

Abstract
Evolutionary changes in organismal traits may occur gradually or suddenly. Until recently, however, there has been little direct information about how phenotypic changes are related to the rate and nature of underlying changes in genotype. Technological advances enabling whole-genome and whole-population sequencing coupled with experiments that watch evolution in action have brought new precision and insights to studies of mutation rates and genome evolution. Here, we discuss the evolutionary forces and ecological processes that govern genome dynamics in various laboratory systems in the context of relevant population genetic theory, and we relate these findings to evolution in natural populations.
Introduction
Evolutionary and ecological questions that could once be approached only by comparative or theoretical methods are increasingly amenable to direct study. In evolution experiments, populations of organisms are maintained in controlled environments where changes in genotype and phenotype can be monitored over timescales spanning many tens, hundreds, or even thousands of generations1,2. Bringing evolution into the laboratory has several advantages, including the ability to generate a “fossil” record for later study and to test the predictability of evolution across replicate populations. Studies of microbes also benefit from rapid generation times and the viability of frozen organisms, which can be revived to allow an ancestor to compete head-to-head against its own descendants, or to replay evolution starting from various past states to investigate whether a particular outcome was contingent on some prior event.
How many and what types of genetic changes accumulate in evolving populations over time? The field of population genetics has developed a sophisticated mathematical framework for describing rates of evolutionary change in terms of the fundamental processes of mutation, recombination, genetic drift, and natural selection3. This theory guides a general understanding of evolutionary regimes and dynamics, but specific outcomes in any given biological system may also critically depend on the molecular details of a particular genome and how it encodes metabolic, regulatory, and developmental pathways. Both perspectives are necessary for unravelling contentious issues in evolutionary biology related to rates of sequence evolution, such as the relative importance of adaptive and non-adaptive processes and whether the predominant tempo of evolutionary change is gradual or episodic.
Recent advances in DNA sequencing technologies have made it possible, for the first time, to identify genetic changes between ancestral and derived organisms on a whole-genome scale for any species4,5. We begin this review by examining some of the previously hidden details that whole-genome and whole-population sequencing are revealing about evolution in even the simplest laboratory scenarios. Then, we discuss genetic dynamics in experiments that add back various components of the complexity present in the natural world. We focus primarily on asexual microbial systems where most studies with extensive genome sequencing have been conducted to date. But, we also discuss multicellular eukaryotes and experiments in which sexual recombination plays a role, where genomic data is increasingly becoming available.
Mutation rates
Most evolution experiments begin with clonal or inbred populations of a model organism so that there is a homogenous, well-characterized genetic starting point. Therefore, knowing the rates at which new mutations arise and lead to genetic and phenotypic diversity within a population is a useful starting point for understanding evolutionary dynamics. MUTATION ACCUMULATION (MA) experiments allow one to estimate the intrinsic rates and effects of new mutations by repeatedly imposing POPULATION BOTTLENECKS of one or a few randomly chosen breeding individuals to minimize selection that would otherwise favour some variants6(Fig. 1a). Under these special conditions, one can simply count the number of genetic changes present in independently evolved genomes after a known number of generations to estimate the spontaneous mutation rate (Box 1). Recently, classic long-term mutation accumulation studies with model organisms — including Saccharomyces cerevisiae7, Arabidopsis thaliana8. Drosophila melanogaster9, and Caenorhabditis elegans10 — have been revisited using whole-genome sequencing to estimate spontaneous mutation rates. New MA studies of microbes have also been performed with this express aim11–13.
Figure 1. Types of evolution experiments.
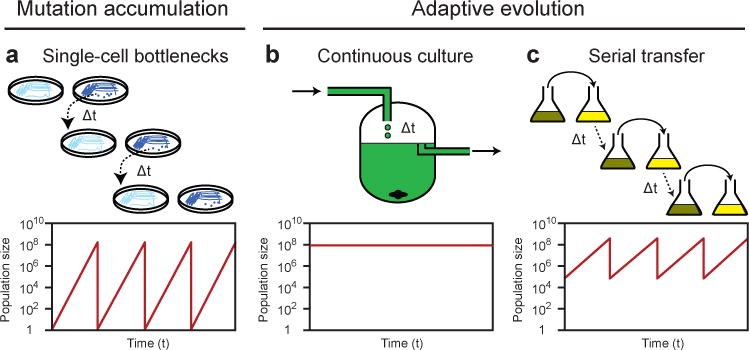
There are three main ways that populations are propagated in evolution experiments, and they lead to different types of genetic dynamics. The mechanics of how populations are maintained in each setup are illustrated for microbes (top panels), with representative changes in population sizes over time also shown for each procedure (bottom panels). Analogous procedures exist for multicellular organisms, although population sizes are generally much smaller. a | Mutation accumulation. Frequent and deliberate population bottlenecks through one or a few randomly chosen breeding individuals, as accomplished by picking colonies of microorganisms that grow from single cells on agar plates, purge genetic diversity and lead to the fixation of arbitrary mutations without respect to their effects on fitness. b | Continuous culture. Maintaining organisms in populations where there is a constant inflow of nutrients and an outflow of random individuals and waste, as occurs in a chemostat, leads to adaptive evolution and genetic diversity within populations that typically maintain a nearly constant size. c | Serial transfer. Batch growth, where a fraction of the population is periodically transferred to fresh media and allowed to regrow until the limiting nutrient is exhausted, also leads to adaptive evolution because ample genetic diversity is maintained through each transfer. Alternatively, transfers can be made prior to nutrient depletion, thereby allowing perpetual population growth. A second, cryptic type of population bottleneck occurs during adaptive evolution experiments (b and c) as a consequence of selective sweeps, especially in asexual populations, that drive out competing lineages and thereby reduce genetic diversity.
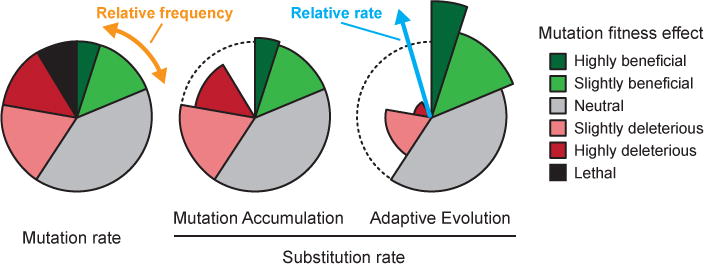
Box 1. Mutation rates versus substitution rates.
It is important to distinguish between the rate at which spontaneous mutations occur and the rate at which genetic changes accumulate in a surviving lineage. The MUTATION RATE reflects the probability of a change in genome sequence between a parent and its offspring. It is the compound result of unrepaired DNA damage, polymerase errors, intragenomic recombination events, movements of transposable elements, and other molecular processes that introduce errors during the transmission of genetic information. However, only those mutations in lineages that persist — typically in the face of selection — contribute to the SUBSTITUTION RATE measured by whole-genome sequencing. The failure to carefully distinguish between these two types of rates is a persistent cause of confusion and misconceptions about whether mutations are random. In the same vein, the frequency of a mutant allele in a population generally does not equal the rate at which the corresponding mutational event occurs.
Mutations can be broadly categorized as beneficial, neutral, deleterious, or lethal with respect to their effects on BIOLOGICAL FITNESS88. In a variety of organisms, many or most new mutations are thought to be neutral or nearly so, and deleterious mutations greatly outnumber beneficial mutations under most circumstances89. Some mutations may change the magnitude or even sign of the fitness effects of other mutations, a phenomenon known as epistasis90. Nevertheless, each new mutation in an evolving lineage can be classified into one of these categories depending on its fitness effect at the time and in the genetic context in which it appears. In this review, we discuss two kinds of evolution experiments where these different categories of mutations make very different contributions to the substitution rate.
In MUTATION ACCUMULATION (MA) experiments, populations are continually forced through a bottleneck of one or a few breeding individuals, so the probability that any given mutation survives is essentially random and independent of its fitness effect. Thus, all mutations – except lethal or extremely deleterious ones – accumulate at rates close to their underlying mutation rates (shown as dashed curve) in surviving lineages (see the figure, part b). The overall fraction of mutations in these highly unfavourable categories is usually thought to be small, and it is therefore common to equate substitution rates and mutation rates in MA experiments, although this will slightly underestimate the true mutation rate. The ultimate MA experiment is to sequence large numbers of parents and their offspring to avoid changes in environmental or genetic factors that might affect mutation rates during a longer experiment91.
In ADAPTIVE EVOLUTION (AE) experiments, by contrast, beneficial mutations typically drive the genetic dynamics. The substitution rate of beneficial mutations exceeds the actual mutation rate (shown as dashed curve) for this category because lineages with these rare mutations increase in frequency as they outcompete their ancestors and lineages with other mutations. Deleterious mutations, by contrast, are underrepresented in AE experiments because they are usually purged by selection, although slightly deleterious mutations can sometimes hitchhike with beneficial ones. The nature of competition between genetically diverged lineages will affect the extent of mutational diversity in a population, but the expected rate of accumulation of neutral mutations in any one surviving lineage will still equal the underlying mutation rate for this category.
The overarching conclusion of these experiments is that spontaneous mutation rates are usually very low. MA experiments with bacteria11–13 and single-celled eukaryotes7,13 typically find that the rate of single base mutations is on the order of 10−10 to 10−9 per base pair per replication. With typical genome sizes in these organisms on the order of 106 to 107 bp, these rates correspond to just one point mutation every few hundred to several thousand cell divisions, which is in reasonable agreement with earlier estimates for DNA-based microbes from reporter gene assays14. Rates of point mutations in multicellular eukaryotes8–10 are on the order of 0.05 to 1.0 per generation across the entire protein-coding portions of these genomes13,15, which is still quite low given the much longer generation times and the multiple cell divisions in the germ line between generations in these organisms. Other types of mutations, such as insertions and deletions of one or a few bases, typically have a lower rate than single-base changes but vary more between species and with sequence context7. Other types of mutations, such as insertions of mobile DNA elements and large-scale chromosomal rearrangements, are more difficult to identify from short-read DNA sequencing data and have not yet been systematically examined in MA experiments.
