

Introduction
The 21st century is considered by many as the era of genomic medicine. Indeed, fascinating advances have been achieved both in terms of high-throughput technologies and biological insight, leading to a deeper understanding of human disease, improved diagnostic and prognostic tools and better therapeutic approaches. In cardiology, extensive work over decades has led to the identification of genetic and environmental factors contributing to disease pathogenesis. In hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) alone, hundreds of mutations in over 20 and 30 genes, respectively, have been identified to date, with approximately half of them discovered during the past 5 years [1–5]. A variety of genetic variations, in an even greater number of genes, have been associated with variations in the pathological phenotype or in response to treatments [6–10]. These genetic findings not only promote scientific knowledge, but also serve as the basis for pre-symptomatic and diagnostic genetic testing, revealing promising new therapeutic targets. However, genetics alone does not suffice to explain the full pathological spectrum and variable prognosis of these complex multifactorial heart diseases while available therapies are far from ideal. The latest advances in genomics are now opening the way to a new era for molecular cardiology with novel concepts and molecules meriting in-depth exploration. Through epigenetic, microRNA (miRNA) and modifier gene studies, all three basic levels of biological information namely, DNA, RNA and proteins, are being revisited, and exciting new discoveries are promising to change the way we view and combat heart disease. Epigenetics in molecular cardiology is the focus of this review.
The emerging role of epigenetic modifications
Epigenetics is one of the most rapidly expanding fields in biology with over 2100 publications recorded in PubMed (search term ‘epigenetics’, limits: title/abstract, search date 10 November 2011) (Fig. 1). The recent characterization of a human DNA methylome at single nucleotide resolution, the discovery of the CpG island shores, the finding of new histone variants and modifications and the unveiling of genome-wide nucleosome positioning maps highlight the accelerating speed of discovery over the past three years. Understanding the epigenetic mechanisms, their interactions and alterations in health and disease promises to make a significant contribution to the cardiology clinic [11–13]. Epigenetic modifications refer to changes in chromosomal components that do not include alterations in the nucleotide sequence of the genome or result in heritable regulation of gene expression [14]. The control of gene transcription in the eukaryotic nucleus is highly dependent on chromatin condensation, which represents the DNA compaction levels and determines the transcriptional status of a genetic locus (Fig. 2). An open chromatin structure allows for the binding of transcription factors, leading to gene activation and up-regulation of the encoded protein, whilst transcriptionally inactive regions are contained in more condensed chromatin structures where transcription factor binding is inhibited [15]. DNA methylation at CpG dinucleotides and histone modifications such as methylation and acetylation are two well-characterized epigenetic control mechanisms known to participate in chromatin remodeling and thus, regulate gene transcription.
Figure 1. “The first publication on epigenetics was in 1964, yet progress has been slow until the advent of high throughput technologies in the early 21st century. As of 10th November 2011 more than 2,100 publications with the term “epigenetics” in their title or abstract were available in PubMed (blue bars). However, the role of epigenetics in molecular cardiology is only starting to emerge (search terms “epigenetics heart” in title or abstract (red bars)).”.
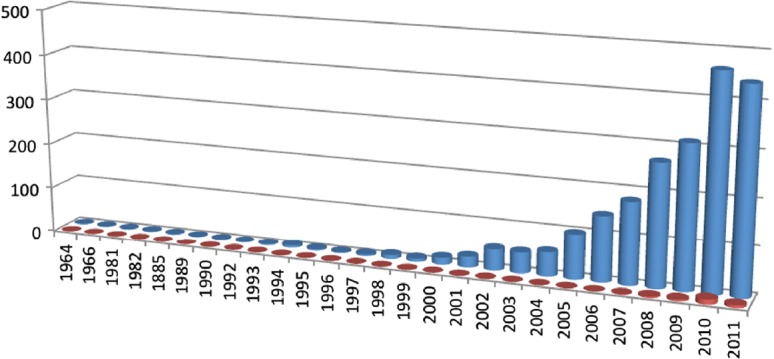
Figure 2. Epigenetic modifications are located on the chromatin and regulate transcriptional activity. DNA methylation at CpG nucleotides triggers chromatin remodeling, ultimately resulting in gene silencing, which is often associated with disease pathogenesis. Histone modifications such as acetylation, methylation, phosphorylation etc. co-determine the transcriptional status of the respective genomic region, holding a critical role in various cell responses.
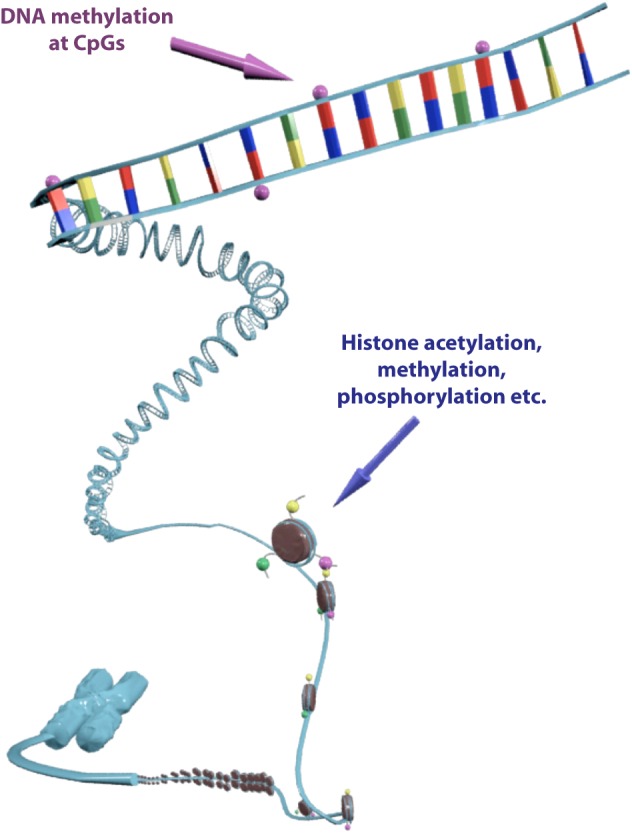
Mechanisms of epigenetic modifications
DNA methylation
Methylation of CpG dinucleotides is associated with gene silencing [16]. Methylated CpGs act as docking sites for methyl binding proteins (MBPs) such as methyl CpG binding protein 2 (MeCP2), which has the ability to oligomerize through the DNA in order to recruit chromatin remodeling complexes that, in turn, cause chromatin condensation and gene inactivation [17,18] (Fig. 3). Differential DNA methylation has been associated with multifactorial and complex diseases including cancer [19] and schizophrenia [20], while recent data suggests a role in cardiomyopathy and heart failure, as discussed below.
Figure 3. Epigenetic modifications and transcriptional activity. Genes that are transcriptionally inactive are assembled in a compact chromatin structure with methylated DNA, deacetylated histones, and histone H3 methylated in lysine 9 and 79 residues. In contrast, transcriptional activation is characterized by an open chromatin structure composed of undermethylated DNA, histone H3 acetylated on lysine 9 and 14 residues, histone H3 methylated on lysine 4 residues, and histone H4 acetylated on lysine 5 and 12 residues.

Histone modifications
The interaction of genomic DNA with histones results in the formation of nucleosomes, the basic structural units of chromatin that contain two copies of each of the histones H2A, H2B, H3 and H4 [21]. This allows for tight compacting of the eukaryotic genome that renders it inaccessible to transcription factors (Fig. 3). Histones undergo a variety of modifications, mainly targeting amino acid residues of the N-terminal tails that protrude from the chromatin fiber. The histone tails are formed by 15 to 38 amino acids from each histone N-terminus, providing a platform for post-translational modifications that act as modulators of the underlying DNA expression [22]. These modifications include acetylation, methylation, ubiquitination, sumoylation and ADP-ribosylation of lysine (K) residues, as well as methylation of arginine (R), and phosphorylation of serine (S) and threonine (T) [23]. Among these different modifications, histone acetylation and methylation have been studied the most in the context of cardiomyopathies and heart failure, and will therefore be described in detail.
Histone acetylation
Histone acetylation is an epigenetic modification of great significance for the regulation of gene expression, as shown in the case of cell differentiation, cell cycle progression, DNA repair, and other cellular processes [24]. Acetylation neutralizes the positive charge of lysine residues, leading to chromatin decondensation and an open conformation that enables the transcriptional activation of that DNA region [25]. Histone acetylation is a dynamic process, mediated by the opposing actions of two enzyme families, namely histone acetyl transferases (HATs) and histone deacetylases (HDACs) [25].
