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Martin S, Parton RG. Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol. 2006;7:373–378. doi: 10.1038/nrm1912. [DOI] [PubMed] [Google Scholar]
- 137.Moradpour D, Englert C, Wakita T, Wands JR. Characterization of cell lines allowing tightly regulated expression of hepatitis C virus core protein. Virology. 1996;222:51–63. doi: 10.1006/viro.1996.0397. [DOI] [PubMed] [Google Scholar]
- 138.Rouillé Y, Helle F, Delgrange D, Roingeard P, Voisset C, Blanchard E, Belouzard S, McKeating J, Patel AH, Maertens G, et al. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J Virol. 2006;80:2832–2841. doi: 10.1128/JVI.80.6.2832-2841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 140.Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, Miyamura T, Koike K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol. 1997;78(Pt 7):1527–1531. doi: 10.1099/0022-1317-78-7-1527. [DOI] [PubMed] [Google Scholar]
- 141.Olofsson SO, Boström P, Andersson L, Rutberg M, Perman J, Borén J. Lipid droplets as dynamic organelles connecting storage and efflux of lipids. Biochim Biophys Acta. 2009;1791:448–458. doi: 10.1016/j.bbalip.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 142.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci USA. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thomssen R, Bonk S, Propfe C, Heermann KH, Köchel HG, Uy A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med Microbiol Immunol. 1992;181:293–300. doi: 10.1007/BF00198849. [DOI] [PubMed] [Google Scholar]
- 144.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol. 2008;82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nahmias Y, Goldwasser J, Casali M, van Poll D, Wakita T, Chung RT, Yarmush ML. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology. 2008;47:1437–1445. doi: 10.1002/hep.22197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gastaminza P, Kapadia SB, Chisari FV. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol. 2006;80:11074–11081. doi: 10.1128/JVI.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tao W, Xu C, Ding Q, Li R, Xiang Y, Chung J, Zhong J. A single point mutation in E2 enhances hepatitis C virus infectivity and alters lipoprotein association of viral particles. Virology. 2009;395:67–76. doi: 10.1016/j.virol.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 148.Icard V, Diaz O, Scholtes C, Perrin-Cocon L, Ramière C, Bartenschlager R, Penin F, Lotteau V, André P. Secretion of hepatitis C virus envelope glycoproteins depends on assembly of apolipoprotein B positive lipoproteins. PLoS One. 2009;4:e4233. doi: 10.1371/journal.pone.0004233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nourbakhsh M, Douglas DN, Pu CH, Lewis JT, Kawahara T, Lisboa LF, Wei E, Asthana S, Quiroga AD, Law LM, et al. Arylacetamide deacetylase: a novel host factor with important roles in the lipolysis of cellular triacylglycerol stores, VLDL assembly and HCV production. J Hepatol. 2013;59:336–343. doi: 10.1016/j.jhep.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 150.Fazio S, Linton MF, Swift LL. The cell biology and physiologic relevance of ApoE recycling. Trends Cardiovasc Med. 2000;10:23–30. doi: 10.1016/s1050-1738(00)00033-5. [DOI] [PubMed] [Google Scholar]
- 151.Chang KS, Jiang J, Cai Z, Luo G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J Virol. 2007;81:13783–13793. doi: 10.1128/JVI.01091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jiang J, Luo G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol. 2009;83:12680–12691. doi: 10.1128/JVI.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Long G, Hiet MS, Windisch MP, Lee JY, Lohmann V, Bartenschlager R. Mouse hepatic cells support assembly of infectious hepatitis C virus particles. Gastroenterology. 2011;141:1057–1066. doi: 10.1053/j.gastro.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 154.Hueging K, Doepke M, Vieyres G, Bankwitz D, Frentzen A, Doerrbecker J, Gumz F, Haid S, Wölk B, Kaderali L, et al. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell-to-cell transmission by contributing to a postenvelopment step of assembly. J Virol. 2014;88:1433–1446. doi: 10.1128/JVI.01815-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Benga WJ, Krieger SE, Dimitrova M, Zeisel MB, Parnot M, Lupberger J, Hildt E, Luo G, McLauchlan J, Baumert TF, et al. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology. 2010;51:43–53. doi: 10.1002/hep.23278. [DOI] [PubMed] [Google Scholar]