Mutation rates can change over evolutionary time, so it is instructive to understand how genetic and environmental factors impact mutation rates. In particular, hypermutator lineages with elevated mutation rates and highly biased mutational spectra may arise when mutations cause a loss of normal DNA repair or proofreading activities16. MA lineages derived from a Salmonella hypermutator strain had a 30-fold increase in point mutation rates compared to a wild-type strain, and 91% of the resulting base substitutions were G:C→T:A transversions — a bias consistent with misincorporation of oxidized guanine bases during DNA replication11. Another MA study found that an Escherichia coli strain defective in mismatch repair experienced 138 times the wild-type mutation rate with 70% A:T→G:C transitions12.
Chemical mutagens are the main environmental factor that has been examined with MA experiments to date. At the extreme level of mutation that can be achieved in experimental evolution studies of mutagenized bacteriophage17,18 — for example, mutating ~1% of the bases in the T7 genome17 — one can begin to ask questions about what sites in a genome must be unchanged to remain viable. RNA viruses, such as HIV, typically have high mutation rates on the order of 1 per genome per generation19, and experiments with mutagens have sought to test whether therapies that increase that rate further could lead to a mutational meltdown and the extinction of viral populations in infected individuals20. More generally, we anticipate that the sequencing of MA lines will be used to enable a more precise version of the Ames test21 to define the mutagenicity of potential carcinogens in the near future.
It is important to caution that laboratory MA experiments may not precisely match the mutation rates or spectra experienced under natural conditions, where organisms are often nutritionally deprived or otherwise stressed. For example, bacteria in the human gastrointestinal tract are thought to achieve only about 1 generation per day in this complex mixture of nutrients and biotic interactions22, while bacteria in the soil probably undergo prolonged starvation and far fewer generations on average. Similarly, plants may experience extreme temperatures, damage from predation, and increased UV exposure in nature8. These and other environmental and genetic factors will likely be examined in future MA experiments.
Adaptive evolution
Now we will consider evolutionary dynamics in experiments where differential survival and reproduction lead to the preferential accumulation of genetic variants that are better adapted to their environment. The simplest ADAPTIVE EVOLUTION (AE) experiments maintain populations derived from a single ancestral genotype in a uniform environment, such that selection pressures either remain constant or fluctuate in a controlled way. This situation can be achieved in continuous culture where replenishment of resources and removal of individuals happens at a constant rate (Fig. 1b) or by periodic serial transfer of a fraction of the population to a new microcosm with fresh resources (Fig. 1c). In AE experiments, selection for mutations with beneficial effects drives the evolutionary dynamics (Box 1). These dynamics are often visualized in terms of successive steps as populations climb ridges and peaks in a FITNESS LANDSCAPE23. Phenotypic evolution within a population may involve either gradual optimization or discontinuous innovation (Box 2), or perhaps some mix of both. For example, a discontinuous change – such as the ability to survive a previously lethal stress or grow on a new resource – might be followed by a period of gradual refinement of that new ability.
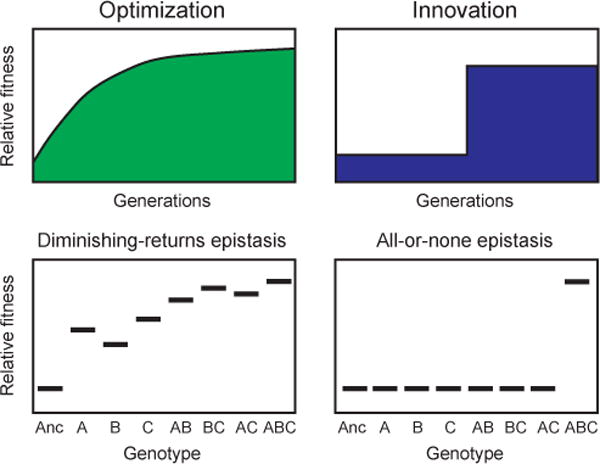
Box 2. Adaptive evolution: optimization versus innovation.
When new beneficial mutations optimize the overall performance of existing genetic, metabolic, and developmental networks during an adaptive evolution experiment, the fitness of organisms tends to improve gradually over time. In some experimental systems, interactions between the fitness effects of these mutations typically exhibit DIMINISHING-RETURNS EPISTASIS92–94. That is, each beneficial mutation increases fitness to a lesser extent in the presence of the other beneficial mutations than it would if it appeared alone in the ancestral genetic background. Typical beneficial mutations in the optimization regime tweak gene expression levels, adjust regulatory interactions, or alter metabolic fluxes. In some cases, these adjustments may be beneficial simply because they reduce the expression, and hence energetic costs, of unused functions. The early beneficial mutations of largest effect are often in global control “hubs” of networks, while later mutations often target the “spokes” of specific pathways36,85,90,95. As these networks and pathways become more finely tuned to a particular environment, it becomes more difficult to improve the overall system performance in a single mutational step. This form of gradual evolution is often associated with “microevolutionary” change.
More sudden and dramatic changes also sometimes occur during evolution experiments, in particular when beneficial mutations produce innovations that allow the organism to occupy a new ecological niche28. This character of adaptive evolution may involve, for example, the generation of a new connection or activity in a cellular network. Innovations may arise from mutations that exhibit ALL-OR-NONE EPISTASIS72. That is, several mutations may first occur that, on their own, have little or no effect on the trait, but this evolved genetic background is required for some keystone mutation to produce the phenotypic novelty. Models of RNA folding and regulatory circuits have been used to explore the abstract properties of these so-called “neutral networks” and how they can promote the evolution of novelty96. This form of evolution, where new phenotypes appear suddenly, is sometimes associated with “macroevolutionary” change, and it can give rise to new opportunities for further adaptation and diversification97.
Of course, multiple processes may be intertwined and their timescales overlap. For example, some innovations may only be possible after a period of random exploration by mutation and genetic drift, as assumed in models of neutral networks. Alternatively, an innovation may have been enabled by earlier beneficial mutations that arose during an epoch of optimization for a different function, a kind of adaptive preadaptation. In this case, the large fitness gain obtained by co-opting these mutations for the innovation may dwarf the fitness gains during the earlier period of optimization. Note, however, that even during optimization, mutations of large effect can give a step-like appearance to fitness trajectories if measured with high resolution on short timescales98,99.
Optimization regime
With asexual organisms, the simplest situation occurs when the rate of appearance of beneficial mutations is low enough relative to the fitness advantages of new beneficial mutations and the population size such that there is effectively only one beneficial mutation present at a time. This mutation, if it survives stochastic loss by genetic drift while it is still rare, will begin a SELECTIVE SWEEP whereby its frequency increases until it reaches GENETIC FIXATION in the population (that is, the mutant completely replaces its ancestor) before another beneficial mutation becomes established (Fig. 2a). These dynamics have been called PERIODIC SELECTION after classic experiments that inferred sweeps of beneficial mutations in E. coli populations24.
Figure 2. | Genetic dynamics in evolution experiments.
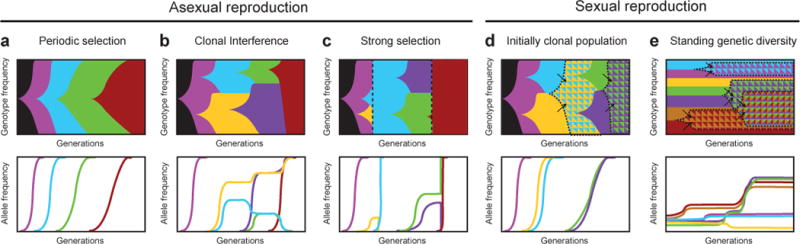
Five scenarios are illustrated using Muller plots (panels at left), which show the frequencies of different genotypes over time as shaded wedges69. As new mutations appear, they are linked with mutations that previously arose in their predecessors. When there is sexual reproduction, existing mutational variants may also be recombined to produce new genotypes (as indicated by arrows pointing to multiply shaded regions with dashed boundaries). The frequencies of different alleles (mutational variants) within the population, as would be measured by metagenomic sequencing, are also shown for each scenario (panels at right). a | Periodic selection. If the rate at which new beneficial mutations appear is low and the fitness benefit of each mutation large, then only one mutation will usually sweep through the population at a time. These dynamics cause near step-like trajectories for fitness, phenotypic traits, and the number of beneficial mutations that accumulate over time. Successive sweeps typically take longer as the expected marginal benefit of a later mutation decreases if evolution is in the optimization regime, as pictured. b | Clonal interference. If the supply rate of beneficial mutations is higher, because either the population size or overall mutation rate is increased, then multiple beneficial mutations may arise before one of them achieves fixation. In asexual populations, competition between the contending mutations slows their progress toward fixation, allowing time for additional beneficial mutations to occur and giving rise to more complex trajectories for fitness and mutation number. c | Strong selection. If strong selection is imposed periodically, in ways that may even be lethal to most of the population (dashed vertical lines), then only one or a few genotypes may persist, and they can then quickly achieve fixation after this selection-induced bottleneck. This scenario can lead to large and sudden changes in a phenotype such as resistance to an antibiotic or stress. d | Sexual reproduction in an initially clonal population. As new beneficial mutations arise, they can be recombined into the same genetic background, rather than only competing with one another as in asexual populations. Thus, sex may lead to more rapid genetic evolution and adaptation. e | Sexual reproduction with standing genetic diversity. Shuffling of the genetic diversity initially present in a population may generate fitter genotypes faster than waiting for new beneficial mutations. Even so, if many different combinations of existing alleles give similar benefits, then no one allele will necessarily sweep to fixation on the timescale of the experiment.