Histone acetyl transferases (HATs) catalyze the transfer of an acetyl group to the amino group of the lysine side chains [26]. There are two major classes of HATs, type A and type B; type B enzymes acetylate free histones (H4 at K5, K12 and H3) whilst type A enzymes acetylate multiple sites within the N-terminal tails and are a more complex group of three enzyme families (GNAT, MYST, CBP/300) [27].
Histone deacetylases (HDACs) reverse histone acetylation, restoring the positive charge of lysines and potentially stabilizing the local chromatin structure, a function consistent with HDACs being predominantly transcriptional repressors [26]. Mammalian genomes encode eleven proteins with a highly conserved deacetylase domain, which can be classified into four families (class I, IIa, IIb and IV), and another group of deacetylases, sirtuins, which are sometimes referred to as class III HDACs [26].
Histone methylation
Histone methylation is a covalent modification commonly occurring in lysine (K) and arginine (R) residues of histones H3 (K4, 9, 27, 36, 79; R2, 17, 26) and H4 (K20; R3). It is defined as the transfer of one or more methyl groups to the amino group of lysine and arginine side chains, and plays a key role in a number of biological functions including X-chromosome inactivation, heterochromatin (a transcriptionally inactive form of chromatin) formation and transcriptional regulation [28]. Histone methylation and demethylation reactions are catalyzed by the enzymes histone methyl transferases (HMTs; EHMT2 and SUV39H1-2) [29] and histone demethylases (HDMs; KDM1A or LSD1, KDM4C or JMJD2C) [30], respectively.
Lysine residues K4, 9, 27, 36, 79 of H3 and K20 of H4 can be mono-, di- or tri-methylated by histone lysine methyl transferases (HKMTs) [31], a subtype of HMTs. The first HKMT identified was SUV39H1, which selectively catalyzes methylation of H3K9 and contains an evolutionary conserved SET domain with catalytic activity [32]. SET-containing HKMTs exhibit high specificity regarding both the exact lysine residue and the level of methylation they catalyze, possibly due to their structure variability within the SET domains and in the pre-SET and post-SET sequences [33]. The vast majority of HKMTs contain a SET domain, with the exception of DOT1L which catalyzes the methylation of H3K79 within the histone core [34].
Arginine residues can be mono- or di-methylated by protein arginine methyl transferases (PRMTs, a subtype of HMTs) [35], 11 of which have been described in humans, with 8 of them exhibiting catalytic activity [36,37]. PRMTs are classified based on their end product; type I enzymes (PRMT-1, -3, -6, -8, CARM1/PRMT4) catalyze mono- and asymmetrical di-methylation and type II enzymes (PRMT-5, -7, FBXO11) mediate mono- and symmetrical di-methylation of guanidino groups of arginine residues [38]. PRMTs -1, -4, -5, -6 are associated with histone arginine methylation [37,39].
The demethylation reactions of lysine and arginine residues are catalyzed by enzymes which exhibit high specificity, such as that of the recently identified histone demethylases (HDMs) [25]. Lysine demethylases include LSD1, which demethylates H3K4 and H3K9, and many Jumonji-domain containing proteins (KDM4A-D or JMJD2A-D, KDM5A-D or JARID1A-D) targeting the trimethylated lysine residues [40]. Less is known about arginine demethylation, and preliminary findings suggesting that the Jumonji-domain containing protein JMJD6 can catalyze the reaction on H3R2 and H4R3 [41].
Both gene repression and activation have been associated with histone methylation and are dependent on which lysine and arginine residues are methylated, as well as the degree of methylation [28]. For instance, heterochromatin protein 1 contains a chromodomain through which it binds the trimethylated lysine residue 9 of H3 (H3K9me3), causing heterochromatin formation and gene silencing [42]. On the contrary, the human bromodomain PHD finger transcription factor (BPTF) recognizes and binds di- or tri-methylated lysine residue 4 of H3 (H3K4me3/me2) through its plant homeodomain (PHD) finger. This enables the recruitment of the nucleosome remodeling factor NURF, leading to gene activation [43,44]. Another aspect of the effects of histone methylation in transcription regulation may be provided by the “histone code” hypothesis; histone modifications function complementary to form marks on chromatin, which are read by proteins capable of triggering distinct downstream events upon their recruitment to chromatin [45].
Epigenetic modifications in cardiac disease
DNA methylation
As previously stated in this review, an increasing amount of data support a role for DNA methylation in cardiac disease. In the case of cardiomyopathies, three angiogenesis-related genes have already been identified as differentially methylated in patients with ischemic and idiopathic end-stage cardiomyopathy; the hypomethylation of the angiomotin-like 2 gene (AMOTL2) and hypermethylation of the 5’ promoter region in the platelet/endothelial cell adhesion molecule gene (PECAM1) led to decreased expression of both, whereas hypermethylation within the gene body of Rho GTPase-activating protein 24 (ARHGAP24) favored its expression [46]. AMOTL2 binds angiostatin and inhibits endothelial cell migration and tube formation [47], while ARHGAP24 regulates tube formation [48] and PECAM1 functions as an adhesion molecule in endothelial intracellular junctions [49]. The altered expression of the three genes in end-stage heart failure may reflect adaptive or maladaptive angiogenic processes in disease pathogenesis [46] and requires further investigation to fully decipher their role in cardiomyopathies. Of note, an abnormality in the vasculature of the diseased myocardium, microvascular dysfunction, that presents with intramural coronary arteriole remodeling and has been associated with recurrent ischemia, is one of the clinical manifestations of HCM [50]. This process is a consequence of hypertrophic alterations in the HCM heart and has proven to be a predictor of disease progression and death [51] thereby suggesting a significant role for angiogenic adaptations in cardiac disease.
In the context of elucidating the molecular mechanisms underlying heart failure, sarcoplasmic reticulum Ca2 +-ATPases (SERCA2a or ATP2A2) and their regulation via tumor necrosis factor alpha (TNFα) have been investigated (Fig. 4). This selection was based on the key role of ATP2A2 in normal cardiac function and heart failure [52–54], as well as the high levels of TNFα in the failing human myocardium [55]. TNFα-treated HL1 murine atrial cardiomyocytes exhibited decreased Atp2a2 expression, increased CpG methylation of the gene's promoter, along with elevated levels of the DNA (cytosine-5-)-methyltransferase 1 (Dnmt1). These findings combined suggest that methylation plays an important role in the negative transcriptional regulation of Atp2a2, and therefore inhibition of Atp2a2 hypermethylation may provide a novel therapeutic strategy against heart failure, if these results are confirmed in vivo [56].
Figure 4. Diverse effects of epigenetic modifications in cardiac hypertrophy and HF. DNA methylation in specific genes contribute to HF, whilst histone acetylation and methylation are implicated in pro- and anti-hypertrophic responses and HF.

Histone acetylation
The acetylation of specific histones has been associated with numerous aspects of cardiac function, ranging from the early stage of heart development to the establishment of pathological cardiac hypertrophy. During cardiac development, the pattern of gene expression is closely linked with histone modifications such as histone H3 and H4 acetylation. For example, the promoters of -myosin heavy chain (-Mhc or Myh6), -myosin heavy chain (-Mhc or Myh7), atrial natriuretic peptide (Anp or Nppa) and B-type natriuretic peptide (Bnp or Nppb) genes were found to be significantly hyperacetylated in cases of increased transcriptional activation of the corresponding genes during cardiac development, and this has been attributed to histone H3 (H3K9, H3K14) and histone H4 acetylation [57].
A deeper insight into histone acetylation/deacetylation mechanisms reveals intriguing details related to the enzymes catalyzing these functions, with respect to the cardiac function. Specifically, class IIa HDACs (HDAC-4, -5, -7, -9), unlike other HDACs, exhibit restricted expression patterns; HDAC-4, -5 and -9, for instance, are highly enriched in skeletal muscles, the heart and the brain [58,59]. These HDACs bear large N-terminal extensions that contain conserved binding sites for the transcription factor myocyte enhancer factor 2 (MEF2), also showing highest expression in skeletal muscle, the heart and the brain [60,61] and the 14-3-3 chaperone, rendering them signal-responsive. The phosphorylation of class IIa HDACs regulates the levels of these enzymes inside the nucleus and consequently affects the balance of HATs and HDACs; the equilibrium of HATs and HDACs is crucial for gene expression and regulation in general, and also applies to myogenesis activation. Phosphorylation of class II HDACs results in them exiting the nucleus via binding 14-3-3 chaperone [62–65], and thereby being dissociated from MEF2, which can now bind p300 and trigger myogenic transcriptional activation through MyoD [66,67]. HDAC5 on the other hand, when phosphorylated at a different serine residue (Ser 280), binds and represses MEF2 to silence MFE2-dependent gene transcription programs that control cell differentiation and cell growth [68]. These findings pinpoint that, it is not only the type of modification, but also the specific amino acids being modified that are the key factors in understanding the epigenetic mechanisms of cardiac function and disease development.