- 156.Merz A, Long G, Hiet MS, Brügger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J Biol Chem. 2011;286:3018–3032. doi: 10.1074/jbc.M110.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Meunier JC, Russell RS, Engle RE, Faulk KN, Purcell RH, Emerson SU. Apolipoprotein c1 association with hepatitis C virus. J Virol. 2008;82:9647–9656. doi: 10.1128/JVI.00914-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Aizaki H, Morikawa K, Fukasawa M, Hara H, Inoue Y, Tani H, Saito K, Nishijima M, Hanada K, Matsuura Y, et al. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J Virol. 2008;82:5715–5724. doi: 10.1128/JVI.02530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yamamoto M, Aizaki H, Fukasawa M, Teraoka T, Miyamura T, Wakita T, Suzuki T. Structural requirements of virion-associated cholesterol for infectivity, buoyant density and apolipoprotein association of hepatitis C virus. J Gen Virol. 2011;92:2082–2087. doi: 10.1099/vir.0.032391-0. [DOI] [PubMed] [Google Scholar]
- 160.Grove J, Nielsen S, Zhong J, Bassendine MF, Drummer HE, Balfe P, McKeating JA. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J Virol. 2008;82:12020–12029. doi: 10.1128/JVI.01569-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Prentoe J, Jensen TB, Meuleman P, Serre SB, Scheel TK, Leroux-Roels G, Gottwein JM, Bukh J. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J Virol. 2011;85:2224–2234. doi: 10.1128/JVI.01594-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Koutsoudakis G, Dragun J, Pérez-Del-Pulgar S, Coto-Llerena M, Mensa L, Crespo G, González P, Navasa M, Forns X. Interplay between basic residues of hepatitis C virus glycoprotein E2 with viral receptors, neutralizing antibodies and lipoproteins. PLoS One. 2012;7:e52651. doi: 10.1371/journal.pone.0052651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Thomssen R, Bonk S, Thiele A. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med Microbiol Immunol. 1993;182:329–334. doi: 10.1007/BF00191948. [DOI] [PubMed] [Google Scholar]
- 164.Felmlee DJ, Fauvelle C, Heydmann L, Hiet MS, Fofana I, Bartenschlager R, Stoll-Keller F, Zeisel MB, Fafi-Kremer S, Baumert TF. Hepatitis C Virus Liver Transplantation Escape Variant Is Characterized by Both Enhanced Triglyceride-Rich Lipoprotein Association and Sensitivity to Apoe Antibodies. J Hepatol. 2013;58:S468–S468. [Google Scholar]
- 165.Fafi-Kremer S, Fofana I, Soulier E, Carolla P, Meuleman P, Leroux-Roels G, Patel AH, Cosset FL, Pessaux P, Doffoël M, et al. Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J Exp Med. 2010;207:2019–2031. doi: 10.1084/jem.20090766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Molina S, Castet V, Pichard-Garcia L, Wychowski C, Meurs E, Pascussi JM, Sureau C, Fabre JM, Sacunha A, Larrey D, et al. Serum-derived hepatitis C virus infection of primary human hepatocytes is tetraspanin CD81 dependent. J Virol. 2008;82:569–574. doi: 10.1128/JVI.01443-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tan YJ, Lim SP, Ng P, Goh PY, Lim SG, Tan YH, Hong W. CD81 engineered with endocytotic signals mediates HCV cell entry: implications for receptor usage by HCV in vivo. Virology. 2003;308:250–269. doi: 10.1016/s0042-6822(02)00136-8. [DOI] [PubMed] [Google Scholar]
- 168.Meuleman P, Hesselgesser J, Paulson M, Vanwolleghem T, Desombere I, Reiser H, Leroux-Roels G. Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology. 2008;48:1761–1768. doi: 10.1002/hep.22547. [DOI] [PubMed] [Google Scholar]
- 169.Lacek K, Vercauteren K, Grzyb K, Naddeo M, Verhoye L, Słowikowski MP, Fafi-Kremer S, Patel AH, Baumert TF, Folgori A, et al. Novel human SR-BI antibodies prevent infection and dissemination of HCV in vitro and in humanized mice. J Hepatol. 2012;57:17–23. doi: 10.1016/j.jhep.2012.02.018. [DOI] [PubMed] [Google Scholar]