In fact, however, genetic dynamics in evolution experiments rarely, if ever, seem to be in this simple regime. Owing to the many possible routes for adaptation, the rate at which beneficial mutations appear is typically high enough that before one beneficial mutation can sweep to fixation another appears in a separate lineage (Fig. 2b). In asexual populations, competition between these alternative beneficial mutations means that the rate at which any one of these spreads through the population is slowed because it must displace more-fit competitors rather than just its ancestor25. This effect is called CLONAL INTERFERENCE, and the resulting genetic dynamics have been observed in several studies, most notably by deeply sequencing entire yeast populations at frequent intervals to follow the frequencies of many new mutations26.
Genetic dynamics become even more complex when considering that neutral and deleterious mutations continually happen alongside the beneficial mutations discussed so far. In large populations, deleterious mutations would rarely reach high frequency on their own, and neutral mutations would do so only over very long timescales. However, neutral and even deleterious mutations can rapidly “hitchhike” to prominence when they occur in the same genome as a beneficial mutation. The interplay of all of the factors discussed so far can also give rise to apparently contradictory observations. For example, the rate of genomic change in an Escherichia coli population in the long-term evolution experiment (LTEE) was surprisingly constant and clock-like – a signature sometimes taken as evidence of neutral evolution – even though most mutations that fixed in the population were beneficial27 (Box 3).
Box 3. Identifying adaptive mutations.
In molecular evolution and comparative genomics studies, the ratio of synonymous to nonsynonymous base substitutions (dN/dS) in a protein-coding gene is commonly used to test whether it has been subject to positive or negative (purifying) selection. There are rarely enough base substitutions in an evolution experiment to apply this test on a per-gene basis. However, because so few mutations accumulate, it is also highly unlikely that the same gene would change in several independently evolved genomes unless these variants had been enriched by selection. Therefore, such genetic parallelism provides a strong signal that the mutations were beneficial. Depending on the phenotypic effect that is required for adaptation, this parallelism may occur at the level of an individual nucleotide or codon, a specific gene, or some step in a particular metabolic or regulatory pathway27,63,72,100–102. More complex patterns of covariation — such as a mutation in either one gene or another, but not both — in multiple independently evolved genomes can be used to identify new genes involved in the same pathway39,103.
However, “hotspots” that experience unusually high rates of spontaneous mutations relative to the rest of a genome can also lead to genetic parallelism for mutations that are only slightly beneficial or not adaptive at all104–106. Therefore, the “gold standard” for establishing whether a particular mutation is adaptive is to use a genome editing or genetic exchange method to make an ISOGENIC CONSTRUCT that differs from another strain by only the single mutation of interest. Then, one can either test for a change in a trait known to be related to fitness or compete the two organisms under the conditions that prevailed during the evolution experiment to determine whether the mutation is beneficial, neutral, or deleterious.
In longer evolution experiments, after many mutations have accumulated, one must consider several potential complications when interpreting the results of these measurements. First, some mutations may be adaptive only in the context of the genetic background in which they arose, owing to interactions with mutations that occurred earlier in that lineage90. In this case, it might be cleaner to “deconstruct” the mutation by removing it from an evolved genotype, where one would then expect fitness to decrease. Again, however, there is the potential for interactions with mutations that evolved after the focal mutation, which might alter its measured fitness effect. Second, ecological interactions may affect fitness measurements. These often take the form of negative-frequency dependence, where a genotype has an advantage over some competitor only when it is rare within the population56. In other cases, non-transitive interactions may arise such that an evolved genotype is more fit than its immediate progenitor, yet it is less fit than some earlier ancestor that it never encountered because they were present at different times in the evolving population107.
Innovation regime
Some experiments have observed qualitatively new, often “game-changing” abilities appear that have the hallmarks of evolutionary innovations28. Some innovations may require only a single mutation. For example, whole-genome sequencing of experimentally evolved Myxococcus xanthus strains found that 14 mutations were substituted after 1000 generations in a liquid medium, during which time social motility and the capacity to form fruiting bodies were lost, but only one mutation was involved in the subsequent restoration of those functions29. That key mutation did not revert any of the previous mutations but occurred instead in a previously uncharacterized small RNA30. The experimentally evolved transition of a chimeric Ralstonia solanacearum strain from a plant pathogen into a symbiont able to colonize root nodules also required only a single mutation in hrpG, which encodes a protein that regulates the expression of several virulence factors31. It is also likely that some examples of yeast that evolved a new multicellular “snowflake” phenotype needed only single adaptive mutations32.
The evolution of new metabolic capabilities was studied in several early experiments with microorganisms33. These organisms often gained the ability to utilize new compounds as nutrient sources by successive mutations in genes that activated their transcription under new conditions, increased their overall expression levels, or altered their substrate specificities. In these examples, the relevant mutations each conferred a direct advantage in terms of using the new resource, but this need not be the case. The LTEE uses a glucose-limited medium, but citrate – another nutrient that E. coli generally cannot use – has also been present throughout the experiment. The ability to utilize this abundant but untapped resource evolved after 30,000 generations (~15 years), and in only one of twelve replicate populations34. This innovation was difficult because it was contingent on one or more early “potentiating” mutations that had to be present in the GENETIC BACKGROUND for the key “actualizing” mutation to establish in the population. The latter occurred by a chromosomal duplication that rewired gene expression by placing a new transcriptional promoter upstream of a previously silent citrate transporter gene to give the Cit+ phenotype35. The earlier potentiating mutations did not confer any immediate advantage with respect to using citrate, but they may have been beneficial with respect to growth on glucose. If so, it is possible that other populations in the LTEE have also become potentiated and might evolve the Cit+ phenotype, although no others have done so even after > 50,000 generations.
Stressful environments
An important “dial” that can be adjusted in evolution experiments is the STRENGTH OF SELECTION, particularly if improving some phenotypic property is the goal. In one limit, selection may be so strong that it becomes a genetic screen where only rare mutants with extreme, perhaps innovative phenotypes can survive the stress (Fig. 2c). When selection is less stringent, more genetic diversity can usually be sustained, allowing more opportunity for populations to optimize by exploring alternative mutational paths. Indeed, divergent paths have been described in most evolution experiments with microorganisms, and these dynamics have been reconstructed in certain cases36. A potential disadvantage of a weak selection strategy, if the goal is to maximize phenotypic change, is that those mutations conferring the highest tolerance to stresses such as organic solvent tolerance37, temperature38,39, radiation tolerance40, or antibiotic pressures41 may not be favoured under this strategy. On the other hand, if selection is too strong, then an evolving population might be driven towards a “quick fix” – a local peak in the fitness landscape – that renders an even better solution less accessible. These concerns can potentially be balanced by constructing connected microenvironments with a gradient of conditions, such that organisms can colonize and exploit new resources in previously inhospitable regions by gaining new mutations that give them greater tolerance41.
The impact of clonal interference depends on how closely matched the most beneficial that occurs in a population is to the next-most beneficial mutation. Stressful environments can sometimes help to separate the most-beneficial mutations from the less-beneficial mutations. For example, a study of bacteriophage ΦX174 manipulated how harsh the environment was by restricting CaCl2 (which is required for efficient attachment to the E. coli host) and monitored genome sequence diversity over time42. It showed that clonal interference was more prevalent in benign environments, where more beneficial mutations of similar effect were evidently available, and that this led to slower overall rates of genetic change.
Second-order selection for evolvability
When there is genetic diversity within evolving populations for long periods of time, there is the opportunity for selection to operate not only on the immediate effects of mutations or new combinations of alleles, but also on how those new genotypes differ in their capability to further evolve (i.e., their evolvability). In particular, prior mutational steps in a path on the fitness landscape may affect evolvability in at least two main ways. They may alter mutation rates (and/or recombination rates for organisms that are capable of sexual reproduction or horizontal gene transfer), or they may lead to differences in epistatic interactions with potential further mutations (Box 2).
Mutation rates
It is fairly common for some populations in AE experiments to become dominated by hypermutators with elevated mutation rates43–45. How do hypermutators invade populations? The immediate effect of an elevated mutation rate on fitness is invariably negative, on average, because more mutations are deleterious than beneficial (Box 1). However, the initial spontaneous mutation rate is typically very low, as discussed earlier, so that the fitness cost of producing mutations at even a 100-fold higher rate is very small. Furthermore, new hypermutator variants arise frequently because loss-of-function mutations in many genes give this phenotype. Given the balance between their high rate of occurrence and small fitness costs, hypermutators might exist at frequencies of 10−6 to 10−4 in experimental populations of E. coli46,47. The hypermutator subpopulation, though, has increased evolvability because it has a much higher per capita chance of producing the next highly beneficial mutation, or multiple beneficial mutations, than a non-mutator competitor (Fig. 3a). Thus, despite their slight fitness costs, hypermutators can sometimes hitchhike to fixation with the beneficial mutations they generate48.
Figure 3. | Second-order selection for evolvability.