Modifications in histone acetylation have been linked to cardiac hypertrophy, with studies implicating both HATs and HDACs activity (Fig. 4). More specifically, the activity of HATs seems to have a positive role in hypertrophy, whilst the activity of HDACs has been implicated in both pro-hypertrophic and anti-hypertrophic pathways.
HATs appear to play a significant role in cardiac hypertrophy, as exemplified by the histone acetyl transferase activity of the transcriptional co-activators CREB binding protein (CBP) and p300. Overexpression of CBP or p300 in cardiomyocytes resulted in hypertrophy, whereas overexpression of mutant CBP and p300 lacking HAT activity did not. Consistent with these findings, the pro-hypetrophic phenylephrine (PE) directly enhanced CBP and p300 HAT activity, whereas the use of antisense or dominant negative mutant constructs inhibiting CBP and p300 in PE-induced hypertrophy, resulting in a loss of the hypertrophic effect [69]. Overall, the HAT activity of these proteins appears to be directly implicated in cardiac hypertrophy. The pro-hypertrophic properties of p300 were also demonstrated in a study exploiting the effects of a dietary compound in rats. The polyphenol curcumin, which exhibits CBP/p300 specific HAT inhibitory function, prevented hypertrophic changes and heart failure in the myocardium of heart failure rat models. The action of this non-toxic compound included inhibition of the hypertrophy-induced acetylation levels of the hypertrophy-responsive transcription factor GATA4 and disruption of the p300/GATA4 complex in rat cardiomyocytes. Curcumin could therefore be a suitable candidate for further investigation, with a therapeutic potential in human heart failure [70].
More data, along with conflicting findings, are available in relation to the role of HDACs in cardiac disease. For example, class IIa HDACs (HDAC-4, -5, -7, -9) have been shown to suppress cardiac hypertrophy, and mice lacking HDAC5 and 9 exhibit profoundly enlarged hearts in response to cardiac stress signals [60,61]. Furthermore, in the absence of the HDAC4 -14-3-3 chaperone interaction, the enzyme can enter the nucleus and associate with HDAC3 and MEF2, ultimately resulting in MEF2-dependent transcription repression [71] and thereby preventing the MEF2 activated fetal cardiac gene program associated with cardiac hypertrophy [72,73].
In agreement with this data, CaMK signaling seems to act through the HDAC-MEF2 pathway to promote myogenesis. It has been reported for both yeast and mammalian cells, that CaMK signaling stimulates HDAC5 binding to 14-3-3 chaperone, leading to HDAC-MEF2 complex disruption and activation of pro-hypertrophic gene expression [63]. In accordance with this data, the CaMKII isoform, which is mainly expressed in neonatal cardiomyocytes and is upregulated in neonatal and adult rat models with isoproterenol-induced hypertrophy, was reported to act via the HDAC4-MEF2 complex as well [74].
Of note, HDAC4 has been identified as a major downstream target of the CaMKII isoform during cardiac hypertrophy [75,76] whilst CaMKII was also shown to be activated in hypertrophy, inducing DCM and HF [77]. However, unlike CaMKII , which is only expressed in diseased hearts, therapeutic targeting of and isoforms may be accompanied by serious side effects since both isoforms are constitutively expressed in normal adult hearts [78–80].
On the contrary, the positive role of HDAC activity in hypertrophy is broadly supported by several studies reporting the HDAC inhibitors protective role against cardiac hypertrophy in animal models. In order to clarify the exact role of HDACs in cardiac hypertrophy, the effects of HDAC inhibitors in response to hypertrophic stimuli was assessed in primary rat cardiomyocytes. In response to hypertrophic agonists (PE, E1), the HDAC inhibitor (TSA, NaB)-treated cells exhibited dose-dependent inhibition of the induced hypertrophic response, as shown by Nppa, Myh6 and Myh7 expression, and sarcomere organization. Further elucidating the underlying mechanism, HDAC inhibitors were reported to specifically interfere with an activator (PE) of the Nppa promoter, in order to block Nppa expression [81].
The anti-hypertrophic role of HDAC inhibitors was further demonstrated in vivo. An endogenous HDAC inhibitor, the nuclear homeodomain protein Hop, has been implicated in HDAC-dependent transcription regulation in cardiac hypertrophy, via the serum response factor (SRF). Transgenic mice overexpressing Hop exhibited severe cardiac hypertrophy, which was prevented by TSA (a HDAC class I inhibitor) treatment, whereas both Hop knockout and transgenic mice overexpressing a Hop mutant incapable of interacting with HDACs (HopH2), did not present with cardiac hypertrophy. The ability of Hop to repress SRF-dependent transcription via HDAC inhibition, along with Hop being capable of recruiting HDAC activity (in the form of Hop-HDAC2 containing complexes), as determined by cell-based assays, suggest an endogenous cardiomyocyte molecular program that normally functions to suppress hypertrophic responses [82].
Moreover, the effects of non-specific and HDAC class I-specific inhibitors were assessed in mice and rats with induced cardiac hypertrophy by chronic treatment with angiotensin II or aortic banding (AB). Interestingly, all inhibitors prevented hypertrophic adaptations of the myocardium in response to the hypertrophic stimuli, with the class I-specific inhibitor doing so to a greater extent. In addition, non-specific HDAC inhibitors were reportedly capable of reversing pre-existing cardiac hypertrophy up to a certain point [83]. These findings indicate a regenerative role of HDAC inhibitors, in addition to the protective one, which could have a significant therapeutic potential for HCM patients.
Evaluating the broader spectrum of HDAC inhibitor's potential anti-hypertrophic effect, inhibitor-treated animal models following thoracic AB, were assessed for systolic function along with hypertrophy-induced remodeling, both short-term and long-term. In mice with pressure overload-induced cardiac hypertrophy, treatment with broad spectrum HDAC inhibitors (TSA, Scriptaid) resulted in decreased hypertrophic response in a dose-dependent manner. Cardiomyocyte growth inhibition was achieved at both 3 and 9 weeks (corresponding to 10–12 human years), without signs of cell death or apoptosis and diminished fibrosis. Moreover, preservation of systolic function and LV intrinsic contractility was reported, possibly resulting from blocking the pathological switch from adult Myh6 to fetal Myh7 expression [84]. These findings suggest a significant clinical potential of HDAC inhibitor treatment, as demonstrated by their effectiveness against hypertrophy, and the increased tolerance during long-term treatment [85].
The tolerance of HDAC inhibitors was an inclusion criterion for another study where the anti-hypertrophic potential of the HDAC class I-specific inhibitors API-D, MS27-275 and PXD-101 was assessed in cardiomyocytes. An apicidin derivative (API-D) was selected for in vivo assessment due to its high efficacy/toxicity ratio in neonatal rat cardiomyocytes with PE-induced hypertrophic response, and its pharmacokinetics. API-D exhibited anti-hypertrophic activity in a thoracic aortic constriction (TAC)-induced pressure overload mouse model both short and long-term. After 1 week of TAC, API-D treated mice showed decreased cardiac hypertrophy and fetal gene expression (Nppa, Myh7, Sk Akt), and after 9 weeks cardiac hypertrophy (LV/BW ratio, LV mass index), cardiac function (electrocardiogram, hemodynamic parameters) and fibrosis were significantly improved compared to TAC untreated mice. Overall, API-D seems a promising therapy against cardiac hypertrophy and heart failure [86].
A pro-hypertrophic role has also been attributed to class I HDAC2 independent of Hop [82]. Specifically, HDAC2 knockout mice exhibited resistance to hypertrophic stimuli, possibly as a result of increased expression of the inositol polyphosphate-5 phosphatase (Inpp5f) gene, and inactivation of Akt and Pdk1, resulting in constitutive activation of glycogen synthase kinase () [87]. In agreement with these findings, HDAC2 transgenic mice presented with augmented hypertrophy and inactivation, further supporting the potential of HDACs as therapeutic targets.