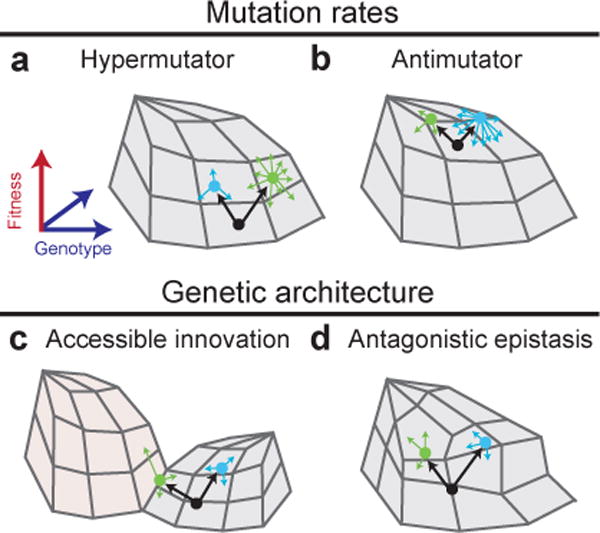
The success of a new mutation or a new combination of alleles may depend on its effect on the rate or fitness benefits of subsequent mutations, in addition to its immediate effect on fitness. Several scenarios are illustrated as alternative mutational paths in fitness landscapes. Genotypes are represented by circles; thick arrows represent initial mutations that generate a new genotype, and thin arrows represent subsequent mutational paths that are available to the genotypes; the mutation rate of a genotype is reflected by the number of thin arrows projecting from it. a | A fitness landscape favouring a hypermutator is shown. From a progenitor with a low ancestral mutation rate (blue), a variant that causes an elevated mutation rate (green) can sometimes take over an asexual population because it has a higher per-capita probability of generating beneficial mutations. Access to these opportunities may outweigh the immediate fitness cost of an increased genetic load. b | A fitness landscape favouring an antimutator is shown. In the longer term, as evolution approaches a local optimum and there are fewer beneficial mutations available, genotypes with lower mutation rates may evolve and be favoured because they have a reduced genetic load. c | A fitness landscape promoting an accessible innovation is shown. Starting from the same progenitor genotype (black), two mutants may have different probabilities of eventual success owing to differences in their evolvability. Here, one mutation (green) makes it possible for a subsequent mutation to invade an open niche, while the other mutation (blue) does not. d | A fitness landscape with antagonistic epistasis is shown. Even in the same niche, one beneficial mutation (blue) may constrain opportunities for further fitness gain more than another (green) because of antagonistic epistatic interactions. In essence, some beneficial mutations may lead to cul-de-sacs in the fitness landscape, allowing other beneficial mutations that do not limit further adaptation to prevail, provided they coexist for enough time. If evolution can be replayed many times starting with the two different genotypes, then an over- or under-representation of mutations in specific genes would provide a signature of such epistatic effects.
In the long term, elevated mutation rates are not without evolutionary risk. Opportunities for mutations that greatly improve fitness will eventually run out in the optimization regime. Then, it may be beneficial to compensate for a hypermutator defect, and become less evolvable, in order to lower the GENETIC LOAD49 (Fig. 3b). Indeed, both mutation-rate scenarios have been observed in an E. coli LTEE population50. After thousands of generations, a mutT mutation that caused a roughly 150-fold increase in mutation rates spread through the population, presumably by hitchhiking with one or more beneficial mutations. Later, parallel mutations in the mutY gene arose in independent lineages that roughly halved the mutation rate, apparently by knocking out a mechanism by which misincorporations of oxidized nucleotides during DNA replication (caused by the original mutT defect) were misrepaired. The resulting reduction in genetic load was estimated to be ~0.5%. That value appears similar in magnitude to other beneficial mutations that drove adaptation late in the LTEE during, whereas some beneficial mutations substituted early in the experiment had much larger fitness effects27. In an experiment with yeast, several populations that started as hypermutators also evolved lower mutation rates and, as a result, reduced genetic loads51. In natural populations, comparative evidence indicates that complete reversion of a hypermutator to an ancestral mutation rate can occur by horizontal gene transfer of an intact gene from a non-mutator52. In addition to affecting mutation rates, hypermutators also generally change the spectrum of different types of mutations, and those differences might also influence a lineage’s evolvability.
Genetic architecture
Because fitness landscapes are complex and may have multiple peaks, some mutational paths may lead to “dead-ends” with no, or at least fewer, opportunities to further improve. In other cases, certain mutations may open up new opportunities for evolution that could not be accessed if other routes were taken. The term GENETIC ARCHITECTURE refers to how genotypes, and mutations that alter genotypes, map onto changes in phenotypes and fitness; hence, differences in evolvability can result from mutations that change the genetic architecture of an organism. We have already seen how the evolution of citrate utilization depended on a “potentiated” genetic background, which could be said to have evolved greater evolvability, in the section on innovations34,35 (Fig. 3c).
A more subtle change in evolvability involved two genotypes that competed early in the history of another E. coli population from the LTEE53. One of these genotypes prevailed despite having a significantly lower fitness. By replaying evolution many times from these two different starting points, it was demonstrated that the “eventual winners” reproducibly gained more fitness over time than did the “eventual losers", such that this seemingly unexpected outcome in the original LTEE population was, in fact, the more likely outcome (Fig. 3d). Genome sequencing showed that the eventual winners often experienced subsequent mutations in a gene, spoT, where mutations were never observed in the eventual losers. The SpoT protein is a regulator of the stringent response. Reconstruction of this mutation in the two genetic backgrounds showed that it was highly beneficial in the winners, but did not significantly affect the fitness of the losers (Box 3). It remains to be determined exactly why the earlier mutations that distinguish the losers from the winners reduced their evolutionary potential, and how important such epistatic cul-de-sacs are in natural populations.
Eco-evolutionary dynamics
In The Origin of Species54, Darwin memorably envisioned a “tangled bank” where organisms were “dependent upon each other in so complex a manner” as an outcome of natural selection. To this point, we have largely ignored ecological interactions beyond “scramble” competition for limiting resources. Even in simple laboratory environments, evolution can lead to NICHE CONSTRUCTION55 that enables diverged lineages of organisms to coexist stably for long periods of time. Other experiments have examined evolution in environments with multiple resources or multiple interacting species. In both cases, METAGENOMIC SEQUENCING of DNA isolated from whole-population or whole-community samples, rather than from individual clones, is yielding new insights into the dynamic interactions between distinct ecotypes.
Multiple nutrients
The well-shaken flask environment of the LTEE nominally has a single niche, with a low concentration of glucose limiting growth. However, one of these E. coli populations gave rise to two distinct ecotypes, first noticed as small (S) and large (L) colony morphotypes after 6,000 generations, and these types coexisted for at least 30,000 generations56. The two types exhibit NEGATIVE FREQUENCY DEPENDENCE, such that each has a fitness advantage and can invade the other type when rare57. In this case, the balance leading to stable coexistence results from the L type growing faster on glucose and the S type having better growth on metabolic by-products58. The genetic and physiological bases of these differences are subjects of on-going investigation57,58.
Metagenomic sequencing of another LTEE population revealed transient diversification59. Mutations in genes related to acetate utilization repeatedly arose, but they never persisted for more than a few thousand generations or reached high frequencies. Acetate is a by-product excreted by E. coli during growth on glucose. As the glucose runs out, the acetate is then taken up and utilized by cells. This cross-feeding opportunity acetate specialists suggests that other LTEE populations might also be on the cusp of evolving more complex ecologies.
In both of these cases from the LTEE, continued evolution after diversification feeds back on the ecological interactions and their stability. In the case of the S and L polymorphism, the L types continually encroached upon the niche occupied by the S type; if the S type had stopped evolving, it would have been driven extinct57. In the acetate case, descendants derived from the main population apparently displaced the acetate specialists multiple times, but repeatedly gave rise to new mutants that later reinvaded this niche59. Understanding the effects of newly evolved ecologies on evolutionary dynamics is an area worthy of theoretical and empirical investigation.
Complex ecologies also evolve in closed cultures where the nutrients are exhausted and not renewed. In these cultures, the viable cell population declines over time, as starvation takes its toll. However, not all genotypes die at the same rate, and the survivors are enriched for “GASP” mutants60,61 – named for their Growth Advantage in Stationary Phase – that exploit by-products of dead and dying cells. Mutations that confer a GASP phenotype have been identified in rpoS, which encodes the alternative “starvation” sigma factor, and other genes encoding high-affinity amino-acid transporters; all of them enhance the cell’s ability to obtain and use amino acids for carbon and energy61. In addition, genomic analyses reveal frequent gene-amplification variants among the survivors61. It appears that copy-number variants are generated at a high rate during starvation, and those that result in extra copies of genes whose products prove useful during starvation can then proliferate.
Substantial diversity also evolves in glucose-limited continuous-culture chemostats62,63, even without the feast-famine seasonality in resource abundance that occurs in serial transfer studies. Metagenomic sequencing of populations propagated in chemostats at two different dilution rates found more genetic and phenotypic diversity in populations that were evolved at slow dilution rates than in those that were evolved at high dilution rates64, which possibly indicates a higher mutation rate at the lower growth rate, more distinct strategies for dealing with this challenge, or some combination of the two. Diversification in this system is driven by regulatory mutations that lead to different balances in the trade-off between stress resistance and nutrient utilization, including rpoS mutations64.
Diversity can be encouraged experimentally by providing a mixture of nutrients to create multiple niches. Evolution experiments in which E. coli populations are serially propagated in a mixture of glucose and acetate reliably give rise to two strategies: glucose specialist “slow-switchers” that shift slowly from using glucose to acetate and “fast-switchers” that utilize acetate earlier65. Metagenomic sequencing of many populations showed clear molecular signatures of parallel evolution within each ecotype, and suggests that some mutations in one type forced evolution in the other type to follow certain mutational pathways66.
Spatial gradients
An alternative approach to generating multiple niches is to establish spatial heterogeneity that results in distinct microenvironments67,68. Pseudomonas fluorescens rapidly evolves into three ecotypes — recognizable by their distinct colony morphologies — that populate different physical zones within unshaken flasks due to heterogeneity in the availability of oxygen68. Diversification also occurred when Burkholderia cenocepacia evolved under daily selection for the abilities both to disperse and the ability to then colonize a new surface as a biofilm69. Here, too, three distinct colony morphotypes rapidly emerged. However, metagenomic sequencing revealed more complex genetic dynamics that were not apparent from the time course of phenotypic diversity. Evolved genotypes with a “studded” colony morphotype gave rise to new versions of all three types, which then drove extinct lineages that had previously exploited other niches. In essence, the studded type appears to be a “source” population that continuously generates new variants that periodically displace the current lineages in secondary niches that become evolutionary “sinks” (Fig. 4a).
Figure 4. | Ecological and coevolutionary dynamics.
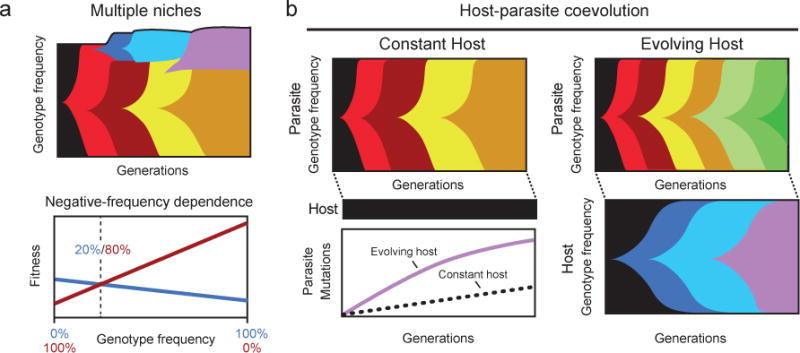
Examples of genetic diversification and dynamics that are driven by ecological interactions within and between species. a | Multiple niches stabilize genetic diversity that evolves within one species. A new lineage colonizes an open niche, increasing the total population size, as shown here when the red ecotype using the primary niche gives rise to the blue ecotype that expands into an open niche. Negative frequency dependence in the fitness of these ecotypes allows their coexistence. In some cases, one lineage may be a source population that can evolve to recolonize the other niche and displace the lineage that previously occupied that niche, as shown here when the later, yellow ecotype from the primary niche gives rise to the violet ecotype that invades the second niche, displacing the previous occupant. b | In host-parasite co-evolution experiments, either hosts and parasites can be allowed to evolve over time or one partner — in this case, the host — can be kept unchanged by continually replenishing its population from a non-evolving stock. Genetic and phenotypic evolution typically occurs at higher rates when the partners co-evolve in response to one other — a phenomenon that is often referred to as “Red Queen” dynamics.
Coevolution
Theory predicts, and comparative studies support, “Red Queen” dynamics during host-parasite coevolution, in which the rate of genome evolution is accelerated, especially in those genes that encode parasite-invasion or host-protection factors. Studies of P. fluorescens and its phage Φ2 have been used to examine the genome-wide dynamics of allele frequencies in replicate phage populations70. The experimental setup allowed the investigation of an interesting contrast: in one treatment the host coevolved with the parasite (Fig. 4b), whereas in another the parasite evolved while the host genotype was kept constant by repeatedly reviving the ancestral strain from a frozen stock (Fig. 4c). As predicted, the phage genome evolved faster in the coevolution treatment than when the host was not allowed to coevolve. By continually providing new opportunities for adaptive mutations of large effect, coevolution also leads to second-order selection for hypermutators in this system71.
Another study of host-pathogen coevolution, using E. coli and a virulent (i.e., exclusively lytic) variant of phage λ, integrates many of the concepts discussed so far72. In some replicate populations, the phage evolved the ability to infect hosts through a new cell-surface receptor. This innovation was contingent on the bacterial population evolving along a certain mutational pathway, one that initially reduced (but did not eliminate) expression of the original receptor; it was also important that later host mutations did not result in the loss of the channel that the phage uses to cross the cell’s inner membrane. Moreover, the innovation in the phage required several prior mutations in the gene encoding its tail fiber; these prior mutations apparently spread because they improved the phage’s ability to infect through the original receptor. In short, the complex interplay between ecological interactions and genetic contingency determined the evolutionary outcomes in these coevolving populations.
Sexual reproduction
Whole-genome studies of the genetic dynamics of adaptive evolution in multicellular animals and plants face different challenges than studies of microbes. Most multicellular organisms can or must reproduce sexually and are diploid, so they have two different copies of each chromosome. Recombination, usually during meiosis, in which DNA sequences are exchanged between homologous chromosomes, breaks the linkage between mutations and their genetic background and produces new combinations of alleles. Many classical population-genetics models assume recombination, and certain inferential procedures (e.g., distinguishing beneficial driver mutations from linked hitchhikers) may prove to be easier in sexual systems.
In populations where adaptation is driven by new beneficial mutations, recombination that brings those mutations together into the same genome may outpace the de novo appearance of successive beneficial mutations in any one lineage73. Perhaps at least partly for this reason, even bacteria have mechanisms — including conjugation, transduction, and transformation — that allow parasexual recombination of alleles between lineages. In experimental E. coli populations where new beneficial mutations drive adaptation, adding recombination has been shown to alleviate clonal interference and accelerate adaptation under some circumstances74 (Fig. 2d). However, most experiments with multicellular organisms do not (and often cannot) begin with a clonal population and then wait for new beneficial mutations to arise. Instead, they usually begin with substantial genetic diversity, such as typically exists as standing variation in natural populations. Under these conditions, the initial genotypic diversity and the new types generated by recombining alleles from across the genome are the primary sources of the genetic variation available for adaptation, at least in the short term (Fig. 2e).
The effects of these issues on the tempo and mode of genome evolution are just beginning to be explored in experiments with sexual multicellular organisms. Whole-genome sequencing of outbred Drosophila melanogaster populations selected for accelerated development (shorter time from egg to adult) over 600 generations found that no alleles, either those initially present or that arose de novo, had swept to fixation75. However, a number of parallel changes occurred in the distribution of allele frequencies and levels of homozygosity across each chromosome, indicating that selection had repeatedly enriched certain variants. These “soft” sweeps of existing alleles may have been incomplete because the experiment was too short or, alternatively, because different allelic combinations resulted in similar levels of phenotypic improvement. By analogy to clonal interference, the second possibility suggests a sort of “sexual interference” where alternative allelic combinations that produce similar benefits impede any given sweep to fixation, and thereby maintain genetic diversity for longer than would otherwise be expected.
In another study, inbred lines of D. melanogaster were pooled, and their offspring evolved in increasingly hypoxic environments for >200 generations76. Individuals from endpoint populations could survive at low oxygen levels that were lethal to their ancestors. Genomic sequencing of the evolved populations found numerous apparently complete fixations of alleles, as indicated by the depletion of genetic diversity in some chromosomal regions. There are several potential explanations for the difference between this study and the one on accelerated development. One possibility is that the stronger selection for survival may have caused more extreme population bottlenecks. Another possibility is that hypoxia-tolerance may involve fewer genetic loci than development time. A third possible explanation is that this experiment began with a set of inbred lines, whereas the experiment on development time used outbred lines that presumably harboured much more initial genetic diversity.
In an even longer experiment with D. melanogaster, flies were propagated for >50 years in complete darkness, and they have also been studied by whole-genome sequencing77. However, it is difficult to draw conclusions from these data because the experiment suffered an extinction of control lineages and historical DNA samples are not available. In future studies, the planned preservation of time series of samples for genomic analysis should provide additional insights into the tempo and mode of genetic change in animal and plant populations.
Perspective
We end by briefly commenting on other systems where whole-genome sequencing is likely to be applied to understand evolutionary dynamics in the near future. First, more genetics, systems biology, and synthetic biology studies may be unwitting evolution experiments than is commonly appreciated. In microbiology, there is a growing realization that strains once believed to be isogenic are not: additional mutations, beyond those deliberately introduced and studied, have accumulated over their history78,79. Furthermore, one often introduces single genetic changes, or defined combinations of changes, in a reference genome in these types of studies. This genetic manipulation may be accomplished in various ways: by spontaneous mutation, through the use of a mutagen, by some means of genetic exchange, by genome editing technologies, or by some combination of these strategies. These manipulations may, either occasionally or typically, cause unintended mutations, in addition to the desired changes80–82. These considerations will also apply to the synthesis and large-scale editing of genomes83,84, where mutations may occur and perhaps even be favoured during these iterative processes. Such secondary mutations may need to be removed in order to accurately infer the effects of the intended genetic manipulations.
At the same time, it will be very interesting to see what similarities and differences emerge between the dynamics of genome evolution in laboratory experiments and in natural settings, including medically relevant ones such as during microbial infections and tumor progression. For example, one can sequence genomes of bacteria sampled at multiple points over the course of chronic infections, including samples stored in the past. This approach has recently been applied to Pseudomonas aeruginosa sampled over the course of multiple decades as the bacteria evolved in the airways of individuals with cystic fibrosis85. It has also been used to identify adaptive mutations that arose during a local outbreak of Burkholderia dolosa among people with cystic fibrosis86. To do so, whole-genome sequences were first used to reconstruct the transmission history between host individuals, and then analyses were performed to identify genes in the pathogen that underwent parallel changes that implied adaptation to the host environment86. The genes thus identified include some related to therapeutic interventions (antibiotic resistance) and host immune responses (cell-surface antigens), as well as others not previously known to be important for these infections. Elucidating genome dynamics has similarly proven to be key for understanding many observations regarding the progression of neoplastic tumors87. These types of studies will undoubtedly lead to important advances in identifying specific genes and mutations that contribute to disease and resistance to treatment. Future studies might also reveal the extent to which ecological interactions and differences in evolvability within these genetically diverse cell populations affect disease outcomes.
Online Summary.
New DNA sequencing technologies are being used to characterize whole-genome and whole-population dynamics during laboratory evolution experiments at a new level of resolution. Population genetic theory, including differences between asexual and sexual modes of reproduction, is crucial to interpreting these data.
Spontaneous mutation rates caused by DNA replication and repair errors can be measured with great precision by sequencing mutation accumulation experiments, in which parallel lineages are propagated through bottlenecks so that evolution is effectively random with respect to mutational effects on fitness.