Another molecular mechanism underlying maladaptive cardiac remodeling, autophagy [88], seems to implicate class I HDACs. Specifically, mice having undergone transverse aortic constriction and exhibiting moderate pressure overload-induced cardiac hypertrophy showed increased autophagy (measured by LCII levels). This was correlated with the steady-state cardiac mass () and was abolished by TSA treatment. Administration of HDAC inhibitors and RNAi knockdown of essential autophagy effectors (Atg5, Beclin 1) both resulted in abrogation of PE-induced autophagic and hypertrophic responses in cardiomyocytes. These findings pointed to HDAC2 as a PE-induced autophagy mediator. Moreover, a transgenic mouse model overexpressing Beclin 1 and subjected to long-term pressure overload stress, presented with cardiac hypertrophy and impaired systolic function, both of which were significantly restored by TSA treatment [89], in which was in agreement with previous data proposing a regenerative role for HDAC inhibitors [83]. Overall, HDAC2 appears to hold an essential role in autophagy during pathological cardiac remodeling, providing a possible molecular mechanism for the pro-hypertrophic activity of HDACs and accentuating the therapeutic potential of HDAC inhibitors [89].
In summary, there is strong evidence to implicate HATs and HDACs in cardiac pathophysiology. However, extensive work is still needed to reconcile the seemingly contradictory findings, to fully decipher the role of the aforementioned HATs and HDACs, as well as to explore and characterize the role of the remaining members of these protein families.
Histone methylation
Histone methylation is implicated in a number of physiological and pathological processes, including cardiac hypertrophy and heart failure (Fig. 4). Interestingly, histone methylation pro-hypertrophic, as well as, anti-hypertrophic pathways have been described in several studies. A representative example involves interleukin-18 (IL-18), which functions as a pro-hypertrophic stimulus and which was reported to affect the level of H3K9 trimethylation in genes deregulated during cardiac hypertrophy. Specifically, when IL-18 was used to induce hypertrophy in H9c2 rat cardiomyocytes, along with increased size, increased expression of Nppa, desmin (Des), skeletal -actin (Acta-1), and the Myh genes was also induced. Epigenetic studies showed elevated H3K9me3 levels in bulk chromatin but decreased H3K9me3 of the upregulated genes as a result of IL-18 treatment, which was abolished when cells were co-treated with the HDAC inhibitor CBHA. Regarding the Myh genes canonical switch, an antisense RNA encoded by the Myh6 and Myh7 intergenic region was shown to repress Myh7, when Myh6 was expressed [90]. H3K9me3 in the respective region was elevated in response to IL-18, and was ultimately associated with gene repression of Myh6 and the intergenic region, favoring Myh7 expression. Of note, IL-18 also increased H3S10 phosphorylation, but had minimum effect on H3K9 acetylation levels, that were in line with transcriptional activity [91].
The anti-hypertrophic effect of H3K9me3 has also been demonstrated in a study utilizing Kdm4a (or Jmjd2a) loss- and gain-of-function mouse models [92]. The lysine demethylase Kdm4a decreased H3K9 trimethylation of the biomechanical stress sensor FHL1 resulting in its transcriptional activation, thereby enabling hypertrophic responses to pressure overload/cardiac stress through the mitogen-activated protein kinase (MAPK) pathway. It therefore appears that KDM4A is another contributor to cardiac hypertrophy, consistently with its observed overexpression in the hearts of human HCM patients [92].
In a different example, the increased expression of natriuretic peptide genes (Nppa, Nppb) in H9c2 rat cardiomyocytes subjected to a hypertrophic stimulus (endothelin 1), was associated with increased dimethylation of H3K4 in the proximity of their promoter regions. Moreover, the repressor element 1-silencing transcription factor (REST), whose inhibition is sufficient to trigger cardiac hypertrophy, was found to repress the transcription of Nppa and Nppb in H9c2 rat cardiomyocytes. REST can recruit various histone modifying enzymes, including HDACs and HMT KDM1A and thereby, its gene transcription regulation function in cardiac hypertrophy might be mediated in part by the enzymes’ opposing actions [93]. Another set of findings further supported the implication of H3K4me3 in cardiac function and pathology, demonstrating that its loss results in destabilization of the gene expression program and the respective physiological functions in adult mouse models. In order to assess the role of H3K4me3 in fully differentiated, non-dividing cells, a transgenic mouse model with an inducible ablation of the PAX interacting protein1 Paxip1 (Ptip) was used. Paxip1 is an essential component of the Mll3/2 (Kmt2c/d) complex and H3K4me3 methylation. Following Paxip1 ablation, H3K4me3 was decreased, causing downregulation of Kcnip2 a gene that has been related to arrythmogenesis and is reported to be downregulated in the human heart failure [94]. Importantly, these changes in H3K4me3 led to changes in the electrophysiology of the heart (including increase in intracellular calcium and systolic function), as well as susceptibility to premature ventricular beats which can lead to lethal ventricular arrhythmias.
Genome-wide approaches have also been used for the global evaluation of histone methylation profiles in relation to cardiac hypertrophy. Through such a study, using rats with induced cardiac hypertrophy and congestive heart failure, it was shown that H3K4me3 and H3K9me3 were the histone modifications most affected in heart failure. Follow up chromatin immunoprecipitation experiments in human specimens from patients with retained LV ejection fraction (HighEF) or DCM-related CHF revealed CHF- and HighEF- specific gene clusters throughout the genome, that were significantly overpopulated with cardiac function related genes (P<0.05, Fisher's exact test). Although many of these cardiac function-related pathways were shared between the two gene clusters, the specific genes in each group of clusters (CHF or HighEF) were distinct. Interestingly, some genes located close to H3K4me3 and H3K9me3 in humans encode components of signaling pathways related to cardiac functions, such as intracellular calcium concentration and muscle contraction (RYR2, CACNA2D1, CACNB2 associated with H3K4me3) [95]. Despite these valuable discoveries, further analysis is required to fully decipher the role of histone methylation in disease pathogenesis and clarify issues, such as the fact that the transcription levels of these genes did not differ significantly in the CHF state. Additionally, as previously proposed [45] specific methylation marks might not be solely responsible for the transcriptional status of the neighboring genetic loci, thus the presence of H3K4me3 does not always trigger gene activation, as expected [43,44].
In addition to H3K4 and H3K9 methylation, H3K79me has also been associated with heart disease. Mouse models with cardiac specific ablation of Dot1L, the gene encoding for the HKMT Dot1L which catalyzes H3K79 methylation in mammals, presented with a cardiac phenotype that resembled DCM. Moreover, loss of H3K9 di- and trimethylation was observed in the mutant cardiomyocytes indicating gene repression [96]. A DCM related gene, namely dystrophin (Dmd), was found downregulated, the dystrophin-glycoprotein complex (DGC) destabilized and its components ( sarcoglycan Sgca, dystroglycan Sgcb) degraded, thus negatively affecting cardiomyocyte viability. Interestingly, the Dmd region was shown to be enriched in H3K79me, an effect abolished by Dot1L deficiency, whilst chromatin immunoprecipitation experiments in myoblasts confirmed that Dot1L directly targets Dmd and regulates its transcription. As a proof of concept, the pathological phenotype in the knockout animals was rescued by postnatal mini Dmd delivery and expression, presenting with restored cardiac function as measured by ECG and echocardiogram analysis. These findings not only demonstrate that the Dmd contribution to the DCM phenotype is associated with H3K79 methylation, but importantly they highlight the importance epigenetic modifications can have even in seemingly well-established molecular mechanisms, such as the DGC complex formation.
The epigenetic studies on heart disease reveal a substantial number of modifications affecting its development and progression; however, the data available to date describe only a small fraction of these modifications. Extensive work is required to evaluate all the different molecules involved in epigenetic regulation as well as their effect on the various cardiac function related proteins. The results will need to be translated to humans and considered in the light of different environments, ethnic backgrounds, genders etc. Once the “cardio-epigenome” has been established the clinical implications of its disturbances will be easier to appreciate and combat. The discovery of disease-specific epigenetic signatures could potentially facilitate diagnosis and especially prognosis, while providing new therapeutic targets (HDACs).
Epigenetic therapies
Pharmacoepigenetics
Powered by the expanding field of epigenetics, the novel discipline of pharmacoepigenetics is starting to emerge, aiming to reveal new therapeutic targets among the components of the epigenetic machinery and to decipher the epigenetic mechanism affecting interindividual drug response variability [97]. Thereby, the establishment of the “cardio-epigenome” would pave the way to the development of targeted epigenetic therapies. To date, successfully developed epigenetic drugs mostly apply to cancer treatment and include DNA methylation inhibitors (nucleoside analogs 5-aza cytidine, 5-aza-2’deoxycytidine, zebularine), histone deacetylase inhibitors (suberoylanilide hydroxamic acid SAHA) and histone methylation inhibitors (3-Deazaneplanocin DZNep) [98]. Although these therapeutic strategies could be potentially applied to cardiac disease, once their targets are identified and confirmed, their effectiveness is accompanied by several limitations. For instance, the existing anti-tumor demethylating agents lack specificity leading to a global effect, possibly compromising genomic stability, indicating that methylation site-specific drug design is required. In addition, however promising the effects of HDAC inhibition towards cardiac hypertrophy inhibition and cardiac function improvement may be, HDAC inhibitors target both histone and non-histone protein acetylation and as such, they are involved in many cellular processes. Closing up to cardiac disease, HDAC inhibitors were also shown to affect sarcomeric protein acetylation implicated in myofilament contractility [99], suggesting that their specificity may as well be a matter of discussion. Although therapies for cardiac diseases have not yet been designed to target epigenetic processes, existing drugs, such as statins, already seem to exploit some of these mechanisms. Several studies on the mechanisms of action of statins have demonstrated inhibition of HDAC, which implies reduced deacetylation and potentially increased transcription. However, other studies have shown an opposite action for statins, where gene expression is reduced [100].
Epigenetics, stem cells and regenerative medicine
In addition to pharmacoepigenomics, another possible application of epigenetics to therapeutics is related to the rapidly evolving field of stem cell therapies and regenerative medicine. Stem cell-based therapy is a promising option for the treatment of cardiac diseases such as heart failure [101]. However, current understanding on the best ways to utilize stem cells therapeutically is limited largely because the full set of molecular mechanisms regulating their functions are unclear. For example, although the treatment of patients with acute myocardial infarction with bone marrow-derived progenitor cells results in an overall improvement of cardiac function, the effects on ejection fraction appear to be modest [102]. One reason for the limited effects might be the low capacity of the injected cells to differentiate into vascular cells and cardiomyocytes. Therefore, the understanding of the mechanisms regulating lineage commitment is important to programme adult cells to specific cardiovascular lineages. Epigenetic control mechanisms play a key regulatory role in the lineage commitment of stem/progenitor cells in the cardiovascular system. These mechanisms include DNA and histone modifications, which modulate the chromatin structure, thereby regulating access of transcription factors. In particular, the modification of histone acetylation and methylation, which is controlled by families of histone acetylases and deacetylases as well as methyltransferases and demethylases, respectively, controls stem cell maintenance, differentiation, and function. Appropriate differentiation requires a differential regulation of pluripotency genes and lineage-committed genes. Pluripotency genes are silenced during differentiation while lineage-committed genes often show bivalent modification in embryonic stem cells. Upon differentiation the lineage-committed gene needs to be de-repressed, whereas other lineages need to be suppressed [103]. The manipulation of enzymes involved in epigenetic control may be useful in facilitating the reprogramming of cells for stem cell therapy (Fig. 5). Chromatin-modifying factors are closely associated with transcriptional networks such as the homeodomain protein Nkx2.5, the factor MEF2c, the zinc finger domain protein GATA-4, and the T-box transcription factor Tbx5. The epigenetic control of these cardiac transcription factors as well as their targets, by chromatin modifying enzymes is involved in proper cardiac differentiation and development [104]. In adult cells, the combination of the three transcription factors Gata4, Mef2c, and Tbx5 alone was recently shown to directly reprogram fibroblasts into functional cardiac myocytes [105], and it remains to be determined whether the addition of epigenetic modulators indeed facilitates reprogramming of adult cells.
Figure 5. The cardiogenic factors turn cardiac genes on during differentiation of pluripotent stem cells into cardiomyocytes lineages. The transcription factors, Gata4, Tbx5 and Nkx2.5, cannot access the DNA unless the chromatin modifying protein Baf60c is present. When all four are introduced, Baf60c helps open up the condensed chromatin, and allows Gata4, Tbx5 and Nkx2.5 to work together and activate cardiac gene expression. In addition, pharmacological treatment with histone deacetylase inhibitors (e.g. TSA) or with the DNA demethylating agent 5-azacytidine induces cardiogenesis, presumably by increasing the transcription of the same cardiogenic factors. The pluripotency genes are turned on in stem cells and they are turned off during differentiation to cardiomyocytes. The specific lineage committed genes are turned off at stem cells and some of them have to be turned on, whilst some others need to be turned off depending on the cell fate decision.

Srivastava's group has shown that the fibroblast induced cardiomyocytes (iCMs), have indeed gained a cardiomyocyte-like chromatin state. More specifically, the promoter regions of the cardiac-specific genes Actn2, Ryr2, and Tnnt2 were analyzed for enrichment of trimethylated histone H3 of lysine 27 (H3K27me3) and lysine 4 (H3K4me3), which mark transcriptionally inactive or active chromatin, respectively [23]. The analysis was performed in cardiac fibroblasts, iCMs, and neonatal cardiac cells by chromatin immunoprecipitation, followed by qPCR. After reprogramming, H3K27me3 was significantly depleted at the promoters of all the genes analyzed in iCMs, reaching levels comparable to those in cardiac cells, whereas H3K4me3 increased on the promoter regions of Actn2 and Tnnt2 in iCMs, as compared with cardiac fibroblasts. Ryr2 had similar levels of H3K4me3 in iCMs as in fibroblasts, suggesting that its activation reflects the resolution of a “bivalent” chromatin mark [106]. These results suggested that cardiac fibroblast-derived iCMs gained a chromatin status similar to cardiomyocytes at least in some cardiac specific genes. Intriguingly, H3K27me3 levels were higher in tail-tip fibroblasts than cardiac fibroblasts on all three genes analyzed and, despite a significant reduction upon reprogramming of iCMs, remained somewhat higher than in cardiac cells and cardiac fibroblast-derived iCMs.
The DNA methylation status of specific loci also reflects the stability of the reprogramming event from cardiac fibroblasts to iCMs. The promoter regions of 2 cardiomyocyte-specific genes, Nppa and Myh6 were analyzed by bisulfite genomic sequencing in cardiac fibroblasts, iCMs, and neonatal cardiomyocytes. Both promoter regions were hypermethylated in cardiac fibroblasts, as expected from the cardiomyocyte-specific expression of these genes, but were comparatively demethylated in iCMs, similar to neonatal cardiomyocytes. These results indicated that reprogramming by Gata4, Mef2c, and Tbx5 induced epigenetic resetting of the fibroblast genome to a cardiomyocyte-like state [105].
The finding that overexpression of the chromatin remodeling enzyme Baf60c potentiates the function of the transcription factors Gata4, Nkx2.5, and Tbx5 to induce transdifferentiation of mouse mesoderm to cardiac myocytes is another example of how the modulation of chromatin remodeling may be useful to drive cardiac regeneration [107]. Most likely, in amniotes, an additional layer of tissue-specific gene regulation, through Baf60c, has been superimposed on cardiac transcriptional regulators. Consistent with loss-of-function experiments, Gata4 was found to be a key factor in initiating the cardiac program, whereas Tbx5 was required for full differentiation into contracting cardiomyocytes [108]. Unlike other contexts where DNA-binding factors were sufficient for transdifferentiation [109], Baf60c was required to potentiate the function of Gata4 and Tbx5, partly by allowing binding of Gata4 to cardiac-related genetic loci (Fig. 5). These findings reveal a novel mechanism for tissue-specific gene expression regulation by chromatin remodeling complexes.
To enhance cardiac differentiation several pharmacological compounds have been examined. 5-Azacytidine, an inhibitor of DNA methylation, promotes cardiac differentiation in a time-dependent manner in embryonic stem cells and adult mesenchymal stem cells [110]. In addition, inhibition of HDAC by TSA promotes cardiac differentiation by increased expression of acetylated GATA4, NKx2.5, and MEF2c [111]. However, conflicting data are often obtained in studies pharmacologically interfering with histone modifications. The reason for this may be, at least in part, the different responses of cells at different states of differentiation. Additionally, most if not all histone-modifying enzymes have multiple targets and often directly control both transcription factor activities and signaling pathways. Therefore, prior to targeting epigenetic enzymes for modulating cell fate decisions, their exact molecular mechanisms need to be characterized in full.