By contrast, adaptive evolution experiments provide insights into the genetic basis and dynamics of adaptation. Genomic analyses show that adaptation rarely occurs by periodic sweeps of single beneficial mutations through asexual populations. Rather, divergent paths involving multiple mutations are often explored before any one evolved type can displace its competitors.
The long-term fates of mutations may depend not only on their immediate effects on competitive fitness but also on second-order selection for evolvability. This process requires sufficient population sizes and genetic diversity that multi-step mutational pathways are explored, and it may be mediated either by mutation rates or epistasis.
Coevolutionary interactions can promote genetic diversity within populations and accelerate the rate of genomic evolution. Complex ecological interactions, such as cross-feeding, often evolve even in simple environments.
New combinations of alleles derived from standing genetic diversity rather than de novo mutations can drive genetic dynamics during adaptation in sexually reproducing populations.
Laboratory evolution experiments provide a framework for interpreting genome dynamics observed during the evolution of microbial pathogens and cancers in human disease.
Acknowledgments
We thank the editor and three reviewers for helpful suggestions. We acknowledge support from the US National Institutes of Health (R00-GM087550 to J.E.B.), the US National Science Foundation (DEB-1019989 to R.E.L.), and the BEACON Center for the Study of Evolution in Action (NSF Cooperative Agreement DBI-0939454 to J.E.B. and R.E.L.).
Glossary terms
- ADAPTIVE EVOLUTION
Evolution under conditions where surviving organisms accumulate genetic changes that lead to a fitness advantage over their progenitors.
- ALL-OR-NONE EPISTASIS
Interactions among mutations such that an entire set of mutations is required to confer a fitness advantage or new trait; no subset that lacks one of these mutations has the advantage or an intermediate form of the relevant trait.
- BIOLOGICAL FITNESS
A quantitative measure of the contribution of a specific organism or genotype to future generations due to differential survival reproduction, or both associated with its phenotype fitness is often expressed relative to other organisms or genotypes.
- CLONAL INTERFERENCE
Competition between lineages with different beneficial mutations in asexual populations, which slows the rate at which any particular allele fixes in the population relative to a freely recombining population.
- DIMINISHING-RETURNS EPISTASIS
Interactions among mutations such that the combined effect of the mutations on fitness or some other trait is less than expected from their individual contributions.
- FITNESS LANDSCAPE
The visualization of the genotype-to-fitness mapping for an organism where the height of a position on the map represents the fitness of that genotype and the location is a reduced-dimensional projection of possible genotypes. An evolutionary trajectory of genetic changes can be visualized as a “walk” and adaptation as a “climb” in the fitness landscape.
- GENETIC ARCHITECTURE
Properties of an organism including its metabolic, regulatory, and developmental pathways, that determine how new mutations affect phenotypes and fitness.
- GENETIC BACKGROUND
The genotype of an organism i.e. its complete genome sequence or the alleles that distinguish it from other organisms.
- GENETIC FIXATION
The point at which an allele has completely displaced ancestral and competitor types. That is the allele is present in every surviving individual in the population.
- GENETIC LOAD
Indirect fitness cost to an organism caused by producing offspring with mutations that either reduce their fitness or are lethal.
- ISOGENIC CONSTRUCT
Organism produced in the laboratory using various genetic tools, with defined genetic differences from a reference organism. Used to study the effects on fitness and other phenotypic traits of single mutations or combinations of mutations.
- LTEE
Long-term evolution experiment with E. coli that has surpassed 25 years and 55,000 generations in duration.
- METAGENOMIC SEQUENCING
The sequencing DNA fragments that are randomly derived from a population containing a mixture of many genotypes.
- MUTATION ACCUMULATION
A type of evolution experiment in which populations are deliberately forced through a bottleneck of one or a few breeding individuals, which allows non-lethal mutations to accumulate with little or no filtering by natural selection.
- MUTATION RATE
The rate at which new genetic mutations spontaneously occur during the replication and transmission of genetic information from parent to offspring.
- NEGATIVE-FREQUENCY DEPENDENCE
An allele (or trait) that undergoes a decline in fitness as it becomes more common in a population. If the allele confers an advantage when it is rare but is disadvantageous when common then a genetic polymorphism is stably maintained.
- NICHE CONSTRUCTION
Production of a new resource or other ecological opportunity that is caused by the actions or evolution of organisms.
- PERIODIC SELECTION
The phenomenon whereby successive beneficial mutations sweep completely through an evolving population. Other mutations that are linked but not beneficial, can hitchhike with the beneficial driver.
- POPULATION BOTTLENECKS
Reductions in population size that typically also reduce genetic diversity. Bottlenecks can be deliberately imposed as in a mutation accumulation experiment. Cryptic bottlenecks also arise as a consequence of selective sweeps, especially in asexual populations, that drive out competing lineages and thus reduce genetic diversity.
- SELECTIVE SWEEP
Increase in the frequency of an advantageous allele within a population as it displaces ancestral and competitor alleles.
- STRENGTH OF SELECTION
Benefit of accessible beneficial mutations relative to current mean population fitness. Sweeps of new genotypes generally occur more rapidly and less diversity builds up within a population, under stronger selection.
- SUBSTITUTION RATE
The rate at which new mutations accumulate in an evolving lineage over time, which typically depends on both the mutation rate and the effects of natural selection.
Biographies
Author biographies
Jeffrey E. Barrick received his Ph.D. from Yale University in Molecular Biophysics and Biochemistry. He was a postdoctoral fellow at Michigan State University in the laboratories of Dr. Richard E. Lenski and Dr. Charles A. Ofria. He is an assistant professor in the Department of Molecular Biosciences at the University of Texas at Austin. His current research interests are in using synthetic biology approaches to alter the evolutionary potential of microorganisms. Laboratory website: http://barricklab.org.
Richard E. Lenski received his Ph.D. from the University of North Carolina, Chapel Hill, in Zoology. He was a postdoctoral fellow at the University of Massachusetts, Amherst, in the lab of Dr. Bruce R. Levin. He is the Hannah Professor of Microbial Ecology at Michigan State University. His research includes an on-going 25-year evolution experiment with E. coli that examines the phenotypic and genomic dynamics of adaptation and divergence. Laboratory website: http://myxo.css.msu.edu.
Footnotes
Copyright permissions
None.
Competing interests statement
The authors declare no competing financial interests.
Further information
Long-term E. coli evolution experiment: http://myxo.css.msu.edu/ecoli/
The US National Science Foundation BEACON Center for the Study of Evolution in Action: http://beacon-center.org/
Supplementary information
None.
Contributor Information
Jeffrey E. Barrick, Email: jbarrick@cm.utexas.edu.
Richard E. Lenski, Email: lenski@msu.edu.
References
- 1.Garland T, Rose MR, editors. Experimental Evolution: Concepts, Methods, and Applications of Selection Experiments. Univ. of California Press; 2009. [Google Scholar]
- 2.Kawecki TJ, et al. Experimental evolution. Trends Ecol Evol. 2012;27:547–560. doi: 10.1016/j.tree.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Hartl DL, Clark AG. Principles of Population Genetics. Sinauer; Sunderland, MA: 2007. [Google Scholar]
- 4.Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genom Human Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 5.Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing. Hum Mol Genet. 2010;19:R227–R240. doi: 10.1093/hmg/ddq416. [DOI] [PubMed] [Google Scholar]
- 6.Halligan DL, Keightley PD. Spontaneous mutation accumulation studies in evolutionary genetics. Annu Rev Ecol Evol Syst. 2009;40:151–172. [Google Scholar]
- 7.Lynch M, et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A. 2008;105:9272–9277. doi: 10.1073/pnas.0803466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ossowski S, et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327:92–94. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keightley PD, et al. Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genome Res. 2009;19:1195–1201. doi: 10.1101/gr.091231.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denver DR, et al. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci U S A. 2009;106:16310–16314. doi: 10.1073/pnas.0904895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lind PA, Andersson DI. Whole-genome mutational biases in bacteria. Proc Natl Acad Sci U S A. 2008;105:17878–17883. doi: 10.1073/pnas.0804445105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee H, Popodi E, Tang H, Foster PL. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A. 2012;109:E2774–E2783. doi: 10.1073/pnas.1210309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M. Drift-barrier hypothesis and mutation-rate evolution. Proc Natl Acad Sci U S A. 2012;109:18488–18492. doi: 10.1073/pnas.1216223109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drake JW. Spontaneous mutation. Annu Rev Genet. 1991;25:125–146. doi: 10.1146/annurev.ge.25.120191.001013. [DOI] [PubMed] [Google Scholar]
- 15.Lynch M. The Origins of Genome Architecture. Sinauer Associates Inc; 2007. [Google Scholar]
- 16.Friedberg EC, et al. DNA Repair and Mutagenesis. American Society for Microbiology Press; 2006. [Google Scholar]
- 17.Bull JJ, Badgett MR, Rokyta D, Molineux IJ. Experimental evolution yields hundreds of mutations in a functional viral genome. J Mol Evol. 2003;57:241–248. doi: 10.1007/s00239-003-2470-1. [DOI] [PubMed] [Google Scholar]