Cutting-edge technologies in epigenomics
Research in epigenetics is accelerating considerably due to rapidly evolving, cutting-edge technologies. Chromatin immunoprecipitation (ChIP) represents a powerful method for studying protein-DNA interactions in vivo, and has been widely used in epigenetics research involving both DNA methylation and histone modifications. The advent of microarrays led to the development of ChIP-on-Chip techniques that allowed whole genome studies of protein-DNA interactions, such as the mapping of histone marks at the genome-wide or global promoter level [112] (Fig. 6).
Figure 6. Graphic representation of the ChIP-on-chip method. ChIP-on-chip combines Chromatin Immunoprecipitation with microarray technologies. In the initial step of this method proteins are cross-linked on the DNA usually with gentle formaldehyde fixation that is reversible. Then, the cells are lysed and the DNA is sheared by sonication or nuclease treatment. This results in double-stranded DNA fragments normally less than 1 kb in length. The DNA/protein complexes can be immunoprecipitated with appropriate antibodies against histone modifications (or against methylated cytosines in the Methylated DNA ChIP assay-MeDIP). After reversing the cross-linking and a step of amplification, the immunoprecipitation as well as the input DNA can be differentially labeled and hybridized to genomic microarrays (ChIP-on chip).

More recently, the revolution of next-generation sequencing technologies is transforming once more the field of epigenetics, with the introduction of ChIP-sequencing which enables the identification of epigenetic modification sites through direct counting of ChIP sequence tags. This provides a number of advantages compared to pre-existing methods, including greater genomic coverage and applicability to any genome for which the genomic sequence is available [113].
A closely related method called MeDNA IP, or MeDIP, has also become very useful in studying DNA methylation [114]. In MeDIP, DNA regions containing methylated cytosines at densities above the typical sparse locations across the genome are immunoprecipitated using antibodies that recognize 5-methyl-cytosine. The DNA is subsequently isolated and analyzed using microarrays (MeDNA IP-on-Chip) or next-generation sequencing (MeDNA IP sequencing or MeDIP sequencing) [115]. The essential requirements of successful genome-wide ChIP and MeDNA IP epigenetic studies are high-quality antibodies, high-quality and robust protocols, and comprehensive data analysis.
Another approach is the differential chromatin scanning (DCS) method, which offers a more qualitative approach to effectively isolating genome fragments with differential epigenetic regulation. This method was first developed by Kaneda et al. [116] and couples ChIP to subtraction polymerase chain reaction (PCR). More specifically, DNA fragments bound to acetylated (Ac) histones are purified by immunoprecipitation and subjected to adaptor ligation and PCR amplification (Fig. 7).
Figure 7. The differential chromatin scanning (DCS) method. DNA fragments bound to acetylated (Ac) histones from the tester cells (cells treated with HDAC inhibitors) are purified by immunoprecipitation and subjected to TAG adaptor ligation (green bars) and PCR amplification. The tester DNA is then digested with XmaI, ligated to the first subtraction adaptor (red bars), and annealed with an excess amount of the driver DNA. Given that only the tester specific fragments self-anneal, PCR with the first subtraction primer selectively amplifies these fragments. The products are subjected to a second round of subtraction PCR with the second subtraction adaptor and primer to ensure the fidelity of the subtraction.

Conclusions
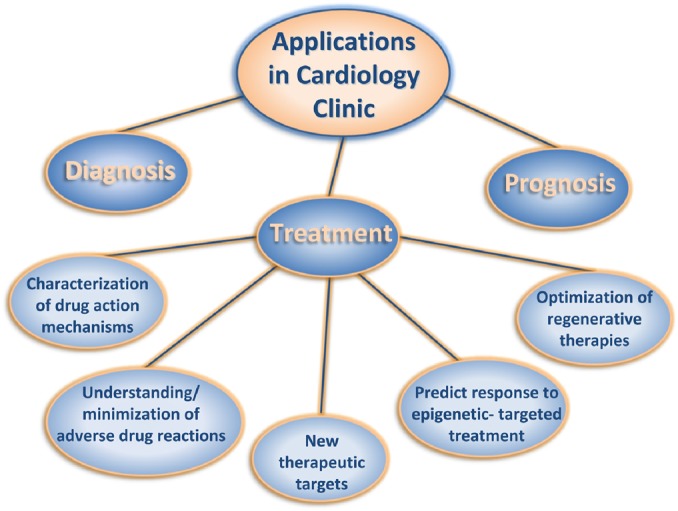
Ever since the discovery of the double helix and the role of genes in human physiology and pathology, the efforts of the international scientific community have focused on reading the entire human genome, mapping its genes and deciphering their roles. These efforts culminated with the completion of the Human Genome Project (HGP) which, for many, signified the transition to a new era. Indeed the HGP enabled a broad range of breathtaking scientific advances that have already begun to improve diagnostics and treatments [117–123]. However, the true benefits for clinical medicine have thus far been modest and well below initial expectations [124,125]. Several reasons for this lie in the inherent complexity of the human genome, which is proving significantly more profound than anticipated. New technologies, experimental strategies and computational approaches are unveiling new aspects of genome regulation, which in turn fuel hopes for greater biomedical achievements. Epigenetics is one of the next frontiers in molecular cardiology, and the rapid accumulation of new knowledge is anticipated to contribute to the cardiology clinic, but not without a great deal of effort. Deeper understanding of the biology of cardiac disease through better designed, multi-ethnic, large scale, prospective cohort studies with careful consideration being given to the genome, environment and phenotype relationships (comprehensive cataloguing of genomic data, development of new technologies and deeper understanding of fundamental biological principles) [126–131]. A comprehensive translation of research findings to clinically useful diagnostic/prognostic/therapeutic tools (Fig. 8) is also important, as is the effective incorporation of these genomic tools into routine clinical practice through easy-to-use, sensitive, specific, accurate, reproducible and cost-effective molecular tests. Clear guidelines regarding when to perform such tests, how to interpret test results and what treatment protocols should be followed thereafter, including the education of healthcare professionals, patients and the public.
Figure 8. Applications of epigenetics in the cardiology clinic including diagnosis, prognosis and treatment.
The past decade established a strong basis for understanding cardiac disease while the coming decade promises to bring genomic medicine one step closer to the heart disease patient. The elucidation of the complex role of epigenetic modifications in cardiac disease and the establishment of pharmacogenetics, as well as pharmacoepigenetics, will be the next milestones in this fascinating and exciting endeavor.
Acknowledgements
We would like to thank the “katArt-e Creative Group & Design Lab” (www.katart-e.com) for their assistance with the graphics of Fig. 1. The authors are supported by grants from the European Community's Seventh Framework Programme FP7/2007–2013 under grant agreement #HEALTH-F2-2009-241526, “EUTrigTreat”, the Hellenic Cardiological Society and the John S. Latsis Public Benefit Foundation.
References
- [1].Frey N, Luedde M, Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol. 2012;9(2):91–100. doi: 10.1038/nrcardio.2011.159. [DOI] [PubMed] [Google Scholar]
- [2].Girolami F, et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010;55(14):1444–1453. doi: 10.1016/j.jacc.2009.11.062. [DOI] [PubMed] [Google Scholar]
- [3].Olivotto I, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2008;83(6):630–638. doi: 10.4065/83.6.630. [DOI] [PubMed] [Google Scholar]
- [4].Torricelli F, et al. Prevalence and clinical profile of troponin T mutations among patients with hypertrophic cardiomyopathy in tuscany. Am J Cardiol. 2003;92(11):1358–1362. doi: 10.1016/j.amjcard.2003.08.031. [DOI] [PubMed] [Google Scholar]
- [5].Landstrom AP, Ackerman MJ. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circ Heart Fail. 2010;122(23):2441–2449. doi: 10.1161/CIRCULATIONAHA.110.954446. discussion 2450 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Belus A, et al. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J Physiol. 2008;586(Pt 15):3639–3644. doi: 10.1113/jphysiol.2008.155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Taegtmeyer AB, Barton PJ, Yacoub MH. Genetic association studies: personalized medicine in cardiac transplantation. Nat Clin Pract Cardiovasc Med. 2006;3(2):58–59. doi: 10.1038/ncpcardio0453. [DOI] [PubMed] [Google Scholar]
- [8].Taegtmeyer AB, et al. Effect of adenosine monophosphate deaminase-1 C34T allele on the requirement for donor inotropic support and on the incidence of early graft dysfunction after cardiac transplantation. Am J Cardiol. 2009;103(10):1457–1462. doi: 10.1016/j.amjcard.2009.01.360. [DOI] [PubMed] [Google Scholar]
- [9].Taegtmeyer AB, et al. Effect of ABCB1 genotype on pre- and post-cardiac transplantation plasma lipid concentrations. J Cardiovasc Transl Res. 2011;4(3):304–312. doi: 10.1007/s12265-011-9269-z. [DOI] [PubMed] [Google Scholar]
- [10].Yuen AH, et al. Association of improved cardiac function in donors with C34T mutation of the AMP deaminase 1 gene. Nucleosides Nucleotides Nucleic Acids. 2005;24(4):275–277. doi: 10.1081/NCN-59709. [DOI] [PubMed] [Google Scholar]
- [11].Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- [12].Feinberg AP. Epigenomics reveals a functional genome anatomy and a new approach to common disease. Nat Biotechnol. 2010;28(10):1049–1052. doi: 10.1038/nbt1010-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bernstein BE, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28(10):1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Egger G, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- [15].Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128(4):787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- [16].Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9(6):465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- [17].Nikitina T, et al. Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol Cell Biol. 2007;27(3):864–877. doi: 10.1128/MCB.01593-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ng HH, et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet. 1999;23(1):58–61. doi: 10.1038/12659. [DOI] [PubMed] [Google Scholar]
- [19].Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- [20].Mill J, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82(3):696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Luger K, et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- [22].Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol. 2004;14(14):R546-51. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- [23].Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- [24].Kouzarides T. Histone acetylases and deacetylases in cell proliferation. Curr Opin Genet Dev. 1999;9(1):40–48. doi: 10.1016/s0959-437x(99)80006-9. [DOI] [PubMed] [Google Scholar]
- [25].Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hodawadekar SC, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene. 2007;26(37):5528–5540. doi: 10.1038/sj.onc.1210619. [DOI] [PubMed] [Google Scholar]
- [28].Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- [29].Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev. 2002;12(2):198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- [30].Agger K, et al. The emerging functions of histone demethylases. Curr Opin Genet Dev. 2008;18(2):159–168. doi: 10.1016/j.gde.2007.12.003. [DOI] [PubMed] [Google Scholar]
- [31].Sims 3rd RJ, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet. 2003;19(11):629–639. doi: 10.1016/j.tig.2003.09.007. [DOI] [PubMed] [Google Scholar]
- [32].Rea S, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406(6796):593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- [33].Upadhyay AK, Cheng X. Dynamics of histone lysine methylation: structures of methyl writers and erasers. Prog Drug Res. 2011;67:107–124. doi: 10.1007/978-3-7643-8989-5_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011;123(19):2145–2156. doi: 10.1161/CIRCULATIONAHA.110.956839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15(18):2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- [36].Bedford MT. Arginine methylation at a glance. J Cell Sci. 2007;120(Pt 24):4243–4246. doi: 10.1242/jcs.019885. [DOI] [PubMed] [Google Scholar]
- [37].Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33(1):1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Herrmann F, et al. Human protein arginine methyltransferases in vivo–distinct properties of eight canonical members of the PRMT family. J Cell Sci. 2009;122(Pt 5):667–677. doi: 10.1242/jcs.039933. [DOI] [PubMed] [Google Scholar]
- [39].Wolf SS. The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell Mol Life Sci. 2009;66(13):2109–2121. doi: 10.1007/s00018-009-0010-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet. 2007;8(11):829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- [41].Chang B, et al. JMJD6 is a histone arginine demethylase. Science. 2007;318(5849):444–447. doi: 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- [42].Lachner M, et al. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410(6824):116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- [43].Wysocka J, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442(7098):86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- [44].Li H, et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442(7098):91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- [46].Movassagh M, et al. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PLoS One. 2010;5(1):e8564. doi: 10.1371/journal.pone.0008564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bratt A, et al. Angiomotin belongs to a novel protein family with conserved coiled-coil and PDZ binding domains. Gene. 2002;298(1):69–77. doi: 10.1016/s0378-1119(02)00928-9. [DOI] [PubMed] [Google Scholar]
- [48].Su ZJ, et al. A vascular cell-restricted RhoGAP, p73RhoGAP, is a key regulator of angiogenesis. Proc Natl Acad Sci USA. 2004;101(33):12212–12217. doi: 10.1073/pnas.0404631101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Woodfin A, Voisin MB, Nourshargh S. PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol. 2007;27(12):2514–2523. doi: 10.1161/ATVBAHA.107.151456. [DOI] [PubMed] [Google Scholar]
- [50].Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356(8):830–840. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- [51].Cecchi F, et al. Coronary Microvascular Dysfunction and Prognosis in Hypertrophic Cardiomyopathy. New England Journal of Medicine. 2003;349(11):1027–1035. doi: 10.1056/NEJMoa025050. [DOI] [PubMed] [Google Scholar]
- [52].Meyer M, et al. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92(4):778–784. doi: 10.1161/01.cir.92.4.778. [DOI] [PubMed] [Google Scholar]
- [53].Inesi G, Prasad AM, Pilankatta R. The Ca2+ ATPase of cardiac sarcoplasmic reticulum: Physiological role and relevance to diseases. Biochem Biophys Res Commun. 2008;369(1):182–187. doi: 10.1016/j.bbrc.2007.11.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zarain-Herzberg A, et al. Decreased expression of cardiac sarcoplasmic reticulum Ca(2+)-pump ATPase in congestive heart failure due to myocardial infarction. Mol Cell Biochem. 1996;163–164:285–290. doi: 10.1007/BF00408669. [DOI] [PubMed] [Google Scholar]
- [55].Levine B, et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323(4):236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- [56].Kao YH, et al. Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit Care Med. 2010;38(1):217–222. doi: 10.1097/CCM.0b013e3181b4a854. [DOI] [PubMed] [Google Scholar]
- [57].Mathiyalagan P, et al. Cardiac ventricular chambers are epigenetically distinguishable. Cell Cycle. 2010;9(3):612–617. doi: 10.4161/cc.9.3.10612. [DOI] [PubMed] [Google Scholar]
- [58].Zhang CL, et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110(4):479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chang S, et al. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24(19):8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lyons GE, et al. Expression of mef2 genes in the mouse central nervous system suggests a role in neuronal maturation. J Neurosci. 1995;15(8):5727–5738. doi: 10.1523/JNEUROSCI.15-08-05727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Edmondson DG, et al. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development. 1994;120(5):1251–1263. doi: 10.1242/dev.120.5.1251. [DOI] [PubMed] [Google Scholar]
- [62].Vega RB, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24(19):8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci USA. 2000;97(26):14400–14405. doi: 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lu J, et al. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA. 2000;97(8):4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Passier R, et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000;105(10):1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Molkentin JD, et al. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell. 1995;83(7):1125–1136. doi: 10.1016/0092-8674(95)90139-6. [DOI] [PubMed] [Google Scholar]
- [67].Lu J, et al. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000;6(2):233–244. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- [68].Ha CH, et al. PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci USA. 2010;107(35):15467–15472. doi: 10.1073/pnas.1000462107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Gusterson RJ, et al. The transcriptional co-activators CREB-binding protein (CBP) and p300 play a critical role in cardiac hypertrophy that is dependent on their histone acetyltransferase activity. J Biol Chem. 2003;278(9):6838–6847. doi: 10.1074/jbc.M211762200. [DOI] [PubMed] [Google Scholar]