- 18.Domingo-Calap P, Cuevas JM, Sanjuán R. The fitness effects of random mutations in single-stranded DNA and RNA bacteriophages. PLoS Genet. 2009;5:e1000742. doi: 10.1371/journal.pgen.1000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drake JW, Charlesworth B, Charlesworth D, Crow JF. Rates of spontaneous mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Springman R, Keller T, Molineux IJ, Bull JJ. Evolution at a high imposed mutation rate: adaptation obscures the load in phage T7. Genetics. 2010;184:221–232. doi: 10.1534/genetics.109.108803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ames BN, Durston WE, Yamasaki E, Lee FD. Carcinogens are mutagens: a simple test system combining liver homogenates for activation and bacteria for detection. Proc Natl Acad Sci U S A. 1973;70:2281–2285. doi: 10.1073/pnas.70.8.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savageau MA. Escherichia coli habitats, cell types, and molecular mechanisms of gene control. Am Nat. 1983;122:732–744. [Google Scholar]
- 23.Orr HA. Fitness and its role in evolutionary genetics. Nat Rev Genet. 2009;10:531–539. doi: 10.1038/nrg2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atwood KC, Schneider LK, Ryan FJ. Periodic selection in Escherichia coli. Proc Natl Acad Sci U S A. 1951;37:146–155. doi: 10.1073/pnas.37.3.146. This study is a classic early demonstration of adaptive evolution in experimental populations of bacteria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fogle CA, Nagle JL, Desai MM. Clonal interference, multiple mutations and adaptation in large asexual populations. Genetics. 2008;180:2163–2173. doi: 10.1534/genetics.108.090019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lang GI, et al. Pervasive genetic hitchhiking and clonal interference in 40 evolving yeast populations. Nature. 2013;500:571–574. doi: 10.1038/nature12344. This paper presents the most detailed analysis to date of the dynamics of mutations in asexual populations, including the effects of clonal interference, by metagenomic sequencing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrick JE, et al. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 2009;461:1243–1247. doi: 10.1038/nature08480. This paper describes the first application of whole-genome sequencing to the LTEE and includes discussions of genetic parallelism, changes in mutation rates, and evolution in the optimization regime. [DOI] [PubMed] [Google Scholar]
- 28.Wagner A. The Origins of Evolutionary Innovations. Oxford Univ. Press; New York: 2011. [Google Scholar]
- 29.Velicer GJ, et al. Comprehensive mutation identification in an evolved bacterial cooperator and its cheating ancestor. Proc Natl Acad Sci U S A. 2006;103:8107–8112. doi: 10.1073/pnas.0510740103. This study was the first to use whole-genome sequencing to discover the genetic basis of an innovative change in an organism’s lifestyle that occurred during an evolution experiment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu YTN, Yuan X, Velicer GJ. Adaptive evolution of an sRNA that controls Myxococcus development. Science. 2010;328:993. doi: 10.1126/science.1187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchetti M, et al. Experimental evolution of a plant pathogen into a legume symbiont. PLoS Biol. 2010;8:e1000280. doi: 10.1371/journal.pbio.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ratcliff WC, Denison RF, Borrello M, Travisano M. Experimental evolution of multicellularity. Proc Natl Acad Sci U S A. 2012;109:1595–600. doi: 10.1073/pnas.1115323109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mortlock RP, editor. Microorganisms as model systems for studying evolution. Plenum Press; New York: 1984. [Google Scholar]
- 34.Blount ZD, Borland CZ, Lenski RE. Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. Proc Natl Acad Sci U S A. 2008;105:7899–7906. doi: 10.1073/pnas.0803151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blount ZD, Barrick JE, Davidson CJ, Lenski RE. Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature. 2012;489:513–518. doi: 10.1038/nature11514. This paper descibes the genetic basis of the evolution of citrate utilization in the LTEE including the potentiation, actualization, and refinement stages of this innovation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conrad TM, Lewis NE, Palsson BO. Microbial laboratory evolution in the era of genome-scale science. Mol Syst Biol. 2011;7:509. doi: 10.1038/msb.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minty JJ, et al. Evolution combined with genomic study elucidates genetic bases of isobutanol tolerance in Escherichia coli. Microbial Cell Fact. 2011;10:18. doi: 10.1186/1475-2859-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blaby IK, et al. Experimental evolution of a facultative thermophile from a mesophilic ancestor. Appl Environ Microbiol. 2012;78:144–155. doi: 10.1128/AEM.05773-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tenaillon O, et al. The molecular diversity of adaptive convergence. Science. 2012;335:457–461. doi: 10.1126/science.1212986. This study employed whole-genome sequencing of a large number of independently evolved populations to examine the diversity of alternative genetic pathways leading to improved fitness at high temperature. [DOI] [PubMed] [Google Scholar]
- 40.Harris DR, et al. Directed evolution of ionizing radiation resistance in Escherichia coli. J Bacteriol. 2009;191:5240–5252. doi: 10.1128/JB.00502-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Q, et al. Acceleration of emergence of bacterial antibiotic resistance in connected microenvironments. Science. 2011;333:1764–1767. doi: 10.1126/science.1208747. This study showed that migration between populations living in environments with different selection strengths can speed up adaptation. [DOI] [PubMed] [Google Scholar]
- 42.Pepin KM, Wichman HA. Experimental evolution and genome sequencing reveal variation in levels of clonal interference in large populations of bacteriophage phiX174. BMC Evol Biol. 2008;8:85. doi: 10.1186/1471-2148-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sniegowski PD, Gerrish PJ, Lenski RE. Evolution of high mutation rates in experimental populations of E. coli. Nature. 1997;387:703–705. doi: 10.1038/42701. [DOI] [PubMed] [Google Scholar]
- 44.Brown CT, et al. Whole genome sequencing and phenotypic analysis of mutations found in Bacillus subtilis following evolution under relaxed selection for sporulation. Appl Env Microbiol. 2011;77:6867–6877. doi: 10.1128/AEM.05272-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aguilar C, et al. Genetic changes during a laboratory adaptive evolution process that allowed fast growth in glucose to an Escherichia coli strain lacking the major glucose transport system. BMC Genomics. 2012;13:385. doi: 10.1186/1471-2164-13-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mao EF, Lane L, Lee J, Miller JH. Proliferation of mutators in a cell population. J Bact. 1997;179:417–422. doi: 10.1128/jb.179.2.417-422.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lenski RE. Phenotypic and genomic evolution during a 20,000-generation experiment with the bacterium Escherichia coli. Plant Breeding Rev. 2004;24:225–265. [Google Scholar]
- 48.Desai MM, Fisher DS. The balance between mutators and nonmutators in asexual populations. Genetics. 2011;188:997–1014. doi: 10.1534/genetics.111.128116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sniegowski PD, Gerrish PJ, Johnson T, Shaver A. The evolution of mutation rates: separating causes from consequences. BioEssays. 2000;22:1057–1066. doi: 10.1002/1521-1878(200012)22:12<1057::AID-BIES3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 50.Wielgoss S, et al. Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load. Proc Natl Acad Sci U S A. 2013;110:222–227. doi: 10.1073/pnas.1219574110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonald MJ, Hsieh YY, Yu YH, Chang SL, Leu JY. The evolution of low mutation rates in experimental mutator populations of Saccharomyces cerevisiae. Curr Biol. 2012;22:1235–40. doi: 10.1016/j.cub.2012.04.056. [DOI] [PubMed] [Google Scholar]
- 52.Denamur E, et al. Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell. 2000;103:711–721. doi: 10.1016/s0092-8674(00)00175-6. [DOI] [PubMed] [Google Scholar]
- 53.Woods RJ, et al. Second-order selection for evolvability in a large Escherichia coli population. Science. 2011;331:1433–1436. doi: 10.1126/science.1198914. This study used replay experiments to demonstrate that antagonistic epistasis can lead some genotypes toward an adaptive cul-de-sac and eventual extinction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Darwin C. The Origin of Species by Means of Natural Selection. John Murray; 1859. [Google Scholar]
- 55.Laland KN, Odling-Smee FJ, Feldman MW. Evolutionary consequences of niche construction and their implications for ecology. Proc Natl Acad Sci U S A. 1999;96:10242–10247. doi: 10.1073/pnas.96.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rozen DE, Schneider D, Lenski RE. Long-term experimental evolution in Escherichia coli. XIII Phylogenetic history of a balanced polymorphism. J Mol Evol. 2005;61:171–180. doi: 10.1007/s00239-004-0322-2. [DOI] [PubMed] [Google Scholar]
- 57.Le Gac M, Plucain J, Hindré T, Lenski RE, Schneider D. Ecological and evolutionary dynamics of coexisting lineages during a long-term experiment with Escherichia coli. Proc Natl Acad Sci U S A. 2012;109:9487–9492. doi: 10.1073/pnas.1207091109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rozen DE, Philippe N, Arjan De Visser J, Lenski RE, Schneider D. Death and cannibalism in a seasonal environment facilitate bacterial coexistence. Ecol Lett. 2009;12:34–44. doi: 10.1111/j.1461-0248.2008.01257.x. [DOI] [PubMed] [Google Scholar]
- 59.Barrick JE, Lenski RE. Genome-wide mutational diversity in an evolving population of Escherichia coli. Cold Spring Harbor Symp Quant Biol. 2009;74:119–129. doi: 10.1101/sqb.2009.74.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zambrano MM, Siegele DA, Almirón M, Tormo A, Kolter R. Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science. 1993;259:1757–60. doi: 10.1126/science.7681219. [DOI] [PubMed] [Google Scholar]
- 61.Finkel SE. Long-term survival during stationary phase: evolution and the GASP phenotype. Nat Rev Microbiol. 2006;4:113–120. doi: 10.1038/nrmicro1340. [DOI] [PubMed] [Google Scholar]
- 62.Treves DS, Manning S, Adams J. Repeated evolution of an acetate-crossfeeding polymorphism in long-term populations of Escherichia coli. Mol Biol Evol. 1998;15:789–797. doi: 10.1093/oxfordjournals.molbev.a025984. [DOI] [PubMed] [Google Scholar]
- 63.Maharjan R, Seeto S, Notley-McRobb L, Ferenci T. Clonal adaptive radiation in a constant environment. Science. 2006;313:514–517. doi: 10.1126/science.1129865. [DOI] [PubMed] [Google Scholar]