- [70].Morimoto T, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. 2008;118(3):868–878. doi: 10.1172/JCI33160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Grozinger CM, Schreiber SL. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci USA. 2000;97(14):7835–7840. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27(1):40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- [73].Lowes BD, et al. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med. 2002;346(18):1357–1365. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- [74].Li C, et al. The deltaA isoform of calmodulin kinase II mediates pathological cardiac hypertrophy by interfering with the HDAC4-MEF2 signaling pathway. Biochem Biophys Res Commun. 2011;409(1):125–130. doi: 10.1016/j.bbrc.2011.04.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Backs J, et al. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116(7):1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Little GH, et al. Nuclear calcium/calmodulin-dependent protein kinase IIdelta preferentially transmits signals to histone deacetylase 4 in cardiac cells. J Biol Chem. 2007;282(10):7219–7231. doi: 10.1074/jbc.M604281200. [DOI] [PubMed] [Google Scholar]
- [77].Zhang T, et al. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92(8):912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- [78].Backs J, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA. 2009;106(7):2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bossuyt J, et al. Ca2+/calmodulin-dependent protein kinase IIdelta and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ Res. 2008;102(6):695–702. doi: 10.1161/CIRCRESAHA.107.169755. [DOI] [PubMed] [Google Scholar]
- [80].Ling H, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119(5):1230–1240. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Antos CL, et al. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem. 2003;278(31):28930–28937. doi: 10.1074/jbc.M303113200. [DOI] [PubMed] [Google Scholar]
- [82].Kook H, et al. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J Clin Invest. 2003;112(6):863–871. doi: 10.1172/JCI19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kee HJ, et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation. 2006;113(1):51–59. doi: 10.1161/CIRCULATIONAHA.105.559724. [DOI] [PubMed] [Google Scholar]
- [84].Olivotto I, et al. The many faces of hypertrophic cardiomyopathy: from developmental biology to clinical practice. J Cardiovasc Transl Res. 2009;2(4):349–367. doi: 10.1007/s12265-009-9137-2. [DOI] [PubMed] [Google Scholar]
- [85].Kong Y, et al. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation. 2006;113(22):2579–2588. doi: 10.1161/CIRCULATIONAHA.106.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Gallo P, et al. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc Res. 2008;80(3):416–424. doi: 10.1093/cvr/cvn215. [DOI] [PubMed] [Google Scholar]
- [87].Trivedi CM, et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007;13(3):324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- [88].Zhu H, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117(7):1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Cao DJ, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci USA. 2011;108(10):4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Haddad F, et al. Role of antisense RNA in coordinating cardiac myosin heavy chain gene switching. J Biol Chem. 2003;278(39):37132–36138. doi: 10.1074/jbc.M305911200. [DOI] [PubMed] [Google Scholar]
- [91].Majumdar G, et al. Epigenetic regulation of cardiac muscle-specific genes in H9c2 cells by Interleukin-18 and histone deacetylase inhibitor m-carboxycinnamic acid bis-hydroxamide. Mol Cell Biochem. 2008;312(1–2):47–60. doi: 10.1007/s11010-008-9720-x. [DOI] [PubMed] [Google Scholar]
- [92].Zhang QJ, et al. The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J Clin Invest. 2011;121(6):2447–2456. doi: 10.1172/JCI46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Bingham AJ, et al. The repressor element 1-silencing transcription factor regulates heart-specific gene expression using multiple chromatin-modifying complexes. Mol Cell Biol. 2007;27(11):4082–4092. doi: 10.1128/MCB.00269-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Stein AB, et al. Loss of H3K4 methylation destabilizes gene expression patterns and physiological functions in adult murine cardiomyocytes. J Clin Invest. 2011;121(7):2641–2650. doi: 10.1172/JCI44641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kaneda R, et al. Genome-wide histone methylation profile for heart failure. Genes Cells. 2009;14(1):69–77. doi: 10.1111/j.1365-2443.2008.01252.x. [DOI] [PubMed] [Google Scholar]
- [96].Nguyen AT, et al. DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev. 2011;25(3):263–274. doi: 10.1101/gad.2018511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Peedicayil J. Pharmacoepigenetics and pharmacoepigenomics. Pharmacogenomics. 2008;9(12):1785–1786. doi: 10.2217/14622416.9.12.1785. [DOI] [PubMed] [Google Scholar]
- [98].Gal-Yam EN, et al. Cancer epigenetics: modifications, screening, and therapy. Annu Rev Med. 2008;59:267–280. doi: 10.1146/annurev.med.59.061606.095816. [DOI] [PubMed] [Google Scholar]
- [99].Gupta MP, et al. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem. 2008;283(15):10135–10146. doi: 10.1074/jbc.M710277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ordovas JM, Smith CE. Epigenetics and cardiovascular disease. Nat Rev Cardiol. 7(9):510–519. doi: 10.1038/nrcardio.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Yacoub M, Suzuki K, Rosenthal N. The future of regenerative therapy in patients with chronic heart failure. Nat Clin Pract Cardiovasc Med. 2006;3(Suppl 1):S133-5. doi: 10.1038/ncpcardio0401. [DOI] [PubMed] [Google Scholar]
- [102].Assmus B, et al. Clinical outcome 2 years after intracoronary administration of bone marrow-derived progenitor cells in acute myocardial infarction. Circ Heart Fail. 3(1):89–96. doi: 10.1161/CIRCHEARTFAILURE.108.843243. [DOI] [PubMed] [Google Scholar]
- [103].Ohtani K, Dimmeler S. Epigenetic regulation of cardiovascular differentiation. Cardiovasc Res. 90(3):404–412. doi: 10.1093/cvr/cvr019. [DOI] [PubMed] [Google Scholar]
- [104].Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10(5):467–474. doi: 10.1038/nm0504-467. [DOI] [PubMed] [Google Scholar]
- [105].Ieda M, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 142(3):375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- [107].Takeuchi JK, Bruneau BG. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature. 2009;459(7247):708–711. doi: 10.1038/nature08039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313(5795):1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Zhou Q, et al. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455(7213):627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Fukuda K, Yuasa S. Stem cells as a source of regenerative cardiomyocytes. Circ Res. 2006;98(8):1002–1013. doi: 10.1161/01.RES.0000218272.18669.6e. [DOI] [PubMed] [Google Scholar]
- [111].Karamboulas C, et al. HDAC activity regulates entry of mesoderm cells into the cardiac muscle lineage. J Cell Sci. 2006;119(Pt 20):4305–4314. doi: 10.1242/jcs.03185. [DOI] [PubMed] [Google Scholar]
- [112].Huebert DJ, et al. Genome-wide analysis of histone modifications by ChIP-on-chip. Methods. 2006;40(4):365–369. doi: 10.1016/j.ymeth.2006.07.032. [DOI] [PubMed] [Google Scholar]
- [113].Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Weber M, Schubeler D. Genomic patterns of DNA methylation: targets and function of an epigenetic mark. Curr Opin Cell Biol. 2007;19(3):273–280. doi: 10.1016/j.ceb.2007.04.011. [DOI] [PubMed] [Google Scholar]
- [115].Morahan JM, et al. A genome-wide analysis of brain DNA methylation identifies new candidate genes for sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10(5–6):418–429. doi: 10.3109/17482960802635397. [DOI] [PubMed] [Google Scholar]
- [116].Kaneda R, et al. High-throughput screening of genome fragments bound to differentially acetylated histones. Genes Cells. 2004;9(12):1167–1174. doi: 10.1111/j.1365-2443.2004.00804.x. [DOI] [PubMed] [Google Scholar]
- [117].Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- [118].Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- [119].Dietz HC. New therapeutic approaches to mendelian disorders. N Engl J Med. 2010;363(9):852–863. doi: 10.1056/NEJMra0907180. [DOI] [PubMed] [Google Scholar]
- [120].Brooke BS, et al. Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N Engl J Med. 2008;358(26):2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Bignell GR, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463(7283):893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Finishing the euchromatic sequence of the human genome, Nature. 2004; 431: 7011, 931–945. [DOI] [PubMed]
- [123].Frueh FW, et al. Pharmacogenomic biomarker information in drug labels approved by the United States food and drug administration: prevalence of related drug use. Pharmacotherapy. 2008;28(8):992–998. doi: 10.1592/phco.28.8.992. [DOI] [PubMed] [Google Scholar]
- [124].Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470(7333):187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- [125].Collins F. Has the revolution arrived? Nature. 2010;464(7289):674–675. doi: 10.1038/464674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Manolio TA, Collins R. Enhancing the feasibility of large cohort studies. JAMA. 2010;304(20):2290–2291. doi: 10.1001/jama.2010.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Manolio TA, Bailey-Wilson JE, Collins FS. Genes, environment and the value of prospective cohort studies. Nat Rev Genet. 2006;7(10):812–820. doi: 10.1038/nrg1919. [DOI] [PubMed] [Google Scholar]
- [128].Thorisson GA, Stein LD. The SNP Consortium website: past, present and future. Nucleic Acids Res. 2003;31(1):124–127. doi: 10.1093/nar/gkg052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Altshuler DM, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467(7311):52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].A map of human genome variation from population-scale sequencing, Nature. 2010; 467: 7319, 1061–1073. [DOI] [PMC free article] [PubMed]