- 64.Maharjan RP, et al. The multiplicity of divergence mechanisms in a single evolving population. Genome Biol. 2012;13:R41. doi: 10.1186/gb-2012-13-6-r41. This paper used whole-genome sequencing to uncover extensive genetic diversity in populations evolving in chemostats. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saxer G, Doebeli M, Travisano M. The repeatability of adaptive radiation during long-term experimental evolution of Escherichia coli in a multiple nutrient environment. PLoS ONE. 2010;5:e14184. doi: 10.1371/journal.pone.0014184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herron MD, Doebeli M. Parallel evolutionary dynamics of adaptive diversification in Escherichia coli. PLoS Biol. 2013;11:e1001490. doi: 10.1371/journal.pbio.1001490. This study examined whole-population genetic dynamics in a system that diversified into two ecotypes that coexisted owing to different strategies for nutrient utilization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korona R, Nakatsu CH, Forney LJ, Lenski RE. Evidence for multiple adaptive peaks from populations of bacteria evolving in a structured habitat. Proc Natl Acad Sci U S A. 1994;91:9037–9041. doi: 10.1073/pnas.91.19.9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rainey PB, Travisano M. Adaptive radiation in a heterogeneous environment. Nature. 1998;394:69–72. doi: 10.1038/27900. [DOI] [PubMed] [Google Scholar]
- 69.Traverse CC, Mayo-Smith LM, Poltak SR, Cooper VS. Tangled bank of experimentally evolved Burkholderia biofilms reflects selection during chronic infections. Proc Natl Acad Sci U S A. 2013;110:E250–E259. doi: 10.1073/pnas.1207025110. This study characterized the mutational pathways by which ecotypes from a primary niche evolved to displace ecotypes established in other niches. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paterson S, et al. Antagonistic coevolution accelerates molecular evolution. Nature. 2010;464:275–278. doi: 10.1038/nature08798. This study used whole-genome sequencing of a bacteriophage to observe “Red Queen” dynamics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pal C, Maciá MD, Oliver A, Schachar I, Buckling A. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature. 2007;450:1079–1081. doi: 10.1038/nature06350. [DOI] [PubMed] [Google Scholar]
- 72.Meyer JR, et al. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science. 2012;335:428–432. doi: 10.1126/science.1214449. This paper characterized multistep genetic pathways that led to an innovative host-receptor shift in bacteriophage populations, and showed that the shift was contingent on the evolutionary trajectory of the bacterial host populations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Muller H. Genetic aspects of sex. Am Nat. 1932;66:118–138. [Google Scholar]
- 74.Cooper TF. Recombination speeds adaptation by reducing competition between beneficial mutations in populations of Escherichia coli. PLoS Biol. 2007;5:e225. doi: 10.1371/journal.pbio.0050225. This study used knowledge of the genetic targets of selection to show that plasmid-mediated recombination accelerated adaptive evolution by overcoming clonal interference. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burke MK. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature. 2010;467:587–590. doi: 10.1038/nature09352. This paper presented one of the first analyses based on whole-genome sequencing to characterize experimentally evolved populations of a sexual multicellular organism. [DOI] [PubMed] [Google Scholar]
- 76.Zhou D. Experimental selection of hypoxia-tolerant Drosophila melanogaster. Proc Natl Acad Sci U S A. 2011;108:2349–2354. doi: 10.1073/pnas.1010643108. This paper used extensive sequencing to identify targets of selection in another set of experimentally evolved populations of a sexual multicellular organism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Izutsu M, et al. Genome features of “Dark-fly”, a Drosophila line reared long-term in a dark environment. PLoS ONE. 2012;7:e33288. doi: 10.1371/journal.pone.0033288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ferenci T, et al. Genomic sequencing reveals regulatory mutations and recombinational events in the widely used MC4100 lineage of Escherichia coli K-12. J Bacteriol. 2009;191:4025–4029. doi: 10.1128/JB.00118-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nahku R, et al. Stock culture heterogeneity rather than new mutational variation complicates short-term cell physiology studies of Escherichia coli K-12 MG1655 in continuous culture. Microbiology. 2011;157:2604–2610. doi: 10.1099/mic.0.050658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hobman JL, et al. Comparative genomic hybridization detects secondary chromosomal deletions in Escherichia coli K-12 MG1655 mutants and highlights instability in the flhDC region. J Bacteriol. 2007;189:8786–8792. doi: 10.1128/JB.00977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pósfai G, et al. Emergent properties of reduced-genome Escherichia coli. Science. 2006;312:1044–1046. doi: 10.1126/science.1126439. [DOI] [PubMed] [Google Scholar]
- 82.Studier FW, Daegelen P, Lenski RE, Maslov S, Kim JF. Understanding the differences between genome sequences of Escherichia coli B strains REL606 and BL21(DE3) and comparison of the E coli B and K-12 genomes. J Mol Biol. 2009;394:653–680. doi: 10.1016/j.jmb.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 83.Gibson DG, et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329:52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- 84.Isaacs FJ, et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang L, et al. Evolutionary dynamics of bacteria in a human host environment. Proc Natl Acad Sci USA. 2011;108:7481–7486. doi: 10.1073/pnas.1018249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lieberman TD, et al. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet. 2011;43:1275–1280. doi: 10.1038/ng.997. This study used whole-genome sequencing to investigate adaptation of a bacterial pathogen during a multi-decade outbreak in a human population; although not based on an experiment, it relied on inferential approaches similar to those used in experimental evolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sprouffske K, Merlo LMF, Gerrish PJ, Maley CC, Sniegowski PD. Cancer in light of experimental evolution. Curr Biol. 2012;22:R762–R771. doi: 10.1016/j.cub.2012.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Loewe L, Hill WG. The population genetics of mutations: good, bad and indifferent. Philos Trans R Soc Lond B Biol Sci. 2010;365:1153–1167. doi: 10.1098/rstb.2009.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eyre-Walker A, Keightley PD. The distribution of fitness effects of new mutations. Nat Rev Genet. 2007;8:610–618. doi: 10.1038/nrg2146. [DOI] [PubMed] [Google Scholar]
- 90.De Visser JAGM, Cooper TF, Elena SF. The causes of epistasis. Philos Trans R Soc Lond B Biol Sci. 2011;278:3617–3624. doi: 10.1098/rspb.2011.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kondrashov FA, Kondrashov AS. Measurements of spontaneous rates of mutations in the recent past and the near future. Philos Trans R Soc Lond B Biol Sci. 2010;365:1169–1176. doi: 10.1098/rstb.2009.0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Khan AI, Dinh DM, Schneider D, Lenski RE, Cooper TF. Negative epistasis between beneficial mutations in an evolving bacterial population. Science. 2011;332:1193–1196. doi: 10.1126/science.1203801. [DOI] [PubMed] [Google Scholar]
- 93.Chou HH, Chiu HC, Delaney NF, Segrè D, Marx CJ. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science. 2011;332:1190–1192. doi: 10.1126/science.1203799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kvitek DJ, Sherlock G. Reciprocal sign epistasis between frequently experimentally evolved adaptive mutations causes a rugged fitness landscape. PLoS Genet. 2011;7:e1002056. doi: 10.1371/journal.pgen.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Philippe N, Crozat E, Lenski RE, Schneider D. Evolution of global regulatory networks during a long-term experiment with Escherichia coli. BioEssays. 2007;29:846–860. doi: 10.1002/bies.20629. [DOI] [PubMed] [Google Scholar]
- 96.Wagner A. Neutralism and selectionism: a network-based reconciliation. Nat Rev Genet. 2008;9:965–974. doi: 10.1038/nrg2473. [DOI] [PubMed] [Google Scholar]
- 97.Heard S, Hauser D. Key evolutionary innovations and their ecological mechanisms. Historical Biol. 1995;10:151–173. [Google Scholar]
- 98.Lenski RE, Travisano M. Dynamics of adaptation and diversification: a 10,000-generation experiment with bacterial populations. Proc Natl Acad Sci U S A. 1994;91:6808–6814. doi: 10.1073/pnas.91.15.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Carroll SM, Marx CJ. Evolution after introduction of a novel metabolic pathway consistently leads to restoration of wild-type physiology. PLoS Genet. 2013;9:e1003427. doi: 10.1371/journal.pgen.1003427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wichman HA, Badgett MR, Scott LA, Boulianne CM, Bull JJ. Different trajectories of parallel evolution during viral adaptation. Science. 1999;285:422–424. doi: 10.1126/science.285.5426.422. This paper was one of the first to use whole-genome sequencing to examine the nature of genetic changes during an adaptive evolution experiment. [DOI] [PubMed] [Google Scholar]
- 101.Woods R, Schneider D, Winkworth CL, Riley MA, Lenski RE. Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc Natl Acad Sci U S A. 2006;103:9107–9112. doi: 10.1073/pnas.0602917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bull JJ, et al. Exceptional convergent evolution in a virus. Genetics. 1997;147:1497–1507. doi: 10.1093/genetics/147.4.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Crozat E, et al. Parallel genetic and phenotypic evolution of DNA superhelicity in experimental populations of Escherichia coli. Mol Biol Evol. 2010;27:2113–2128. doi: 10.1093/molbev/msq099. [DOI] [PubMed] [Google Scholar]
- 104.Jerome JP, et al. Standing genetic variation in contingency loci drives the rapid adaptation of Campylobacter jejuni to a novel host. PLoS ONE. 2011;6:e16399. doi: 10.1371/journal.pone.0016399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee MC, Marx CJ. Repeated, selection-driven genome reduction of accessory genes in experimental populations. PLoS Genet. 2012;8:e1002651. doi: 10.1371/journal.pgen.1002651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cooper VS, Schneider D, Blot M, Lenski RE. Mechanisms causing rapid and parallel losses of ribose catabolism in evolving populations of Escherichia coli B. J Bacteriol. 2001;183:2834–2841. doi: 10.1128/JB.183.9.2834-2841.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Paquin CE, Adams J. Relative fitness can decrease in evolving asexual populations of S. cerevisiae. Nature. 1983;306:368–370. doi: 10.1038/306368a0. [DOI] [PubMed] [Google Scholar]